DOI:
10.1039/D4SC08612K
(Edge Article)
Chem. Sci., 2025,
16, 10701-10713
3MMLCT excited states of luminescent binuclear PdII complexes: excited state inner-sphere electron-transfer reactions and application†
Received
21st December 2024
, Accepted 4th May 2025
First published on 8th May 2025
Abstract
Compared with PtII analogues that exhibit unique stimulus-induced switching luminescence properties and novel material applications, the properties and reactivity of the 3MMLCT excited state of PdII complexes in solutions are under-developed. Here, we prepared a series of binuclear cyclometalated PdII complexes with short intramolecular Pd–Pd distances of 2.79–2.89 Å and luminescent 3MMLCT excited states in solutions at 298 K (emission quantum yield and radiative decay rate constant up to 0.70 and 2 × 105 s−1, respectively). Their photophysical properties have been examined by femtosecond time-resolved absorption spectroscopy, and the 1e oxidation products of binuclear PdII complexes have been studied by electron paramagnetic resonance spectroscopy and computational studies. Density functional theory (DFT) and time-dependent DFT (TDDFT) calculations show that changing the C-deprotonated aryl pyridine (C^N) ligand to the strong σ-donor aryl N-heterocyclic carbene (C^C*) ligand significantly increases the energy level of the metal centered (3dd) excited state. The binuclear PdII complex with a redox-active formamidinate bridging ligand reacts with benzyl bromide to immediately generate PdII–PdIII–Br species upon light irradiation. Quenching and time-resolved absorption experiments show that the PdII–3MMLCT excited state reacts with alkyl bromides via an inner-sphere electron transfer pathway. These binuclear PdII complexes were examined as organic light-emitting diode (OLED) emitters and photocatalysts for C–C bond formation reactions.
Introduction
The metal–metal-to-ligand charge transfer [also defined as MMLCT or (dσ* → π*)] excited state of d8 metal complexes, taking the PtII–3MMLCT excited state as an example, exhibits unique photophysical properties and reactivity.1 This type of excited state usually exhibits luminescence properties that can be reversibly switched by external stimulus,2 as well as a large radiative decay rate constant (kr ≈ 105–106 s−1) and low emission energy.3 In addition, due to the presence of axial vacant coordination sites, the 3MMLCT excited state is reactive towards the activation of C–X bonds through the inner-sphere atom transfer or electron transfer mechanism.4 We are interested in developing the photochemistry of binuclear PdII complexes through the 3MMLCT excited state, which reacts with C–X bonds to produce reactive PdIII species (PdIII–PdII–X or X–PdIII–PdIII–X). Compared to Pt analogues, these PdIII–PdII–X or X–PdIII–PdIII–X species are rarely studied and reported. PdIII species are generally reactive and unstable, and can be easily reduced back to PdII,5 suggesting that luminescent binuclear PdII complexes with long-lived 3MMLCT excited states can be more reactive photocatalysts. In the literature, only a few binuclear PdII complexes with long-lived luminescent 3MMLCT excited states in solution have been reported.6 The small ligand-field splitting of the PdII ion results in a low-lying metal-centered 3dd excited state that is more thermally accessible than in the PtII ion, thus allowing efficient non-radiative decay through electron population in the antibonding 4dx2−y2 orbital.7 To destabilize the dd excited state, researchers have employed strong-field C-deprotonated tridentate or tetradentate cyclometalated ligands to form luminescent PdII complexes,8 or used σ-donating diarylacetylide bridging ligands to construct discrete binuclear PdII complexes.6a
We describe here the synthesis, photophysical and photochemical properties of a series of binuclear PdII complexes (1–7) and two PtII counterparts (Ref-Pt1 and Ref-Pt53c). Complexes 1 and 3–7 have short intramolecular Pd–Pd distances of 2.79–2.89 Å. Binuclear PdII complexes with cyclometalated [C^C*] (aryl N-heterocyclic carbene) ligands exhibit strong 3MMLCT emissions in dilute solution, solid state and PMMA thin films. Based on DFT/TDDFT calculations, two factors affecting the photophysical properties of binuclear PdII complexes include (1) the relative energy of the 3dd excited state compared to the lowest 3MMLCT state, which is closely related to the non-radiative decay process (knr); (2) radiative decay process, affected by the spin–orbit coupling constant of Pd atoms. Upon photo-irradiation, the binuclear PdII complex with DPF bridging ligand (DPF = N,N'-diphenylformamidinate) activates alkyl bromides through the inner-sphere type electron transfer pathway, to produce PdII–PdIII–Br species. A binuclear PdII complex in this work was also shown to be a photosensitizer or photocatalyst for C–C bond coupling reactions. OLEDs fabricated using binuclear PdII complexes exhibit high maximum luminance and external quantum efficiency (EQE) up to 104![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cd m−2 and 22.9%, respectively.
000 cd m−2 and 22.9%, respectively.
Results and discussion
Synthesis and characterization
The structures of the binuclear PdII complexes (1–7) and PtII analogues (Ref-Pt1 and Ref-Pt5) are shown in Fig. 1. Their syntheses and characterization data are given in the ESI.† Complexes 1 and 2 were prepared by the reaction of [Pd(C^N)(μ-OAc)]2 (C^N = 2-[1,1′-biphenyl]-3-yl-4-phenylpyridine) with pyrazole (Pz) ligands and NaOMe in THF. The PtII analogue Ref-Pt1 was similarly prepared from [Pt(C^N)(μ-Cl)]2. These complexes were purified by crystallization to give a mixture of cis and trans isomers (Fig. 1a), as shown by 1H NMR (see the ESI†). Complex 3 was prepared by treating [Pd(C^N)(μ-Cl)]2 with DPF ligand and KOtBu in N,N-dimethylformamide (DMF), while 4–7 and Ref-Pt5 were synthesized by a one-pot reaction; the [M(C^C*)(μ-Cl)]2 (M = Pd or Pt) was generated in situ through transmetalation between silver carbene intermediate and Pd(COD)Cl2 or Pt(COD)Cl2. The products were purified by column chromatography to give complexes 3–7 and Ref-Pt5 as pure trans isomers in moderate yields (15–40%). All binuclear PdII complexes were obtained as air-stable light green to orange solids, and Ref-Pt1 and Ref-Pt5 are red solids. In the absence of light, complex 5 shows higher stability than complex 1 in aerated CH2Cl2 and toluene; the former is stable for at least a week while the latter decomposed after one day (Fig. S7 and S8†).
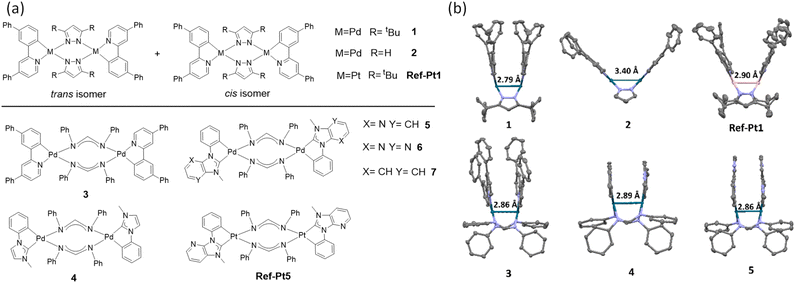 |
| | Fig. 1 (a) Chemical structures of binuclear PdII and PtII complexes in this work. (b) Crystal structures of complexes 1–5 and Ref-Pt1 (hydrogen atoms are omitted for clarity). | |
The crystal structures of complexes 1–5 and Ref-Pt1 are shown in Fig. 1b. Complex 2 shows a boat-shaped conformation, similar to that reported for [(ppy)Pd(μ-Pz)]2 (Hppy = 2-phenylpyridine) with the same unsubstituted Pz ligand.9 The other complexes adopt a “double decker” conformation in which the bridging ligand is orthogonal to the plane containing the Pd/Pt atom and the cyclometalated ligand. From their crystal structures, 2 and Ref-Pt1 exhibit the cis configuration. On the other hand, complex 1 shows the trans configuration in its crystal structure. As the steric bulk of the bridging Pz ligand increases, the Pd–N(Pz)–N(Pz) angle decreases from 119.8° in 2 to 108.5° in 1, resulting in a shortening of the Pd–Pd distance from 3.40 Å in 2 to 2.79 Å in 1. A similar finding was reported by Ma et al. in analogous binuclear PtII complexes.10Ref-Pt1, which has the same ligand scaffold as 1, also exhibits a close Pt–Pt distance of 2.90 Å. Complexes 3–5 with bridging DPF ligands have short intramolecular Pd–Pd distances of 2.86–2.89 Å and are in the trans configuration.
