
Mechanochemical synthesis of iron aluminyl complexes†
Fabian
Kallmeier
 ,
Aidan J. R.
Matthews
,
Gareth R.
Nelmes
,
Nina R.
Lawson
and
Jamie
Hicks
*
,
Aidan J. R.
Matthews
,
Gareth R.
Nelmes
,
Nina R.
Lawson
and
Jamie
Hicks
*
Research School of Chemistry, Australian National University, ACT 2601, Australia. E-mail: jamie.hicks@anu.edu.au
First published on 11th July 2024
Abstract
A series of iron aluminyl complexes have been synthesised in good crystalline yields from reactions between bulky diamido aluminium iodide complexes and K[Fe(CO)2Cp] in the solid state. The series of metal–metal bonded complexes have been characterised by X-ray crystallography and were investigated using density functional theory to probe the effects of ligand substitution on the Al–Fe bond.
Mechanochemistry has been deemed a future direction of ‘green chemistry’.1 This is because reactions can be performed solvent-free (or near solvent-free), cheaper, safer, and often with less energy input than traditional solution-state chemistry.1,2 Mechanochemistry is already widely used in many areas of chemistry3 but has only recently started to make an impact in main group synthesis.4–11 Some examples of note include Kubota, Ito and co-workers 2021 report that Grignard reagents can be synthesised mechanochemically in high yields (Fig. 1, I).12 Furthermore, the Grignard ‘paste’ isolated from the mill reacted mechanochemically with a range of electrophiles typical of Grignard reagents. In 2022, Harder and co-workers showed that magnesium radical complexes could be synthesised by the mechanochemical reduction of magnesium iodide precursor complexes with potassium-coated KI (II).13,14 Last year, García, García-Álvarez and co-workers reported a fast, scalable and solvent-free method to access the commonly named “Lappert's heavier tetrylenes” E[N(SiMe3)2]2 (E = Ge, Sn, Pb) by a mechanochemical synthesis (III).15 Earlier this year Yamashita and co-workers also reported that non-solvated dialkylaluminyl anion can be isolated from the mechanochemical reduction of the dialkylaluminium iodide with an Na/K alloy (IV).16
 | ||
| Fig. 1 Selected main group mechanochemical reactions. Ar = aryl, TMS = trimethylsilyl, R/R1/R2 = various organic groups. | ||
In terms of using mechanochemical reactions in the synthesis of metal–metal bonded compounds, results in the literature are scarce. Harder and co-workers recently showed that Mg(I)–Mg(I) bonded dimers can be synthesised through the mechanochemical reduction of β-diketiminate magnesium iodide compounds (Fig. 1, V).17 However, the group proposed that the Mg–Mg bond was likely formed upon extraction into C6D6via radical dimerisation. In this work, we build on these recent reports showing that mechanochemical synthesis can be used in the preparation of heterobimetallic complexes with unsupported metal–metal bonds.
