
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide hydrogels as slow-release formulations of protein therapeutics: case study of asparaginase-loaded hydrogels†
Hue
Vu‡
a,
Evelien
Peeters‡
bc,
Kenneth
Hofkens
bc,
Katrien
Vandemeulebroecke
cde,
Sara
T'Sas
bc,
Charlotte
Martin
f,
Steven
Ballet
 f,
Richard
Hoogenboom
g,
Steven
Goossens
bc,
Tim
Lammens
cde,
Maaike
Van Trimpont
*bc and
Annemieke
Madder
*a
f,
Richard
Hoogenboom
g,
Steven
Goossens
bc,
Tim
Lammens
cde,
Maaike
Van Trimpont
*bc and
Annemieke
Madder
*a
aOrganic and Biomimetic Chemistry Research Group, Ghent University, 9000 Ghent, Belgium. E-mail: annemieke.madder@ugent.be
bResearch Unit for Translational Research in Oncology, Department Diagnostic Sciences, Ghent University, 9000 Ghent, Belgium. E-mail: steven.goossens@ugent.be
cCancer Research Institute Ghent (CRIG), 9000 Ghent, Belgium
dDepartment of Internal Medicine and Pediatrics, Ghent University, 9000 Ghent, Belgium
eDepartment of Pediatric Hematology-Oncology and Stem Cell Transplantation, Ghent University Hospital, 9000 Ghent, Belgium
fResearch Group of Organic Chemistry, Vrije Universiteit Brussel, B-1050 Brussels, Belgium
gSupramolecular Chemistry Group, Centre of Macromolecular Chemistry (CMaC), Department of Organic and Macromolecular Chemistry, Ghent University, 9000 Ghent, Belgium
First published on 11th June 2025
Abstract
In this study, hexamer peptide-based hydrogels were loaded with different model protein cargos and the release profiles investigated to explore the balance between injectability and loading capacity permitting the release of a therapeutically relevant dose. We demonstrate that the release of protein cargos from our hexamer peptide hydrogels depends on the stability of the hydrogel network, the mobility of the cargo to diffuse out of the network, and the interaction between the hydrogel network and the cargo. For the first time, our peptide hydrogels were used to develop an injectable sustained release formulation of a therapeutic enzyme, namely Erwinase®, an FDA-approved asparaginase for the treatment of acute lymphoblastic leukemia. We show that the current hexamer peptide-based hydrogels allow sufficient protein loading and sustained release of the fully active asparaginase enzyme both in vitro and in vivo. Altogether, this study describes how peptide hydrogels can be exploited to provide injectable slow-release formulations of biologics, including enzyme therapeutics, to enhance their clinical applicability.
1. Introduction
High dosage and repeated administration of drugs are often required in conventional medicine, as many drugs are quickly eliminated from the body either via metabolization or via excretion, which often leads to complicated adverse effects and consequentially poor compliance or treatment outcome.1 A sustained release drug delivery system that provides a prolonged therapeutic effect can minimize the need for frequent drug administrations, which is especially desirable for cases involving painful injections or complex surgical procedures. This becomes particularly relevant in the case of protein therapeutics, as they often exhibit short serum half-lives lasting only minutes to hours,2–4 entailing the need for more parenteral injections with shorter intervals between doses. It is therefore particularly beneficial for protein therapeutics to be formulated in suitable delivery systems that not only allow their sustained release in vivo but also provide protection from enzymatic degradation. Moreover, this opens up possibilities for the development of localized depot formulations.Hydrogels, which consist of a three-dimensional network of hydrophilic (bio)polymers, are able to encapsulate a variety of therapeutic molecules within the interstices of their networks and have been recognized as promising biomaterials for sustained drug release.4 However, many biodegradable synthetic polymer-based hydrogels feature disadvantages such as (i) monomer and degradation product toxicity,5 (ii) uncontrollable polymer swelling in vivo that can cause pain in the host,6 and (iii) non-uniform networks that lead to variable release properties.7 In contrast, self-assembling peptide hydrogels composed of natural amino acids have been reported to be one of the most interesting alternative drug delivery vehicles to tackle those problems.8,9 Additionally, these peptide hydrogels possess interesting properties ideal for drug-delivery applications such as injectability, tunable mechanical as well as release properties, ease of synthesis and high purity based on a defined peptide component.8–10
Since the first short peptide hydrogelator, EAK16 or Ac-AEAEAKAKAEAEAKAK-NH2, was reported in 1993 by Zhang et al.,11 a number of β-sheet forming oligopeptides have been introduced, usually containing amino acids with an alternating distribution of polar and non-polar blocks, providing amphiphilicity to the peptides and driving the self-assembly process.12–14 In 2015, we reported the first, short, hexamer peptide gelator, H-FEFQFK-OH, as a result of conscientious amino acid selection.15 The arrangement of hydrophilic and hydrophobic side chains on opposite sides of the peptide backbone leads to the formation of β-strands, whose alignment results in β-sheet secondary structures. These are stabilized by hydrogen bonding between neighboring amide groups on the backbone and a combination of hydrophobic, aromatic, and coulombic interactions between the side chain groups of different peptide molecules. β-Sheet bilayers are formed upon hydrophobic collapse and their elongation results in fiber formation with subsequent entanglement under increased concentration and/or ionic strength leading to the establishment of hydrogel networks (Fig. 1A).16 Further optimization regarding the pH17 and the stability of the hydrogel formulations led to the introduction of other related sequences, most notably H-FEFQFK-NH2 (1) and H-FQFQFK-NH2 (2), which allow syringe-delivered formulations of different therapeutic small molecules (e.g. morphines, opioid peptides…) that can be released over a period of 1–2 days upon subcutaneous injection in mice.15–18 Recently, we successfully encapsulated a human PD-L1 nanobody, resulting in a sustained release and tumor-uptake, up to 72 hours post subcutaneous injection in a melanoma mouse model.17,19,20 Thus, this collection of short peptide hydrogelators can be considered a suitable and biocompatible platform for the encapsulation of a broad spectrum of therapeutics.
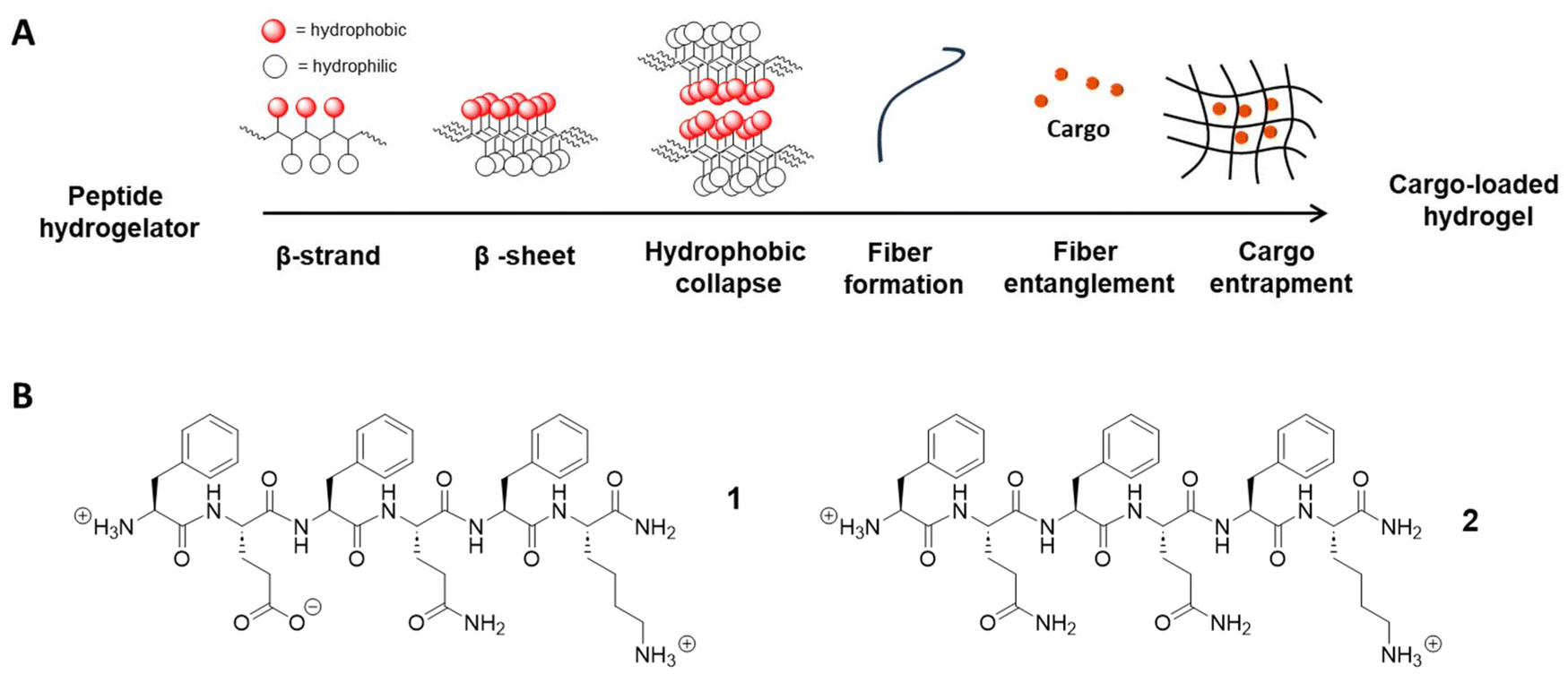 | ||
| Fig. 1 Self-assembling peptide hydrogels. (A) Illustration of the self-assembly mechanism and gelation process that leads to the entrapment of cargos under suitable external stimuli inside the fiber network. (B) Structure of hexamer hydrogelator peptides 1 and 2, all containing alternating hydrophobic and hydrophilic residues, forming hydrogel via β-sheet assembly. | ||
In the present study, we further explored the versatile applications of this hydrogel system for the delivery of proteins by systematically studying the loading capacity of the gels, as well as a multitude of factors affecting release of various encapsulated protein cargos. This knowledge will facilitate the development of novel optimized hydrogel formulations. Although encapsulation and sustained release of bioactive proteins using hydrogel materials have been well explored20,21 to the best of our knowledge, there have not been previous reports on peptide-based hydrogel formulations of therapeutic enzymes. As it is important to ensure the stability and activity of enzymes in order to maintain their therapeutic effectiveness, it is a critical challenge to find optimal hydrogel formulations for their in vivo delivery.22–24 Previous attempts have been mostly focused on the use of polymer-based hydrogels, either natural,25 synthetic,26 or semi-synthetic.27 These systems, however, often result in formulations with low encapsulation efficiency, altered enzyme activity due to unwanted chemical conjugation, and potential toxicity from the polymer precursors or byproducts of the polymer degradation. In this regard, peptide hydrogels, especially those from short sequences, are advantageous as their building blocks are natural amino acids. Also, polymer-based hydrogels, both natural and synthetic, even though are designed to be biocompatible, may still trigger an immune response, leading to inflammation or rejection in the host organism. It is known that host response to biomaterials is strongly affected by factors such as size, shape, stiffness, and chemistry.35 Most biomaterials, including polyethylene glycol (PEG), polyethylene, and crosslinked collagen induce a classical immune response called the foreign body response (FBR), resulting in an unresolved chronic inflammation and fibrous encapsulation of the materials.28 The current hydrogel systems are based on short self-assembling peptide sequences containing only natural amino acids, and fully degrade within only several days, therefore providing a safe platform for delivery of bio-therapeutics.