Absorption and emission spectroscopy
The electronic absorption spectra of binuclear PdII complexes, Ref-Pt1, and Ref-Pt5 are shown in Fig. 2a, b and S5.† The spectral data are summarized in Table 1. In CH2Cl2, complexes 1, 2, and Ref–Pt1 show an intense absorption band with λmax at 268–279 nm (ε > 104 M−1 cm−1) and a less intense absorption band at 356–385 nm (ε = 5.8 − 7.3 × 103 M−1 cm−1). Both bands were assigned to (π–π*) singlet intraligand (1IL) transitions localized on the cyclometalated C^N ligand. For 1, there is an additional weak absorption band with λmax at 433 nm (ε = 2.8 × 103 M−1 cm−1), and this band is blue-shifted relative to Ref-Pt1 (λmax = 518 nm). These low-energy absorption bands are attributed to the [dσ*(Pd2/Pt2) → π*(C^N)] 1MMLCT transition, as shown by the DFT calculations (vide infra). For 3–7, the intense absorption bands at 242–358 nm (ε > 104 M−1 cm−1) were assigned to (π–π*) 1IL transition of the C^N/C^C* or DPF ligands.
 |
| | Fig. 2 Absorption spectra of (a) complexes 1, 2, and Ref-Pt1, (b) complexes 4–7 and Ref-Pt5 in CH2Cl2 at RT. Emission spectra of (c) complexes 1 and Ref-Pt1, (d) complexes 5–7 and Ref-Pt5 in deoxygenated CH2Cl2 (2 × 10−5 M) at RT. Emission spectra of (e) complexes 1 and (f) 5 in various solvents. Asterisk (*) indicates Raman scattering of excitation light. | |
Table 1 UV-visible absorption and emission data of complexes 1–7, Ref-Pt1, and Ref-Pt5 in CH2Cl2, solid state and 2 wt% PMMA thin films
|
|
UV-vis absorptiona |
Emission at 298 K |
|
λ
max [nm] (ε [103 M−1 cm−1]) |
λ
em [nm] (τd [μs]; Φem [%]; kri [103 s−1]; knrj [104 s−1]) |
| In deoxygenated CH2Cl2b |
Solid statec |
2 wt% in PMMAc |
|
At a concentration of 1 × 10−5 M.
At a concentration of 2 × 10−5 M.
Measured in air.
Emission lifetime.
Emission quantum yield was calculated using [Ru(bpy)3](PF6)2 as reference (bpy = 2,2′-bipyridyl, in MeCN, Φem: 0.062, λem = 619 nm).
Emission quantum yield was obtained by integrating sphere.
Emission quantum yield was calculated using [Pt(tptbp)] as reference (H2tptbp = tetraphenyltetrabenzoporphyrin, in toluene, Φem: 0.51, λem = 770 nm).
Weighted average lifetime for biexponential decay.
Radiative decay rate constant estimated from the equation kr = Φem/τ.
Non-radiative decay rate constant estimated from the equation knr = (1 − Φem)/τ.
|
|
1
|
269 (102.1), 365 (sh, 5.9), 433 (br, 2.8) |
609 (0.30; 0.016e; 0.53; 333) |
612 (4.8; 30; 63; 15) |
592 (7.1; 3.0; 4.2; 14), 736 (sh) |
|
2
|
269 (102.2), 356 (sh, 5.8) |
Non-emissive |
497 (sh), 521 (0.11; <0.1; n.d.; n.d.) |
479, 506 (0.29; <0.1; n.d.; n.d.) |
|
3
|
268 (119.7), 358 (24.9), 463 (br, 2.0) |
595 (0.88; 2.0f; 23; 111) |
585 (6.3; 38; 60; 9.8) |
583 (9.8; 2.0; 2.0; 10) |
|
4
|
242 (sh, 75.5), 293 (38.9), 332 (50.7) |
Non-emissive |
589 (63.9; 70; 11; 0.47) |
579 (30.8; 29; 9.4; 2.3) |
|
5
|
286 (55.7), 338 (40.2), 410 (br, 5.0) |
532 (0.45; 3.0f; 67; 216) |
550 (6.2; 45; 73; 8.9) |
519 (7.4; 47; 64; 7.2) |
|
6
|
286 (sh, 50.5), 307 (59.0), 335 (sh, 38.8), 450 (br, 4.7) |
576 (5.9; 61f;103; 6.6) |
566 (0.25; 5.0; 200; 380) |
559 (6.7; 67; 100; 4.9) |
|
7
|
281 (50.1), 337 (47.8), 400 (5.5) |
555 (0.062; 0.15e; 24; 1610) |
510 (3.8h; 27; 71; 19) |
541 (12.9; 58; 45; 3.3) |
|
Ref-Pt1
|
279 (86.9), 385 (7.3), 460 (4.9), 518 (4.5) |
806 (0.14; 8.7g; 621; 652) |
661 (0.88; 46; 523; 61) |
663(0.76; 31; 408; 91) |
|
Ref-Pt5
|
288 (58.6), 328 (sh, 43.6), 472 (br, 8.1) |
619 (1.4; 62f; 443; 27) |
628 (1.4; 52; 371; 34) |
607 (1.7; 99; 582; 0.59) |
Complexes 3 and 5–7 show a broad shoulder band tailing to 500 nm (ε = 2.0 − 5.5 × 103 M−1 cm−1), which was assigned to a mixed [π(DPF) → π*(C^N/C^C*)] 1LLCT (major) and [dσ*(Pd2) → π*(C^N/C^C*)] 1MMLCT (minor) transitions (see DFT calculations). The energy of these low-energy absorption bands follows the order 7 > 5 > 6, which is related to the energy level of the π* orbitals of the C^C* ligand. The high energy level of π* orbitals of the non-conjugated C^C* ligand of 4 causes its 1LLCT/1MMLCT transitions to occur in the region <400 nm. The low-energy absorption bands (λmax > 400 nm) of 1 and 5 show hypsochromic shifts of up to 13 nm with increasing solvent polarity (Fig. S6 and Table S7†), further indicating the charge transfer nature of these absorption bands.
All complexes, except 2 and 4, show emission in deoxygenated CH2Cl2 at room temperature (RT) (Fig. 2c, d and Table 1). For the binuclear PdII complex, an unstructured emission band is observed with λmax at 532–609 nm and τ in the sub-microsecond regime. The λmax of 1 (609 nm) undergoes a hypsochromic shift from its PtII counterpart Ref-Pt1 (806 nm), consistent with the spectral assignment of the emission of 1 to [dσ*(Pd2) → π*(C^N)] 3MMLCT excited state. The kr of 1 with τ of 0.30 μs and Φem of <0.01 is small, about 102 s−1. Replacing the bridging Pz ligands to DPF ligands increases the Φem and kr, as observed in 3 (Φem: 0.02; kr: 2.3 × 104 s−1). On the other hand, the Φem varies greatly (0.0015–0.61) for binuclear PdII complexes with Pd–NHC bonds (5–7). Both complexes 5 and 7 have lower Φem than 6, which is mainly due to significant non-radiative decay in solution at RT (knr: 2.2 × 106 s−1 [5], 1.6 × 107 s−1 [7] vs. 6.6 × 104 s−1 [6]). The emissions of complexes 3 and 5–7 were assigned to a mixed [dσ*(Pd2) → π*(C^N/C^C*)] 3MMLCT and [π(DPF) → π*(C^N/C^C*)] 3LLCT excited state. As the solvent polarity increases from toluene, through CH2Cl2, CHCl3 and EA to MeCN, the emission λmax of 1 shows a hypsochromic shift of up to 11 nm (Fig. 2e). In contrast, the emission λmax of 5 is bathochromic shifted by up to 7 nm (Fig. 2f). These shifts suggest that the differences in dipole changes between the ground and excited states of 1 and 5 are opposite. The emission of complex 1 is too weak to be detected in MeCN, and the Фem of complex 5 drops from 0.043 in toluene to 0.007 in MeCN (Table S7†).
Complexes 1 and 3–7 show broad emission bands with λmax at 510–612 nm in 2 wt% PMMA thin films and in the solid state at RT (Fig. S9 and S10†). In contrast, the emission band of 2 shows a vibronic structure with the first vibronic peak at 479–497 nm attributed to the [π–π*] 3IL excited state. Complexes 1 and 3–7 have Φem spanning from 0.05 to 0.70, and τ in the microsecond regime, revealing large kr from 104 s−1 to 2 × 105 s−1, all of which are significantly larger than the kr of 2 (≤103 s−1) and other reported PdII complexes with ligand-centered excited states.11
Variable-temperature emission decay lifetime
The binuclear PdII complexes (1, 3, 5, and 7) exhibit weak emissions in dilute solutions at 298 K with low Фem (≤0.03) and large knr values (1.1 − 16.1 × 106 s−1). In this regard, the variation of τobs over a large temperature range provides information on the radiationless decay process of the emitting excited state(s) and could be modelled by eqn (1):12| |  | (1) |
where k0 is the temperature-independent decay rate of the decay process, A is the pre-exponential factor of the Arrhenius term that contributes to the non-radiative process, Ea is the activation energy, and kB is the Boltzmann constant. To gain insight into the non-radiative processes of the binuclear Pd complexes, the temperature-dependent emission lifetimes of complexes 1, 3, and 5–7 were measured in deoxygenated toluene. As shown in Fig. 3, using eqn (1), the kobs values fit well with 1/T. Complexes 1 and 3 have similarly large A values, about 1012 s−1 (Table 2), but the Ea value of 3 (2989 cm−1) is slightly larger than that of 1 (2605 cm−1). This is consistent with the higher Фem and smaller knr of 3 compared to 1 in dilute solutions (Table 1). In particular, the A values for 1 and 3 are approximately 1011–1014 s−1, suggesting the existence of an activated and rate-determining surface crossing mechanism to reach higher-lying dark electronic state based on previous studies on luminescent d6 and d8 metal complexes.13 Although the A values of 5 (2.81 × 1010 s−1) and 7 (1.56 × 1010 s−1) are about two orders of magnitude smaller than those of 1 and 3, the Ea values of 5 (2088 cm−1) and 7 (1520 cm−1) make the thermal deactivation process non-negligible. Despite having a similar Ea value to complex 5, the pre-exponential term of complex 6 is significantly smaller (A = 1.73 × 108 s−1), which allows moderately efficient emission to occur at RT. The kinetic parameters of complexes 1, 3, 5, and 7 (Table 2) reveal an overwhelming contribution (92–97%) of the temperature-dependent non-radiative decay rate knr(T) to kobs at 298 K. For 6, only 8.2% of the kobs come from the knr(T) term. This is in sharp contrast to complexes 1, 3, 5, and 7, whose excited states undergo significant thermal populated deactivation processes, resulting in these four complexes having lower Фem and large knr in dilute solutions at 298 K.
