
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomimetic double network hydrogels of chondroitin sulfate and synthetic polypeptides for cartilage tissue engineering†
Juhi
Singh
 ,
Jacob C.
Kadir
,
Jason D.
Orlando
and
Stefanie A.
Sydlik
*
,
Jacob C.
Kadir
,
Jason D.
Orlando
and
Stefanie A.
Sydlik
*
Department of Chemistry, Carnegie Mellon University, 4400 Fifth Avenue, Pittsburgh, PA 15213, USA. E-mail: ssydlik@andrew.cmu.edu; juhis@andrew.cmu.edu; jkadir@andrew.cmu.edu; jorlando@andrew.cmu.edu
First published on 23rd June 2025
Abstract
Articular cartilage defects are common, and the progressive deterioration of cartilage frequently results in the onset of osteoarthritis. However, the intrinsic regenerative capacity of articular cartilage is minimal. Synthetic therapeutic solutions for treating cartilage damage are being developed. However, current scaffolds and hydrogels employed in cartilage tissue engineering face limitations in promoting cellular activity and providing sufficient load-bearing strength. This is primarily due to suboptimal crosslinking methods for the synthetic scaffolds composed of natural proteins and glycosaminoglycans (GAGs). Synthetic polypeptides, owing to their customizable reactive functional groups, present an exciting opportunity to enhance crosslinking through both physical and chemical approaches. This study introduces a strategy for the development of injectable, shape-adaptive double network hydrogels that closely replicate the structural integrity and mechanical properties of native cartilage. These hydrogels are composed of photocrosslinkable GAGs, specifically methacrylated chondroitin sulfate A (CSMA), combined with a synthetic polypeptide, poly(L-lysine) (PLL). By varying the degree of polymerization (DP) of PLL and weight percentage of PLL in the composition, the hydrogels can be optimized for desired material properties. Varying DP of PLLs varies the molecular weight between crosslinks, thus leading to tunable rigidity (yield strength, ultimate compression strength, storage modulus) and toughness. We further this tunability through the integration of photoresponsive components, enabling controlled, non-invasive post-injection modifications. Initial testing indicates that these double network hydrogels exhibit significantly improved mechanical strength compared to hydrogels formed solely from CSMA, positioning them as strong candidates for minimally invasive cartilage defect repair. This innovative method offers the potential to accelerate recovery, restore joint function, and improve patients’ overall quality of life.
1. Introduction
Cartilage injuries impact millions of patients further affecting their quality of life.1 The cartilage tissue has limited self-regeneration and intrinsic repair ability owing to limited blood and neuron supply.2 This further complicates recovery wherein cartilage rarely recovers and, in many cases, leads to a degradation cascade resulting in osteoarthritis characterized by defects of articular cartilage.3 The primary issue with osteoarthritis is cartilage damage, wherein inflammation and enzymatic activity lead to further joint damage which impact joint movement requiring total joint replacement.3 Current clinical approaches for cartilage regeneration involve strategies such as microfracture,4 osteochondral allograft5 and autografts,6 and autologous chondrocyte implantation.7,8 These strategies are invasive in nature often leading to inadequate healing and mechanical mismatch of grafts with that of the cartilage.9 To address the limitations of current approaches, tissue regeneration strategies that integrate cell therapy with synthetic materials, designed to replicate the mechanical properties of cartilage are essential.Injectable hydrogels offer a minimally invasive approach for the management of cartilage defects.10 The design principle for injectable hydrogels involves the use of polymer components that can mimic the extracellular matrix of natural cartilage in terms of chemical composition and mechanical properties.11 Thus, injectable hydrogels derived from natural polysaccharides and proteins have emerged as promising alternatives to traditional cartilage regeneration methods. To this end hydrogels based on natural polysaccharides, proteins as well as synthetic polymers have been studied with various in vivo and ex vivo gelation mechanisms. A pioneering study reported the development of a chondroitin sulfate (CS)-based hydrogel designed for cartilage regeneration. In this approach, CS was chemically functionalized with methacrylate and aldehyde groups, creating two functional arms: one for covalent bonding with a biomaterial scaffold and the other for anchoring to the tissue surface.12 Another study integrated hyperbranched multifunctional poly(ethylene glycol) with thiolated CS to develop injectable hydrogels. This system was designed as a stem cell delivery platform to address the challenges of weak mechanical properties and rapid degradation typically associated with CS hydrogels.13 Another leading study focused on the development of a variety of photosensitive hyaluronic acid hydrogels to promote chondrogenesis.14–16 Hydrogels based solely on GAGs suffer from limited mechanical strength and viscoelastic properties, lower by an order of magnitude compared to native cartilage. Composite hydrogels comprising of GAGs along with synthetic toughening agents as an alternative to collagen fibers found in the native cartilage could be the solution.
Hydrogels that rely primarily on physical crosslinking for gelation often exhibit low crosslinking density, which compromises their rigidity, however, are flexible and stretchable owing to reversibility of physical crosslinks. This results in reduced storage and loss moduli, as well as lower yield and ultimate compressive strength. A notable example is the development of injectable and shear-reversible alginate hydrogels, achieved through a combination of cell-based crosslinking and ionic crosslinking techniques for cartilage regeneration. The hydrogels exhibit shear reversible gelation, however the storage modulus is limited to 0.01–0.2 kPa.17 Alternatively, hydrogels which exhibit covalent crosslinking upon external stimuli exposure have been designed by incorporating photoreactive groups. However, hydrogels relying solely on covalent crosslinks yield hydrogels with improved rigidity but are brittle in nature due to low flexibility. A common approach is functionalizing CS with crosslinkable moieties such as catechol or methacrylate which would enable covalent crosslinking to form hydrogel matrix.18,19
Thus, double network hydrogels which combine a fine balance of physical and chemical crosslinks could offer the perfect blend of toughness and robustness required to mimic the cartilage. For instance, a double-network hydrogel composed of sulfated alginate functionalized with catechol and methacrylate, combined with methacrylated chitosan, was reported. The hydrogel crosslinked through UV irradiation and the addition of an oxidizing agent, forming a robust double network. This type of hydrogels can demonstrate a compressive strength up to 400 kPa, five times greater than hydrogels made with catechol-functionalized alginate and chitosan in the presence of an oxidizer alone.20 Another example of hybrid photocrosslinkable hydrogel combining methacrylated hyaluronic acid (HA) and o-nitrobenzyl (NB)-grafted HA demonstrated enhanced mechanical performance (storage modulus – 6–12 kPa compared to 1–2 kPa of individual gels) via double-network mechanisms. Rapid crosslinking (within 4–6 seconds) was achieved through radical polymerization, while aldehyde groups photogenerated from the NB moieties efficiently reacted with amino groups present in gelatin or on surrounding cartilage surfaces.21
Incorporation of naturally derived proteins into GAG hydrogels have been reported. For example, polysaccharide-protein-based scaffolds such as HA/CS/gelatin,22 cellulose/peptide,23 CS/collagen,24 HA/silk fibroin,25 and CS/gelatin.26 These scaffolds exhibit compressive strengths ranging from 10 to 130 kPa and storage moduli between 0.1 and 10 kPa. Natural proteins, however, are limited in terms of density of reactive functional groups responsible for physical or chemical crosslinks leading to slow gelation and limited mechanical properties. Moreover, these proteins are expensive and difficult to obtain in large quantities.
Natural cartilage is majorly composed of proteoglycans (core proteins linked with glycosaminoglycans (GAGs)), hyaluronan, collagen, water and chondrocytes.27 Water is responsible for cartilage resilience, collagen for tensile strength and chondrocytes for maintaining and repairing the cartilage.27 The GAGs such as CS and keratan sulfate are involved in chondrocyte signaling pathways further promoting chondrogenesis by binding integrins that increase TGF-β1 expression leading to production of hyaluronic acid and type II collagen.28 Collagen fibers in the native cartilage are responsible for toughening of GAG scaffold thus enhancing the longevity and lubricity of the tissues.27,29
Synthetic polypeptides offer an alternative to natural proteins with wide tunability in terms of reactive functional groups and their density. Synthetic polypeptides prepared through ring opening polymerizations such as poly(L-lysine) (PLL) and poly (L-glutamic acid) have been widely studied for tissue regeneration and antibacterial properties. Varying the degree of polymerization and thus the chain length offers tunability in terms of density of reactive functional groups. The chain length can be used to manipulate the molecular weight between crosslinks and further dictates hydrogel properties: shorter molecular weight between the crosslinks leads to higher rigidity while larger molecular weight between the crosslinks lead to higher flexibility.
In this work, we propose the design of a double network composite hydrogel based on chondroitin sulfate and PLL. CS is chosen owing to its presence in the natural cartilage and ability to promote chondrogenesis. PLL is chosen as the synthetic alternative for collagen present in cartilage responsible for toughness. Methacrylated CS is employed for this study as it allows for light responsive covalent crosslinking, thereby giving surgeons control to initiate crosslinking. The covalent crosslinking within CSMA chains is expected to provide rigidity to the system. Incorporation of PLL chains is expected to establish physical crosslinks resulting in enhanced flexibility and toughness of the composite hydrogels. It is hypothesized that incorporation of PLLs with lower degree of polymerization (DP) will yield composite hydrogels with a shorter molecular weight between the crosslinks and thus higher rigidity (yield strength, ultimate compression strength, storage modulus) while those with higher DPs will yield tougher hydrogels.
2. Results & discussion
2.1. Rationale for material design
Cartilage is a specialized tissue with unique chemical and structural composition responsible for its resilience, elasticity and compressive strength along with bioactivity. Structurally, cartilage comprises of about 65–80% water by weight majorly responsible for viscoelastic property and the extracellular matrix composed of collagen fibers, proteoglycans and hyaluronic acid. An ideal injectable hydrogel should be viscous in nature such that it can be injected at the defect site in a minimally invasive manner, followed by stimuli responsive gelation in less than a minute.11 The formed hydrogel should also match the viscoelastic and mechanical load bearing properties of the native cartilage around the defect to avoid separation due to mechanical mismatch.11 The hydrogel in combination with cells should have the ability to promote chondrocyte growth and differentiation leading to reduced healing time.11 The hydrogel should also have suitable porosity and interconnectivity to enable cell migration and efficient exchange of nutrients and waste.11,30To mimic the structure and function of cartilage, double network hydrogels composed of proteoglycan chondroitin sulfate (CS) and synthetic polypeptide poly(L-lysine) (PLL) is proposed. CS is incorporated owing to its presence in natural cartilage and its ability to promote chondrogenesis. Natural proteins are rich in lysine, which provides important crosslinking, for example to stabilize the collagen triple helix.31 Thus, we selected PLL to provide toughness to the hydrogel matrix in similar manner as collagen fibers in the cartilage.