Here, we explored the possibility of utilizing the current peptide hydrogel system for sustained release of bioactive enzymes and applied the generated knowledge in a case study on a sustained-release formulation of Erwinase, an FDA-approved asparaginase enzyme derived from Erwinia chrysantemi for the treatment of acute lymphoblastic leukemia (ALL). Due to the downregulation and/or loss of asparagine synthetase (ASNS), leukemic cells have a limited capacity for endogenous asparagine synthesis, rendering them dependent on extracellular asparagine for survival.29–31 Enzymatic depletion of circulating asparagine with asparaginases therefore selectively induces cell death in leukemic cells.29–31 Erwinase is currently used in the clinic as second-line therapy for patients with ALL who have developed allergies and/or hypersensitivity towards the first-line product, a PEGylated – and thus long-acting – formulation of E. coli-derived asparaginase.29–31 However, its administration is associated with a plethora of problems. One important limitation is the limited in vivo half-life, approximately 10 hours in mice and 15 hours in patients.29,32,33 As a result, patients will need frequent visits to the clinic to maintain sufficient L-asparaginase activity in their serum to preserve the efficacy of their asparagine deprivation therapy. Furthermore, the glutaminase co-activity of the bacterial-derived asparaginases currently used in the clinic is the cause of undesired toxicities, such as pancreatitis and neurotoxicity.32,34 As a result of its very short half-life and to extend its activity, an excessive bolus injection of Erwinase has to be administered, which is accompanied by stronger toxic side effects.23,26,30 Using our hydrogel platform, we evaluated whether peptide-based hydrogels would allow a slow sustained-release of Erwinase from a high loading gel formulation and allow a longer-lasting plasma concentration of the enzyme, which could lead to improved quality of life of patients with ALL receiving Erwinase treatment.
2. Materials and methods
2.1. Reagents
The hydrogelators H-FEFQFK-NH2 (1) and H-FQFQFK-NH2 (2) (TFA salts) were bought from Royobiotech Co., Ltd (Shanghai, China). Glacial acetic acid was from Sigma-Aldrich whereas acetonitrile (HPLC grade) was purchased from Chem-Lab. Analytical grade AG1-X8 anion exchange resin (100–200 mesh, chloride form) was from Bio-Rad (Belgium). Water used in all in vitro experiments was purified with a Millipore system (18.2 MΩ cm). Phosphate-buffered saline (PBS, 1× and 10×) and UltraPure water used for in vivo experiments purchased from Thermo Fisher Scientific (Invitrogen, USA). Bovine serum albumin (BSA) and avidin were purchased from Sigma-Aldrich (Belgium) whereas neutravidin and HRP-conjugated streptavidin is from Thermo Scientific (Belgium). Erwinase® (crisantaspase) was obtained through the pharmacy of Ghent University Hospital as a lyophilized powder, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 IU per vial. All proteins were received as lyophilized powders and were reconstituted with PBS 2× to the required concentration except for HRP-conjugated streptavidin which was supplied as a 1.25 mg mL−1 solution. Microbial transglutaminase (mTG), supplied as powder formulation from Bindly, was reconstituted in PBS buffer and filtered with 0.22 μm filter.
000 IU per vial. All proteins were received as lyophilized powders and were reconstituted with PBS 2× to the required concentration except for HRP-conjugated streptavidin which was supplied as a 1.25 mg mL−1 solution. Microbial transglutaminase (mTG), supplied as powder formulation from Bindly, was reconstituted in PBS buffer and filtered with 0.22 μm filter.
2.2. Counter ion exchange
To conduct the exchange from trifluoroacetate to acetate salt, a high excess of 1500 mg of the ion-exchange resin was prepared in a 50 mL Steriflip unit. The resin was successively washed three times with 1.6 M acetic acid (25 × 3 mL), followed by three times with 0.16 M acetic acid (25 × 3 mL). 125 mg of the peptide was dissolved in 25 mL of miliQ water and poured on the resin. After 2 hours of rotary stirring, the peptide-resin suspension was filtered, and the liquid phase recovered. The resin was washed twice with 2 × 5 mL of miliQ water. The solutions were pooled for freeze-drying. The peptide (acetate salt) was collected as fluffy white powder.2.3. Hydrogel preparation
First, a stock solution of each hydrogelator peptide was prepared in Milli-Q or UltraPure water to have 4% w/v final peptide concentration. The gel was then prepared by adding an equal volume of PBS 2× (with or without protein cargoes) to an equal volume of peptide stock solution and mixed by pipetting while avoiding bubble formation (2% w/v gel). The samples were left to rest overnight at room temperature (in case of model cargos) or at 4 °C (in case of enzymes and therapeutic cargos).2.4. Rheology measurement
The rheological properties of the peptide hydrogels were measured on an Anton Paar MCR 301 modular rheometer equipped with a Peltier plate and evaporation blocker accessories. The measurements were performed in a parallel–plate mode (diameter: 25 mm) at 25 °C. 500 μL of hydrogel were prepared as described in 2.3 and withdrawn in a 1 mL syringe and left overnight at RT. 350 μL of the prepared hydrogel was then applied to the plate from the syringe (without needle). Gels were left for 2 minutes before measurements. The dynamic strain sweep (from 0.01% to 500% strain, 1 Hz oscillation) was previously conducted to determine the linear viscoelastic regime. For the shear-recovery experiment, a dynamic time sweep (1 Hz and 1% strain) was performed for 60 seconds after loading a gel, followed by the application of 50% strain for 30 seconds to shear-thin the gel. Then, the strain was decreased to 1%, and the dynamic time sweep was performed again for another 60 seconds. Such a shear recovery experiment was performed for three cycles at 25 °C and repeated three times. The Anton Paar Rheometer software was used for analysis.2.5. In vitro release studies
The in vitro release behavior of BSA, avidin, neutravidin, and Erwinase from the hydrogels was evaluated by determining the cumulative release value at each timepoint and the cumulative release curve was drawn as the function of time. Briefly, the protein-loaded gels were prepared first by mixing 50 μL cargo solution in PBS 2× with 50 μL peptide 4% w/v solution in an Eppendorf tube (1.5 mL), as described in 2.3. At the start of the release experiment, 400 μL of a PBS 1× solution (10 mM, pH 7.4) was carefully added on top of each gel (= timepoint 0). No significant swelling of the hydrogels was observed in all cases. At regular timepoints, the supernatant was homogenized and aliquots of 20 μL were collected and replaced by equal amounts of fresh PBS. Subsequently, these samples were analyzed with reversed phase high-performance liquid chromatography (RP-HPLC) (see ESI†), where the area under the curve (AUC) value was determined for each sample. Hence, the cumulative doses released (Mt) could be calculated by making use of a calibration curve. Finally, the cumulative protein release percentage was determined over time, considering the initial encapsulated dose (Mi), using eqn (1).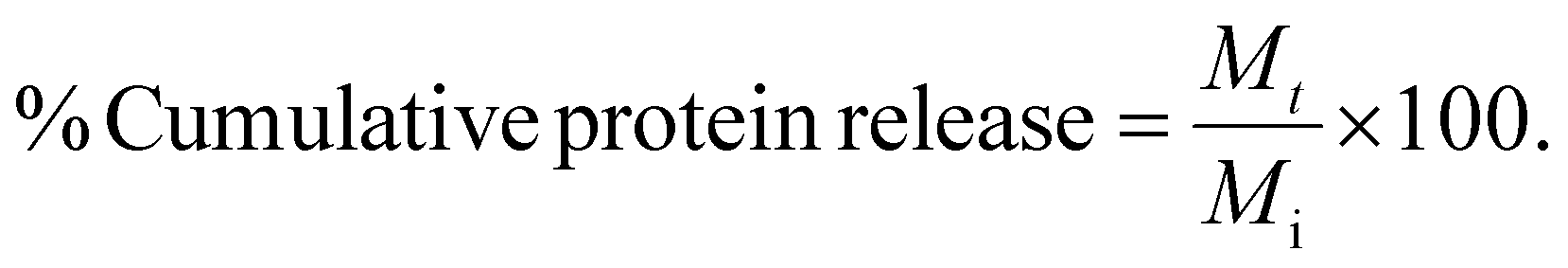 | (1) |
2.6. Horseradish peroxidase (HRP) enzyme activity determination
The HRP-conjugated streptavidin is from Thermo Scientific (Product code: 434323). The specific isoenzyme used in this product is not explicitly stated in the product information. It is likely a mixture of HRP isoenzymes, potentially with a higher proportion of the most abundant isoenzyme, C1A, which has a pI value of 7.2.Hydrogels made up of peptide 2 (2% w/v) loaded with HRP-streptavidin enzyme (5 μg mL−1 final concentration) were prepared following the above procedure (section 2.3). For the determination of enzyme activity in release buffer over time, 500 μL of a PBS 1× solution (10 mM, pH 7.4) was carefully added on top of each gel (= timepoint 0). At regular timepoints (1, 3, 6, 8, 24, 48 hours), the supernatant was homogenized and aliquots of 10 μL were collected and replaced by equal amounts of fresh PBS. The release samples were stored at 4 °C until the time of measurement (see ESI†).