 |
| | Fig. 3 Plots of the temperature dependence of the excited-state decay rate constant (kobs) of (a) complexes 1 (5 × 10−5 M), 3, 5, and 7 (2 × 10−5 M) and (b) 6 (2 × 10−5 M) in deoxygenated toluene. | |
Table 2 Kinetic parameters for excited-state deactivation
| Complex |
k
0 [s−1] |
A
1 [s−1] |
E
a [cm−1] |
|
1
|
144852 |
1.08 × 1012 |
2605 |
|
3
|
37601 |
2.05 × 1012 |
2989 |
|
5
|
96000 |
2.81 × 1010 |
2088 |
|
6
|
125769 |
1.73 × 108 |
1994 |
|
7
|
406039 |
1.56 × 1010 |
1520 |
Time-resolved absorption and emission spectroscopy
Complexes 1 and 5 were selected as representative examples to study the excited-state dynamics of binuclear PdII complexes by femtosecond/nanosecond time-resolved absorption difference (fs/ns-TA) and femtosecond time-resolved fluorescence (fs-TRF) spectroscopy. In toluene, different excited state kinetics were observed for the two complexes. The initial TA (∼0.8 ps) of 1 has two excited-state absorption (ESA) signals at 509 and 610 nm that decay throughout the delay time (0.8 ps–2.5 ns; Fig. S11a†). The kinetics at 509 nm showed two ps decay time constants of 1.1 and 8.8 ps (Fig. S11b†); the former is consistent with TRF decay (τTRF: 1.0 ps; Fig. S12b†). For 5, its initial TA (∼0.9 ps) is characterized by two ESA signals at 462 and 625 nm (Fig. 4a). The spectral evolution of 5 occurs in three stages. The early phase occurs with a time constant of 2.0 ps and involves an ESA growth at 462 nm. This 2.0 ps time constant is consistent with the TRF decay (τTRF: 2.5 ps; Fig. 4c and d). In the second stage, with a time constant of 13.8 ps, the TA rises at 462 and 560 nm, with the former undergoing an 18 nm redshift (Fig. 4b). After the second stage of TA evolution, the fs-TA signals undergo a long-lived decay process on the ns timescale to give a TA spectrum consistent with the respective ns-TA spectrum (Fig. S13b†), which can be attributed to the formation of T1 state. Based on these time-resolved spectroscopic measurements, the excited state cascades of complexes 1 and 5 were constructed (Fig. S14†). Upon excitation, the emission λmax of the S1 excited state is at 521 nm for 1 (Fig. S12a†) and 486 nm for 5 (Fig. 4c), both of which are depleted by S1-to-triplet (Tn or T1) intersystem crossing (ISC) with an ultrafast time constant (1.1 ps for 1; 2.0 ps for 5). The formed triplet excited state undergoes an internal conversion from Tn to T1 or vibrational cooling of T1 with time constants of 8.8 ps for 1 and 13.8 ps for 5. This is followed by radiative decay to the ground state in a sub-microsecond timescale (Table 1).
 |
| | Fig. 4 (a) and (b) fs-TA spectra of 5 at selected time delays; (c) fs-TRF spectra and (d) its kinetic trace of 5 in toluene. | |
Electrochemistry
The electrochemical properties of complexes 1–7, Ref-Pt1, and Ref-Pt5 were investigated by cyclic voltammetry (CV). To avoid undesirable side reactions between oxidized species and coordinating solvent molecules, the non-coordinating solvent CH2Cl2 was used for anodic scanning.14 Cathodic scanning was performed in DMF due to its wide electrochemical window for reduction. The CV curves and electrochemical data are shown in Fig. 5, S15† and Table 3, respectively. Complex 1 exhibits a reversible oxidation couple with E1/2 at 0.86 V vs. SCE, while 2 shows an irreversible oxidation wave with Epa at 1.22 V vs. SCE. This is consistent with the HOMO of 1 being destabilized by metal–metal interactions. Complexes 3–7 show reversible/quasi-reversible oxidation couple with E1/2 at 0.72–0.87 V vs. SCE. Compared to their PtII analogues Ref-Pt1 and Ref-Pt5, the E1/2 values of 1 and 5 are anodically shifted by 0.42 and 0.29 V, respectively (Fig. 5). Based on the DFT calculations (vide infra), the oxidation process of complex 1 is assigned to be metal-centered, whereas the oxidation process of complex 5 occurs on the DPF ligand. For the reduction process, complexes 1, 3, 6, and Ref-Pt1 exhibit two reversible reduction couples with E1/2 ranging from −1.68 to −1.79 and −1.82 to −2.04 V vs. SCE, respectively, while 2 exhibits one reversible reduction couple with E1/2 at −1.70 V vs. SCE. The reduction process of complexes 4, 5, 7, and Ref-Pt5 is irreversible with Epc spanning from −2.11 to −2.73 V vs. SCE. The reduction potentials of binuclear PdII complexes 1–7 vary with the cyclometalated ligands. Also pairs of binuclear complexes with the same cyclometalated and bridging ligands have similar reduction potentials, such as complex 1 (E1/2 = −1.72 vs. SCE) and Ref-Pt1 (E1/2 = −1.68 vs. SCE), or complex 5 (Epc = −2.17 V vs. SCE) and Ref-Pt5 (E1/2 = −2.11 V vs. SCE). This result shows that the reduction of the binuclear PdII complex in this study is mainly ligand centered.
 |
| | Fig. 5 Cyclic voltammograms of selected complexes in CH2Cl2 for anodic sweeps and in DMF for cathodic sweeps with 0.1 M NBu4PF6 as electrolyte. E1/2 (Cp2Fe+/0) was recorded in the range of 0.43–0.52 V vs. SCE in CH2Cl2 or DMF. | |
Table 3 Electrochemical dataa and excited-state redox properties
| Complex |
E(M+/M0) [V] |
E(M0/M−, M−/M2−) [V] |
E
0–0
[V] |
E(M+/*M0)f [V] |
E(*M0/M−)f [V] |
|
Measured in CH2Cl2 for anodic sweeps and DMF for cathodic sweeps with 0.1 M NBu4PF6 as supporting electrolyte at a scan rate of 100 mV s−1; E1/2 (Cp2Fe+/0) is recorded at the range of 0.43–0.52 V vs. SCE in CH2Cl2 or DMF.
Values refer to E1/2versus SCE.
Value refers to the anodic peak versus SCE.
Value refers to the cathodic peak versus SCE.
Approximate zero–zero excitation energy, E0–0, was estimated from the emission onset at 298 K in CH2Cl2.