2.2. Synthesis and formulation of the double network hydrogels
To impart injectability and stimuli-responsive gelation to the hydrogels, CS is functionalized with methacrylate groups to yield chondroitin sulfate methacrylate (CSMA). This undergoes vinyl free radical polymerization upon UV-Visible light exposure. CSMA is synthesized through an esterification reaction using methacrylic anhydride wherein hydroxyl groups of CS act as nucleophiles and attack the carbonyl in methacrylic anhydride. CSMA with degree of substitution 0.5–0.8 (as determined by 1H-NMR) is synthesized (Fig. S2 and S3†) used to formulate the hydrogels. PLL, which is the second component of the hydrogels, is synthesized by ring-opening polymerization of L-lysine(Z)–NCA (Fig. S4†) followed by deprotection and dialysis. For CSMA–PLL hydrogels, covalent crosslinking of CSMA is expected to form the first network of the double network composite hydrogels, while physical crosslinks among PLL chains form the second network (Scheme 1). | ||
| Scheme 1 Illustrative representation of the double network hydrogel formed by CSMA–PLL. CSMA forms the covalently crosslinked network upon light exposure while PLL forms the physical crosslinks via hydrogen bonding. Tuning the PLL chain length is hypothesized to tune the flexibility and rigidity of hydrogels. | ||
The composition of CSMA–PLL hydrogels is described in Table 1. The weight percentage (%w/w) of water is maintained at 87–89% for all hydrogels to mimic that of the cartilage. The (%w/w) of CSMA is around 8%. To initiate photocrosslinking upon light exposure, 0.05% lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) is added to the hydrogels which generates free radicals initiating the crosslinking. In case of PLL incorporation, the degree of polymerization of PLL is varied to be DP15, DP35 and DP50 to assess the impact of molecular weight on viscoelasticity and mechanical properties. The %w/w of PLL is varied (0, 2.5 and 5%) to assess the impact of PLL concentration on the composite hydrogel performance. Structure–property relationships evaluate the effect of PLL incorporation, PLL DP and %w/w PLL on viscoelasticity (storage modulus, loss modulus, dynamic viscosity, yield stress and strain at break point), structural morphology determined by SEM, compression strength in freshly prepared and swollen states, viscoelasticity upon enzymatic degradation and human mesenchymal stem cell cytocompatibility.
| CSMA–PLL hydrogel composition | CSMA | CSMA–PLL 2.5 | CSMA–PLL 5.0 |
|---|---|---|---|
| Chondroitin sulfate methacrylate (CSMA) | 8.00% | 8.00% | 8.00% |
| Poly(L-lysine) (PLL) | 0.00% | 2.50% | 5.00% |
| LAP | 0.05% | 0.05% | 0.05% |
| Water | 92.00% | 89.00% | 87.00% |
2.3. PLL Incorporation improves the viscoelastic properties of CSMA hydrogels
CSMA–PLL hydrogels formulated by mixing varying weight percentages of CSMA and PLL are evaluated for changes in viscoelastic properties upon light exposure using a photorheology setup (Fig. S11A†). Real-time photorheology determines the extent of light-dependent crosslinking. The samples are subjected to UV light (365 nm) with intensity of 100 mW cm−2 for 200 s which amounts to 20 J of light dose. Upon light exposure, the photoinitiator LAP generates free radicals primarily from the cleavage of the benzoyl–phosphinate bond, creating a phenyl radical and a radical derived from the trimethylbenzoyl group. These radicals then initiate free radical vinyl polymerization reactions with the methacrylate groups of CSMA, leading to crosslinking of the hydrogels. The corresponding changes in the storage and loss modulus due to crosslinking are recorded with respect to time for all the formulations.The first step of photorheological experiment involves exposing the hydrogels to 1% oscillatory strain for 30 s, which disrupts the weak intermolecular attractive forces (Fig. 1A). Dynamic viscosity is evaluated at this time which gives an idea about the injectability of the hydrogels. Hydrogels with dynamic viscosity lower than 10 Pa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ·s are injectable using a syringe. CSMA–PLL hydrogels exhibit dynamic viscosity ranging from 0.03–0.4 Pa·s (Fig. S11B†), which is higher than neat CSMA hydrogels (0.024 Pa·s). A green horizontal line at 0.024 Pa·s represents the properties of neat CSMA hydrogel without any PLL incorporation. This suggests PLL incorporation leads to increase in dynamic viscosity, possibly due to covalent and non-covalent interactions between the CSMA and PLL polymer chains. The extent of interaction, however, is also dictated by the %w/w of PLL and PLL chain length evident by variation in dynamic viscosity. Decrease in PLL %w/w leads to decreased viscosity for formulations with PLL DP15. However, the dynamic viscosity remains comparable to CSMA for DP35 and an increase in viscosity is observed for PLL DP50.
·s are injectable using a syringe. CSMA–PLL hydrogels exhibit dynamic viscosity ranging from 0.03–0.4 Pa·s (Fig. S11B†), which is higher than neat CSMA hydrogels (0.024 Pa·s). A green horizontal line at 0.024 Pa·s represents the properties of neat CSMA hydrogel without any PLL incorporation. This suggests PLL incorporation leads to increase in dynamic viscosity, possibly due to covalent and non-covalent interactions between the CSMA and PLL polymer chains. The extent of interaction, however, is also dictated by the %w/w of PLL and PLL chain length evident by variation in dynamic viscosity. Decrease in PLL %w/w leads to decreased viscosity for formulations with PLL DP15. However, the dynamic viscosity remains comparable to CSMA for DP35 and an increase in viscosity is observed for PLL DP50.
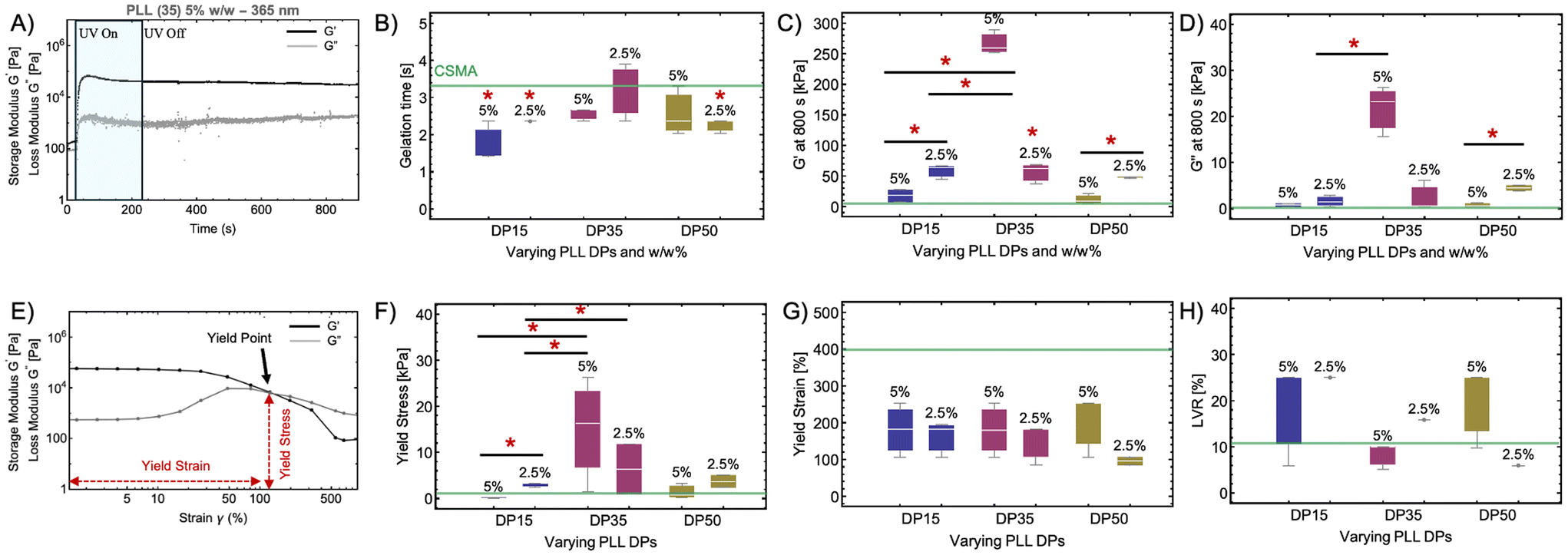 | ||
| Fig. 1 Viscoelastic properties of CSMA–PLL hydrogels upon photocuring with UV light (365 nm). (A) Representative rheogram of CSMA–PLL hydrogel with PLL DP35 (5% w/w) depicting the increase in storage (G′) and loss modulus (G′′) upon light exposure (365 nm). (B) Gelation time for the CSMA–PLL hydrogels with varying PLL chain lengths (DP) and PLL weight percentage compared to neat CSMA hydrogels. Comparison of (C) G′ and (D) G′′ of CSMA–PLL hydrogels. (E) Representative amplitude sweep curve depicting yield stress, yield strain and yield point. Comparison of (F) yield stress; (G) yield strain (%) and (H) linear viscoelastic range (LVR) obtained from the amplitude sweeps of CSMA–PLL hydrogels with varying PLL chain lengths (DP) and PLL weight percentage. The box plot represents the 25th and 75th percentiles as the edges of the box, with the median indicated by the line inside the box. The error bars in the box plots correspond to the interquartile range (IQR), n = 3. | ||
After the first step, the samples are exposed to UV light (365 nm) for 200 s, amounting to 20 J light dose and the changes in storage and loss modulus are recorded as depicted in the representative rheogram (Fig. 1A). The storage (G′) and loss modulus (G′′) are then allowed to stabilize for 670 s to allow for complete crosslinking to occur. Gelation time is the time required for the hydrogels to crosslink and reach a point where G′ becomes equal to the G′′. All CSMA–PLL hydrogels exhibit gelation times ranging from 1–4 s (0.1–0.4 J), comparable to, or in most cases lower than CSMA hydrogels.
After crosslinking, all CSMA–PLL hydrogels exhibit storage and loss moduli higher than neat CSMA hydrogels. The G′ for all CSMA–PLL hydrogels after photocuring ranges from 50–300 kPa which is the same order of magnitude as that of the natural human cartilage (G′ – 300 kPa).32,33 The G′′ ranges from 1–30 kPa. Both G′ and G′′ for CSMA–PLL formulations are higher than that of neat CSMA hydrogels suggesting the toughening effect of incorporated PLL chains. The hydrogel with 5% w/w incorporation of PLL DP35 exhibits exceptionally high G′ and G′′ when compared to other formulations. This can be attributed to the synergistic physical and covalent interactions at play between CSMA and PLL DP35 chains. Conversely, for DP15 and DP50, hydrogels with 5% w/w PLL incorporation exhibit lower G′ and G′′ compared to 2.5%. This suggests increasing PLL content is inversely proportional to G′ and G′′ for hydrogels with PLL DP15 and DP50.
Hydrogels with 5% PLL DP35 also exhibit an interconnected pore network with larger pore size (100–180 μm, Fig. S18†) as opposed to all other CSMA–PLL hydrogels with smaller pore size (0.7–25 μm) (Fig. 3). A well-defined, highly interconnected porous structure in case of PLL DP35 facilitates efficient stress distribution, preventing localized stress concentrations that could lead to premature failure. Hydrogels with an optimal pore network, such as those with DP35, exhibit a balanced crosslinking density and polymer chain mobility, allowing for both structural reinforcement and flexibility.34,35 This results in a higher storage modulus, as the network can effectively store elastic energy during deformation, and a higher loss modulus, indicating the material's ability to dissipate energy as heat or molecular reorganization.34,35 The presence of interconnected pores also enhances viscoelastic energy dissipation, enabling the hydrogel to withstand repeated loading and unloading cycles without significant degradation.34,35 In contrast, a dense, tightly crosslinked network (e.g., DP15) may limit energy dissipation, leading to brittle behavior, whereas a loosely crosslinked, highly porous structure (e.g., DP50) may compromise mechanical integrity, reducing both storage and loss modulus. Thus, an optimized interconnected pore network is essential for achieving superior mechanical resilience and durability under physiological loading conditions.
The formulations were then subjected to an amplitude sweep (1–1000% strain, 10 Hz) after photocuring to evaluate changes in viscoelastic properties upon oscillatory strain application (Fig. 1E). The strain % at which the G′ and G′′ crossover is defined as the yield point, whereas the yield stress is defined as G′ at break (yield point). Yield strain is the strain at which the hydrogel transitions from an elastic regime to a plastic or flow regime. All CSMA–PLL hydrogels exhibit yield stress (0.03–30 kPa) comparable or higher than neat CSMA hydrogels (Fig. 1F). Hydrogels with PLL DP35 incorporation exhibit comparatively higher median yield stress (20–30 kPa) suggesting stronger internal crosslinks or network interactions, making it more resistant to deformation before yielding. In terms of yield strain (Fig. 1G), all CSMA–PLL hydrogels exhibit median yield strain (80–250%) lower than that of neat CSMA hydrogels (400%). This suggests CSMA–PLL hydrogels exhibit reduced chain mobility because of denser crosslinking. Higher yield stress and lower yield strain compared to CSMA can be attributed to additional interactions (Scheme 2) between CSMA and PLL chains in the CSMA–PLL hydrogels.
 | ||
| Scheme 2 Illustrative representation of the interactions within the CSMA–PLL hydrogel, highlighting (A) polymer chain entanglement, (B) electrostatic interactions between negatively charged CSMA and positively charged PLL, and (C and D) covalent interactions, including vinyl free radical polymerization and Michael addition of PLL onto the CSMA vinyl groups. | ||
The linear viscoelastic region (LVR) is the range of strains or stresses within which a material's properties are independent of the applied stress or strain. In this region, the stress and strain have a linear relationship. All hydrogels except those incorporated with PLL DP35 (5% w/w) and DP50 (2.5% w/w) exhibit LVR higher than neat CSMA hydrogel (11%) (Fig. 1H). A negative correlation between PLL chain length (DP) and LVR is observed for hydrogels incorporated with 2.5% w/w PLL suggesting increased crosslinking density or stronger intermolecular interactions within the hydrogel network, contributing to better resistance against deformation. Hydrogels with PLL DP35 (5% w/w) exhibits higher G′, G′′ and yield stress but lower LVR owing to rigid matrix due to higher crosslinking density.