For the qualitative determination of enzyme activity while being encapsulated within the hydrogel matrix, the enzyme-loaded hydrogel of peptide 2 was injected into a vial containing a mixture of 3,3′,5,5′-tetramethylbenzidine (TMB) substrate and hydrogen peroxide (1-Step Ultra TMB ELISA Substrate Solutions, Thermo Scientific) and the color development was recorded. Alternatively, the TMB substrate solution was added directly on top of the priorly prepared enzyme-loaded hydrogel.
2.7. In vivo release assay – pharmacokinetic (PK) study
Prior to the PK study, we tested different needle sizes (21G, 25G, and 27G) for the injection of the hydrogel samples. While all needle sizes allowed a smooth flow of hydrogel which instantly reformed after model injection in vitro (Fig. S1, see ESI†), the use of a 27G needle resulted in less back flow when injecting in mice without anesthesia. Therefore, 27G needles were used for all samples in the PK study. Healthy female wild-type C57BL/6J mice (Charles River; n = 60) were subcutaneously injected in the flank (150 μL per injection, 12 mice per group) with a single bolus injection of Erwinase (5000 IU kg−1) formulated in peptide hydrogels or PBS buffer. Hydrogels without Erwinase were similarly injected as negative controls. At 8 different timepoints (1, 4, 8 hours and 1, 2, 3, 5 and 7 days), peripheral blood (50 μL per mouse) was collected in EDTA-covered blood collection tubes and the samples were centrifuged at 2000 rcf at 4 °C for 10 minutes to obtain plasma which was then stored at −80 °C until needed for activity measurements (see ESI Table S1†). During the experiment, mice were monitored for clinical signs of toxicity and their body weight was carefully monitored. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Ghent University and approved by the Animal Ethics Committee of the Faculty of Medicine and Health Sciences of Ghent University.2.8. Asparaginase enzyme activity determination
The release samples from the in vitro release assays and the plasma samples from the in vivo PK study were subjected to the enzymatic assay to determine asparaginase activity. For the in vitro release samples, aliquots of 20 μL in the release buffer were mixed with 80 μL of Tris-BSA 5% and stored at −80 °C until the time of measurement. Plasma asparaginase activity was quantified by incubating the samples with an excess amount of L-aspartic acid β-hydroxamate (AHA; Sigma-Aldrich A6508) at 37 °C. L-Asparaginase hydrolyses AHA to L-aspartic acid and hydroxylamine, which was detected at 690 nm with a SpectraMax M3 (molecular devices) spectrophotometer, after condensation with 8-hydroxyquinoline (Merck 820261) and oxidation to indo-oxine. Detailed procedures can be found in the ESI.†2.9. Transglutaminase enzyme activity determination
The release samples containing microbial transglutaminase (mTG) from the in vitro release assays (10 μL per sample) were quantified by incubating the samples with the reactivity mixture containing N-benzyloxycarbonyl-L-glutaminylglycine (Z-Gln-Gly) and hydroxylamine (140 μL) (Zedira, Z009) at 37 °C in a 96 well plate. After 10 minutes of incubation, a stop solution containing iron(III) was added to each well (150 μL per well). In the presence of mTG, hydroxylamine is incorporated into Z-Gln-Gly to form Z-glutamyl(γ-hydroxamate)-glycine which develops a colored complex with iron(III). The absorbance signal was detected at 525 nm with DropSense 96 (Trinean) spectrophotometer. Detailed procedures can be found in the ESI.†2.10. Statistical analysis
In vitro experiments were carried out in duplicate or triplicate and were presented with standard deviation (SD). In vivo experiments were done in sample size of 6–12 mice per group and the data were presented with standard error mean (SEM). The data were evaluated using Prism 8.0 (GraphPad software). Experimental data of free versus encapsulated enzyme was analyzed by two-way ANOVA and followed by Dunnet's multiple comparison test. An unpaired t-test was employed to find the significance of serum asparaginase levels between treatments or between specific time points. In all cases, p values < 0.05 were considered significant.3. Results and discussion
We previously demonstrated that peptides H-FEFQFK-NH2 (1) and H-FQFQFK-NH2 (2) serve as a safe matrix to encapsulate and release a variety of chemical cargos including morphine, fluorescein sodium, ciprofloxacin, and opioid peptides in a controlled manner.15–18 In previous works, these hydrogelators have been demonstrated to be biodegradable and exhibit good biocompatibility, since no significant cytotoxicity (as 2% w/v gel, 100 μM) was observed.18 Moreover, they are prepared under mild physiological conditions, thereby generating a highly aqueous self-supporting hydrogel network with convenient thixotropic properties, making them a suitable formulation for injection-based delivery of therapeutics, particularly beneficial in the case of protein biologics as has been demonstrated in the case of nanobodies.16,19 The current study investigates in detail the loading capacity of different members of this peptide hydrogel platform and factors that govern the release of a series of encapsulated proteins in order to better characterize and understand the system in detail.3.1. Protein loading capacity and hydrogel characterization
BSA was chosen as a model protein cargo to study the loading capacity and injectability of the final hydrogel formulations. Although it has been demonstrated in several studies that electrostatic interactions between encapsulated cargos and the hydrogel networks can significantly affect the release profiles of the cargos,35,36 the effect of electrostatic interactions on the initial loading of the cargos and characteristics of the loaded hydrogels have not been systematically explored. The relatively low pI (4.7) of BSA suggests that at pH higher than 4.7, BSA bears an overall negative charge,37 leading to potential strong electrostatic interactions between the protein molecules and the positively charged hydrogel fibers of 1 and 2.Cargos can be loaded to hydrogel matrices via different methods, most commonly through either post-loading or in situ loading.38 The post-loading technique refers to the immersion of the preliminary formed hydrogel into a drug solution for a certain time period to allow the cargos to diffuse into the hydrogel matrix. The loading efficiency is highly dependent on the size of the protein cargo and porosity of the peptide hydrogels. Besides, as the matrix can already undergo partial disentanglement during the incubation period, particularly in the case of short peptide hydrogelators, we did not apply this method to the currently investigated peptide hydrogels. Instead, the in situ loading method, often used for hydrophilic drugs, was employed. In the previous works, the aqueous solutions containing the drug of interest were added to the hydrogelator powder, followed by repeated vortex, sonication, and centrifugation steps for complete solubilization. In this work, the proteins were loaded into the hydrogels via mixing a fiber-rich solution (a very viscous 4%w/v peptide solution) of the peptide hydrogelators in water with the protein solution in buffer. This, due to the salting-out effect, results in spontaneous formation of physical crosslinks between the peptide molecules and entanglement of the fibers, leading to the instant formation of a self-supporting hydrogel and the entrapment of the cargos within the fiber network.39 In this way, as solubilization of the peptide is conducted prior to the mixing with cargos, the vigorous vortexing and ultrasonication step that might affect the stability and activity of certain sensitive proteins,40 can be avoided.