Estimation of approximate excited-state redox potentials: E(M+/*M0) = E(M+/M0) − E0–0 (V vs. SCE), E(*M0/M−) = E(M0/M−) + E0–0 (V vs. SCE).
|
|
1
|
0.86b |
−1.72, −1.84b |
2.26 |
−1.40 |
0.42 |
|
2
|
1.22c |
−1.70b |
— |
— |
— |
|
3
|
0.86b |
−1.79, −2.04b |
2.40 |
−1.54 |
0.67 |
|
4
|
0.72b |
−2.73d |
— |
— |
— |
|
5
|
0.81b |
−2.17d |
2.69 |
−1.88 |
0.52 |
|
6
|
0.87b |
−1.68, −1.82b |
2.45 |
−1.58 |
0.80 |
|
7
|
0.78b |
−2.27d |
2.67 |
−1.89 |
0.40 |
|
Ref-Pt1
|
0.44b |
−1.68, −1.83b |
1.83 |
−1.39 |
0.15 |
|
Ref-Pt5
|
0.52b |
−2.11b |
2.25 |
−1.73 |
0.10 |
DFT/TDDFT calculations
DFT/TDDFT calculations were performed on the binuclear PdII complex 1 and its PtII counterpart Ref-Pt1 to study their electronic structures in the ground and excited states. Because both trans and cis isomers were observed in the 1H NMR spectra of 1 and Ref-Pt1, calculations were performed for both isomers. A small energy difference (ranging from 0.04 to 0.06 eV) was observed between the cis and trans isomers (Table S8†). Since calculations for the cis and trans isomers show no significant differences, only the complexes with the trans geometry are described here. The optimized geometries of 1 and Ref-Pt1 match well with their crystal structures, as shown in Fig. 6a, where the intramolecular Pd–Pd and Pt–Pt distances are calculated to be 2.83 Å and 2.87 Å, respectively. In the calculated absorption spectrum (Fig. 6b), the lowest transition band is located at 462 nm for 1 and 530 nm for Ref-Pt1, both close to the experimental values (λmax: 440 nm for 1, 518 nm for Ref-Pt1). As the distance between two metal atoms is less than the sum of their van der Waals radii, the overlap between the two valence ndz2 (n = 4 for Pd and 5 for Pt) orbitals will result in the formation of a bonding (ndσ) and an antibonding (ndσ*) orbital. In the MO diagrams shown in Fig. 6c, the HOMOs of both 1 and Ref-Pt1 consist of the ndσ* orbital between two metals, while the LUMO is mainly located at the C^N ligand. The calculated S1 states of 1 and Ref-Pt1 are derived from the HOMO (ndσ*) to LUMO (π*) transition. The 4dσ* orbital energy level of complex 1 is lower than the 5dσ* orbital of Ref-Pt1, which causes the HOMO–LUMO gap of complex 1 to increase and the lowest absorption band to blue-shift. According to the calculation results, the lowest absorption bands (S0 → S1) of 1 and Ref-Pt1 are attributed to the 1MMLCT transition, and the calculated oscillator strengths are 0.02 and 0.04, respectively.
 |
| | Fig. 6 (a) Optimized structures of 1 and Ref-Pt1 at S0 and T1 state. (b) Calculated absorption spectra of 1 and Ref-Pt1 based on the optimized S0 structure. (c) Calculated MO diagrams of 1 and Ref-Pt1 at T1 state. (d) Calculated ZFS and SOC coupling of 1 and Ref-Pt1 at T1 state. | |
The triplet excited state T1 of 1 and Ref-Pt1 was also calculated. Calculations show that the M–M distance in the T1 state of 1 is shortened to 2.61 Å and that of Ref-Pt1 is shortened to 2.65 Å (Fig. 6a). This is due to the M–M bonding interaction formed by excitation of electrons from the antibonding dσ* orbital to the ligand π* orbital. The calculated emission wavelength of the 3MMLCT excited state of 1 is 632 nm, which is consistent with the experimentally observed emission λmax at 609 nm.
The phosphorescent radiative decay process of 1 and Ref-Pt1 was evaluated by calculating the values of zero-field splitting (ZFS) and SOC (〈Sm|HSO|T1〉). As shown in Fig. 6d, the calculated ZFS of Ref-Pt1 (39 cm−1) is larger than that of 1 (3.8 cm−1). The ZFS value is closely related to the SOC constant, which is 1412 and 4000 cm−1 for Pd and Pt, respectively. The contribution of metal orbitals in the singlet and triplet excited states also affects ZFS. Compared to Ref-Pt1, complex 1 exhibits a reduced contribution of PdII-4d orbitals in the MMLCT excited state due to the low-lying PdII-4d orbitals, resulting in a smaller ZFS in the PdII complex. For Ref-Pt1, the SOC between S2 and T1z, S4 and T1y, S6 and T1z states mainly contributes to the T1 → S0 transition (the T1x, T1y, and T1z refer to three T1 substates), with calculated values (SOC) of 404, 467 and 952 cm−1, respectively (Fig. 6d). For complex 1, the T1 → S0 transition is mainly contributed by the SOC between S6 and T1x states, with a value of 270 cm−1, which is smaller than that of Ref-Pt1.
DFT/TDDFT calculations were also performed on 5 to examine the effect of DPF ligands on the excited state. In the calculated absorption spectrum (Fig. 7a), the S0 → S1 transition of 5 is located at 418 nm, which is consistent with the experimental observation (λmax: 410 nm). As shown in the MO diagram (Fig. 7c), the HOMO of 5 consists of π orbitals of the DPF ligand (82%) and the LUMO is mainly located at the C^C* ligand. The 4dσ* orbital is located in the H-2 orbital. The S1 state of 5 is mainly contributed by the HOMO → LUMO transition, and the H-2 → LUMO transition contributes moderately. Therefore, the lowest absorption band of 5 was assigned to a mixture of 1LLCT/1MMLCT transitions. In the T1 excited state, the Pd–Pd distance is significantly shortened (Fig. 7b), which lifts the H-2 orbital (4dσ*) to the HOMO. The T1 → S0 transition of 5 is mainly contributed by the HOMO → LUMO transition. The HOMO of complex 5 at T1 state contains contributions from both the ligand and the metal, so the T1 state was assigned as a mixed 3MMLCT/3LLCT excited state.
 |
| | Fig. 7 (a) Calculated absorption spectrum of 5 (red line) based on the optimized structure, and the experimental absorption spectrum (black line). (b) Optimized structures of complex 5 at ground state (S0) and triplet excited state (T1). (c) Calculated MOs diagram of 5 at S0 (left) and T1 (right). (d) Calculated 3MMLCT and 3dd excited state energy of complexes 1, 5, 6, and Ref-Pt1. Note that due to mixing, the state labels may not strictly apply. | |
Since the ligand field splitting of PdII ions is smaller relative to PtII ions, the thermal population of the metal centered (3dd) excited state provides a quenching pathway for the emissive 3MMLCT excited state of the binuclear PdII complex. In this regard, the relative energies between the 3MMLCT and 3dd excited states of complexes 1 and Ref-Pt1 were calculated (Fig. 7d, S18 and S19†). For complex 1, the T1 state consists of 90% 3MMLCT and 10% 3dd characters. The 3dd character also has a major contribution to the T2 state and the T1–T2 energy difference is calculated to be 0.27 eV. In comparison, the 3dd character does not contribute to the T1 state of Ref-Pt1, but has a major contribution to the T8 state, which has an energy 1.4 eV higher than the T1 state. These results indicate that the excited-state deactivation process of 1 through the 3dd excited state is thermally more feasible relative to Ref-Pt1, leading to a faster non-radiative decay process of 1. For complexes 5 and 6, the excited states with dominant 3dd character are the T5 states at 2.92 and 2.77 eV, respectively (Fig. S20 and S21†). These energies are significantly higher than the T2 state energy of complex 1, showing that the incorporation of strong σ-donating NHC ligands can destabilize the 3dd excited state.15 The calculated T1–T5 energy differences of complexes 5 and 6 are 0.66 eV and 0.69 eV, respectively (Fig. 7d), indicating the deactivation pathway through the 3dd state of complexes 5 and 6 is less feasible than for complex 1.
Oxidation of complexes 1 and 5
The UV-vis absorption spectra of 1 (Fig. 8a) and 5 (Fig. 8b) were recorded before and after the addition of the one-electron oxidant tris(4-bromophenyl)ammoniumyl hexachloroantimonate (TBPA)SbCl6 (E1/2 = 1.05 V vs. SCE). For both complexes, a spectral evolution with attenuation of the low-energy absorption band (λmax = 410–440 nm) and the emergence of new absorption features in the lower energy region (λ = 500–1000 nm) is observed. The spectral transformation is similar to that observed for the two complexes during spectroelectrochemical oxidation (Fig. S16†), although the latter could not recover to its original state due to the instability of the oxidized species under spectroelectrochemical conditions. EPR spectra of reaction mixtures of complexes 1 and 5 with (TBPA)SbCl6 were recorded at 100 K to identify the electronic configuration of the 1e-oxidized species. For the reaction of complex 1 with (TBPA)SbCl6, spectral simulations gave axial g values with g⊥ = 2.240 and g∥ = 1.996, and 105Pd hyperfine structures with A⊥ = 53 G and A∥ = 45 G (Fig. 8c), showing that the SOMO has dominant dz2 character. The EPR spectrum is similar to that reported for [PdIIPdIII(μ-dpb)4]+.16 In contrast, the EPR spectrum of 5 after adding (TBPA)SbCl6 showed a typical organic radical signal (g = 2.00) (Fig. 8d), pointing to the ligand-based SOMO.17 Therefore, the chemical oxidation of 1 with (TBPA)SbCl6 produces [(PdII–PdIII)(Pz−)2]3+ (1+) species, while the chemical oxidation of 5 produces [(PdII–PdII)(DPF0.5−)2]3+ (5+) species. The formation of 5+ was also observed in the photoredox reaction of 5 with 4-(methoxycarbonyl)-N-methylpyridinium hexafluorophosphate. The excited-state redox potential E(M+/*M0) of 5 is −1.88 V vs. SCE (Table 3); therefore, the excited state of 5 can react with 4-(methoxycarbonyl)-N-methylpyridinium hexafluorophosphate (Ered = −0.78 V vs. SCE)18 through the electron-transfer mechanism (eqn (2)). We recorded ns-TA spectra at 1 μs after| |  | (2) |
laser excitation of a deoxygenated MeCN/toluene (1:9 v/v) solution containing 5 and 4-(methoxycarbonyl)-N-methylpyridinium hexafluorophosphate. The spectrum shows absorption bands at 520 and 820 nm (Fig. 8e), similar to the absorption characteristics of 5+ produced by oxidation of 5 with (TBPA)SbCl6 (Fig. 8b). The high-energy absorption band at 398 nm is consistent with the reported spectrum of 4-(methoxycarbonyl)-N-methylpyridinyl radical.18 The 820 nm absorption band follows a bi-exponential decay kinetics with lifetimes of 12.2 and 48.8 μs. Attempts to detect the formation of 1+ under similar photoredox conditions were unsuccessful because the excited state of complex 1 was significantly quenched in MeCN.