To assure the clinical applicability of the hydrogels, photocuring is also assessed with UV-Vis light (350–550 nm) at 150 mW cm−2 for 200 s amounting to 30 J of light dose (Fig. S12†). In case of exposure to UV-Vis light, CSMA hydrogel requires about 6 s (0.6 J) for gelation while all other hydrogels exhibit lower gelation times of 1–5 s (0.1–0.5 J). Hydrogels cured under UV-Vis light exhibit significantly higher storage modulus (G′), ranging from 50 to 200 kPa. This enhancement is likely attributed to the increased crosslinking density resulting from the higher light dose applied during the curing process. All CSMA–PLL hydrogels exhibit G′ and G′′ higher than neat CSMA hydrogel. The general trend observed is higher median G′ and G′′ for hydrogels with 2.5% wt PLL compared to 5% PLL. Like the UV-cured hydrogels, CSMA–PLL hydrogels demonstrate higher yield stress (ranging from 1 to 10 kPa) compared to neat CSMA. However, they exhibit lower yield strain in comparison to neat CSMA hydrogels. The LVR of UV-Vis light-cured hydrogels shows a trend opposite to that of UV-cured hydrogels. Specifically, an increase in LVR is observed with increasing PLL DP for both 2.5% and 5% PLL concentrations.
The enhanced storage modulus, loss modulus, and yield strength observed in CSMA–PLL hydrogels photocured with a UV-Vis light combination can be attributed to several factors. First, the applied light dose in these cases was 30 J, 1.5 times higher than that used for UV light alone, likely leading to increased crosslinking density. Another key factor is the penetration depth of UV versus UV-Vis light. Given the 200-micron thickness of the hydrogel samples, UV light efficiently penetrates only about 50 microns, whereas UV-Vis light can reach 100–200 microns, leading to a more uniformly crosslinked network.36,37 This increased crosslinking density contributes to the higher storage modulus, loss modulus, and yield strength observed with UV-Vis curing.
2.4. CSMA–PLL hydrogels exhibit a combination of physical and chemical crosslinking
The double network of hydrogels which provides it with tough and robust nature occurs due to the combination of physical and chemical crosslinking between the components. Similarly, in the case of CSMA–PLL hydrogels, it's a combination of covalent and non-covalent interactions. The non-covalent interactions involve polymer entanglement and electrostatic forces (Scheme 2). Physical entanglement is anticipated between the PLL chains, CSMA chains as well as between the CSMA and PLL chains. The positively charged groups of PLL will likely interact with the negatively charged carboxylates of CSMA, resulting in electrostatic interactions leading to physical crosslinks.There are two types of covalently crosslinked networks. The first type is formed via free-radical polymerization of the methacrylate groups on CSMA chains.38,39 This reaction is initiated by the generation of free radicals upon exposure to UV or UV-Vis light, leading to polymerization through the vinyl groups of the CSMA methacrylate (Scheme 2).
The second type of covalent crosslink arises from the Michael addition reaction, where the primary amine groups of PLL act as nucleophiles and add to the vinyl bonds of the CSMA methacrylate. To confirm this Michael addition between CSMA and PLL, small molecules mimicking the repeating units of CSMA and PLL were mixed in a deuterated solvent and monitored for changes in the methacrylate peaks (Fig. 2A).
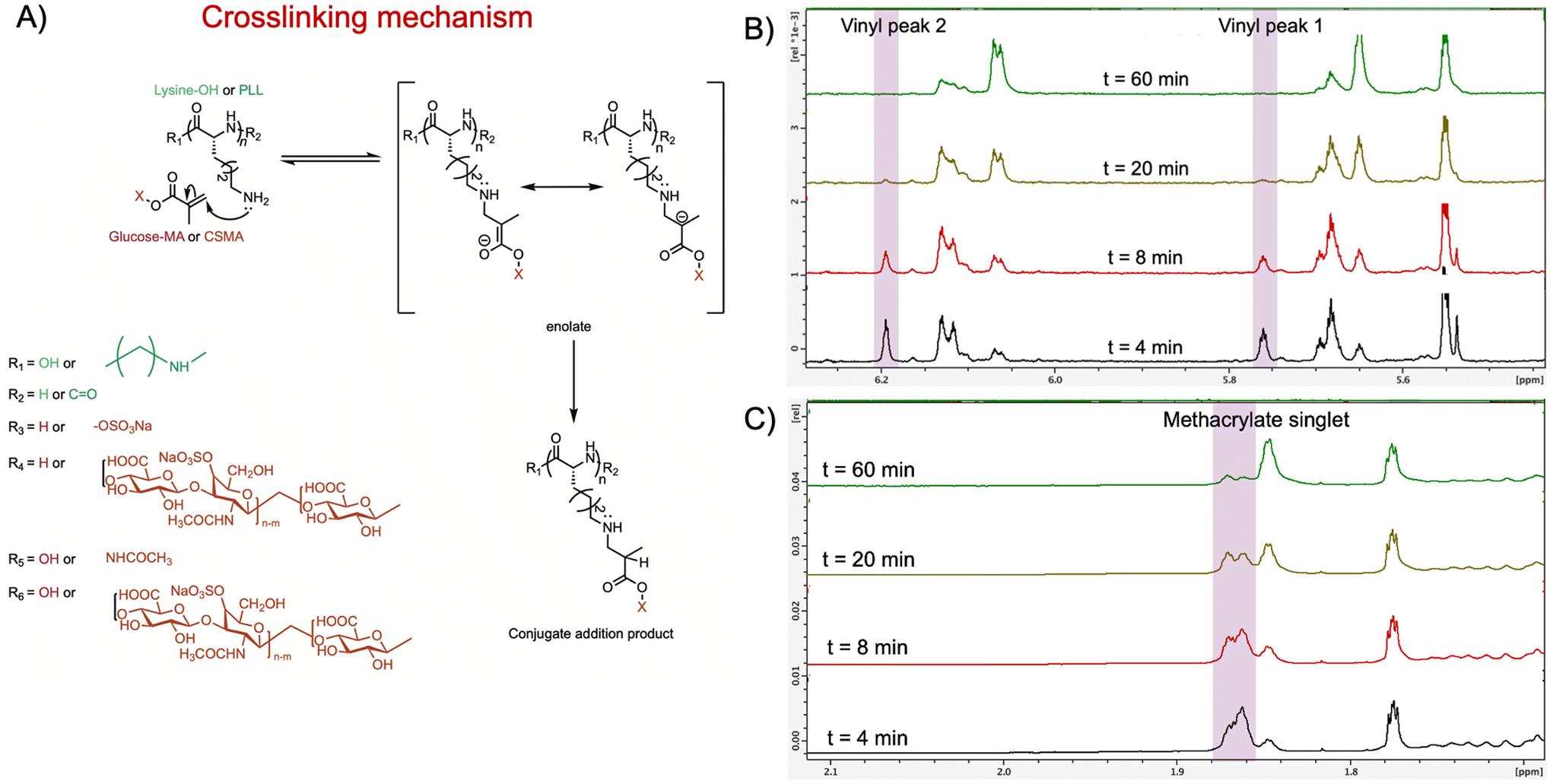 | ||
| Fig. 2 (A) Proposed mechanism illustrating the Michael addition reaction between PLL and the vinyl groups of CSMA. The primary amine of PLL performs a nucleophilic attack on the vinyl bond of CSMA, facilitating the addition. (B) Temporal changes in the vinyl proton signals of methacrylated compounds (e.g., glucose–MA or CSMA) monitored over 60 min upon interaction with primary amine-containing molecules (e.g., lysine–OH or PLL). (C) Evolution of the methyl group singlet peak in methacrylated compounds over a 60 min period, observed upon reaction with primary amine-containing molecules. | ||
D-Glucose functionalized with methacrylate (glucose methacrylate), serving as a mimic for CSMA, is reacted with L-lysine, which represented PLL. L-Lysine is added in excess to a solution of glucose–methacrylate in the presence of DMF as an internal standard. The vinyl proton signals (5.75 ppm and 6.19 ppm) and the methyl proton signal (1.86 ppm) from the methacrylate groups were monitored over a period of 60 min using 1H-NMR spectroscopy.
At the 4 min time point after mixing glucose–methacrylate with L-lysine, the vinyl proton and methyl proton signals were clearly observed. Over time, the intensity of the vinyl proton signals diminished, disappearing completely by the 60 min mark (Fig. 2B). This indicates that the vinyl groups reacted with the primary amines of L-lysine to form a conjugate addition product, as evidenced by new peaks appearing at 6.1 ppm and 5.7 ppm.
Similarly, the signal intensity for the methyl proton decreased, and the singlet began splitting, accompanied by the emergence of a new peak at 1.85 ppm (Fig. 2C). This new peak corresponds to the methyl group of the methacrylate, which shifted upfield due to the absence of the adjacent vinyl group. The splitting of the peak is attributed to the introduction of a new hydrogen atom adjacent to the carbon bonded to the methyl group in the conjugate addition product.
The viscoelastic and mechanical properties of CSMA–PLL hydrogels result from a dynamic interplay of covalent and non-covalent interactions between the CSMA and PLL chains. The covalent bonds, including those formed through free-radical polymerization and Michael addition, provide structural integrity and robustness to the network. Meanwhile, non-covalent interactions such as electrostatic forces and polymer entanglements contribute to the flexibility and adaptability of the hydrogel. This synergistic combination of interactions enables the hydrogel to exhibit both strength and flexibility, making it a promising material for applications in tissue engineering and regenerative medicine.
2.5. Interplay of PLL chain length and concentration in the CSMA–PLL hydrogels impacts the microarchitecture of the hydrogels
The microarchitecture of the hydrogels, often represented by pore size and pore area is crucial for replicating the native cartilage environment, as it directly influences nutrient diffusion, cellular infiltration, and mechanical resilience. However, a delicate balance must be maintained between structural integrity and biological properties as larger pores often lead to reduced mechanical properties but improved biological properties.40 Thus, determining structural morphology and pore structure of hydrogels is essential to ensure a balance between mechanical support and biological functionality, essential for effective cartilage repair and regeneration.The microstructural analysis of CSMA–PLL hydrogels is performed using scanning electron microscopy (SEM) (Fig. 3). The influence of increasing molecular weight between crosslinks (PLL chain length) and PLL concentration on the microarchitecture of the hydrogels is assessed. Evolution of pore structure and network density with increasing PLL chain length (left to right, A to C) and PLL weight percentage in the hydrogels (bottom to top, D to F) is presented (Fig. 3).
 | ||
| Fig. 3 SEM micrographs of freeze dried CSMA hydrogels incorporated with 5% w/w of PLL and varying PLL chain length (A) DP15; (B) DP35; and (C) DP50. SEM micrographs of freeze dried CSMA hydrogels incorporated with 2.5% w/w of PLL and varying PLL chain length (D) DP15; (E) DP35; and (F) DP50. (G) SEM micrograph of freeze dried neat CSMA hydrogel. Increasing molecular weight between crosslinks leads to reduced pore size. Similarly increasing PLL concentration (% w/w) leads to hydrogel network with smaller pores compared to that of CSMA. | ||
Panels A–C and D–F demonstrate that longer PLL chains lead to high molecular weight between crosslinks that produce a more uniform and dense pore structure, indicative of limited polymer flexibility and restricted entanglement. As the chain length decreases, the pores become larger and more irregularly shaped, resulting in a hydrogel network that appears to have increased porosity. This is likely due to enhanced chain mobility and greater electrostatic interactions between PLL and CSMA, leading to less compact crosslinking.
Increasing the PLL concentration enhances crosslinking density leading to increased pore frequency (Fig. S19A†) and reduces pore size (Fig. S19B†). At low PLL weight percentages, the network exhibits sparse crosslinking, resulting in larger pores (0.7–50 μm2, Fig. S19B†), comparable to CSMA (0.7–65 μm2, Fig. S19B†) (Fig. 3G). However, at higher concentrations, the increased number of electrostatic and covalent interactions between PLL and CSMA promotes a denser and more cohesive hydrogel matrix leading to smaller pores (0.7–25 μm, Fig. S20†). This is observed for hydrogels incorporated with all types of PLL chain lengths except PLL DP35 which exhibits relatively larger pores (2–180 μm, Fig. S18 and S20†). For mammalian cell diffusion, a pore size generally considered optimal is around 50–150 micrometer.41 Thus, based on the microarchitecture, CSMA–PLL hydrogels with 2.5% w/w PLL incorporation are suitable for cell diffusion and movement. Specifically, hydrogels with PLL DP15 and DP35 are the closest to neat CSMA hydrogel.