A range of BSA-loaded hydrogels formulated with peptide hydrogelators 1 and 2 (2%w/v) were prepared to obtain 0, 2.5, 25 and 50 mg mL−1 final concentrations of the BSA protein. Two different buffers, Tris and PBS, were tested as they are the most commonly used in protein reconstitution and physiological hydrogel formulations. The physical appearance of the resulting hydrogels was monitored, with changes in the opacity upon increasing concentration of the protein (Fig. 2A). While there was no significant difference in the appearance of the hydrogels formulated in Tris and PBS, the BSA concentration and nature of the peptide strongly affected the characteristics of the resulting hydrogels. In the case of hydrogels from peptide 1 (net charge +1), aggregation (white cloudy appearance) started to appear non-homogenously in the hydrogels when BSA was loaded at 25 mg mL−1, while in the case of hydrogels from peptide 2 (net charge +2), aggregation was already observed at loading concentrations of 2.5 mg mL−1 BSA. In addition, we observed a link between cargo loading, aggregation capacity and loss of gelation. More specifically, aggregation correlated with loss of gelation as part of the gel started to flow at 25 mg mL−1 BSA, and a complete loss of gel behavior was observed at 50 mg mL−1 concentration (Fig. S2, ESI†). Shear-recovery rheological measurements of BSA-loaded hydrogels of 1 confirmed this, as at 25 mg mL−1 and 50 mg mL−1 loading, the difference between the storage moduli (G′) and loss moduli (G′′) reduced significantly, suggesting the loss of gel-like behavior (Fig. 2B). In contrast, at 2.5 mg mL−1 BSA loading, there was no apparent change in visual appearance and mechanical properties (strength and shear-thinning behavior) of the gel as compared to the unloaded hydrogel of 1. Overall, in contrast to what was reported earlier in studies with small molecule cargos,36 above a certain concentration, the presence of BSA cargo strongly affects the emergent properties of the hydrogels. It is possible that certain interactions, most likely electrostatic attraction, between protein molecules and peptide fibers, resulted in the localization of the cargo on the surface of pre-assembled peptide fibers, thereby increasing the fiber's hydrophobicity and leading to fiber aggregation together with the loss of gel-like state. The higher net charge of peptide 2 presumably results in a stronger attractive force between BSA molecules and the peptide fibers, resulting in aggregation at lower cargo loading. Similar observations have been reported with the positively charged MAX8 peptide hydrogel where negatively charged proteins were shown to be absorbed to hydrogel, limiting their diffusion out of the gel.35 It was also noted that cargos that bear overall positive charge would preferably stay in the aqueous interface between the fibers of these materials and easily diffuse out of the network.35 We therefore speculate that positively charged cargos would likely result in less aggregation, due to less surface absorption, and thus higher loading upon formulation with the current peptide systems.
 | ||
| Fig. 2 The BSA loading capacity of peptide hydrogels. (A) Visual observation through vial inversion test of hydrogels of 1 and 2 loaded with BSA at varying concentrations from 0–50 mg mL−1. Two different physiological buffer systems, PBS and Tris, were tested; (B) rheological measurements of the hydrogels of 1 loaded with 0 (black), 2.5 (green), 25 (red), and 50 (purple) mg mL−1 BSA. Storage moduli (G′, darker colors) and loss moduli (G′′, lighter colors) were recorded in a shear-recovery experiment at constant frequency of 1 Hz. The shear step was performed at 50% strain for 30 seconds, followed by 60 seconds at 1% strain, and the process was repeated 3 times. In the images corresponding to materials at each loading concentration, white arrows indicate hydrogel region, red arrows indicate the fiber aggregation, and blue arrows indicate the flowing aqueous region. | ||
3.2. In vitro release of model protein cargos
It has been reported before that the release of cargos, including small molecules36 and model proteins,35 is highly dependent on the electrostatic interactions between the encapsulated cargos and the peptide network. Hydrogels with positively charged fibers can strongly retain negatively charged cargos, slowing down their release and potentially resulting in the fact that cargos are not fully released from the network. On the other hand, in case of positively charged cargos, the same hydrogels can induce an electrostatic repulsion force, accelerating the release of the cargo. Previous release studies from the current peptide platform have yielded similar observations. Particularly, the significant difference in the rate of release between the H-Dmt-DArg-Phe-Phe-NH2 opioid tetrapeptide,41 and the H-Ala-Ala-Gly-Ser-Ala-Trp-Tyr-Gly-Thr-Leu-Tyr-Glu-Tyr-Asp-β3hTyr-NH2 peptide41 from the hydrogels of peptide 2 was noted. The tetrapeptide bears an overall positive charge of +1 and a calculated pI value of 11.7 while the pentadecapeptide has a net charge of −1 and the calculated pI value of 4.07 (Table 1), thereby experiencing different electrostatic forces from the positively charged fibers of peptide 2. On the other hand, the absence of strong electrostatic retention forces, as a result of a near-neutrality pI value, together with the small size of the PD-L1 sdAb K2 nanobody19 afforded a quick release, only slightly slower than that of the positively charged opioid peptide (Table 1).| Cargos | MW (kDa) | Theoretical pIb | Net charge at pH 7 | Cumulative release from hydrogel of 1 | Cumulative release from hydrogel of 2 |
|---|---|---|---|---|---|
| a Current work. b pI and MW values were previously reported in literature or calculated using a computing pI/MW tool at ExPASy Server. | |||||
| H-Dmt-DArg-Phe-Phe-NH241 |
0.66 | 11.7 | Positive | 90% | 90% |
| H-Ala-Ala-Gly-Ser-Ala-Trp-Tyr-Gly-Thr-Leu-Tyr-Glu-Tyr-Asp-β3hTyr-NH2 peptide41 | 1.74 | 4.1 | Negative | <5% | Not studied |
| PD-L1 sdAb K219 | 14 | 6.03 | ∼Neutral | 80% | Not studied |
| BSAa | 66 | 4.8 | Negative | <5% | 78% |
| Avidina | 68 | 8.9 | Positive | 40% | 80% |
| Neutravidina | 60 | 6.3 | ∼Neutral | 10% | 50% |
To further investigate this for the protein-loaded hydrogels of 1 and 2 (at 2%w/v) and to study how this translates into the formulation development, we selected BSA, avidin, and neutravidin as model protein cargos. These proteins have similar molecular size (approximately 65 kDa), yet different isoelectric points (pI BSA ∼ 4.5, pI neutravidin ∼ 6.3 and pI avidin ∼ 10.5) (Table 1). We loaded 100 μg of each protein into 2 mg of hydrogelator peptide (to give a 2%w/v hydrogel at 1 mg mL−1 final protein concentration) via the in situ loading method (vide supra). All formulations resulted in strong and homogeneous gels as qualitatively assessed by the tube inversion test (Fig. S3, see ESI†). A slight opacity was observed in the case of BSA-loading of hydrogel 2, however, without apparent fiber aggregation or loss of gel-like behavior (Fig. S3, see ESI†). A PBS solution (pH 7.4), serving as a physiologically relevant release medium, was gently added on top of the gels. No apparent swelling of the hydrogels was observed. This can be attributed to the reduced solubility in PBS and the dense network of fibers that are highly entangled as the result of the salting-out effect.42–44 To assess the released protein amounts, samples were collected from the supernatant after 1, 3, 6, 8, 24, and 48 hours and analyzed by analytical RP-HPLC. As illustrated in Fig. 3, the results showed that BSA (overall negative charge) was completely retained with no detectable release from the hydrogel of peptide 2, whereas a cumulative release of approximately 10% of neutravidin (overall neutral charge) and 40% of the avidin (overall positive charge) were observed from the hydrogel formulations with peptide 2. This clearly showed that electrostatic interactions occur between the protein molecules and the peptide fibers that significantly affect their release from the hydrogels. It should be noted that the size of the protein cargos affects diffusivity of the molecules, therefore also partially influencing the release rates, which is most clearly observed with electrically neutral cargos (i.e., neutravidin and PD-L1 sdAb K2 nanobody).45 This can be seen in the difference between the release rates of neutravidin and the nanobody (Table 1). When looking at the release profiles of the proteins from the hydrogels of peptide 1, the release of all three proteins was observed with much higher rates as compared to the formulations with peptide 2. Here, the trends were less clear possibly due to (i) the lower net charge of peptide 1, leading to less significant electrostatic interactions and (ii) the erosion of the hydrogel network of peptide 1 as indicated by the loss of hydrogel volume over time, which was not observed in the case of hydrogel 2 (Fig. S3†). This suggests that the observed release profiles reflect the combined effect of the electrostatic interactions as well as the gradual disintegration of the hydrogel surface. This is often observed in the case of supramolecular hydrogels and particularly in the case of short peptide hydrogelators.8