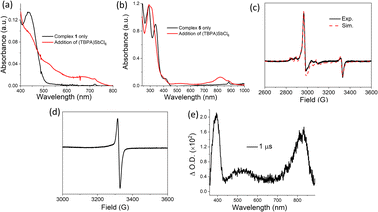 |
| | Fig. 8 UV-visible absorption spectra of (a) complex 1 and (b) complex 5 before and after the addition of 1 equiv. (TBPA)SbCl6 in a mixed CH2Cl2/MeCN solution (2:1 v/v) at −40 °C. (c) X-band EPR spectrum (solid, black) of the reaction mixture of 1 (3 mM) and 0.5 equiv. (TBPA)SbCl6 in frozen CH2Cl2 at 100 K; spectral simulation (dash, red) revealed that the spectrum comprises 75% PdII–PdIII species (1+) and 25% organic radicals. (d) X-band EPR spectrum of the reaction mixture of 5 (3 mM) and 0.5 equiv. (TBPA)SbCl6 in frozen CH2Cl2 at 100 K. (e) ns-TA spectrum recorded at 1 μs after flashing a deoxygenated MeCN/toluene (1:9 v/v) solution of complex 5 (2 × 105 s−1) and 4-(methoxycarbonyl)-N-methylpyridinium hexafluorophosphate (0.001 M) at RT. | |
Quenching and photochemical studies of complex 5 with alkyl bromides
The quenching of 5's phosphorescence by various alkyl bromides was studied in deoxygenated toluene (Fig. S22†). The quenching rate constants (kq) are listed in Table 4. The kq value of CBr4 is almost diffusion-controlled, whereas the kq values for allyl bromide and CH2Br2 are much smaller, about 106 M−1 s−1. Plots of logkq values versus Ered and BDE values of alkyl bromides are shown in Fig. 9a and b. The kq values correlate better with Ered than with BDE values. In order to obtain more spectroscopic evidence of the photoreaction mechanism between 5 and alkyl bromide, the ns-TA spectrum of laser flash photolysis (the excitation wavelength = 355 nm) of 5 and benzyl bromide (0.3 M) in deoxygenated toluene was recorded (Fig. 9c). Under this condition, the excited state of complex 5 is completely quenched by benzyl bromide within 4 μs. Therefore, the absorption bands at 457 and 552 nm observed at 4 μs after the laser flash should originate from photoreaction products. Referring to the absorption characteristics of Br2 (390 nm),19 Br− (<300 nm),20 Br2−˙ (360/700 nm),21 Br3− (265 nm)22 and 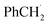 (318/307 nm),23 we exclude that the 457 nm and 552 nm absorption bands originate from the above species. At a high concentration of benzyl bromide (3 M), the excited state of 5 was completely quenched within 0.2 μs, and absorption bands at 457 and 552 nm were observed within 10 ns after the laser flash (Fig. S23†).
(318/307 nm),23 we exclude that the 457 nm and 552 nm absorption bands originate from the above species. At a high concentration of benzyl bromide (3 M), the excited state of 5 was completely quenched within 0.2 μs, and absorption bands at 457 and 552 nm were observed within 10 ns after the laser flash (Fig. S23†).
Table 4 Quenching rate constants of complex 5 by various alkyl bromides in deoxygenated toluene at RT
| Alkyl bromide |
E
red
[V] |
BDEb [kcal mol−1] |
k
q [dm3 mol−1 s−1] |
|
Onset values of the cathodic peak in cyclic voltammogram (measured in DMF solution with 0.1 M NBu4PF6vs. SCE).
Values of bond dissociation energy for the C–Br bond.
|
| CBr4 |
−0.41 |
50.8 |
7.17 × 109 |
| Ethyl tribromoacetate |
−0.49 |
58.9 |
2.55 × 109 |
| Phenacyl bromide |
−0.97 |
64.8 |
9.00 × 108 |
| Bromodiphenyl methane |
−1.08 |
— |
1.52 × 108 |
| Benzyl bromide |
−1.30 |
57.1 |
1.57 × 107 |
| Allyl bromide |
−1.60 |
55.7 |
6.56 × 106 |
| CH2Br2 |
−1.77 |
66.0 |
1.06 × 106 |
 |
| | Fig. 9 (a) Correlation of the reduction potentials of alkyl bromides with logkq of 5. (b) Correlation of the BDE values of alkyl bromides with log(kq/n) of 5; n corresponds to the number of bromine atoms in alkyl bromides. (c) ns-TA spectra of complex 5 (2 × 10−5 M) in the presence of benzyl bromide (0.3 M) in deoxygenated toluene at RT. (d) ns-TA spectra of complex 5 (2 × 10−5 M) in the presence of 4-(methoxycarbonyl)-N-methylpyridinium hexafluorophosphate (0.001 M) and NBu4Br (0.05 mM) in deoxygenated MeCN/toluene (1:9) at RT. (e) Decay of the TA signal at 820 nm for the photolysis reaction of complex 5 and 4-(methoxycarbonyl)-N-methylpyridinium hexafluorophosphate with and without NBu4Br. (f) Decay of the TA signal at 457 nm for the photolysis reaction of complex 5 with 4-(methoxycarbonyl)-N-methylpyridinium hexafluorophosphate and NBu4Br. (g) Dependence of decay rates of 5+ on the concentrations of NBu4Br. | |
Absorption bands at 457 and 552 nm were also detected by laser flash photolysis of complex 5 in the presence of 4-(methoxycarbonyl)-N-methylpyridinium hexafluorophosphate and NBu4Br. As shown in Fig. 9d, the TA spectrum at 300 ns after laser flash shows the characteristic absorption band of 5+ at 820 nm. At later time after the laser flash (2–10 μs), an increase in the absorption bands at 457 and 552 nm is observed, with a concomitant decay at 820 nm, following a bi-exponential kinetics with τ1 of 5.2 and τ2 of 44.9 μs; the former lifetime can be related to the growth lifetime (3.0 μs) extracted from the kinetics at 457 nm (Fig. 9f), showing a reaction between 5+ and Br− (eqn (3)). Increasing the NBu4Br concentration further
| |  | (3) |
reduces
τ1, and the pseudo-first-order rate constant (
kobs) is linearly related to the NBu
4Br concentration, from which the second-order rate constant (
k2) could be determined to be 4.5 × 10
9 M
−1 s
−1 (
Fig. 9g).
To further identify the photoreaction products, the EPR spectra of the photolysis mixture of 5 and benzyl bromide in frozen benzene were recorded at 100 K. An EPR signal attributable to the combined signal of Pd compounds [Pd] (20%) and organic radicals (80%) was observed (Fig. 10a). Spectral simulations gave the following parameters for the S = 1/2 [Pd] species: g∥ = 2.029, g⊥ = 1.990, A∥ = 8 G, A⊥ = 3 G. The low g-anisotropy (Δg = 0.039) and small deviation of gav (2.0030) from the free electron g value (2.0023) indicate that the unpaired electron of [Pd] is partially distributed on the ligand.24 Furthermore, the order g∥ > g⊥ suggests that the unpaired electron also has some Pd characters residing in dxz/dyz or dx2−y2-based orbitals.24,25 DFT calculations were performed to reveal the electronic structure of 5+ and 5-Br. As shown in Fig. 10b, the calculated spin densities of Pd1 and Pd2 atom in 5+ are the same, both are 0.04. The calculated spin density on the four N1, N2, N3 and N4 atoms of 5+ is much larger than that of the Pd atom, totaling 0.7, showing that the unpaired electron is mainly located at the formamidinate ligand. This calculation result is consistent with the EPR characteristic of 5+ generated by chemical oxidation of 5 with (TBPA)SbCl6 (Fig. 8d). The Pd–Pd distance shrinks from 2.89 Å in 5 to 2.79 Å in 5-Br (Fig. 10c), showing Pd–Pd bonding interactions (though weak) due to the oxidation of Pd atoms. The spin density distribution of 5-Br is mainly localized on Pd and Br atoms, and the values of Pd1, Pd2 and Br atom are 0.14, 0.4 and 0.29, respectively. This significant difference in the spin density population between 5-Br and 5+ is consistent with the different EPR patterns of 5-Br and 5+.