The interplay between PLL chain length and concentration directly impacts the microarchitecture of CSMA–PLL hydrogels. These findings underscore the potential to fine-tune hydrogel properties by modulating these parameters, paving the way for the development of customizable cartilage-mimicking hydrogels with desired mechanical and biological characteristics.
2.6. Shorter PLL molecular weight between crosslinks (lower PLL DP) leads to higher yield and compression strength
Cartilage-mimicking hydrogels are subjected to physiological load-bearing conditions upon injection into cartilage defect sites. Therefore, evaluating the compressive properties of CSMA–PLL hydrogels is crucial to ensure their mechanical durability and functionality under such conditions.The mechanical performance of CSMA–PLL hydrogels under compression is thus assessed using a rheometer. Using this technique, the impact of molecular weight between crosslinks and PLL %w/w on the yield and compression strength of the hydrogels is determined. Hydrogel cubes with surface area ranging from 10–25 mm2 (determined using ImageJ) are compressed at the strain rate of 0.02 mm s−1 and the corresponding load values are noted. Load normalized to hydrogel surface area is defined as compression strength. For compression testing, yield strength is the stress at which a material begins to deform plastically (Fig. 4A and D). Up to this point, the material deforms elastically, meaning it can return to its original shape when the stress is removed. Beyond the yield strength, permanent (plastic) deformation occurs. For cartilage, yield strength indicates the point at which the structure can no longer sustain deformation without irreversible damage. Ultimate compression strength represents the peak stress point on a stress–strain curve in compression testing (Fig. 4A and D).
 | ||
| Fig. 4 Mechanical properties of CSMA–PLL hydrogels upon compression. (A) Representative stress–strain curve depicting yield strength and ultimate compression strength. Comparison of median yield strength for CSMA–PLL hydrogels with varying PLL DP and PLL concentration in: (B) freshly prepared state and (C) swollen state. (D) Representative stress–strain curves for CSMA–PLL hydrogels with varying PLL DP and PLL concentrations. Median ultimate compression strength for CSMA–PLL hydrogels with varying PLL DP and PLL concentration in: (E) freshly prepared state and (F) swollen state. The box plot represents the 25th and 75th percentiles as the edges of the box, with the median indicated by the line inside the box. The error bars in the box plots correspond to the interquartile range (IQR), n = 3. | ||
Distinct mechanical behaviors of CSMA–PLL hydrogels demonstrate the importance of molecular weight between the crosslinks (PLL DP) at 5% w/w PLL incorporation (Fig. 4D). Hydrogels with a higher molecular weight between the crosslinks (e.g., DP50) exhibit a higher strain at yield strength compared to those with lower DP (e.g., DP15 and DP35) (Fig. S21†). This suggests that higher molecular weight between crosslinks lead to improved resistance to compressive deformation. This is likely due to increased non-covalent crosslinking density and enhanced network interactions.
Lower molecular weight between crosslinks results in an increase in yield strength. Yield strength of CSMA–PLL hydrogels in freshly prepared state is significantly affected by both PLL concentration and PLL DP (Fig. 4B). The yield strength of CSMA–PLL hydrogels ranges from 0.25–0.8 MPa, comparable to 0.46 MPa of neat CSMA hydrogels. Hydrogels containing 5% PLL showed consistently higher median yield strength across all DP levels compared to their 2.5% counterparts. For example, at DP35, the yield strength increased by approximately 30% when the concentration was increased from 2.5% to 5%. As the molecular weight between the crosslinks decreases, the yield strength increases – reducing the PLL DP from 50 to 15 enhances the yield strength from 0.25 MPa to 0.8 MPa, demonstrating the significant impact of crosslink density on mechanical performance. This trend highlights the reinforcing effect of PLL, with a higher concentration and lower molecular weight between crosslinks promoting greater initial resistance to mechanical deformation.
Ultimate compression strength (UCS) followed similar trends, with higher PLL concentrations and lower molecular weight between crosslinks (DPs) correlating with increased strength (Fig. 4E). Notably, hydrogels containing 5% PLL with DP15 exhibited the highest ultimate compression strength (∼3.8 MPa), nearly doubling the strength observed for 2.5% PLL with DP15 (∼1.5 MPa). This indicates that increased PLL content enhances load-bearing capacity, likely due to a more robust hydrogel matrix. Increasing the molecular weight between crosslinks significantly compromises the UCS. For instance, as the PLL DP increases from 15 to 50, the UCS decreases approximately threefold, from ∼3.8 MPa for DP15 to ∼1.5 MPa for DP 50.
Comparisons across DPs (DP15, DP35, DP50) and PLL concentrations (2.5% vs. 5%) reveal a synergistic relationship between these variables. While increasing PLL concentration improves mechanical properties across all PLL DP levels, the effect is most pronounced at lower PLL DPs. This observation suggests that both PLL chain length and concentration contribute to hydrogel network reinforcement, with lower DPs enhancing inter-polymer interactions and mechanical integrity.
Cartilage-mimicking CSMA–PLL hydrogels are susceptible to swelling and a subsequent deterioration in mechanical properties when exposed to aqueous physiological fluids in vivo. To evaluate the impact of swelling on compressive mechanical properties, CSMA–PLL hydrogels are tested for yield strength and UCS in their swollen state. A decrease in median yield strength is observed for swollen hydrogels with PLL DP15, while the yield strength remained relatively constant for DP35 and DP50. A similar trend was seen in the UCS of swollen CSMA–PLL hydrogels, where a reduction in median UCS occurred for shorter molecular weights between crosslinks, and an increase was noted for higher molecular weights.
In hydrogels with lower molecular weights between crosslinks (DP15), the network has a higher crosslink density. This increased density restricts the polymer chains’ ability to move freely, making them more susceptible to disruption and weakening when they absorb water.42 The higher crosslink density hinders the hydrogel's ability to accommodate the increased volume and flexibility associated with swelling, leading to a decrease in mechanical strength, particularly yield strength and UCS. In contrast, hydrogels with higher molecular weights between crosslinks (e.g., DP 50) possess longer polymer chains. These chains are more flexible and have a greater ability to distribute stresses across the network.42 When such hydrogels swell, the increased chain length provides enhanced stability and flexibility, allowing the network to maintain its integrity under compressive forces. The longer chains are able to accommodate swelling without significant degradation of mechanical properties, leading to less pronounced reductions in yield strength and UCS.
The improvements in mechanical properties with decreasing PLL DP and increasing concentration can be attributed to matrix rigidity owing to smaller molecular weights between the crosslinks. PLL, with its cationic nature and high molecular weight, likely forms strong electrostatic and covalent interactions within the hydrogel matrix, resulting in improved structural integrity and resistance to deformation (Scheme 2). The linear correlation between mechanical strength and PLL DP/concentration suggests that these parameters can be effectively tuned to meet specific mechanical requirements for cartilage or tissue engineering applications.
2.7. Structural integrity of CSMA–PLL hydrogels is retained after possible aqueous and enzymatic degradation
Given the limited regenerative capacity of damaged cartilage, it is crucial for scaffolds used for cartilage regeneration to maintain their mechanical properties in the long-term (weeks to years).43 Swelling and enzymatic degradation are the primary routes of hydrogel erosion and degradation, which can result in the loss of mechanical integrity. Thus, the impact of swelling and enzyme exposure on the viscoelastic properties of the CSMA–PLL hydrogels is assessed.To quantify swelling of the hydrogels, pre-weighed CSMA–PLL hydrogels are incubated in water at 37 °C for 24 h followed by reweighing to calculate the swelling percentage. The swelling percentage of the hydrogels is found to be dependent on PLL concentration and the molecular weight between the crosslinks (Fig. 5A). For hydrogels with 5% w/w PLL incorporation, the median swelling percentage is highest (360%) for hydrogels with PLL DP15 while it reduces to 164% for DP 35 and 168% for PLL DP 50. However, when the PLL concentration is lowered to 2.5% w/w, swelling percentage remain comparable for hydrogels with PLL DP15 and DP35 and reduces for those with DP50 (406% for DP15, 474% for DP35 and 50% for PLL DP50). This could be attributed to the interplay between crosslink density and chain flexibility. At lower PLL concentrations, the reduced crosslink density might cause longer PLL chains to dominate the network behavior, increasing the free volume and enhancing water uptake. Conversely, at higher PLL concentrations, the dense network structure due to higher crosslink density restricts swelling.
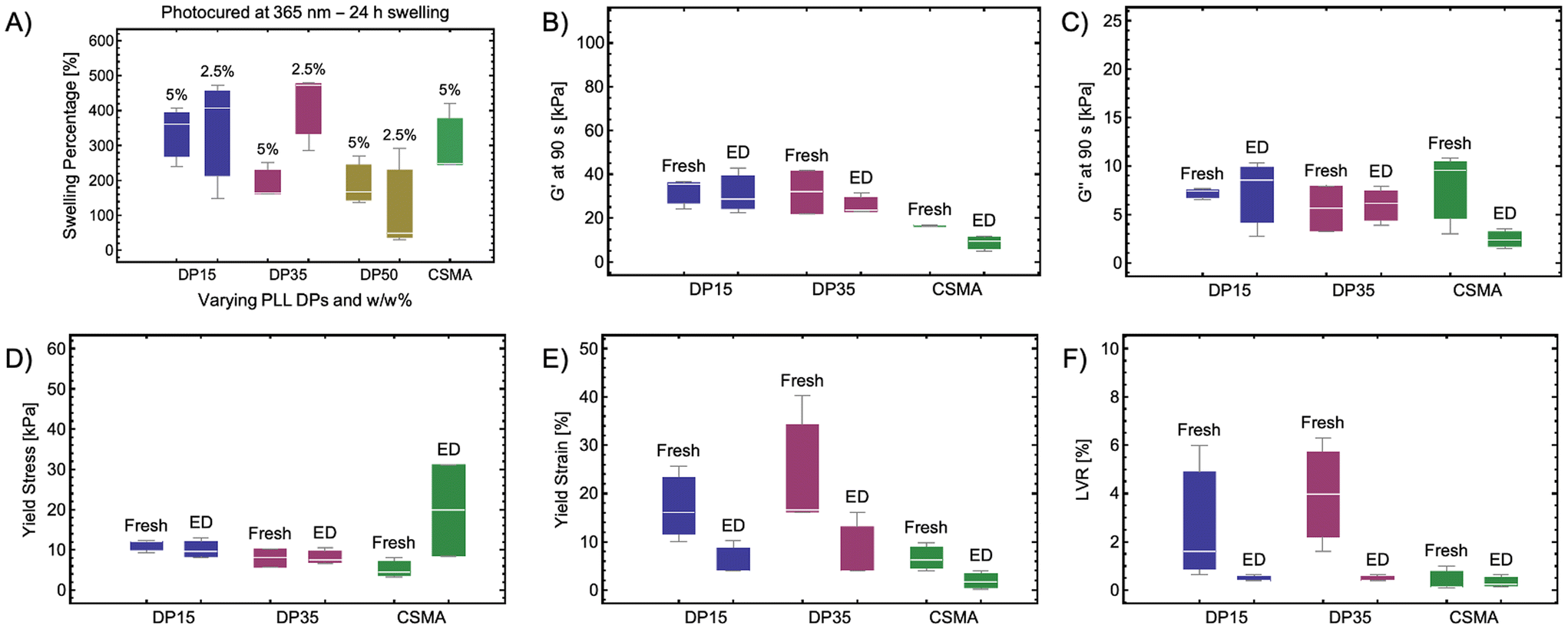 | ||
| Fig. 5 (A) Equilibrium swelling ratio of hydrogels after 24 h of immersion in water, presented as swelling percentage (%). Rheological properties of the hydrogels are used as an indicator of structural biodegradation upon incubation with chondroitinase ABC enzyme in aqueous buffer for 3 weeks. The rheological study assessed storage (G′) and loss modulus (G′′) at 1% strain and 10 Hz frequency followed by amplitude sweep ranging from 0.1–100% strain. Comparison of viscoelastic properties of hydrogels before (Fresh) and after enzyme-based degradation (ED): (B) G′; (C) G′′; (D) yield stress at break; (E) yield strain at break; and (F) linear viscoelastic range (LVR). The box plot represents the 25th and 75th percentiles as the edges of the box, with the median indicated by the line inside the box. The error bars in the box plots correspond to the interquartile range (IQR), n = 3. | ||
The viscoelastic properties of fresh hydrogels and enzymatically degraded (ED) hydrogels are compared to assess the impact of aqueous swelling and degradation. Mechanical properties of hydrogels that rely on structural integrity are used as indicator of structural biodegradation. Chondroitinase ABC is a depolymerizing lyase that cleaves the proteoglycans, chondroitin sulfate in this case, at the glycosidic bond via β-elimination.44 Hydrogels are thus incubated with chondroitinase ABC enzyme in aqueous buffer to evaluate the impact of degradation on mechanical properties. Previous studies have shown that crosslinked CS hydrogels experience a significant decline in viscoelastic properties after 24 h of enzyme exposure. Theoretical estimates indicate that complete degradation of the CS content within the hydrogel could occur in approximately 9 h (Section A.1, ESI†).9 To identify an appropriate experimental timeframe, tests were conducted using CSMA hydrogels incubated for varying durations, followed by rheological evaluation of G′ and G′′. A 3-week timepoint was selected as it represents a relevant period during which noticeable degradation (reduction in G′ and G′′) can be detected, allowing for a meaningful assessment of the hydrogel's long-term structural integrity (Fig. S27, ESI†).