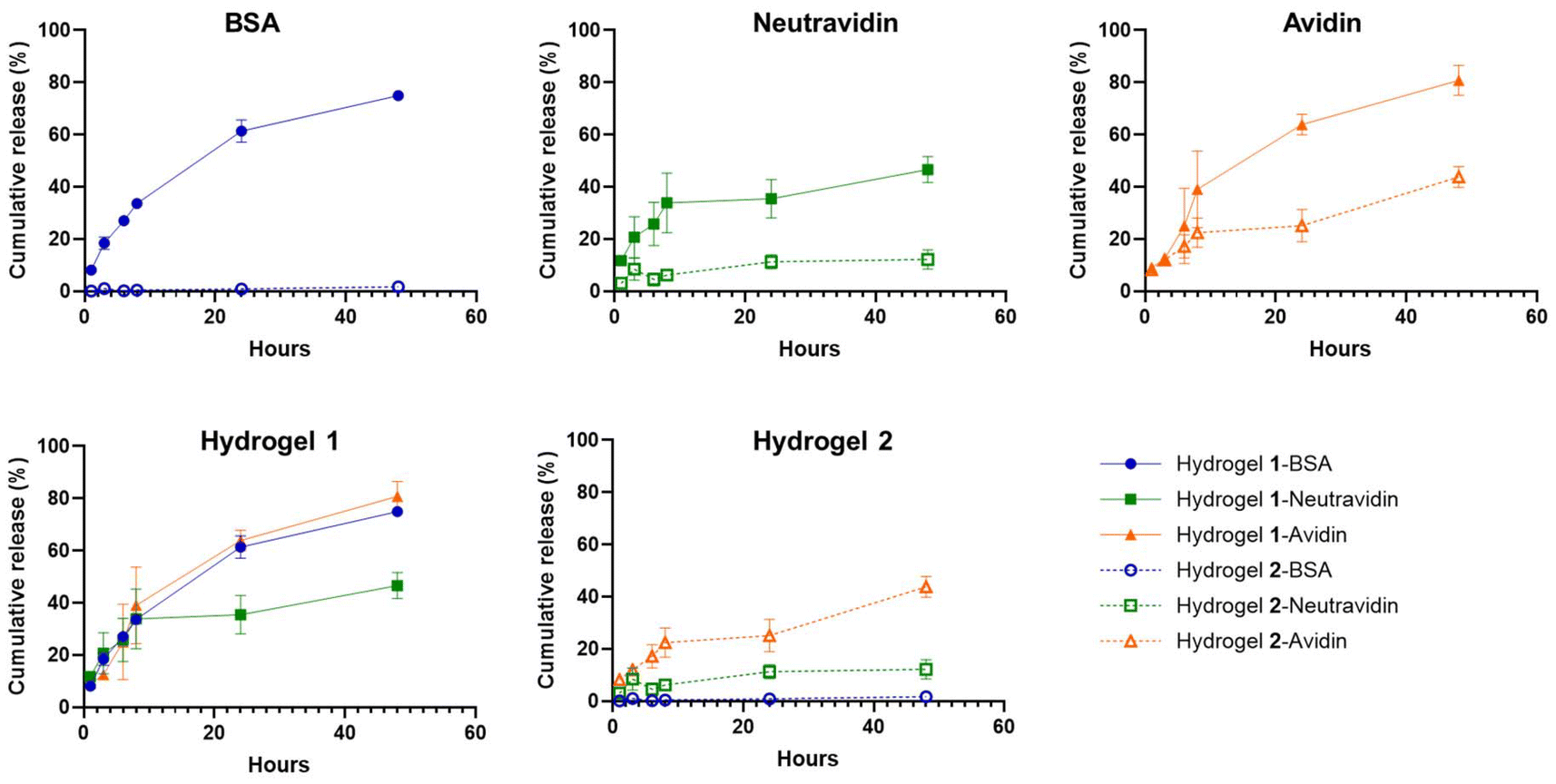 | ||
| Fig. 3 In vitro cumulative release of different model proteins from peptide hydrogels. Solid lines with filled symbols represent the release of proteins from hydrogel of peptide 1, whereas dashed lines with empty symbols correspond to the release of proteins from hydrogel of peptide 2. Dot, square, and triangle symbols correspond to BSA, neutravidin, and avidin, respectively. All hydrogels were formulated at 2% w/v peptide concentration and 1 mg mL−1 (0.1% w/v) loading of protein cargo. Mean values ± SD are represented (n = 2). | ||
Overall, the release window varied with the nature of the loaded proteins and the stability of the hydrogel formulations. Peptide 2 afforded the most stable hydrogels, most likely due to the decreased hydrophilicity upon introduction of glutamine (Q) residues (Fig. S3†). These hydrogels are however not compatible with negatively charged proteins at higher loading (>2.5 mg mL−1). Meanwhile, protein-loaded hydrogels of 1 exhibit gradual erosion in the release buffer, which is likely more profound in the in vivo environment. In such cases, an increase in peptide concentration and/or ionic strength of the buffers,46 or lengthening the peptide sequence41 might help to increase the stability of the hydrogel, making it less susceptible to external stimuli, and thereby mitigating this effect.
3.3. Activity of the released model enzyme cargos
In order to investigate whether a protein encapsulated in such peptide hydrogels can retain its original functionality, we hypothesized that preservation of enzyme activity would provide a good indication of protein stability. A streptavidin-horseradish peroxidase (HRP) protein construct (5 μg mL−1 final concentration) was formulated in a hydrogel made up of peptide 2 (2%w/v, Fig. 4A). The good in vitro mechanical stability of this hydrogel as shown in the previous experiment allows investigating the activity of the enzyme not only upon being released in the buffer, but also while being encapsulated within or bound to the hydrogel matrix. Enzyme activity was verified by direct injection of the HRP-loaded hydrogel to the vial containing the TMB-H2O2 solution. The hydrogel almost instantly colored yellowish – green and later turned into brown as the oxidized product continued to be generated (Fig. 4B and Fig. S4, see ESI†). This observation clearly suggests that the encapsulated enzyme is functionally active while still being entrapped within the hydrogel matrix and/or being bound to the surface of peptide fibers. Furthermore, a blue color was released from the injected hydrogel fragments and quickly intensified in the solution as a qualitative indication of the release of the active enzyme (Fig. 4B). Similar observations were made as the TMB and hydrogen peroxide solutions were added directly on top of the HRP-loaded hydrogel (Fig. S4, see ESI†). To assess the activity of the released enzyme in the buffer, an in vitro release experiment was conducted. For this purpose, aliquots of the release buffers were collected, and concentrations of the active enzyme were determined via end-point reaction with the TMB-H2O2 solution. A gradual increase in the active enzyme concentration, as represented by increasing absorbance at 450 nm, was observed over time (Fig. 4C). | ||
| Fig. 4 Activity of model enzymes upon peptide hydrogel formulation and release. (A) HRP-conjugated streptavidin (5 μg mL−1) loaded into hydrogel composed of H-FQFQFK-NH2 (2) (2%w/v), visual observation via tube inversion test, (B) injected fragments of HRP-conjugated streptavidin loaded hydrogel of 2 are stained green (see red arrows) after addition of the reaction mixture containing TMB substrate and hydrogen peroxide. A blue color can be seen in proximity of the hydrogel fragments and quickly intensified in the solution. Time after adding reaction mixture was indicated. (C) Colorimetric detection of active HRP-conjugated streptavidin in the release buffer from the in vitro release experiment at the indicated time points. Mean values ± SD are represented (n = 3), (D) mTG (8 IU mL−1) loaded into hydrogel 2 (2%w/v), visual observation via tube inversion test, (E) mTG loaded hydrogel of 2 in the solution containing Z-Gln-Gly and hydroxylamine and incubated for 10 min at 37 °C, (F) the enzyme loaded hydrogel fragment stains brown (see red arrow) after addition of Fe(III) solution. Time after adding stop solution was indicated. (G) Colorimetric detection of active mTG in the release buffer from the in vitro release experiment at the indicated time points. Mean values ± SD are represented (n = 2). | ||
To further explore the versatility of the current hydrogel system in enzyme encapsulation and slow release, microbial transglutaminase (mTG), an enzyme commonly used for protein crosslinking, was also formulated together with peptide 2 to afford a clear transparent self-supporting hydrogel (Fig. 4D). Using a similar approach, the hydrogel was injected into the vial containing a solution of the reacting substrate Z-Gln-Gly and hydroxylamine (Fig. 4E). The presence of mTG allows formation of Z-glutamyl(γ-hydroxamate)-glycine which turns into a colored complex with iron(III). As a result, the colorless hydrogel gradually turned brown upon addition of the ion(III) solution (Fig. 4F), indicating the preserved catalytic activity of the encapsulated enzyme. Again, quantitative measurement of enzyme activity from the in vitro release experiment suggested slow release of intact active enzyme (Fig. 4G).
Altogether, although these results do not necessarily correlate to a full preservation of enzyme activity upon hydrogel formulation and release, they provide important insights into the suitability of the currently investigated hydrogel system to accommodate active enzymes, providing a platform for functional protein cargo release while safeguarding in situ activity of the entrapped proteins.
3.4. Peptide hydrogel formulation of Erwinase and the in vitro release profile
After successfully demonstrating the compatibility of the applied hydrogel system for encapsulation, functional preservation and sustained release of model protein (including enzyme) cargos, we applied this knowledge to design an innovative sustained-release formulation of Erwinase. We evaluated whether encapsulation of Erwinase into a hydrogel matrix could allow an extension of the enzyme activity as well as buffer the initial activity peak after administration, thereby potentially lowering the adverse effects. Erwinase, with a pI of 8.6, has a positive charge at physiological pH. We selected both peptides 1 and 2 for hydrogel formulations with Erwinase considering the differences in their stability, electrostatic behavior, and protein release mechanism. Both hydrogel formulations (at 2% w/v) resulted in transparent, self-supporting hydrogels with good shear-thinning property at relatively high loading of Erwinase (1.86 mg mL−1) (Fig. S5-A†).To allow assessment of functionality of the Erwinase-loaded gels, we examined the asparaginase activity in the released samples at multiple time points alongside the in vitro release kinetics of Erwinase from the gels. Enzyme activity was determined spectrophotometrically while the enzyme concentration in the release samples was analyzed by RP-HPLC for cumulative release measurements. As can be seen in Fig. 5A, both hydrogel formulations allow gradual increase of the enzyme activity without significant burst effect. Non-significant differences in the enzyme activity were observed as compared to the non-encapsulated enzyme control after 24 hours in case of the formulation with hydrogel 1 and after 72 hours in case of hydrogel 2, indicating that the enzyme remained functionally intact upon release. The cumulative release (Fig. S5-B†) correlated nicely with the asparaginase activity present in the release buffer.