 |
| | Fig. 10 (a) X-band EPR spectrum and simulation of the reaction mixture of 5 and benzyl bromide in frozen benzene at 100 K after light irradiation for 15 minutes. (b) Optimized structure (left) and spin density population (right) of complex 5+. (c) Optimized structure (left) and spin density population (right) of complex 5-Br. | |
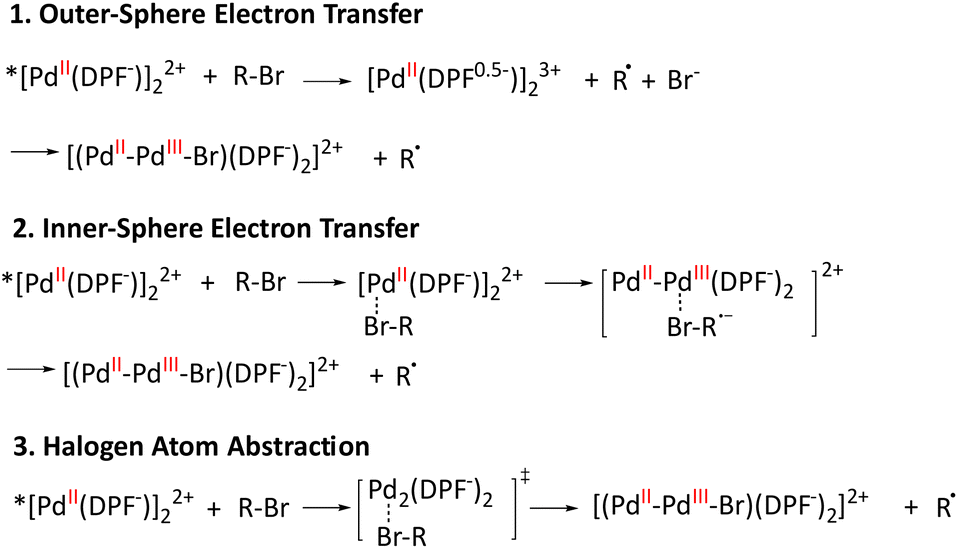
There are three possible reaction mechanisms for the photoreaction of binuclear PdII complex 5 with alkyl bromides, as shown in Scheme 1:26 (1) in the outer-sphere electron transfer mechanism, the alkyl bromide R–Br receives electrons from the excited state of 5 and undergoes C–Br bond cleavage to generate the bromide anion, which is subsequently added to the metal center; (2) the inner-sphere electron transfer reaction involves the formation of caged ion pairs as intermediates prior to the electron transfer process; (3) alternatively, the excited state of 5 can react with alkyl bromides through a halogen atom abstraction mechanism. The linear correlation between the logkq values of 5 and the reduction potentials of the alkyl bromides is inconsistent with the halogen atom abstraction hypothesis, which predicts that the reaction rate depends on the BDE of the alkyl bromide. No formation of 5+ was observed in laser flash photolysis experiments of 5 with benzyl bromide, whereas the direct observation of 5-Br on the nanosecond time scale suggests that the reaction most likely follows an inner-sphere electron transfer mechanism.
 |
| | Scheme 1 Possible reaction mechanisms for the photoreaction of complex 5 with alkyl bromides. | |
Photocatalysis
The bimolecular photoreaction between 5 and alkyl halides prompts us to study the application of 5 as a photocatalyst in the photo-induced intramolecular cyclization of indole/pyrrole (Scheme 2).27 For all substrates examined, 100% substrate conversion and high yields of cyclized products (68–95%) were achieved (Scheme 2). Both electron-rich indoles and electron-deficient indoles perform well in photo-induced cyclization reactions. Notably, the C3 electron-rich indole gave the highest product yield (95%). The conversion efficiency of 1-(3-iodopropyl)-1H-indole to the cyclized product was less satisfactory (68%), probably due to the higher ring strain of the five-membered fused ring than the six-membered fused ring. This photoredox reaction also worked for pyrrole, furnishing the cyclized product in 72% yield.
 |
| | Scheme 2 Photo-induced indoles/pyrroles functionalization by intramolecular cyclization (upper), and aerobic C–C coupling reactions of N-phenyl-1,2,3,4-tetrahydroisoquinoline with acetone (bottom). (a) Reaction conditions: indoles/pyrroles (0.05 mmol), DIPEA (0.10 mmol), complex 5 (2.5 mol%) in 0.56 mL deoxygenated benzene-d6, light (LED lamp, λmax = 400 nm, 24 W), irradiation time: 2 h. (b) Reaction conditions: N-phenyl-1,2,3,4-tetrahydroisoquinoline (0.128 mmol), acetone (0.4 mL), L-proline (0.128 mmol), PC (1 mol%), MeCN (0.4 mL), MeOH (0.8 mL), oxygen, light (LED lamp, λmax = 455 nm, 24 W), irradiation time: 6 h. Product yields and substrate conversion were calculated by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. | |
With E(DIPEA+/DIPEA0) = 0.65 V vs. SCE and Ered of 1-(4-iodobutyl)-1H-indole = −1.68 V vs. SCE, the oxidative quenching of *5 by 1-(4-iodobutyl)-1H-indole is thermodynamically favored by 0.20 V and the reductive quenching of *5 by DIPEA is thermodynamically uphill by 0.13 V. Stern–Volmer quenching experiments showed that 1-(4-iodobutyl)-1H-indole quenches the emission of 5 with a kq of 1.65 × 107 M−1 s−1, while the quenching of the emission of 5 by DIPEA is negligible (Fig. S24†). In addition, we observed a long-lived species (τ > 10 μs) in the ns-TA spectrum of a deoxygenated toluene solution containing 5 and 1-(4-iodobutyl)-1H-indole (Fig. 11b). This shows that the photoreaction is initiated by oxidative quenching of *5 with indole. Photoreaction of 1-(4-iodobutyl)-1H-indole in the presence of TEMPO (2,2,6,6-tetramethyl-1-piperidyloxy) as a scavenger was conducted; the TEMPO-coupling product was obtained in 90% yield (Fig. 11c). In view of the above results, a plausible reaction mechanism was proposed and shown in Fig. S25.† The mixed 3MMLCT/3LLCT excited state of 5 can induce reductive dehalogenation of N-alkylated indole/pyrrole to form an alkyl radical, which subsequently undergoes cyclization reaction to give a benzylic radical. Oxidation of the benzylic radical followed by deprotonation affords the desired cyclized product. DIPEA was used as a sacrificial electron donor to regenerate 5.
 |
| | Fig. 11 (a) ns-TA spectra at 0.01 and 4 μs after flashing a deoxygenated toluene solution of 5 (2 × 10−5 M). (b) ns-TA spectra at 0.01 and 4 μs after flashing a deoxygenated toluene solution containing 5 (2 × 10−5 M) and 1-(4-iodobutyl)-1H-indole (0.1 M). (c) Photoreaction of 1-(4-iodobutyl)-1H-indole in the presence of TEMPO. Product yield was calculated by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. | |
The feasibility of these binuclear PdII complexes as photosensitizers (PC) was also examined in the photochemical generation of singlet oxygen (1O2). As shown in Fig. S26,† the emission of 1O2 was detected upon photoexcitation of binuclear PdII complexes (3, 5, and 7) in aerated CHCl3. Referring to the emission intensity of 1O2 produced with H2tpp as the photosensitizer (Φso = 0.55 in CHCl3) at λmax = 1270 nm,28 it was found that the Φso values of 3, 5, and 7 were 0.19, 0.15 and 0.08, respectively. Subsequently, we examined the photo-induced aerobic C–C coupling reaction of N-phenyl-1,2,3,4-tetrahydroisoquinoline with acetone using these complexes as photosensitizers. This reaction is known to be initiated by the oxidation of amines to iminium ion intermediates via photo-chemically generated 1O2.29 As shown in Scheme 2, complete substrate conversion (100%) and high product yields (81–88%) were achieved. Control experiments showed that in the absence of binuclear PdII complexes, L-proline, O2 or light irradiation, only trace amounts of coupling products were detected.