The viscoelastic properties of CSMA–PLL hydrogels with 2.5% w/w PLL DP15 and DP35 are evaluated and compared to neat CSMA hydrogels. These specific formulations (2.5% w/w PLL DP15 and DP35) are selected for their enhanced viscoelastic performance compared to CSMA and porosity suitable to facilitate mammalian cell diffusion. The G′ (storage modulus) for both these hydrogels exhibit slight decrease in the median (25%), however no significant reduction is observed (Fig. 5B). The G′′ (loss modulus) for these hydrogels exhibit an increase of about 10–15%, not statistically significant (Fig. 5C). In case of CSMA, however, 43% reduction in G′ and 75% reduction in G′′ is observed indicating structural degradation.
All hydrogels exhibit reduction in median yield stress, yield strain and narrowing of linear viscoelastic range upon incubation with the enzyme (Fig. 5D–F). Overall, the yield stress for CSMA–PLL hydrogels remain comparable to their fresh counterparts and that of fresh CSMA suggesting no significant structural degradation due to the enzyme attack. Enzymatically degraded CSMA–PLL formulations and neat CSMA exhibit a 75% decrease in yield strain. This suggests reduction in matrix flexibility or increased brittleness. A similar trend is observed in the case of linear viscoelastic range (LVR%) wherein CSMA–PLL formulations demonstrate a decrease in LVR upon enzyme incubation while still being comparable to that of neat CSMA hydrogels.
The study examines the combined swelling and enzymatic effects on the structural degradation of CSMA–PLL formulations compared to that of CSMA. It is found that the hydrogels undergo structural degradation evident by the slight decrease in G′ and G′′, suggesting reduction in the rigidity of the hydrogels. Decrease in yield stress, yield strain, and LVR suggests reduced flexibility or increased brittleness due to structural degradation. Although enzymatic degradation might not be significant enough to impact the rigidity of the hydrogels, it certainly impacts the elasticity of these hydrogels. This could be attributed to the combined impact of enzymatic degradation and swelling. Enzymatic degradation preferentially breaks down CSMA chains. Since elasticity (related to flexibility and yield strain) depends more on the polymer chain mobility and the crosslinking density, minor enzymatic degradation can compromise the elastic properties without drastically affecting the overall rigidity (G′). Swelling on the other hand causes an expansion of the hydrogel network, increasing chain mobility and reducing the effectiveness of crosslinks. This stretching of crosslinking chains due to swelling likely impacts the elastic recovery of the network related to yield strain and elasticity, with the hydrogel retaining some structural rigidity.
While these findings provide insight into the structural degradation of CSMA–PLL hydrogels under swelling and enzymatic conditions, further studies are required to assess their long-term degradation behavior in physiologically relevant environments. Native cartilage ECM remodeling occurs over an extended timeframe, with collagen turnover taking decades and proteoglycan turnover occurring within years.45–47 In contrast, hydrogel degradation is influenced by polymer composition, crosslinking density, and enzymatic susceptibility, which may not fully replicate the remodeling dynamics of native cartilage. Future work will focus on extended degradation studies to evaluate the impact of prolonged enzymatic exposure and mechanical loading on the hydrogel network, with an emphasis on comparing its degradation profile to native cartilage ECM remodeling. Additionally, in vitro and in vivo assessments will be conducted to investigate the functional longevity of these hydrogels in cartilage–mimetic conditions. Understanding the interplay between enzymatic degradation, swelling effects, and mechanical performance over time will be crucial in optimizing hydrogel formulations for sustained cartilage repair applications and ensuring their clinical relevance.
2.8. CSMA–PLL hydrogels support mesenchymal stem cell viability both on the surface and upon in situ incorporation
To evaluate the impact of PLL concentration and the molecular weight between crosslinks (PLL chain length), adipose-derived human mesenchymal stem cells (AD-hMSCs) are cultured on the hydrogel surface in growth media for a period of 3 weeks. The percentage of metabolically active cells, relative to the control group (cells grown on tissue culture plates, set to 100%), is assessed after 3 weeks using resazurin assay. Cells seeded on CSMA hydrogel demonstrate approximately 100% viability (Fig. 6A), with the cells spreading across both the hydrogel surface and the adjacent plate area (Fig. 6B). | ||
| Fig. 6 Mesenchymal stem cells (hMSCs) seeded both on CSMA–PLL hydrogels and in the surrounding plate area remain viable. (A) Cellular viability (%) of adipose-derived human mesenchymal stem cells (AD-hMSCs) seeded on CSMA–PLL hydrogels after 3 weeks, as determined by resazurin assay. The box plot represents the 25th and 75th percentiles as the edges of the box, with the median indicated by the line inside the box. The error bars in the box plots correspond to the interquartile range (IQR), n = 3. (B) Overlay image of cellular nuclei (blue fluorescence, Hoechst 33342), cytoplasm (green fluorescence, calcein–AM), and dead cells (red fluoroscence, propidium Iodide) for the hMSCs cultured on CSMA hydrogels and plate surface around the hydrogel. (C) Images depict hMSCs cultured on CSMA–PLL hydrogels with varying PLL concentrations and chain lengths, as well as on the surrounding plate surface, showing overlays of cellular nuclei, cytoplasm, and dead cells. | ||
Among the CSMA–PLL hydrogels, those with PLL DP35 exhibit the highest mean cell viability (80–100%), followed by those with PLL DP15 (70–75%, Fig. 6A). The higher viability in case of hydrogels with PLL DP35 can be attributed to the relatively higher pore size (Fig. 3) and rigidity (G′, Fig. 1) compared to other hydrogels. In these cases (PLL DP15 and PLL DP35), PLL concentration does not significantly impact cell viability.
Hydrogels with PLL DP50 show the lowest mean cell viability, with a further reduction observed when PLL concentration is increased to 5% w/w (from 56% viability at 2.5% PLL to 26% at 5%, Fig. 6A). This could be attributed to higher concentrations of positively charged primary amines available for interaction with mammalian cells in case of hydrogels with PLL DP50. This is consistent with the viscoelastic and mechanical properties of hydrogels wherein those with PLL DP50 exhibit lower rigidity (lower physical and chemical crosslinking) compared to that of PLL DP15 indicating a higher concentration of free primary amines. Longer PLL chains (e.g., DP50) are known to exert higher cytotoxic effects due to their strong cationic charge density, which can disrupt cell membranes through electrostatic interactions, leading to cell lysis and apoptosis. This phenomenon is commonly observed in polycationic biomaterials, where excessive positive charge enhances interactions with negatively charged phospholipids, resulting in membrane destabilization.48,49 Osmotic shock has been reported as important contributor to cell death for low molecular weight polycations, whereas for higher molecular weight analogues cell membrane damage due to direct interactions between the polycations and the cell membrane were found to play a role.50 Overall, the reduced viability observed in PLL DP50 hydrogels is likely the result of multiple interrelated factors, including increased cytotoxicity, impaired adhesion, osmotic stress, sequestration of bioactive molecules, and altered mechanical properties. Future studies should investigate surface charge modulation strategies or PLL concentration optimization to mitigate these effects and improve the biocompatibility of long-chain PLL hydrogels for biomedical applications.
CSMA–PLL hydrogels support the attachment and expansion of hMSCs both on the hydrogel surface and the adjacent plate surface, indicating that neither the hydrogel surface nor anything released from the hydrogels are toxic to the cells (Fig. 6C). Because of their lack of toxic leachates coupled with their superior cell viability, neat CSMA and CSMA–PLL hydrogels with 2.5% w/w PLL DP15 and DP35 incorporation are selected for further studies.
For clinical applications targeting cartilage defects, a promising strategy involves encapsulating cells within injectable hydrogels that can be delivered directly to the defect site and subsequently crosslinked on demand through light activation. However, it is crucial to assess the viability of cells encapsulated in the hydrogels, as factors such as light exposure, the crosslinking mechanism, presence of photoinitiator, and direct contact with hydrogel components may contribute to cell toxicity. To assess this, hMSCs are mixed with the hydrogels (neat CSMA, CSMA–PLL with 2.5% w/w PLL DP15 and DP35 incorporation), followed by 30 seconds of light exposure to crosslink the hydrogels. The hydrogels are then incubated with growth media (Fig. 7) and chondrogenic media (Fig. 8) for 3 weeks.
 | ||
| Fig. 7 After 3 weeks of culture in growth media post-encapsulation, hMSCs are distributed throughout the hydrogels. (A) Cellular viability (%) of AD-hMSCs encapsulated in CSMA–PLL hydrogels after 3 weeks of incubation, as determined by resazurin assay. The box plot represents the 25th and 75th percentiles as the edges of the box, with the median indicated by the line inside the box. The error bars in the box plots correspond to the interquartile range (IQR), n = 3. (B) Overlay image of cellular nuclei (blue fluorescence, DAPI, Hoechst 33342), cytoplasm (green fluorescence, GFP, calcein–AM), and dead cells (red fluoroscence, RFP, propidium iodide) for the hMSCs cultured on tissue culture plate surface in growth media. (C) Images show AD-hMSCs encapsulated in CSMA–PLL hydrogels after 3 weeks of culture in growth media, with overlays highlighting cellular nuclei, cytoplasm, and dead cells. | ||
 | ||
| Fig. 8 After three weeks of culture in chondrogenic media post-encapsulation, hMSCs are uniformly distributed throughout the hydrogels. Z-Stack overlays of cellular nuclei (blue fluorescence: DAPI or Hoechst 33342), cytoplasm (green fluorescence: GFP or calcein–AM), and dead cells (red fluorescence: RFP or propidium iodide) reveal consistent viability and spatial distribution. Hydrogels containing 2.5% PLL with DP15 and DP35 exhibit widespread live cell presence throughout the matrix, comparable to the distribution observed in control CSMA hydrogels. | ||
For the hydrogels incubated in growth media, after 3 weeks, the cell viability (%) is determined by using the resazurin assay. Like the ex situ crosslinked hydrogels, CSMA–PLL hydrogels containing hMSCs crosslinked in situ exhibit similar cytocompatibility. CSMA–PLL hydrogels with PLL DP35 and neat CSMA hydrogel exhibit 80–100% cell viability. On the other hand, CSMA–PLL with DP15 exhibit reduced cell viability (20–40%) (Fig. 7A). Since the resazurin assay relies on the diffusion of resazurin into the hydrogel to interact with cells and the subsequent release of resorufin into the media for detection, the lower viability observed in PLL DP15 hydrogels may be attributed to their higher rigidity (higher yield and compression strength due to shorter molecular weight between the crosslinks and narrow pore area distribution), which may hinder resazurin/resorufin diffusion. A highly crosslinked network may alter nutrient diffusion and waste removal dynamics, creating a less favorable microenvironment for cell survival.51,52 To further confirm cell viability, live/dead staining is performed, allowing direct imaging of cellular components.