 | ||
| Fig. 5 Sustained-release hydrogel formulations of Erwinase. (A) Asparaginase activity of the enzyme in release samples from enzyme-loaded hydrogel 1 (blue line, filled circle) and 2 (green line, filled square) as compared to the control sample of Erwinase in PBS (black line, filled triangle). Non-significant differences in the enzyme activity were observed in both cases of loaded hydrogels as compared to the control after 72 hours post release (two-way ANOVA and Dunnet's multiple comparison test; *p < 0.05; ****p < 0.0001; ns, not significant). (B) In vitro cumulative release of peptide hydrogelators from hydrogel 1 loaded with enzyme (blue line, filled circle), without enzyme (blue dotted-line, filled square), hydrogel 2 loaded with the enzyme (green line, empty circle) and without enzyme (green dotted-line, empty square) (all formulated at 2%w/v peptide). In both data sets, mean values ± SD are represented (n = 2). | ||
Release of peptide hydrogelators into the buffer allows a quantitative assessment of hydrogel integrity (Fig. 5B). In the case of hydrogel 1, a cumulative amount of 30% of the total peptide content was detected in the buffer after 1 hour, and only approximately 40% of the gel volume remained after the first 24 hours, after which it stabilized. When it came to gel 2 there was no apparent loss of hydrogel mass over the examined period of 3 days. This suggest that electrostatic repulsion between positively charged peptide fibers and enzyme molecules is the main contributing factor to the release of the enzyme from hydrogel 2 while it is the synergistic effect of electrostatic interaction and matrix surface degradation that governs the release of the enzyme from hydrogel 1. Hence, the release profile of Erwinase can be controlled by selection of the peptide hydrogel. Of note, when comparing the erosion of hydrogel with and without Erwinase in both cases of peptide 1 and 2, the enzyme encapsulation did not affect stability of the formulations.
3.5. Subcutaneous injection of Erwinase-loaded hydrogel and in vivo release study
Next, we evaluated the clinical potential of Erwinase-loaded hydrogels by performing a detailed PK study in healthy C57BL/6 mice. Results (Fig. 6, and Table S1 in ESI†) show that the average plasma asparaginase activity of the mice injected with the non-encapsulated Erwinase control quickly dropped from 502 IU L−1 at 1 hour post administration to below the therapeutic threshold of 100 IU L−1 after 24 hours (69 IU L−1) and became undetectable after 48 hours. Meanwhile, although the Erwinase-loaded hydrogel of 1 initially showed comparable enzyme activity level (785 IU L−1) at 1 hour post injection, a significant increase in activity (2601 IU L−1; p = 0.0087, unpaired t-test) was observed after 4 hours. Clinically relevant asparaginase activity was still detected at 24 hours (159 IU L−1). When it comes to the enzyme-loaded hydrogel of 2, a significantly higher enzyme activity (1249 IU L−1; p = 0.00411, unpaired t-test) as compared to the non-encapsulated control was observed at 1 hour post injection, which slightly dropped at 4 hours but still maintained slightly above the therapeutic threshold after 24 hours post injection (131 IU L−1). For both the enzyme-encapsulated hydrogels of 1 and 2, the asparaginase activity dropped below detectable level after 72 hours. The PK study nicely shows that (i) the hydrogel formulations clearly performed better than the non-encapsulated Erwinase control, (ii) a sustained-release of Erwinase was observed from both enzyme-loaded hydrogel 1 and 2, and (iii) gel 1 has a superior PK profile compared to gel 2 (Fig. 6B). Of note, while residues of both loaded and unloaded hydrogel 2 were still visibly present and subsequently examined after 7 days post injection (Fig. S6, see ESI†), no residue of hydrogel 1 was observed, which is desirable in case of clinical application. Unexpectedly, but importantly, the better biostability of hydrogel 2 did not lead to a more sustained release of Erwinase in vivo as observed in the in vitro experiment, indicating that in vivo behavior cannot be fully predicted based on in vitro release studies. | ||
| Fig. 6 In vivo release study of Erwinase-loaded hydrogels. (A) Visual set-up of in vivo pharmacokinetics experiment, (B) asparaginase activity measurements (IU L−1) in blood plasma samples of mice obtained at 1, 4, 8, 24, 48 and 72 hours after injection of 5000 IU kg−1 Erwinasse, either in PBS or encapsulated in hydrogel 1 or 2. Hydrogels without Erwinase were used as negative controls. Significant differences in enzyme activity were observed between both Erwinase-loaded hydrogels as compared to the positive control Erwinase in PBS (unpaired t-test; *p < 0.05, **p < 0.01). Data is represented as mean values ± SEM (n = 6), (C) bodyweight measurement. No significant bodyweight changes were observed between treatment conditions (two-way ANOVA test; ns, not significant). Data is represented as mean values ± SEM (n = 12). | ||
Toxicity of the Erwinase-loaded hydrogels was evaluated by monitoring the bodyweight loss and visual signs of toxicity (Fig. 6C). As expected, for hydrogel controls and the enzyme-loaded hydrogel of 1 no obvious toxicity or bodyweight loss could be observed, while mice injected with Erwinase, both non-encapsulated and encapsulated in the hydrogel of 2, showed an initial non-significant drop in body weight 48 hours post drug administration,32,34 which then normalized after 96 hours, implying no notable or long-lasting toxicity. Additionally, no substantial elevation of plasma IL-6 cytokine was observed after the injection of hydrogels with or without the encapsulated enzyme (Fig. S7, see ESI†), suggesting the absence of systemic inflammation. Overall, encapsulation of Erwinase in peptide hydrogels prolonged the activity of Erwinase without any remarkable toxicity.
It should be noted that, although the hydrogels maintain their ability for sustained release in an in vivo setting, no extension of activity could be observed compared to intramuscularly injected Erwinase (Fig. S8-A, see ESI†). Encapsulation of Erwinase into the gel did buffer the initial bolus activity, and as such, it is anticipated that the excessive peak of glutaminase activity is lowered. To further explore whether this has a positive impact on toxicity, more in vivo experiments will be necessary with repeated dosing of non-encapsulated versus encapsulated Erwinase.
From the above, it can be concluded that the current Erwinase peptide hydrogel systems expands the options for injectable slow-release delivery of Erwinase. Altogether, the discussed hydrogel-based system provides a simple, cost-effective, biocompatible and biodegradable formulation for sustained release of Erwinase via subcutaneous, intramuscular, or intratumoral injection. To further optimize the Erwinase hydrogel system, following factors can be explored in future endeavors: (i) after injection, local depots are formed at the site of administration that slowly release enzyme via different mechanisms. However, the nature of the local extracellular matrix (ECM) can strongly affect the rate and amount that reaches the vascular compartment. The bio-therapeutic released from the hydrogel depot injected subcutaneously likely undergoes different types of events (such as interactions with the ECM component, uptake by the cells present at the depot and surrounding tissue, interaction with enzymes that could affect the activity of the therapeutics…) before reaching and being absorbed into the blood stream. During all these events, a significant amount of therapeutic can be lost to the surrounding tissue or undergo silent inactivation.47 The injection of hydrogel formulations of biotherapeutics could instead be much more effective for the treatment of solid tumors where local distribution of the release cargos is desired. L-Asparaginase has shown activity in solid tumor models, but clinical applicability is limited due to severe toxicity of long-term treatment. The asparaginase-loaded hydrogel formulations might therefore be useful for other applications beyond subcutaneous injections. (ii) The simultaneous administration of hyaluronidase could potentially help to regulate the surrounding microenvironment to allow better transit of the released therapeutics across the hypodermis space and reach to the blood vessels more efficiently.48 (iii) Finally, Erwinase is positively charged which results in a faster release from the current peptide hydrogels leading to the currently observed two-day therapeutic window. In order to slow down the release of Erwinase from the matrix, efforts can be made to optimize the peptide hydrogelators to allow a better retention of the encapsulated protein (via electrostatic attraction and/or better in vivo stability) while still affording sufficient loading of the cargos.49
4. Conclusions
In conclusion, our hydrogels, formed from amphipathic hexamer peptides H-FEFQFK-NH2 (1) and H-FQFQFK-NH2 (2), offer a suitable platform to develop sustained-release formulations for a variety of protein cargos. The ionic nature of the hydrogelating peptides and encapsulated cargos have a strong influence on the final hydrogel formulation and the release profiles of the protein cargos. By harnessing the electrostatic interaction between the peptide fibers and the cargoes, we can select suitable formulation parameters of the hydrogels for certain cargos of interest which allow sufficiently high loading of proteins without losing thixotropic properties for injectability and afford better control over their release profiles. Importantly, we have shown that enzymes are able to retain their functionality upon encapsulation into our peptide hydrogel system and/or after being released from the hydrogel matrices. This is the first time that a peptide hydrogel system has been utilized to create an injectable formulation of L-asparaginase for the purpose of sustained release, providing alternative options for delivery of the enzyme. Although the circulation time of L-asparaginase is only comparable to the current standard treatment, the initial bolus injection peak was significantly reduced, potentially lowering the side-effect toxicity of treatments with high-doses and/or frequent injections of the enzyme. Future studies are needed to improve the current hour-to-day release window of the L-asparaginase loaded hydrogel formulations, via tuning the erosion of the hydrogel network and/or the interaction between the hydrogel network and the encapsulated cargos. Given the advantages of this system to accommodate high loading of proteins and sustained enzymatic activity, combined with the simple “mix-and-go” procedure yet allowing thixotropic formulation, its application can be extended to create new enzyme therapeutic formulations for different enzyme replacement therapies and cancer treatments.50Author contributions
H. V. – Investigation (sample preparation, in vitro release study, in vivo study), methodology, writing – original draft, writing – review & editing; E. P. – Investigation (sample preparation, in vivo PK study), writing – original draft, writing – review & editing; K. H. – Investigation (in vivo PK study); K. V. – Investigation (in vitro release study); S. T. – Investigation (in vivo PK study); C. M. – funding, writing – review & editing; S. B. – funding, writing – review & editing; R. H. – funding, writing – review & editing; S. G. – conceptualization, methodology, supervision, funding, writing – review & editing; T. L. – conceptualization, methodology, supervision, funding, writing – review & editing; M. V. T. – conceptualization, methodology, investigation (sample preparation, in vivo PK study), funding, writing – review & editing; A. M. – conceptualization, methodology, supervision, resources, funding, writing – review & editing; All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing interests.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work was supported in part by vzw Kinderkankerfonds (grant to T. L.; grant to M. V. T), Kom Op Tegen Kanker (grant to S. G.), Ghent University (BOF Postdoctoral Fellowship to M. V. T.), Cancer Research Institute Ghent (young investigator proof-of-concept grant to M. V. T.) and Fonds Wetenschappelijk Onderzoek (PhD fellowship to E. P. The Research Foundation Flanders (FWO) is further thanked for financial support through project G.0541.19N. C.M. and S. B. thank the Research Foundation Flanders (FWO Vlaanderen) and the Research Council of the VUB for support through the Strategic Research Programme (SRP50 and SRP95). C. M., S. B., and R. H. acknowledge the financial support from the Wetenschappelijke Onderzoeksgemeenschap (WOG) “Supramolecular Chemistry and Materials” of the Research Foundation Flanders (FWO-V).References
- A. Z. Wang, F. Gu, L. Zhang, J. M. Chan, A. Radovic-Moreno, M. R. Shaikh and O. C. Farokhzad, Biofunctionalized Targeted Nanoparticles for Therapeutic Applications, Expert Opin. Biol. Ther., 2008, 1063–1070, DOI:10.1517/14712598.8.8.1063.