Electroluminescence
The high emission quantum yields of 5 and 6 in PMMA thin films prompted us to investigate their potential applications as OLED emitters. Devices with architecture consisting of ITO/HAT-CN (5 nm)/TAPC (40 nm)/CCP (10 nm)/5: PPF (10 nm)/PPF (10 nm)/TmPyPb (40 nm)/LiF (1.2 nm)/Al (100 nm) were fabricated to examine the electroluminescent (EL) properties of 5. In these devices, di-[4-(N,N-ditolyl-amino)-phenyl]cyclohexane (TAPC) and 1,3,5-tri(m-pyrid-3-yl-phenyl)benzene (TmPyPb) were used as the hole-transporting layer (HTL) and electron-transporting layer (ETL), respectively, and 1,4,5,8,9,11-hexaazatriphenylene hexacarbonitrile (HAT-CN) was used as the hole-injecting layer to facilitate the hole injection from ITO to TAPC. Complex 5 was doped in 2,8 bis(diphenylphosphoryl)dibenzo[b,d]furan (PPF) at concentrations ranging from 4 to 16 wt% to form an emissive layer (EML). PPF and CCP (9-phenyl-3,9′-bicarbazole), with triplet energies of 3.0 and 3.1 eV, were used as an exciton-blocking layer to confine the excitons within the EML.30 When the doping concentration is 4 wt%, the EL maximum of 5 is located at 521 nm (Fig. 12a). When the doping concentration is increased to 8 wt%, the EL spectrum of 5 is almost unchanged and is slightly red-shifted to 525 nm at 16 wt%. The device with 4 wt% of 5 achieved a high max. EQE of 22.9% (Fig. 12b), corresponding to a current efficiency (CE) of 71.2 cd A−1 (Table 5). At high luminance of 1000 cd m−2, the EQE and CE of this device slightly dropped to 19.6% and 60.9 cd A−1, respectively. In devices with 8 wt% and 16 wt% doping concentrations of 5, max. EQE values dropped slightly to 21.8% and 20.7%, respectively. Although the PLQY (0.92–0.93) of 5 in PPF is similar at different doping concentrations (Fig. S27†), the luminance and EQE at 1000 cd m−2 of devices with higher concentrations of 5 are lower than those with 4 wt% of 5. The inferior device performance of 5-based devices at higher doping concentrations can be attributed to the strong trapping effect of 5, as shown in Fig. S28.† The HOMO energy level (−5.2 eV) of 5 is 1.5 eV higher than the host PPF (−6.7 eV), causing holes injected from the HTL deeply trapped.
 |
| | Fig. 12 (a) Normalized EL spectra and (b) EQE-luminance characteristics of OLEDs with 5 at various doping concentrations. (c) Normalized EL spectra and (d) EQE-luminance characteristics of OLEDs with 6 at various doping concentrations. | |
Table 5 Key performances of OLEDs with 5 and 6 at various concentrationsa
|
|
L
max
[cd m−2] |
Max. EQEc [%] |
EQEc at 1000 cd m−2 [%] |
Max. CEd [cd A−1] |
Max. PEe [lm W−1] |
CIE coordinatesf (x, y) |
|
Fabricated by vacuum deposition.
Max. luminance.
External quantum efficiency.
Current efficiency.
Power efficiency.
CIE coordinates at 1000 cd m−2.
|
| PPF: 5 (4 wt%) |
18200 |
22.9 |
19.6 |
71.2 |
85.2 |
0.29, 0.57 |
| PPF: 5 (8 wt%) |
12750 |
21.8 |
18.4 |
70.2 |
70.0 |
0.30, 0.57 |
| PPF: 5 (16 wt%) |
11200 |
20.7 |
18.2 |
64.1 |
39.5 |
0.31, 0.58 |
| mCBP: 6 (4 wt%) |
94100 |
21.8 |
18.9 |
76.5 |
61.9 |
0.41, 0.56 |
| mCBP: 6 (8 wt%) |
99100 |
21.2 |
17.9 |
73.8 |
62.3 |
0.42, 0.56 |
| mCBP: 6 (12 wt%) |
104000 |
21.5 |
18.4 |
73.4 |
65.9 |
0.43, 0.55 |
For 6-based OLEDs, EML is formed by doping 6 in mCBP (3,3′-di(9H-carbazol-9-yl)-1,1′-biphenyl) at a concentration of 4 to 12 wt%. As shown in Fig. 12c, the EL spectrum of the 6-based device is unstructured with a peak maximum at 554–560 nm. The max. luminance and max. power efficiency (PE) increase as the doping concentration increases from 4 wt% to 12 wt%, up to 104000 cd m−2 and 69.5 lm W−1, respectively. The max. EQE values (21.2–21.8%) are almost insensitive to the doping concentration (Fig. 12d). The OLED performance of complex 5 is slightly inferior to that of the PtII analogue; the max. EQE of the latter is reported to be over 25%.31 Nevertheless, the max. EQE and max. luminance of the 5-based OLEDs are up to 22.9% and 104000 cd m−2, respectively; these two values are the highest among OLEDs doped with binuclear PdII emitters.6 At high luminance of 1000 cd m−2, the EQE values drop slightly to 17.9–18.9%, corresponding to an efficiency roll-off of 13.3–15.6%.
Conclusion
In this work, binuclear PdII complexes with short intramolecular Pd–Pd distances of 2.79–2.89 Å and 3MMLCT excited state with high Фem (up to 0.7) and large kr (up to 2 × 105 s−1) have been achieved. These binuclear PdII complexes show potential applications as light-emitting dopants for OLEDs and as photocatalysts or photosensitizers for C–C coupling reactions. According to DFT/TDDFT calculations, the energy difference between the 3MMLCT and 3dd excited states of complex 1 is smaller than that of Ref-Pt1. Together with the smaller SOC, complex 1 shows faster non-radiative decay and slower radiative decay processes. The strongly σ-donating (C^C*) ligands of complexes 5 and 6 can significantly destabilize the 3dd excited state. Complex 5 reacts with alkyl bromide through the inner-sphere electron transfer pathway under photo-irradiation, and the Pd-containing product is [(PdII–PdIII–Br)(DPF−)2]2+ (5-Br). Overall, this work provides a comprehensive study of the 3MMLCT excited states of binuclear PdII complexes from basic research to applications.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Chi-Ming Che designed and initiated this research project. Minying Xue synthesized and characterized all complexes in this work, performed the spectroscopic and electrochemical measurements and carried out the photochemical studies. Wai-Pong To provided experimental guidance for this research project. Chi-Ming Che, Qingyun Wan and Minying Xue wrote and revised the manuscript. Yuzhen Zhang grew the single crystals of complex 1 and Ref-Pt1. Gang Cheng performed OLED fabrication and electroluminescence measurements. Zhou Tang carried out the EPR simulations. Lili Du carried out the femtosecond time-resolved absorption and emission spectroscopic measurements. Kam-Hung Low was responsible for X-ray crystal structure determinations. Qingyun Wan was responsible for all the DFT/TDDFT calculations in this work.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge the financial support by the Major Program of Guangdong Basic and Applied Research (2019B030302009), the Science, Technology, and Innovation Commission of Shenzhen Municipality (JCYJ20200109150414471 and JCYJ20180508162429786), the Research Grants Council (17309823) of Hong Kong, and the Hong Kong Quantum AI Lab Limited, AIR @ InnoHK of Hong Kong Government. This work was conducted, in part, using the research computing facilities and advisory services offered by Information Technology Services, The University of Hong Kong.
References
-
(a) M. Yoshida and M. Kato, Coord. Chem. Rev., 2018, 355, 101–115 Search PubMed;
(b) S.-Y. Yang, Y. Chen, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2024, 53, 5366 Search PubMed.
-
(a) M. Han, Y. Tian, Z. Yuan, L. Zhu and B. Ma, Angew. Chem., 2014, 126, 11088 (
Angew. Chem., Int. Ed.
, 2014
, 53
, 10908
) CrossRef;
(b) X.-P. Zhang, J.-F. Mei, J.-C. Lai, C.-H. Li and X.-Z. You, J. Mater. Chem. C, 2015, 3, 2350 RSC;
(c) J. Ni, Y. Zhang, S. Liu and J. Zhang, J. Organomet. Chem., 2024, 1004, 122948 CrossRef CAS;
(d) J. Ni, Q. Zhu, X. Wang, S. Liu and J. Zhang, J. Organomet. Chem., 2024, 1015, 123233 CrossRef CAS.
-
(a) K. T. Ly, R.-W. Chen-Cheng, H.-W. Lin, Y.-J. Shiau, S.-H. Liu, P.-T. Chou, C.-S. Tsao, Y.-C. Huang and Y. Chi, Nat. Photonics, 2017, 11, 63 CrossRef;
(b) K.-H. Kim, J.-L. Liao, S. W. Lee, B. Sim, C.-K. Moon, G.-H. Lee, H. J. Kim, Y. Chi and J.-J. Kim, Adv. Mater., 2016, 28, 2526 CrossRef CAS PubMed;
(c) P. Pinter, J. Soellner and T. Strassner, Eur. J. Inorg. Chem., 2021, 3104 Search PubMed;
(d) H. Leopold, M. Tenne, A. Tronnier, S. Metz, I. Münster, G. Wagenblast and T. Strassner, Angew. Chem., 2016, 128, 16011 (
Angew. Chem., Int. Ed.
, 2016
, 55
, 15779
) Search PubMed;
(e) M. Xue, T.-L. Lam, G. Cheng, W. Liu, K.-H. Low, L. Du, S. Xu, F.-F. Hung, D. L. Phillips and C.-M. Che, Adv. Opt. Mater., 2022, 10, 2200741 CrossRef CAS.
-
(a) V. Sicilia, L. Arnal, S. Fuertes, A. Martín and M. Baya, Inorg. Chem., 2020, 59, 12586 Search PubMed;
(b) V. Sicilia, P. Borja and A. Martín, Inorganics, 2014, 2, 508 CrossRef CAS;
(c) V. Sicilia, M. Baya, P. Borja and A. Martín, Inorg. Chem., 2015, 54, 7316 CrossRef CAS PubMed.