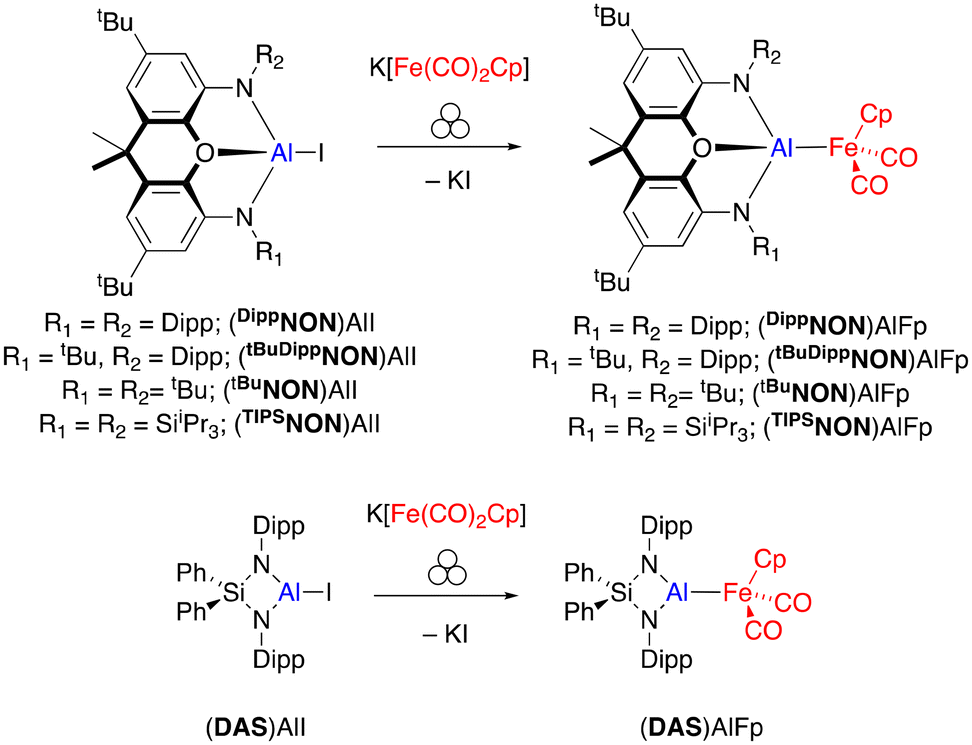
Six diamido aluminium iodide complexes were used as precursors in this work, four bearing xanthene-based diamido ligands ([DippNON]2−, [tBuDippNON]2−, [tBuNON]2−, [TIPSNON]2−), one bearing a diamidosilyl ligand, [DAS]2−, and one bearing two monodentate amido ligands [Dipp(TIPS)N]− for an acyclic comparison (Scheme 1).18 The four xanthene-based ligands all feature the same 2,7-di-tert-butyl-9,9-dimethylxanthene scaffold and vary only by the substituents on the nitrogen donors. (DippNON)AlI and [Dipp(TIPS)N]2AlI have been reported previously,19,20 but (TIPSNON)AlI, (tBuDippNON)AlI, (tBuNON)AlI and (DAS)AlI are reported as part of this work (see ESI† for synthesis and characterising data).
 | ||
| Scheme 1 Mechanochemical synthesis of the five iron aluminyl complexes. | ||
With the range of aluminium iodide compounds in hand, our attention turned to the synthesis of the iron aluminyl complexes. A number of iron aluminyl complexes have previously been reported,21 all synthesised in the solution phase through reactions between bulky aluminium (di)halide complexes and the [Fe(CO)2Cp]− (Fp) anion (either the Na or K salt). Taking inspiration from these previous studies, the reaction between (TIPSNON)AlI and K[Fp] in benzene was investigated. Monitoring the reaction by 1H NMR spectroscopy, it was determined that after stirring the suspension for 16 hours at room temperature, the reaction only saw 51% conversion to the iron aluminyl complex. Stirring the reaction for another 2 days led to complete conversion to the desired iron aluminyl complex (TIPSNON)AlFp. In comparison, the addition of the same reagents (0.25 mmol scale) as solids into an IKA Ultra-Turrax Tube Drive fitted with a 20 mL ball mill tube, 30 stainless steel balls and 258 mg of graphite as flux, saw complete conversion within one hour of milling. Upon workup, this gave (TIPSNON)AlFp in a 79% yield (Scheme 1). A similar procedure was used in the synthesis of (tBuDippNON)AlFp, (tBuNON)AlFp, (DippNON)AlFp and (DAS)AlFp, which upon workup, all gave moderate to good crystalline yields of the iron aluminyl complexes (33–68%). Unfortunately, even after multiple attempts in both solution and the solid state, the reaction between the acyclic precursor [Dipp(TIPS)N]2AlI and K[Fp] led to decomposition in every case, yielding a complex mixture of products by 1H NMR spectroscopy.