- G. W. Ashley, J. Henise, R. Reid and D. V. Santi, Hydrogel Drug Delivery System with Predictable and Tunable Drug Release and Degradation Rates, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(6), 2318–2323, DOI:10.1073/pnas.1215498110.
- D. Jain, S. S. Mahammad, P. P. Singh and R. Kodipyaka, A Review on Parenteral Delivery of Peptides and Proteins, Drug Dev. Ind. Pharm., 2019, 1403–1420, DOI:10.1080/03639045.2019.1628770.
- S. Jacob, A. B. Nair, J. Shah, N. Sreeharsha, S. Gupta and P. Shinu, Emerging Role of Hydrogels in Drug Delivery Systems, Tissue Engineering and Wound Management, Pharmaceutics, 2021, 13(3), 357, DOI:10.3390/pharmaceutics13030357.
- A. M. Hawkins, T. A. Milbrandt, D. A. Puleo and J. Z. Hilt, Synthesis and Analysis of Degradation, Mechanical and Toxicity Properties of Poly(β-Amino Ester) Degradable Hydrogels, Acta Biomater., 2011, 7(5), 1956–1964, DOI:10.1016/j.actbio.2011.01.024.
- H. Kamata, X. Li, U. Il Chung and T. Sakai, Design of Hydrogels for Biomedical Applications, Adv. Healthcare Mater., 2015, 4(16), 2360–2374, DOI:10.1002/adhm.201500076.
- M. C. Georgiadis and M. Kostoglou, On the Optimization of Drug Release from Multi-Laminated Polymer Matrix Devices, J. Controlled Release, 2001, 77(3), 273–285, DOI:10.1016/s0168-3659(01)00510-7.
- C. B. P. Oliveira, V. Gomes, P. M. T. Ferreira, J. A. Martins and P. J. Jervis, Peptide-Based Supramolecular Hydrogels as Drug Delivery Agents: Recent Advances, Gels, 2022, 706, DOI:10.3390/gels8110706.
- M. Nambiar and J. P. Schneider, Peptide Hydrogels for Affinity-Controlled Release of Therapeutic Cargo: Current and Potential Strategies, J. Pept. Sci., 2022, e3377, DOI:10.1002/psc.3377.
- J. Mitrovic, G. Richey, S. Kim and M. O. Guler, Peptide Hydrogels and Nanostructures Controlling Biological Machinery, Langmuir, 2023, 29, 11935–11945, DOI:10.1021/acs.langmuir.3c01269.
- S. Zhang, T. Holmest, C. Lockshin and A. Rich, Spontaneous Assembly of a Self-Complementary Oligopeptide to Form a Stable Macroscopic Membrane, Proc. Natl. Acad. Sci. U. S. A., 1993, 90(8), 3334–3338, DOI:10.1073/pnas.90.8.3334.
- A. Mohammed, A. F. Miller and A. Saiani, 3D Networks from Self-Assembling Ionic-Complementary Octa-Peptides, Macromol. Symp., 2007, 251(1), 88–95, DOI:10.1002/masy.200750512.
- S. Sankar, K. O'Neill, M. Bagot D'Arc, F. Rebeca, M. Buffier, E. Aleksi, M. Fan, N. Matsuda, E. S. Gil and L. Spirio, Clinical Use of the Self-Assembling Peptide RADA16: A Review of Current and Future Trends in Biomedicine, Front. bioeng. biotechnol., 2021, 9, 679525, DOI:10.3389/fbioe.2021.679525.
- A. J. Tompeck, A. U. R. Gajdhar, M. Dowling, S. B. Johnson, P. S. Barie, R. J. Winchell, D. King, T. M. Scalea, L. D. Britt and M. Narayan, A Comprehensive Review of Topical Hemostatic Agents: The Good, the Bad, and the Novel, J. Trauma Acute Care Surg., 2020, 88(1), e1–e21, DOI:10.1097/TA.0000000000002508.
- M. Bibian, J. Mangelschots, J. Gardiner, L. Waddington, M. M. Diaz Acevedo, B. G. De Geest, B. Van Mele, A. Madder, R. Hoogenboom and S. Ballet, Rational Design of a Hexapeptide Hydrogelator for Controlled-Release Drug Delivery, J. Mater. Chem. B, 2015, 3(5), 759–765, 10.1039/c4tb01294a.
- E. Oyen, C. Martin, V. Caveliers, A. Madder, B. Van Mele, R. Hoogenboom, S. Hernot and S. Ballet, In Vivo Imaging of the Stability and Sustained Cargo Release of an Injectable Amphipathic Peptide-Based Hydrogel, Biomacromolecules, 2017, 18(3), 994–1001, DOI:10.1021/acs.biomac.6b01840.
- C. Martin, E. Oyen, J. Mangelschots, M. Bibian, T. Ben Haddou, J. Andrade, J. Gardiner, B. Van Mele, A. Madder, R. Hoogenboom, M. Spetea and S. Ballet, Injectable Peptide Hydrogels for Controlled-Release of Opioids, MedChemComm, 2016, 7(3), 542–549, 10.1039/c5md00440c.
- C. Martin, E. Oyen, Y. Van Wanseele, T. B. Haddou, H. Schmidhammer, J. Andrade, L. Waddington, A. Van Eeckhaut, B. Van Mele, J. Gardiner, R. Hoogenboom, A. Madder, M. Spetea and S. Ballet, Injectable Peptide-Based Hydrogel Formulations for the Extended in Vivo Release of Opioids, Mater. Today Chem., 2017, 3, 49–59, DOI:10.1016/j.mtchem.2017.01.003.
- J. Heremans, R. Maximilian Awad, J. Bridoux, T. Ertveldt, V. Caveliers, A. Madder, R. Hoogenboom, N. Devoogdt, S. Ballet, S. Hernot, K. Breckpot and C. Martin, Sustained Release of a Human PD-L1 Single-Domain Antibody Using Peptide-Based Hydrogels, Eur. J. Pharm. Biopharm., 2024, 196, 114183, DOI:10.1016/j.ejpb.2024.114183.
- Y. Li, F. Wang and H. Cui, Peptide–based Supramolecular Hydrogels for Delivery of Biologics, Bioeng. Transl. Med., 2016, 1(3), 306–322, DOI:10.1002/btm2.10041.
- S. Koutsopoulos, L. D. Unsworth, Y. Nagai and S. Zhang, Controlled Release of Functional Proteins through Designer Self-Assembling Peptide Nanofiber Hydrogel Scaffold, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(12), 4623–4628, DOI:10.1073/pnas.0807506106.
- O. N. Makshakova, L. R. Bogdanova, A. O. Makarova, A. M. Kusova, E. A. Ermakova, M. A. Kazantseva and Y. F. Zuev, κ-Carrageenan Hydrogel as a Matrix for Therapeutic Enzyme Immobilization, Polymers, 2022, 14(19), 4071, DOI:10.3390/polym14194071.
- P. Lu, D. Ruan, M. Huang, M. Tian, K. Zhu, Z. Gan and Z. Xiao, Harnessing the Potential of Hydrogels for Advanced Therapeutic Applications: Current Achievements and Future Directions, Signal Transduction Targeted Ther., 2024, 9, 166, DOI:10.1038/s41392-024-01852-x.
- A. A. Homaei, R. Sariri, F. Vianello and R. Stevanato, Enzyme Immobilization: An Update, J. Biol. Chem., 2013, 185–205, DOI:10.1007/s12154-013-0102-9.