The cells are then stained to visualize cell components: nuclei (blue fluorescence, DAPI channel, Hoechst 33342), cytoplasm (green fluorescence, GFP channel, calcein–AM), and dead cells (red fluorescence, RFP channel, propidium iodide). The stained cells are imaged to observe their distribution within the hydrogel matrix. CSMA–PLL hydrogels with both DP15 and DP35 show live hMSCs distributed throughout the matrix, as indicated by the green fluorescence (Fig. 7C). The cells exhibit a circular morphology, suggesting they have not yet spread within the matrix. In contrast, cells in neat CSMA hydrogels display an elongated morphology, indicating cell expansion (Fig. 7C). This difference may be attributed to the increased rigidity of the CSMA–PLL hydrogels due to the addition of PLL. Previous studies have reported that hMSCs are more likely to remain circular in hydrogels with higher rigidity (G′ around 20 kPa) and spread more easily in hydrogels with lower rigidity.16,53
After 3 weeks, hydrogel-encapsulated cells incubated in chondrogenic media are also stained using the above protocol. Z-Stack images are acquired to study hMSCs distribution within the hydrogel matrix. Neat CSMA and CSMA–PLL hydrogels with both DP15 and DP35 show live hMSCs distributed throughout the matrix, as indicated by the green fluorescence (Fig. 8). The cells display a circular morphology, suggesting limited spreading within the matrix. Calcein–AM staining further confirms that the hydrogels support a comparable number of live cells per unit area, indicating similar levels of cytocompatibility across formulations (Fig. S24†).
2.9. Chondrogenic differentiation of hMSCs in hydrogels
hMSCs encapsulated in hydrogels and cultured for eight weeks in chondrogenic media are stained with Sirius Red F3B to assess collagen production during chondrogenic differentiation. Sirius Red F3B specifically stains collagen by electrostatically binding to lysine and arginine residues, with picric acid enhancing specificity by preventing non-collagenous staining.54 Under polarized light, birefringence confirms the presence of aligned collagen fibers.54,55 Sirius Red F3B is expected to interact with the positively charged lysine moieties of PLL, potentially interfering with collagen deposition by hMSCs undergoing chondrogenic differentiation. However, the absence of birefringence in PLL allows for the clear distinction between oriented collagen fibers, observable under crossed polarizers (Fig. S24†).Pericellular high-chroma red staining was observed in hMSCs cultured on tissue culture plates and within CSMA hydrogels, with collagen deposition aligning along cellular morphology. Birefringence under cross-polarized light further confirms collagen fibril organization. However, CSMA–PLL hydrogels (PLL DP15 and DP35) appeared uniformly red in brightfield and parallel polarizer views, likely due to Sirius Red F3B interacting with PLL lysine groups, making it difficult to distinguish deposited collagen from PLL itself. The uniform staining suggests an even PLL distribution throughout the hydrogel.
Notably, CSMA–PLL hydrogels without hMSCs also stained red under brightfield and parallel polarizers but lacked birefringence (Fig. S24†), unlike hMSC–laden hydrogels (highlighted by yellow arrows, Fig. 9). This confirms that collagen deposition and chondrogenic differentiation occurred in CSMA–PLL hydrogels with encapsulated hMSCs. Quantification of collagen area per cell revealed that hMSCs encapsulated in CSMA and CSMA–PLL (DP35) hydrogels exhibited collagen deposition comparable to that of hMSCs cultured on TCPS. Hydrogel encapsulation of hMSCs significantly suppresses osteogenic differentiation compared to traditional 2D culture on tissue culture plastic. Human mesenchymal stem cells (hMSCs) cultured on tissue culture plastic (TCPS) as a control or encapsulated in CSMA and CSMA–PLL hydrogels are assessed for osteogenic differentiation. After 7 days in growth media, the cells are maintained in chondrogenic differentiation media for an additional 3 weeks. To evaluate endochondral ossification, hMSCs are then stained for alkaline phosphatase (ALP), an osteoblast marker.56 ALP staining, which appears red, is minimal in all hydrogel-encapsulated groups, indicating a lack of endochondral ossification.9 Quantification showed that hMSCs on TCPS exhibited an ALP concentration of approximately 20 μg mL−1, whereas those in CSMA and CSMA–PLL hydrogels had significantly lower levels (Fig. S25†). These findings suggest that hydrogel encapsulation effectively reduces osteogenic differentiation compared to TCPS culture.
 | ||
| Fig. 9 High-magnification brightfield images of Sirius Red F3B-stained hydrogels show hMSCs distributed throughout the matrix after 8 weeks of culture in chondrogenic media, exhibiting high collagen expression. Pericellular, high-chroma red staining indicates collagen production and deposition. True-color images of the same field of view were captured under parallel polarizers (top) and crossed polarizers (bottom). The weak, non-green birefringence observed is characteristic of type II collagen, distinguishing it from type I and type III collagens. | ||
Our findings indicate that the CSMA–PLL hydrogels exhibit promising characteristics for cartilage repair; however, further investigations are necessary to confirm their long-term stability and functional performance. While the current study provides initial insights, future work will focus on conducting in vivo validation and extracellular matrix (ECM) characterization to assess their biological integration and durability. Additionally, key molecular markers such as SOX9, Aggrecan, and Type II Collagen expression will be evaluated to determine the extent of cartilage-specific differentiation. These future studies will provide a more comprehensive understanding of the therapeutic potential of these hydrogels in cartilage regeneration.
3. Conclusion
This study presents a double network hydrogel using synthetic polypeptides in combination with methacrylated chondroitin sulfate. We developed an injectable material capable of filling cartilage defects, providing mechanical support, and enabling in situ crosslinking. These hydrogels can serve as a scaffold for stem cells to promote cartilage regeneration. The incorporation of poly(L-lysine) enhances the storage and loss modulus of the hydrogels, contributing to their improved viscoelastic and mechanical performance.The results indicate that incorporating PLL into CSMA-based hydrogels creates a double network structure characterized by a combination of physical and covalent interactions. While the primary crosslinking mechanism in pure CSMA hydrogels involves free radical polymerization creating covalent bonds between CSMA chains, the inclusion of PLL introduces additional crosslinking mechanisms. In CSMA–PLL hydrogels, physical entanglement and electrostatic interactions occur between the carboxylate groups of CSMA and the positively charged amines of PLL. Furthermore, covalent bonds are formed via Michael addition, where the primary amines of PLL react with the vinyl groups of methacrylates in CSMA. These combined interactions significantly enhance the structural and functional properties of the hydrogels, making them suitable candidates for cartilage tissue engineering.
The CSMA–PLL hydrogels display a dynamic viscosity range of 0.03 to 0.4 Pa·s, making them injectable and suitable for minimally invasive delivery directly at the defect site. Their viscous nature enables them to conform precisely to the shape of the defect, while subsequent light exposure triggers in situ crosslinking. Notably, the hydrogels undergo a rapid transition from a viscous to elastic state in less than 5 seconds upon light activation.
Incorporating PLL into the hydrogels results in enhanced viscoelastic properties, with increased storage modulus, loss modulus, and yield strength, though it reduces the yield strain. The viscoelastic properties of the CSMA–PLL hydrogels, particularly the storage modulus (G′) in the range of 100–300 kPa, closely approximate those of native cartilage. This enhancement in rigidity, however at the expense of flexibility, can be attributed to the increased covalent crosslink density facilitated by PLL.
CSMA–PLL hydrogels exhibit tunable yield and compression strength achieved by varying the PLL %w/w and molecular weight between the crosslinks (PLL chain length DP). Decreasing the PLL DP and increasing its concentration leads to increased compression and yield strength which can be attributed to greater matrix rigidity due to shorter molecular chains between crosslinks. The hydrogels also retain their mechanical properties upon swelling, unlike traditional hydrogels which disintegrate due to swelling. Furthermore, the correlation between mechanical strength and PLL DP/concentration underscores the tunability of these parameters to meet specific mechanical demands for cartilage or tissue engineering applications, making these hydrogels highly adaptable for therapeutic use.
Furthermore, when exposed to chondroitinase ABC enzyme in aqueous media, CSMA–PLL hydrogels maintain their viscoelastic properties, whereas neat CSMA hydrogels exhibit a significant decline in both storage (G′) and loss (G′′) moduli. This highlights the improved structural stability of CSMA–PLL hydrogels under enzymatic degradation, making them promising for long-term applications in cartilage repair.
CSMA–PLL hydrogels demonstrate excellent cytocompatibility, supporting hMSC viability (>80%) and attachment, with minimal cytotoxicity from hydrogel components or leachates. Encapsulated hMSCs remain viable and uniformly distributed within the matrix after 3 weeks of culture in both growth and chondrogenic media. While neat CSMA hydrogels promote greater cell spreading due to lower rigidity, CSMA–PLL hydrogels maintain cells in a circular morphology, reflecting their higher stiffness. CSMA–PLL hydrogels also exhibit signs of chondrogenic differentiation as confirmed by collagen staining and ALP assay. These results highlight the potential of CSMA–PLL hydrogels for cell encapsulation and cartilage regeneration applications. The primary amine groups of PLL offer versatile functionalization opportunities with short peptides or therapeutics, enabling their gradual release at the defect site. This approach would render the hydrogels cell-instructive, fostering cartilage regeneration.
4. Experimental
4.1. Materials
Chondroitin sulfate A sodium salt (90%, 10814-454) and methacrylic anhydride (94%, stabilized with 0.2% 2,4-dimethyl-6-tert-butylphenol) was purchased from Thermo Scientific. Methacrylic acid (stabilized with hydroquinone monomethyl ether), hexylamine, sodium acetate and resazurin sodium salt were purchased from Sigma-Aldrich. Lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) was purchased from TCI chemicals. D-Glucose was purchased from L-lysine (99.1%) was purchased from CHEM-IMPEX INT'L INC. Tris was purchased from G biosciences and bovine serum albumin was purchased from VWR Lifescience. Phosphate-buffered saline (PBS, 10×, pH 7.4, liquid), dichloromethane (DCM) was purchased from Fisher; acetone, methanol and ethanol were purchased from Pharmco. Dimethylformamide (DMF), hydrobromic acid (HBr) and sodium hydroxide pellets (NaOH) were purchased from VWR Chemicals BDH®. DIC and Trifluoroacetic acid (TFA) was purchased from Oakwood chemicals. Pierce SnakeSkin™ Pleated Dialysis Tubing, MWCO 3500 to 10000 was purchased from Thermo Scientific. NMR solvents deuterated water (D2O), dimethyl sulfoxide (DMSO-d6) and chloroform-d (CDCl3) were purchased from Cambridge Isotope Laboratories, Inc.
4.2. Instrumentation and software
A 500 MHz NEO Bruker nuclear magnetic resonance (NMR) spectrometer and TopSpin software were used to acquire NMR spectra of reactants and products. TopSpin was used to extract data from NMR experiments. A Tecan Spark plate reader with SparkControl v2.2 software was used to acquire ultraviolet–visible (UV–Vis) absorption and fluorescence spectra for cytocompatibility experiments. TA instruments Discovery HR-2 hybrid rheometer was used for rheology and axial compression. SEM imaging was performed using FEI Quanta 600 FEG SEM. Fluorescence imaging was performed using an EVOS FL Auto Cell Imaging System (Thermo Fisher Scientific) equipped with a 10×, 0.40 numerical aperture objective. Wolfram Mathematica 13.0.1.0, Mac OS X ARM 64-bit was employed for data analysis and plotting.4.3. Synthesis
PLL(Z) was deprotected to remove the Z protecting group and yield PLL. PLL(Z) (1.15 g) and trifluoroacetic acid (16 mL) were added to a round bottom flask and stirred until the peptide was completely dissolved. Then, HBr (10 mL, 48% v/v in H2O) was added, and the solution was stirred overnight. After overnight stirring, distilled water (15 mL) was added, and the reaction was stirred for another 3 h. Next, the reaction solution was precipitated into ice cold tetrahydrofuran and filtered to give PLL as a white solid. The resulting solid was loaded into dialysis tubing and dialyzed against DI water for 2 days. Then, the dialyzed PLL was frozen to −80 °C and lyophilized for 3 days until dry. This material was used as the free (unconjugated) PLL for hydrogel formulation.