The five iron aluminyl complexes have been characterised by X-ray crystallography, and their solid-state structures are shown in Fig. 2. All five complexes have similar solid-state structures, bearing an unsupported Al–Fe bond between a diamidoaluminium fragment and the Fe(CO)2Cp moiety. However, upon closer inspection, the solid-state structures reveal some interesting trends. Firstly, comparing the four NON-coordinated complexes, the Al–Fe bond length shortens from 2.4356(8) Å in (TIPSNON)AlFp to 2.3860(5)/2.3854(5) Å in (tBuDippNON)AlFp/(DippNON)AlFp, with that of (tBuNON)AlFp in between at 2.4085(11) Å (Table 1).
 | ||
| Fig. 2 Solid-state structures of (DippNON)AlFp (top-left), (tBuDippNON)AlFp (top-right), (tBuNON)AlFp (middle-left), (TIPSNON)AlFp (middle-right) and (DAS)AlFp (bottom) as determined by single-crystal X-ray crystallography. Displacement ellipsoids set at the 50% probability level. Hydrogen atoms have been omitted and selected organic groups have been shown in wireframe for clarity. | ||
| Al–Fe (Å) | Al–O (Å) | O–Al–Fe (°) | N⋯N (Å) | |
|---|---|---|---|---|
| (DippNON)AlFp | 2.3854(5) | 1.9577(11) | 113.23(4) | 3.247(2) |
| (tBuDippNON)AlFp | 2.3860(5) | 1.9204(10) | 110.95(3) | 3.289(2) |
| (tBuNON)AlFp | 2.4087(10) | 1.915(2) | 107.08(7) | 3.308(4) |
| (TIPSNON)AlFp | 2.4356(8) | 1.9142(18) | 101.66(6) | 3.384(4) |
| (DAS)AlFp | 2.2884(7) | — | — |
As the difference in Al–Fe bond length could be a consequence of sterics, a comprehensive analysis of the steric profiles of all five ligands, using both SambVca 2.1 (% buried volume) and AtomAccess (% accessible surface area),22,23 was conducted. Both methods were unanimous in the ordering of the ligands in terms of sterics, finding the following trend: tBuNON < DAS < tBuDippNON < TIPSNON < DippNON (see ESI† for further details). This ordering does appear to correlate with the measured Al–Fe bond length of the complexes; therefore, it can be concluded that sterics alone cannot be responsible for the differences in structures. Returning to the solid-state structures of the four Al–Fe NON-complexes, trends mirroring those of Al–Fe bond length can also be seen in the O–Al bond length, O–Al–Fe angle and N⋯N separation (Table 1). Together, these observations show that moving from (DippNON)AlFp to (TIPSNON)AlFp, the Al centre is being “pulled” further into the tridentate NON-binding pocket (as is seen by the decreasing Al–O length), increasing N⋯N separation and reducing the O–Al–Fe angle.
In an attempt to rationalise the observed trend in terms of electronics, the series of iron aluminyl complexes was investigated using density functional theory (DFT). The geometry of all five complexes was optimised at the PBE0-D3BJ/BS1 level of theory. The optimised geometries are in good agreement with the single-crystal X-ray data, producing the same ordering of Al–Fe bond lengths (Table 2) as observed in the solid-state (see ESI† for further details).24 As expected, all five iron aluminyl complexes display similar electronic structures: in every case, the HOMO and HOMO−1 are NON/DAS ligand-based orbitals, which include electron density from the nitrogen lone pairs. In the four NON complexes, the HOMO−2 corresponds to the Al–Fe σ-bonding orbital, which is a similar energy for (DippNON)AlFp, (tBuDippNON)AlFp and (tBuNON)AlFp (−6.56 to −6.55 eV), but slightly higher for (TIPSNON)AlFp (−6.53 eV) due to the significantly more σ-donating N-bound TIPS groups. This orbital for the (tBuNON)AlFp complex is shown in Fig. 3. In the (DAS)AlFp complex, the equivalent orbital is much lower in energy at −7.87 eV and corresponds to the HOMO−12. In all compounds, three iron-based molecular orbitals that are high in d-character can be located between HOMO−5 and HOMO−13, which are all polarised towards the Al centre.
 | ||
| Fig. 3 Molecular orbital (HOMO−2) of the σ(Fe–Al) bond between (tBuNON)AlFp, showing approximate sigma symmetry along the Al–Fe axis. Hydrogen atoms omitted for clarity, and MO rendered with an isovalue of 0.035 e− Å−3. Aluminium atom depicted in pale pink and iron atom in purple. | ||
| Al–Fe bond length (Å) | Bond index | NPA charge QAl | NPA charge QFe | |
|---|---|---|---|---|
| (TIPSNON)AlFp | 2.390 | 0.373 | +1.821 | −0.443 |
| (tBuNON)AlFp | 2.363 | 0.357 | +1.816 | −0.447 |
| (tBuDippNON)AlFp | 2.352 | 0.350 | +1.840 | −0.430 |
| (DippNON)AlFp | 2.326 | 0.349 | +1.881 | −0.468 |
| (DAS)AlFp | 2.285 | 0.393 | +1.732 | −0.466 |
To gain further insight into the Al–Fe bond, the series was additionally investigated using natural bond orbital (NBO) analysis, with a summary of the findings is presented in Table 2. The NBO corresponding to the σ(Fe–Al) bond is similar in all five complexes, containing significant contributions from the 3d(Fe) and 3s(Al) orbitals and are polarised toward the iron centre (see ESI† for further details). The calculated Al–Fe bond index is similar in all complexes, ranging from 0.35–0.39. This is consistent with that calculated for previously reported iron-aluminyl complexes,21 and therefore shows a significant ionic contribution to the bond. The natural population analysis (NPA) charge on the Al centre (QAl) also follows the same trend as the Al–Fe bond lengths, with the Al centre in (DippNON)AlFp calculated to have the largest positive charge (+1.88) and that in (TIPSNON)AlFp and (tBuNON)AlFp to have the smallest (+1.82) of the four NON complexes (Table 2). The three-coordinate (DAS)AlFp was found to have the lowest QAl of the series at +1.73. In comparison, the charge on Fe (QFe) remains much more consistent across the series (−0.43 to −0.47). (DippNON)AlFp has the largest difference in QAl and QFe and is consequently the NON-complex with the shortest Al–Fe bond. The differences observed in the DAS complex can be attributed to the different coordination environment at aluminium and significantly different N–Al–N bite angle, increasing the Al-s character of the bonding orbital.25
As an interesting final note to this study, in the mechanochemical reaction between (DippNON)AlI and K[Fp], a small (<2%) but reproducible side-product was observed. This was most noticeable during the crystallisation of the (colourless) complex (DippNON)AlFp, which frequently produced a few small, red crystals alongside the primary colourless crystals of (DippNON)AlFp. X-ray crystallographic analysis of this side-product revealed it to be [{(DippNON)AlOC(μ-FeCp)}2], a carbyne-bridged diiron complex with two (DippNON)Al moieties bound to the oxygen atoms of the former carbonyl ligands (Fig. 4). The compound is isostructural to the boron analogue recently reported by us26 and similar to the benzylidyne-bridged diiron complex previously reported by Sitzmann and co-workers.27 It can be envisioned that [{(DippNON)AlOC(μ-FeCp)}2] may be formed from the iron aluminyl complex (DippNON)AlFp, by simple loss of a carbonyl ligand from each iron centre and dimerisation. However, all attempts to convert (DippNON)AlFp into the carbyne complex (refluxing the complex in various solvents and/or irradiation with UV light) either led to no reaction or decomposition of the (DippNON)AlFp. It is therefore proposed that the complex is formed in the milling of (DippNON)AlI and K[Fp], by initial nucleophilic attack of one of the Fp carbonyl ligands on the electrophilic Al centre. Nucleophilic reactivity of the [Fp]− carbonyl ligands has been previously reported,28 and a similar mechanism was proposed for the boron analogue.26
 | ||
| Fig. 4 Solid state structures of [{(DippNON)AlOC(μ-FeCp)}2] as determined by single-crystal X-ray crystallography. Displacement ellipsoids set at the 50% probability level. Hydrogen atoms have been omitted and selected organic groups have been shown in wireframe for clarity. | ||
Conclusions
In conclusion, we have successfully synthesised a series of iron aluminyl complexes via mechanochemical methods using bulky diamido aluminium iodide complexes and K[Fe(CO)2Cp] in the solid state. These complexes were characterised by X-ray crystallography, revealing significant structural insights into the Al–Fe bonding environment. Density functional theory (DFT) calculations further elucidated the electronic structure and bonding interactions, highlighting the influence of ligand substitution on the Al–Fe bond length and the nature of the Al centre. Our findings underscore mechanochemistry as a powerful tool for accessing heterobimetallic complexes with unsupported metal–metal bonds, paving the way for future advancements in main group and transition metal synthesis.Data availability
The data supporting this article have been included as part of the ESI.† Finalised CIFs for all X-ray diffraction structures (2359911–2359919)† have been deposited at the Cambridge Crystallographic Data Centre.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JH would like to thank the Australian Research Council (DP210100454) for funding this work. This research was undertaken with the assistance of resources from the National Computational Infrastructure (NCI Australia), an NCRIS enabled capability supported by the Australian Government.References
- K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145 CrossRef CAS PubMed.
- (a) T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018 CrossRef PubMed; (b) J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13 CrossRef CAS PubMed.
- See for example: (a) X. Liu, Y. Li, L. Zeng, X. Li, N. Chen, S. Bai, H. He, Q. Wang and C. Zhang, Adv. Mater., 2022, 34, 2108327 CrossRef CAS PubMed; (b) A. Krusenbaum, S. Grätz, G. T. Tigineh, L. Borchardt and J. G. Kim, Chem. Soc. Rev., 2022, 51, 2873 RSC; (c) J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080 RSC.
- (a) D. Tan and F. Garcia, Chem. Soc. Rev., 2019, 48, 2274 RSC; (b) N. R. Rightmire and T. P. Hanusa, Dalton Trans., 2016, 45, 2352 RSC.
- D. W. Peters and R. B. Blair, Faraday Discuss., 2014, 170, 83 RSC.
- N. C. Boyde, N. R. Rightmire, T. P. Hanusa and W. W. Brennessel, Inorganics, 2017, 5, 36 CrossRef.
- R. F. Koby, A. M. Doerr, N. R. Rightmire, N. D. Schley, B. K. Long and T. P. Hanusa, Angew. Chem., Int. Ed., 2020, 59, 9542 CrossRef CAS PubMed.
- N. Davison, J. A. Quirk, F. Tuna, D. Collison, C. L. McMullin, H. Michaels, G. H. Morritt, P. G. Waddell, J. A. Gould, M. Freitag, J. A. Dawson and E. Lu, Chem, 2023, 9, 576 CAS.
- Y. Gao, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2023, 62, e202217723 CrossRef CAS PubMed.
- J. V. Nallaparaju, R. Satsi, D. Merzhyievskyi, T. Jarg, R. Aav and D. G. Kananovich, Angew. Chem., Int. Ed., 2024, 63, e202319449 CrossRef CAS PubMed.
- M. Glavinović, M. Krause, L. Yang, J. A. McLeod, L. Liu, K. M. Baines, T. Friščić and J.-P. Lumb, Sci. Adv., 2017, 3, e1700149 CrossRef PubMed.
- R. Takahashi, A. Hu, P. Gao, Y. Gao, Y. Pang, T. Seo, J. Jiang, S. Maeda, H. Takaya, K. Kubota and H. Ito, Nat. Commun., 2021, 12, 6691 CrossRef CAS PubMed.
- D. Jędrzkiewicz, J. Mai, J. Langer, Z. Mathe, N. Patel, S. DeBeer and S. Harder, Angew. Chem., Int. Ed., 2022, 61, e202200511 CrossRef PubMed.
- J. Hicks, M. Juckel, A. Paparo, D. Dange and C. Jones, Organometallics, 2018, 37, 4810 CrossRef CAS.
- J. A. Cabeza, J. F. Reynes, F. García, P. García-Álvarez and R. García-Soriano, Chem. Sci., 2023, 14, 12477 RSC.
- S. Kurumada, R. Yamanashi, K. Sugita, K. Kubota, H. Ito, S. Ikemoto, C. Chen, T. Moriyama, S. Muratsugu, M. Tada, T. Koitaya, T. Ozaki and M. Yamashita, Chem. – Eur. J., 2024, 30, e202303073 CrossRef CAS PubMed.
- D. Jędrzkiewicz, J. Langer and S. Harder, Z. Anorg. Allg. Chem., 2022, 648, e202200138 CrossRef.
- (a) C. A. Cruz, D. J. H. Emslie, C. M. Robertson, L. E. Harrington, H. A. Jenkins and J. F. Britten, Organometallics, 2009, 28, 1891 CrossRef CAS; (b) A. Nicolay, M. S. Ziegler, L. Rochlitz and T. D. Tilley, Polyhedron, 2020, 180, 114420 CrossRef CAS; (c) R. Murugavel, N. Palanisami and R. J. Butcher, J. Organomet. Chem., 2003, 675, 65 CrossRef CAS; (d) B. Shen, L. Ying, J. Chen and Y. Luo, Inorg. Chim. Acta, 2008, 361, 1255 CrossRef CAS.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92 CrossRef CAS PubMed.
- R. A. Jackson, A. J. R. Matthews, P. Vasko, M. F. Mahon, J. Hicks and D. J. Liptrot, Chem. Commun., 2023, 59, 5277 RSC.
- (a) T. Agou, T. Yanagisawa, T. Sasamori and N. Tokitoh, Bull. Chem. Soc. Jpn., 2016, 89, 1184 CrossRef CAS; (b) I. M. Riddlestone, J. Urbano, N. Phillips, M. J. Kelly, D. Vidovic, J. I. Bates, R. Taylor and S. Aldridge, Dalton Trans., 2013, 42, 249 RSC; (c) C. Jones, S. Aldridge, T. Gans-Eichler and A. Stasch, Dalton Trans., 2006, 5357 RSC; (d) T. Yanagisawa, Y. Mizuhata and N. Tokitoh, Heteroat. Chem., 2018, 29, e21465 CrossRef; (e) B. N. Anand, I. Krossing and H. Noth, Inorg. Chem., 1997, 36, 1979 CrossRef CAS PubMed; (f) H. Braunschweig, J. Muller and B. Ganter, Inorg. Chem., 1996, 35, 7443 CrossRef CAS PubMed; (g) I. M. Riddlestone, S. Edmonds, P. A. Kaufman, J. Urbano, J. I. Bates, M. J. Kelly, A. L. Thompson, R. Taylor and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 2551 CrossRef CAS PubMed; (h) R. A. Fischer and T. Priermeier, Organometallics, 1994, 13, 4306 CrossRef CAS; (i) K. Nakaya, A. Ishii and N. Nakata, Mendeleev Commun., 2022, 32, 71 CrossRef CAS; (j) S. Sinhababu, M. R. Radzhabov, J. Telser and N. P. Mankad, J. Am. Chem. Soc., 2022, 144, 3210 CrossRef CAS PubMed.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872 CrossRef CAS PubMed.
- G. K. Gransbury, S. C. Corner, J. G. C. Kragskow, P. Evans, H. M. Yeung, W. J. A. Blackmore, G. F. S. Whitehead, I. J. Vitorica-Yrezabal, N. F. Chilton and D. P. Mills, J. Am. Chem. Soc., 2023, 145, 22814 CrossRef CAS PubMed.
- Y. Minenkov, Å. Singstad, G. Occhipinti and V. R. Jensen, Dalton Trans., 2012, 41, 5526 RSC.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 1702 CrossRef CAS PubMed.
- E. E. Nahon, G. R. Nelmes, P. J. Brothers and J. Hicks, Chem. Commun., 2023, 59, 14281 RSC.
- G. Y. Vollmer, M. W. Wallasch, D. Saurenz, T. R. Eger, H. Bauer, G. Wolmershäuser, M. H. Prosenc and H. Sitzmann, Organometallics, 2015, 34, 644 CrossRef CAS.
- See for example: (a) T. Pugh, N. F. Chilton and R. A. Layfield, Angew. Chem., Int. Ed., 2016, 55, 11082 CrossRef CAS PubMed; (b) M. P. Blake, N. Kaltsoyannis and P. Mountford, Chem. Commun., 2013, 49, 3315 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental and characterisation data, computational and crystallographic details. CCDC 2359911–2359919. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01774a |
| This journal is © The Royal Society of Chemistry 2024 |