-
(a) J. J. Topczewski and M. S. Sanford, Chem. Sci., 2015, 6, 70 RSC;
(b) D. C. Powers and T. Ritter, Acc. Chem. Res., 2012, 45, 840 Search PubMed;
(c) D. C. Powers, M. A. L. Geibel, J. E. M. N. Klein and T. Ritter, J. Am. Chem. Soc., 2009, 131, 17050 Search PubMed;
(d) D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302 CrossRef CAS PubMed.
-
(a) J. Lin, C. Zou, X. Zhang, Q. Gao, S. Suo, Q. Zhuo, X. Chang, M. Xie and W. Lu, Dalton Trans., 2019, 48, 10417 RSC;
(b) L. Qiao, X. Kong, K. Li, L. Yuan, Y. Shen, Y. Zhang and L. Zhou, Adv. Sci., 2024, 2404621 CrossRef CAS PubMed.
- P. K. Chow, C. Ma, W.-P. To, G. S. M. Tong, S.-L. Lai, S. C. F. Kui, W.-M. Kwok and C.-M. Che, Angew. Chem., Int. Ed., 2013, 52, 11775 CrossRef CAS PubMed.
-
(a) Q. Wan, W.-P. To, C. Yang and C.-M. Che, Angew. Chem., 2018, 130, 3143 (
Angew. Chem., Int. Ed.
, 2018
, 57
, 3089
) Search PubMed;
(b) C. Zou, J. Lin, S. Suo, M. Xie, X. Chang and W. Lu, Chem. Commun., 2018, 54, 5319 RSC;
(c) L. Cao, K. Klimes, Y. Ji, T. Fleetham and J. Li, Nat. Photonics, 2019, 15, 185 Search PubMed;
(d) L. Ameri, L. Cao, X. Tan and J. Li, Adv. Mater., 2023, 35, 2208361 Search PubMed.
- J. Pérez, A. Espinosa, J. M. Galiana, E. Pérez, J. L. Serrano, M. A. G. Aranda and M. Insausti, Dalton Trans., 2009, 9625 RSC.
- B. Ma, J. Li, P. I. Djurovich, M. Yousufuddin, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2005, 127, 28 CrossRef CAS PubMed.
-
(a) P.-K. Chow, G. Cheng, G. S. M. Tong, C. Ma, W.-M. Kwok, W.-H. Ang, C. Y.-S. Chung, C. Yang, F. Wang and C.-M. Che, Chem. Sci., 2016, 7, 6083 Search PubMed;
(b) G. Li, J. Zheng, X. Fang, K. Xu, Y.-F. Yang, J. Wu, L. Cao, J. Li and Y. She, Organometallics, 2021, 40, 472 Search PubMed;
(c) G. Li, H. Guo, X. Fang, Y.-F. Yang, Y. Sun, W. Lou, Q. Zhang and Y. She, Chin. J. Chem., 2022, 40, 223 Search PubMed.
- J. Liu, T.-L. Lam, M.-K. Sit, Q. Wan, C. Yang, G. Cheng and C.-M. Che, J. Mater. Chem. C, 2022, 10, 10271 RSC.
- P. S. Wagenknecht and P. C. Ford, Coord. Chem. Rev., 2011, 225, 591 Search PubMed.
-
(a) M. Chaaban, Y.-C. Chi, M. Worku, C. Zhou, H. Lin, S. Lee, A. Ben-Akacha, X. Lin, C. Huang and B. Ma, Inorg. Chem., 2020, 59, 13109 CrossRef CAS PubMed;
(b) S. F. Wang, L. W. Fu, Y.-C. Wei, S.-H. Liu, J.-A. Lin, G.-H. Lee, P.-T. Chou, J.-Z. Huang, C.-I. Wu, Y. Yuan, C.-S. Lee and Y. Chi, Inorg. Chem., 2019, 58, 13892 Search PubMed.
- H. Amouri, Chem. Rev., 2023, 123, 230 CrossRef CAS PubMed.
- C.-L. Yao, L.-P. He, J. D. Korp and J. L. Bear, Inorg. Chem., 1988, 27, 4389 CrossRef CAS.
- J. F. Berry, E. Bill, E. Bothe, F. A. Cotton, N. S. Dalal, S. A. Ibragimov, N. Kaur, C. Y. Liu, C. A. Murillo, S. Nellutla, J. M. North and D. Villagrán, J. Am. Chem. Soc., 2007, 129, 1393 CrossRef CAS PubMed.
- C.-H. Lam, W. K. Tang and V. W.-W. Yam, Inorg. Chem., 2023, 62, 1942 CrossRef CAS PubMed.
- N. Bayliss, A. Cole and B. Green, Aust. J. Chem., 1948, 1, 472 CrossRef CAS.
- M. Hermet, L. Bakás, S. R. Morcelle and D. L. Bernik, Spectrochim. Acta, Part A, 2019, 223, 117266 CrossRef CAS PubMed.
- S. Yamashita, K. Iwamatsu, Y. Maehashi, M. Taguchi, K. Hata, Y. Muroya and Y. Katsumura, RSC Adv., 2015, 5, 25877 RSC.
- M. Dey, S. S. Dhar and M. Kalita, Synth. Commun., 2013, 43, 1734 Search PubMed.
-
(a) J. P. Mittal and E. Hayon, Nat. Phys. Sci., 1972, 240, 20 Search PubMed;
(b) N. A. McAskill and D. F. Sangster, Aust. J. Chem., 1977, 30, 2107 Search PubMed.
- S. H. Eitel, M. Bauer, D. Schweinfurth, N. Deibel, B. Sarkar, H. Kelm, H.-J. Kruger, W. Frey and R. Peters, J. Am. Chem. Soc., 2012, 134, 4683 CrossRef CAS PubMed.
- J. Harmer, C. Finazzo, R. Piskorski, C. Bauer, B. Jaun, E. C. Duin, M. Goenrich, R. K. Thauer, S. V. Doorslaer and A. Schweiger, J. Am. Chem. Soc., 2005, 127, 17744 Search PubMed.
-
(a) D. C. Smith and H. B. Gray, Coord. Chem. Rev., 1990, 100, 169 Search PubMed;
(b) S. J. Atherton and D. M. Roundhill, Inorg. Chem., 1986, 25, 4072 Search PubMed.
- S. J. Kaldas, A. Cannillo, T. McCallum and L. Barriault, Org. Lett., 2015, 17, 2864 CrossRef CAS PubMed.
- F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1993, 22, 113 Search PubMed.
- W.-P. To, Y. Liu, T.-C. Lau and C.-M. Che, Chem.–Eur. J., 2013, 19, 5654 CrossRef CAS PubMed.
-
(a) M. Numata, T. Yasuda and C. Adachi, Chem. Commun., 2015, 51, 9443 RSC;
(b) I. S. Park, K. Matsuo, N. Aizawa and T. Yasuda, Adv. Funct. Mater., 2018, 28, 1802031 Search PubMed.
-
(a) T.-L. Lam, H. Li, K. Tan, Z. Chen, Y.-K. Tang, J. Yang, G. Cheng, L. Dai and C.-M. Che, Small, 2024, 20, 2307393 CrossRef CAS PubMed;
(b) L. Wang, J. Miao, Y. Zhang, C. Wu, H. Huang, X. Wang and C. Yang, Adv. Mater., 2023, 35, 2303066 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Gang
Cheng
a,
Gang
Cheng


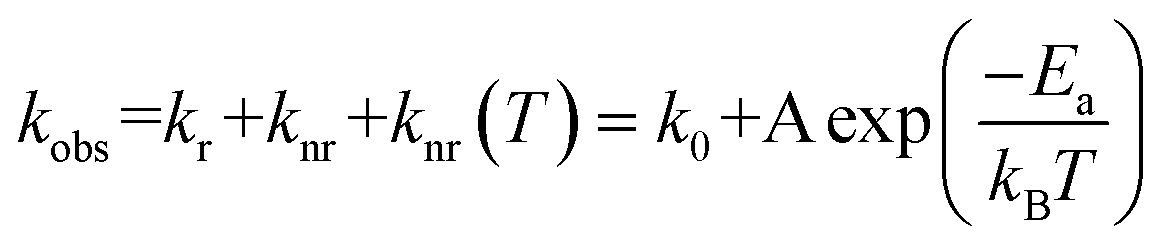







 (318/307 nm),23 we exclude that the 457 nm and 552 nm absorption bands originate from the above species. At a high concentration of benzyl bromide (3 M), the excited state of 5 was completely quenched within 0.2 μs, and absorption bands at 457 and 552 nm were observed within 10 ns after the laser flash (Fig. S23†).
(318/307 nm),23 we exclude that the 457 nm and 552 nm absorption bands originate from the above species. At a high concentration of benzyl bromide (3 M), the excited state of 5 was completely quenched within 0.2 μs, and absorption bands at 457 and 552 nm were observed within 10 ns after the laser flash (Fig. S23†).