4.4. Formulation of CSMA–PLL hydrogels
Hydrogels were prepared at concentrations ranging from 8–13% w/w. The CSMA hydrogel was formulated at 16% w/w in distilled water. PLL solution was prepared at 20% w/w, and the photoinitiator (LAP) at 0.5% w/w. For neat CSMA hydrogels, 8% CSMA and 0.05% LAP were combined with 92% distilled water and vortexed thoroughly. For CSMA–PLL hydrogels, varying w/w percentages of CSMA, PLL, and 0.05% LAP were weighed and mixed accordingly (Table 1).4.5. Real-time photorheometry
Rheological measurements were performed using a TA Instruments Discovery HR-2 hybrid rheometer equipped with a UV-curing accessory, a disposable 8 mm parallel plate (PP8) probe, and a quartz sample holder (Fig. S11A†). Omnicure™ 2000 Light Guide was used as the UV/Visible light source with wavelength ranging from 320–500 nm. The rheometry was performed at UVA 365 nm and UV/Vis light combination (320–500 nm). To achieve a wavelength of 365 nm, the light guide was connected to a collimator (LGC-019-022-07-V) and an optical bandpass filter (365 nm, 15131, Filter BP OD4 10 nm, 25 mm diameter, Edmund Optics). The applied UVA intensity (365 nm) was calibrated to 100 mW cm−2 with an AMTAST UV Radiometer (Model: UVA365). Hydrogels were deposited on the quartz sample holder to cover its surface. The top geometry was lowered, and shear storage modulus (G′) and shear loss modulus (G′′) were acquired. Gelation was defined as the crossover time at which G′ became larger than G′′. The parameters of dynamic oscillatory strain were set as follows: 0.2 mm measuring gap, 1% amplitude, and 10 Hz frequency. The storage modulus (G′) and loss modulus (G′′) were recorded during the initial 30 seconds to assess dynamic viscosity (Phase I), followed by photocuring under UVA irradiation for 200 seconds and continued monitoring until 670 seconds to capture the post-curing plateau (Phase II). An amplitude sweep ranging from 1% to 1000% strain at 1 Hz was then conducted (Phase III), as shown in Fig. S1A in the ESI.†4.6. Determination of crosslinking reaction mechanism
To confirm the Michael addition reaction between primary amines of PLL and vinyl groups of CSMA, small molecular weight monomers similar in structure with the repeat unit of these polymers were employed. L-Lysine (Fig. S13†) was used to represent the monomer of PLL and D-glucose (Fig. S14†) was used to represent the saccharide monomer of CS. D-glucose was methacrylated using DIC coupling with methacrylic acid to obtain glucose methacrylate.Briefly, D-glucose (200 mg, 1.1 mmol, 1 eq.) was dissolved in 10 mL of double-distilled water. Once fully dissolved, 5 mL THF was added. DIC (705 mg, 5.5 mmol, 5 eq.), and methacrylic acid (473 mg, 5.5 mmol eq.) were added to the solution, and the reaction was stirred at room temperature for 24 h. The resulting mixture was filtered to remove the insoluble urea byproduct. The aqueous filtrate was extracted with dichloromethane (DCM) to remove unreacted methacrylic acid. To eliminate any residual DCM, the aqueous phase was subjected to vacuum drying. The resulting glucose–methacrylate (glucose–MA) solution was frozen at −80 °C and lyophilized for 3 days until completely dry. Successful methacrylate substitution on D-glucose was confirmed using 1H-NMR, and the degree of substitution was quantified by 1H-NMR analysis (Fig. S15 and S16†).
To confirm the occurrence of the Michael addition reaction, glucose–MA and L-lysine were reacted in D2O within an NMR tube at room temperature. The changes in the methacrylate peaks of glucose–MA were monitored using 1H-NMR for 60 min, with one scan collected every 2 min. For the initial scan (t = 0), glucose–MA (80 mg) was dissolved in D2O (1.2 mL), and DMF (10 μL) was added as an internal standard before scanning (Reference). Subsequently, 0.6 mL of the reference solution was added, followed by the introduction of L-lysine (8.5 mg, 0.06 mmol) into the tube. The reaction was monitored over a 60 min period, from t = 0 to t = 60 min. The methacrylate peaks (vinyl protons and methyl protons) were integrated to track changes in their concentration relative to the internal standard (DMF).
4.7. Scanning electron microscopy (SEM) to determine structural morphology of the CSMA–PLL hydrogels
Neat CSMA and CSMA–PLL hydrogels were prepared by UV crosslinking 300 mg of hydrogel material in a plastic mold for approximately 5 min. After crosslinking, the hydrogels were removed from the mold and cut into nine equal-sized cubes using a blade. The cubes were frozen at −80 °C and lyophilized for two days to preserve their 3D structure upon drying.The dried hydrogels were mounted on 12.5 mm aluminum SEM sample studs using carbon tape. Prior to imaging, the samples were coated with a 2 nm layer of gold or platinum using a plasma sputter coater (EMS Q300T D), depending on material availability. Imaging was performed with a 30 kV landing voltage using a FEI Quanta 600 FEG SEM. SEM images were captured to analyze the structural morphology of neat CSMA and CSMA–PLL hydrogels and to evaluate the effects of PLL weight percentage (% w/w) and chain length on the hydrogel morphology.
4.8. Determining the swelling behavior of CSMA–PLL hydrogels
Equilibrium swelling was assessed by measuring the weight of hydrogels immersed in a semi-infinite bath of deionized (DI) water for 24 h. Neat CSMA and CSMA–PLL hydrogels were prepared by UV crosslinking 300 mg of hydrogel material in a plastic mold for approximately 5 min. After crosslinking, the hydrogels were removed from the mold and divided into nine equal-sized cubes using a blade.Each hydrogel cube was placed into a pre-weighed scintillation vial, and its initial mass (Wi) was recorded. The hydrogels were then submerged in DI water (2 mL) and allowed to swell to equilibrium over 24 h. After this period, the water was carefully decanted, and any residual water was removed using a micropipette. The swollen mass (Ws) of each hydrogel was measured.
The equilibrium swelling ratio (ESR) was calculated using the following equation:
 | (1) |
4.9. Determining the mechanical properties of CSMA–PLL hydrogels
During testing, hydrogel samples were placed on the bottom geometry, and the top geometry was lowered to a position just above the sample. A pre-force of 0.5 N was applied, followed by controlled lowering of the top geometry at a rate of 0.02 mm s−1. The axial force was monitored as the samples were compressed to 80% strain or until a minimum gap of 0.1 mm was reached. Load was divided by hydrogel area to determine the stress.
After the incubation period, the viscoelastic properties of the hydrogels were measured using a rheometer. The rheological measurements included the shear storage modulus (G′) and shear loss modulus (G′′), which were recorded by lowering the top geometry onto the samples. Dynamic oscillatory strain parameters were set as follows: pre-force of 0.5 N, amplitude of 1%, and frequency of 10 Hz. The storage modulus (G′) and loss modulus (G′′) were initially evaluated over a 100-second period. An amplitude sweep from 1% to 100% strain at 1 Hz was subsequently performed to determine the yield stress, yield strain, and linear viscoelastic range (LVR) of the hydrogels. Identical hydrogel samples that had not been exposed to the enzyme served as fresh controls for comparison with the enzyme-degraded hydrogels.
4.10. Determination of cytocompatibility of CSMA–PLL hydrogels
 | (2) |
Viability was also assessed for hMSCs encapsulated within CSMA hydrogels or CSMA–PLL hydrogels after light-induced gelation. For the formulation of cell–laden hydrogels, the base hydrogel comprised 400 mg of CSMA (16% w/w), 40 μL of a 0.75% LAP solution, and 80 μL of distilled water. hMSCs-loaded CSMA–PLL hydrogels were formulated by mixing 17.5 μL of base hydrogel with 2.5 μL of PLL solution (20% w/w) and 5 μL of cell suspension (106 cells), followed by UVA (365 nm) irradiation for 30 seconds to induce gelation. Neat CSMA hydrogels were formulated by mixing 20 μL of base hydrogel and 5 μL of cell suspension (106 cells), followed by UVA (365 nm) irradiation like other hydrogels. The hMSCs-loaded hydrogels were incubated in 500 μL of culture media at 37 °C for 3 weeks, with media changes performed every 7 days. The study was conducted using both growth media and chondrogenic media. For chondrogenic differentiation, cells were first cultured in growth media for 1 week, after which the media was switched to chondrogenic media at the first media change. Cells were maintained in chondrogenic media for 3 weeks before cytocompatibility assessment. To ensure accurate differentiation, all hMSCs reagents were free from phenol red, as it is known to interfere with differentiation. Resazurin assays were performed as described earlier to determine the percentage of cell viability in the hydrogels.
• DAPI light cube (Thermo Fisher Scientific, no. AMEP4650): excitation at 357/44 nm, emission at 447/60 nm
• GFP light cube (Thermo Fisher Scientific, no. AMEP4651): excitation at 470/22 nm, emission at 510/42 nm
• RFP light cube (Thermo Fisher Scientific, no. AMEP4652): excitation at 531/40 nm, emission at 593/40 nm
Bright-field images were also captured under identical imaging parameters for all samples. These fluorescence images provided a detailed visualization of live, dead, and total cell populations on and within the hydrogels.
Alkaline phosphatase labeling. For alkaline phosphatase (ALP) labeling, the hMSCs were washed with PBS (2×, 15 min each) and fixed in 3.7% formaldehyde for 15 min. Followed by this, the hMSCs were washed with PBS (2×, 15 min each). The SensoLyte pNPP Alkaline Phosphatase Assay Kit Colorimetric from AnaSpec (catalog #AS-72146) was used for quantitative measurement of alkaline phosphatase levels in the samples. 1× assay buffer was prepared by diluting the 10× assay buffer with deionized (DI) water. A standard curve was produced using the provided ALP standard. The standard (10 μg mL−1) was diluted 1
:50 in 1× assay buffer to achieve a 0.2 μg mL−1 (200 ng mL−1) concentration. Serial dilutions were performed to achieve 100, 50, 25, 12.5, 6.25, 3.125, 1.5625, 0.78125 ng mL−1 concentrations, and these solutions were added to wells of a 96 well plate in triplicate with a 50 μL volume. A 50 μL of p-nitrophenyl phosphate (pNPP) solution was added to each well, and the plate was gently shaken for 30 s. After 45 min at room temperature, 50 μL of stop solution was added to each well. The absorbance at 405 nm was measured and plotted against ALP concentration. A best fit linear line was generated.
0.2% Triton X-100 lysis buffer was prepared by adding 10 μL Triton X-100 to 5 mL 1× assay buffer, and vortexed to mix. On day 28 of the experiment, the plate was removed from the incubator for the ALP assay. Media was aspirated and wells were washed twice with 500 μL of 1× assay buffer for 15 min each. A 200 μL lysis buffer was added to each well, and the plate was incubated at 4 °C for 30 min. The plate was sealed with parafilm and sonicated for 10 min, taking care to keep the water level below the top of the wells. The plate was dried off and incubated at −20 °C for 30 min. The liquid was aspirated from each well, transferred to separate 1 mL microcentrifuge tubes, and centrifuged at 2500g and 4 °C for 10 min. The supernatant was aspirated from each tube, taking care to avoid the debris pelleted at the bottom, and was transferred into new wells on the 96 well plate. A 50 μL pNPP solution was added to each well, and the plate was gently shaken for 30 s. After 45 min at room temperature, the absorbance at 405 nm was measured, and the best fit line was used to convert absorbance to concentration of ALP in the samples.
Collagen labeling. Collagen labeling was performed using methods developed and described in the literature.9 The hMSCs were washed with PBS, pH 7.4 (2×, 15 min each) and fixed in 3.7% formaldehyde for 15 min. The hMSCs were washed again in PBS (2×, 15 min each) and exposed to 0.1% sirius red F3B (i.e., Direct Red 80, # AAB21693-06, Alfa Aesar) in saturated picric acid (# P6744-1GA, Sigma-Aldrich) for 1 h. Then they were rinsed twice in 0.5% acetic acid in DI water, washed three times for 30–60 s each with agitation in 100% ethanol. Brightfield images and color images were acquired of the samples both with parallel (0°) and with crossed polarizers (90°). The stained area observed under cross-polarized light (birefringence) was quantified as a percentage of the total hydrogel area using ImageJ (see Section A.2†). This collagen percentage was then normalized to the number of corresponding cells in each group, and the resulting % collagen area per cell was reported.
4.12. Statistical analysis
Data are presented as box plots, with the box edges representing the 25th and 75th percentiles and the median shown as a line within the box. Error bars indicate the interquartile range (IQR), with n = 3. Statistical significance was assessed using the non-parametric Mann–Whitney U test (Wolfram Mathematica 13.0.1.0, Mac OS X ARM 64-bit). A p-value of <0.05 (*) was considered statistically significant. Due to the small sample size (n = 3–5), normality could not be reliably assessed; thus, the Mann–Whitney U test, which does not assume normal distribution and is suitable for small sample sizes, was used.Conflicts of interest
The authors declare no conflict of interest.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This research received support from start-up grants provided by Carnegie Mellon University (S. A. S.). We extend our gratitude to Roberto Gil for granting access to the NMR facilities, which were partially funded by NSF grants # CHE-9808188, CHE-1039870, and CHE-1726525, and to Marcel Bruchez for providing access to the microplate reader and microcentrifuge at Carnegie Mellon University. The authors also acknowledge the use of the Materials Characterization Facility at Carnegie Mellon University, supported by grant MCF-677785, for SEM imaging.References
- K. D. Allen, L. M. Thoma and Y. M. Golightly, Osteoarthritis Cartilage, 2022, 30, 184 CrossRef CAS PubMed.
- C. Madeira, A. Santhagunam, J. B. Salgueiro and J. M. S. Cabral, Trends Biotechnol., 2015, 33, 35 CrossRef CAS PubMed.
- J. Sellam and F. Berenbaum, Nat. Rev. Rheumatol., 2010, 6, 625 CrossRef CAS PubMed.
- J. R. Steadman, W. G. Rodkey, K. K. Briggs and J. J. Rodrigo, Orthopade, 1999, 28, 26 CAS.
- S. Görtz and W. D. Bugbee, Instr. Course Lect., 2007, 56, 469 Search PubMed.
- G. M. Branam and A. Y. Saber, in StatPearls, StatPearls Publishing, Treasure Island (FL), 2024 Search PubMed.
- W. Wei, Y. Ma, X. Yao, W. Zhou, X. Wang, C. Li, J. Lin, Q. He, S. Leptihn and H. Ouyang, Bioact. Mater., 2021, 6, 998 CAS.
- A. J. Gibson, S. M. McDonnell and A. J. Price, Oper. Tech. Orthop., 2006, 16, 262 CrossRef.
- C. Tang, B. D. Holt, Z. M. Wright, A. M. Arnold, A. C. Moy and S. A. Sydlik, J. Mater. Chem. B, 2019, 7, 2442 RSC.
- W. S. Toh, T. C. Lim, M. Kurisawa and M. Spector, Biomaterials, 2012, 33, 3835 CrossRef CAS PubMed.
- C. Celik, V. T. Mogal, J. H. P. Hui, X. J. Loh and W. S. Toh, in Hydrogels: Recent Advances, ed. V. K. Thakur and M. K. Thakur, Springer, Singapore, 2018, pp. 315–337 Search PubMed.
- D.-A. Wang, S. Varghese, B. Sharma, I. Strehin, S. Fermanian, J. Gorham, D. H. Fairbrother, B. Cascio and J. H. Elisseeff, Nat. Mater., 2007, 6, 385 CrossRef CAS PubMed.
- X. Li, Q. Xu, M. Johnson, X. Wang, J. Lyu, Y. Li, S. McMahon, U. Greiser, S. A and W. Wang, Biomater. Sci., 2021, 9, 4139 RSC.
- J. M. Patel, C. Loebel, K. S. Saleh, B. C. Wise, E. D. Bonnevie, L. M. Miller, J. L. Carey, J. A. Burdick and R. L. Mauck, Adv. Healthcare Mater., 2021, 10, 2100315 CrossRef CAS PubMed.
- J. A. Burdick, C. Chung, X. Jia, M. A. Randolph and R. Langer, Biomacromolecules, 2005, 6, 386 CrossRef CAS PubMed.
- I. E. Erickson, S. R. Kestle, K. H. Zellars, M. J. Farrell, M. Kim, J. A. Burdick and R. L. Mauck, Acta Biomater., 2012, 8, 3027 CrossRef CAS PubMed.
- H. Park, S.-W. Kang, B.-S. Kim, D. J. Mooney and K. Y. Lee, Macromol. Biosci., 2009, 9, 895 CrossRef CAS PubMed.
- J. Shin, E. H. Kang, S. Choi, E. J. Jeon, J. H. Cho, D. Kang, H. Lee, I. S. Yun and S.-W. Cho, ACS Biomater. Sci. Eng., 2021, 7, 4230 CrossRef CAS PubMed.
- X. Li, Q. Xu, M. Johnson, X. Wang, J. Lyu, Y. Li, S. McMahon, U. Greiser, S. A and W. Wang, Biomater. Sci., 2021, 9, 4139 RSC.
- P. Baei, H. Daemi, F. Mostafaei, F. Azam Sayahpour, H. Baharvand and M. Baghaban Eslaminejad, Chem. Eng. J., 2021, 418, 129277 CrossRef CAS.
- Y. Hua, H. Xia, L. Jia, J. Zhao, D. Zhao, X. Yan, Y. Zhang, S. Tang, G. Zhou and L. Zhu, et al. , Sci. Adv., 2021, 7, eabg0628 CrossRef CAS PubMed.
- S. Sang, X. Mao, Y. Cao, Z. Liu, Z. Shen, M. Li, W. Jia, Z. Guo, Z. Wang and C. Xiang, et al. , ACS Appl. Mater. Interfaces, 2023, 15, 8895 CrossRef CAS PubMed.
- J. Thomas, N. Gupta, J. P. Joseph, V. Chopra, A. Pal and D. Ghosh, ACS Biomater. Sci. Eng., 2021, 7, 5798 CrossRef CAS PubMed.
- C. E. Kilmer, T. Walimbe, A. Panitch and J. C. Liu, ACS Biomater. Sci. Eng., 2022, 8, 1247 CrossRef CAS PubMed.
- Z. Mao, X. Bi, C. Wu, Y. Zheng, X. Shu, S. Wu, J. Guan and R. O. Ritchie, Adv. Healthcare Mater., 2023, 12, 2201588 CrossRef CAS PubMed.
- Y. Huo, Y. Xu, X. Wu, E. Gao, A. Zhan, Y. Chen, Y. Zhang, Y. Hua, W. Swieszkowski and Y. S. Zhang, et al. , Adv. Sci., 2022, 9, 2202181 CrossRef PubMed.
- A. J. Sophia Fox, A. Bedi and S. A. Rodeo, Sports Health, 2009, 1, 461 CrossRef PubMed.
- M. Bishnoi, A. Jain, P. Hurkat and S. K. Jain, Glycoconjugate J., 2016, 33, 693 CrossRef CAS PubMed.
- F. Burla, Y. Mulla, B. E. Vos, A. Aufderhorst-Roberts and G. H. Koenderink, Nat. Rev. Phys., 2019, 1, 249 CrossRef.
- Z. Cai, Y. Tang, Y. Wei, P. Wang and H. Zhang, Acta Biomater., 2022, 152, 124 CrossRef CAS PubMed.
- A. J. Bailey, R. G. Paul and L. Knott, Mech. Ageing Dev., 1998, 106, 1 CrossRef CAS PubMed.
- M. J. Webber, O. F. Khan, S. A. Sydlik, B. C. Tang and R. Langer, Ann. Biomed. Eng., 2015, 43, 641 CrossRef PubMed.
- A. Getgood, R. Brooks, L. Fortier and N. Rushton, J. Bone Jt. Surg., Br. Vol. B, 2009, 91, 565 CrossRef CAS PubMed.
- S. Taheri, G. Bao, Z. He, S. Mohammadi, H. Ravanbakhsh, L. Lessard, J. Li and L. Mongeau, Adv. Sci., 2022, 9, 2102627 CrossRef CAS PubMed.
- D. Zauchner, M. Z. Müller, M. Horrer, L. Bissig, F. Zhao, P. Fisch, S. S. Lee, M. Zenobi-Wong, R. Müller and X.-H. Qin, Nat. Commun., 2024, 15, 5027 CrossRef CAS PubMed.
- A. R. Jagtap and A. More, Polym. Bull., 2022, 79, 5667 CrossRef CAS.
- K. S. Lim, B. J. Klotz, G. C. J. Lindberg, F. P. W. Melchels, G. J. Hooper, J. Malda, D. Gawlitta and T. B. F. Woodfield, Macromol. Biosci., 2019, 19, 1900098 CrossRef PubMed.
- D.-A. Wang, S. Varghese, B. Sharma, I. Strehin, S. Fermanian, J. Gorham, D. H. Fairbrother, B. Cascio and J. H. Elisseeff, Nat. Mater., 2007, 6, 385 CrossRef CAS PubMed.
- L.-F. Wang, S.-S. Shen and S.-C. Lu, Carbohydr. Polym., 2003, 52, 389 CrossRef CAS.
- Y. Su, J. Zhu, X. Long, L. Zhao, C. Chen and C. Liu, Int. J. Solids Struct., 2023, 264, 112098 CrossRef.
- J. H. Nam, S. Y. Lee, G. Khan and E. S. Park, Arch. Plast. Surg., 2020, 47, 310 CrossRef PubMed.
- G. Hoti, F. Caldera, C. Cecone, A. Rubin Pedrazzo, A. Anceschi, S. L. Appleton, Y. Khazaei Monfared and F. Trotta, Materials, 2021, 14, 478 CrossRef CAS PubMed.
- R. S. Tuan, A. F. Chen and B. A. Klatt, J. Am. Acad. Orthop. Surg., 2013, 21, 303 CrossRef PubMed.
- V. Prabhakar, R. Raman, I. Capila, C. J. Bosques, K. Pojasek and R. Sasisekharan, Biochem. J., 2005, 390, 395 CrossRef CAS PubMed.
- N. Verzijl, J. DeGroot, S. R. Thorpe, R. A. Bank, J. N. Shaw, T. J. Lyons, J. W. J. Bijlsma, F. P. J. G. Lafeber, J. W. Baynes and J. M. TeKoppele, J. Biol. Chem., 2000, 275, 39027 CrossRef CAS PubMed.
- A. Maroudas, M. T. Bayliss, N. Uchitel-Kaushansky, R. Schneiderman and E. Gilav, Arch. Biochem. Biophys., 1998, 350, 61 CrossRef CAS PubMed.
- L. Alcaide-Ruggiero, R. Cugat and J. M. Domínguez, Int. J. Mol. Sci., 2023, 24, 10824 CrossRef CAS PubMed.
- B. D. Monnery, M. Wright, R. Cavill, R. Hoogenboom, S. Shaunak, J. H. G. Steinke and M. Thanou, Int. J. Pharm., 2017, 521, 249 CrossRef CAS PubMed.
- J. Cai, Y. Yue, D. Rui, Y. Zhang, S. Liu and C. Wu, Macromolecules, 2011, 44, 2050 CrossRef CAS.
- Z. Kadlecova, L. Baldi, D. Hacker, F. M. Wurm and H.-A. Klok, Biomacromolecules, 2012, 13, 3127 CrossRef CAS PubMed.
- P. M. Kharkar, K. L. Kiick and A. M. Kloxin, Chem. Soc. Rev., 2013, 42, 7335 RSC.
- L. Bian, C. Hou, E. Tous, R. Rai, R. L. Mauck and J. A. Burdick, Biomaterials, 2013, 34, 413 CrossRef CAS PubMed.
- T. Wang, J. H. Lai and F. Yang, Tissue Eng., Part A, 2016, 22, 1348 CrossRef CAS PubMed.
- L. C. U. Junqueira, G. Bignolas and R. R. Brentani, Histochem. J., 1979, 11, 447 CrossRef CAS PubMed.
- L. C. U. Junqueira, G. Bignolas and R. R. Brentani, Anal. Biochem., 1979, 94, 96 CrossRef CAS PubMed.
- “Calcium phosphate graphene and Ti3C2Tx MXene scaffolds with osteogenic and antibacterial properties – Orlando – 2024 – Journal of Biomedical Materials Research Part B: Applied Biomaterials – Wiley Online Library”, can be found under https://onlinelibrary.wiley.com/doi/full/10.1002/jbm.b.35434, n.d.
- K. E. Eckhart, A. M. Arnold, F. A. Starvaggi and S. A. Sydlik, Biomater. Sci., 2021, 9, 2467 RSC.
- K. E. Eckhart, B. D. Holt, M. G. Laurencin and S. A. Sydlik, Biomater. Sci., 2019, 7, 3876 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5bm00296f |
| This journal is © The Royal Society of Chemistry 2025 |