- O. N. Makshakova, L. R. Bogdanova, A. O. Makarova, A. M. Kusova, E. A. Ermakova, M. A. Kazantseva and Y. F. Zuev, κ-Carrageenan Hydrogel as a Matrix for Therapeutic Enzyme Immobilization, Polymers, 2022, 14(19), 4071, DOI:10.3390/polym14194071.
- M. Flanagan, Q. Gan, S. Sheth, R. Schafer, S. Ruesing, L. E. Winter, K. Toth, S. P. Zustiak and A. M. Montaño, Hydrogel Delivery Device for the In Vitro and In Vivo Sustained Release of Active RhGALNS Enzyme, Pharmaceuticals, 2023, 16(7), 931, DOI:10.3390/ph16070931.
- K. Kamenova, A. Prancheva, S. Stoyanova, L. Radeva, I. P. El Tibi, K. Yoncheva, M. A. Ravutsov, M. K. Marinova, S. P. Simeonov, S. Mitova, R. Eneva, M. M. Zaharieva, H. Najdenski and P. D. Petrov, Functional Hydrogels for Delivery of the Proteolytic Enzyme Serratiopeptidase, Gels, 2024, 10(3), 156, DOI:10.3390/gels10030156.
- T. L. Lopez-Silva, D. G. Leach, A. Azares, I. C. Li, D. G. Woodside and J. D. Hartgerink, Chemical Functionality of Multidomain Peptide Hydrogels Governs Early Host Immune Response, Biomaterials, 2020, 231, 119667, DOI:10.1016/j.biomaterials.2019.119667.
- G. M. Keating, Asparaginase Erwinia Chrysanthemi (Erwinaze®): A Guide to Its Use in Acute Lymphoblastic Leukemia in the USA, BioDrugs, 2013, 27(4), 413–418, DOI:10.1007/s40259-013-0051-4.
- W. L. Salzer, B. L. Asselin, P. V. Plourde, T. Corn and S. P. Hunger, Development of Asparaginase Erwinia Chrysanthemi for the Treatment of Acute Lymphoblastic Leukemia, Ann. N. Y. Acad. Sci., 2014, 1329(1), 81–92, DOI:10.1111/nyas.12496.
- M. Van Trimpont, E. Peeters, Y. De Visser, A. M. Schalk, V. Mondelaers, B. De Moerloose, A. Lavie, T. Lammens, S. Goossens and P. Van Vlierberghe, Novel Insights on the Use of L-Asparaginase as an Efficient and Safe Anti-Cancer Therapy, Cancers, 2022, 14(4), 902, DOI:10.3390/cancers14040902.
- T. Modi and D. Gervais, Improved Pharmacokinetic and Pharmacodynamic Profile of a Novel PEGylated Native Erwinia Chrysanthemi L-Asparaginase, Invest. New Drugs, 2022, 40(1), 21–29, DOI:10.1007/s10637-021-01173-8.
- J. Müller, P. Egyed, D. Erdelyi, K. Kovacs, K. Mudra, S. Szabo, B. Egyed and K. Gabor, Our Experiences with Asparaginase Activity Measurements in Children with Lymphoblastic Diseases, Children, 2023, 10(7), 1160, DOI:10.3390/children10071160.
- H. A. Nguyen, Y. Su and A. Lavie, Design and Characterization of Erwinia Chrysanthemi L-Asparaginase Variants with Diminished L-Glutaminase Activity, J. Biol. Chem., 2016, 291(34), 17664–17676, DOI:10.1074/jbc.M116.728485.
- M. C. Branco, D. J. Pochan, N. J. Wagner and J. P. Schneider, The Effect of Protein Structure on Their Controlled Release from an Injectable Peptide Hydrogel, Biomaterials, 2010, 31(36), 9527–9534, DOI:10.1016/j.biomaterials.2010.08.047.
- B. L. Abraham, E. S. Toriki, N. J. Tucker and B. L. Nilsson, Electrostatic Interactions Regulate the Release of Small Molecules from Supramolecular Hydrogels, J. Mater. Chem. B, 2020, 8(30), 6366–6377, 10.1039/d0tb01157f.
- H. T. M. Phan, S. Bartelt-Hunt, K. B. Rodenhausen, M. Schubert and J. C. Bartz, Investigation of Bovine Serum Albumin (BSA) Attachment onto Self-Assembled Monolayers (SAMs) Using Combinatorial Quartz Crystal Microbalance with Dissipation (QCM-D) and Spectroscopic Ellipsometry (SE), PLoS One, 2015, 10(10), e0141282, DOI:10.1371/journal.pone.0141282.
- A. Onaciu, R. A. Munteanu, A. I. Moldovan, C. S. Moldovan and I. Berindan-Neagoe, Hydrogels Based Drug Delivery Synthesis, Characterization and Administration, Pharmaceutics, 2019, 11(9), 432, DOI:10.3390/pharmaceutics11090432.
- F. Koch, M. Müller, F. König, N. Meyer, J. Gattlen, U. Pieles, K. Peters, B. Kreikemeyer, S. Mathes and S. Saxer, Mechanical Characteristics of Beta Sheet-Forming Peptide Hydrogels Are Dependent on Peptide Sequence, Concentration and Buffer Composition, R. Soc. Open Sci., 2018, 5(3), 171562, DOI:10.1098/rsos.171562.
- A. Vartolomei, I. Calinescu, M. Vinatoru and A. I. Gavrila, A Parameter Study of Ultrasound Assisted Enzymatic Esterification, Sci. Rep., 2022, 12(1), 1421, DOI:10.1038/s41598-022-05551-x.
- J. Heremans, L. Chevillard, M. Mannes, J. Mangialetto, K. Leroy, J. F. White, A. Lamouroux, M. Vinken, J. Gardiner, B. Van Mele, N. Van den Brande, R. Hoogenboom, A. Madder, V. Caveliers, B. Mégarbane, S. Hernot, S. Ballet and C. Martin, Impact of Doubling Peptide Length on in Vivo Hydrogel Stability and Sustained Drug Release, J. Controlled Release, 2022, 350, 514–524, DOI:10.1016/j.jconrel.2022.08.027.
- S. Aleid, M. Wu, R. Li, W. Wang, C. Zhang, L. Zhang and P. Wang, Salting-in Effect of Zwitterionic Polymer Hydrogel Facilitates Atmospheric Water Harvesting, ACS Mater. Lett., 2022, 4(3), 511–520, DOI:10.1021/acsmaterialslett.1c00723.
- Y. Zhan, W. Fu, Y. Xing, X. Ma and C. Chen, Advances in Versatile Anti-Swelling Polymer Hydrogels, Mater. Sci. Eng. Carbon, 2021, 127, 112208, DOI:10.1016/j.msec.2021.112208.
- X. Sun, Y. Mao, Z. Yu, P. Yang and F. Jiang, A Biomimetic “Salting Out—Alignment—Locking” Tactic to Design Strong and Tough Hydrogel, Adv. Mater., 2024, 36(25) DOI:10.1002/adma.202400084.
- J. Li and D. J. Mooney, Designing Hydrogels for Controlled Drug Delivery, Nat. Rev. Mater., 2016, 1(12), 16071, DOI:10.1038/natrevmats.2016.71.
- Y. Feng, M. Taraban and Y. B. Yu, The Effect of Ionic Strength on the Mechanical, Structural and Transport Properties of Peptide Hydrogels, Soft Matter, 2012, 8(46), 11723–11731, 10.1039/c2sm26572a.
- I. M. van der Sluis, L. M. Vrooman, R. Pieters, A. Baruchel, G. Escherich, N. Goulden, V. Mondelaers, J. S. de Toledo, C. Rizzari, L. B. Silverman and J. A. Whitlock, Consensus Expert Recommendations for Identification and Management of Asparaginase Hypersensitivity and Silent Inactivation, Haematologica, 2016, 101(3), 279–285, DOI:10.3324/haematol.2015.137380.
- G. I. Frost, Recombinant Human Hyaluronidase (RHuPH20): An Enabling Platform for Subcutaneous Drug and Fluid Administration, Expert Opin. Drug Delivery, 2007, 4(4), 427–440, DOI:10.1517/17425247.4.4.427.
- F. Villanueva-Flores, A. Zárate-Romero, A. G. Torres and A. Huerta-Saquero, Encapsulation of Asparaginase as a Promising Strategy to Improve in Vivo Drug Performance, Pharmaceutics, 2021, 13(11), 1965, DOI:10.3390/pharmaceutics13111965.
- G. Uzunalli and M. O. Guler, Peptide Gels for Controlled Release of Proteins, Ther. Delivery, 2020, 193–211, DOI:10.4155/tde-2020-0011.
Footnotes |
| † Electronic supplementary information (ESI) available: Reserved-phase high-performance liquid chromatography method and calibration information. HRP-streptavidin release study. Microbial transglutaminase release study. Plasma L-asparaginase activity determination. Injectability of hydrogel with different needle sizes. Visual observation of BSA-loaded hydrogel of 1 (H-FEFQFK-NH2) at different loading concentrations. Visual observation of the real-time activity after release of HRP enzyme from the hydrogel. Pictures of hydrogel residue 7 days post injection in mice. ELISA measurement of the inflammatory cytokine (IL-6) signal in mice sera. Additional information for Fig. 6 including the in vivo pharmacokinetics of Erwinase – hydrogel complexes, with intramuscular Erwinase injection included as control. See DOI: https://doi.org/10.1039/d5bm00138b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |