
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot synthesis of primary phosphines from white phosphorus†
Michael Mendea,
Jose Cammarataa,
Daniel J. Scott *b and
Robert Wolf*a
*b and
Robert Wolf*a
aInstitute of Inorganic Chemistry, University of Regensburg, 93040 Regensburg, Germany. E-mail: robert.wolf@ur.de
bDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AX, UK. E-mail: ds2630@bath.ac.uk
First published on 21st July 2025
Abstract
Aryl and alkyl chlorides are inexpensive and readily accessible, making them ideal reagents for converting white phosphorus (P4) into primary phosphines RPH2. However, methods for achieving this transformation directly, bypassing undesired intermediates like PCl3, have remained elusive. This report describes the ‘one-pot’ synthesis of primary organophosphines from P4 using organotin compounds, including a multi-gram synthesis of PhPH2 featuring efficient recovery of organotin reagents for potential recycling.
White phosphorus (P4) is a crucial industrial feedstock used for the generation of all commercially relevant organophosphorus compounds (OPCs). However, current industrial preparation of these P1 products relies on elaborate multistep processes, where P4 is typically oxidized using hazardous Cl2 gas to give PCl3, which must then be further functionalized towards the desired P1 products in separate steps with concomitant generation of chloride waste (Scheme 1a).1,2 As a result, a major goal for industry and academia is the development of improved, direct functionalisation of P4 towards useful P1 products avoiding hazardous intermediates and wasteful by-products.3 In recent years, several major breakthroughs in this field of chemistry have been reported.4
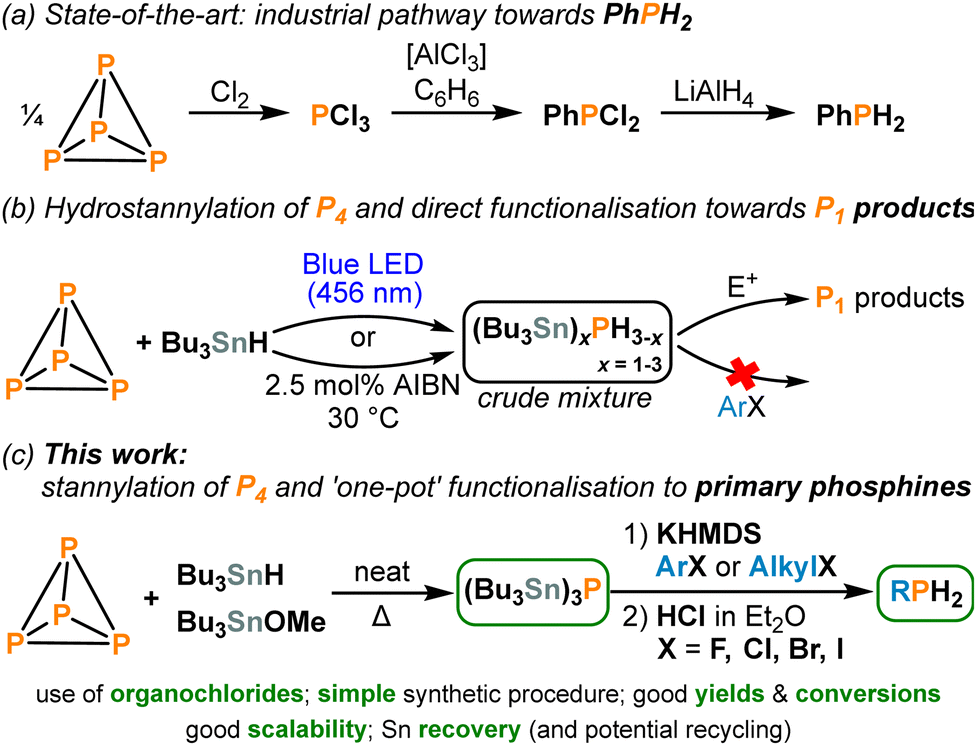 | ||
| Scheme 1 (a) Current industrial pathway for the synthesis of phenylphosphine (PhPH2). (b) Previously reported hydrostannylation of P4 followed by direct functionalisation using electrophiles (E+).4c (c) This work: one-pot transformation of P4 into primary phosphines (RPH2; R = aryl or alkyl). | ||
Phosphines are among the most important phosphorus compounds as they are ubiquitous in inorganic, organometallic and catalytic chemistry.5 Their versatility as ligands stems primarily from their ability to modify the steric and electronic properties of metal centres, which can be tailored by changing the substituents and the structure of the phosphine.5c As the demand for new complexes and catalysts with unique characteristics continues to grow, there is an increasing need for more specialized phosphine ligands. Primary phosphines are key P1 intermediates through which the syntheses of many of these more exotic heteroleptic (and even P-stereogenic) phosphine ligands are achievable.6 Other important applications of primary phosphines can also be found throughout medicinal chemistry, polymer science, carbohydrate modification and macrocycle research.7–10
Recently, we demonstrated that hydrostannylation of P4, using the readily available radical reagent Bu3SnH, can successfully produce a hydrostannylphosphine mixture, leading to various desirable P1 compounds in a convenient ‘one-pot’ manner (see Scheme 1b).4c,11 However, several important limitations affected this reaction. Firstly, the functionalisation of the (Bu3Sn)3−xPHx mixture (x = 0–3), which functions as an in situ “P3−” synthon, using aryl halides was not possible. The use of alkyl chlorides as electrophiles was also not feasible, even though these would be preferable precursors as they constitute the majority of cheap and commercially available organohalides.12 Indeed, using organochlorides in synthesis is often difficult in general due to their relatively strong C–Cl bonds.13 Secondly, the selective addition of only one electrophile per P atom was not achievable (i.e., primary phosphines were not accessible).
Herein, we address both problems and present a straightforward, scalable method for generating primary phosphines from P4, using only inexpensive, readily available terminal reagents and abundant aryl/alkyl chlorides as the key electrophiles (Scheme 1c).
We have recently demonstrated that the phosphine (Bu3Sn)3P can be synthesized directly from P4.4c This simplified alternative exhibits similar reactivity to the (Bu3Sn)3−xPHx mixture, and we anticipated that this chemical simplicity would facilitate selective mono-functionalisation reactions.14 Thus, we began this project by further optimising this transformation. We were pleased to discover that (Bu3Sn)3P can be isolated in excellent yield at mmol scale (3.29 g, 91%) by simply heating the reagents (P4, Bu3SnH, Bu3SnOMe) in the absence of a solvent, without the need for auxiliary radical initiation (Scheme 2a; see also Section S2, ESI†).
 | ||
Scheme 2 (a) Synthesis of (Bu3Sn)3P from P4 under neat conditions. (b) Optimised mono-arylation of (Bu3Sn)3P using PhCl. (c) One-pot transformation of (Bu3Sn)3P into PhPH2. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Spectroscopic yield. Spectroscopic yield. | ||
The arylation of isolated (Bu3Sn)3P was then investigated. In an initial test reaction, combining (Bu3Sn)3P with three equivalents each of potassium bis(trimethylsilyl)amide (KHMDS) and chlorobenzene (PhCl) in toluene resulted in selective transformation into a single phosphorus-containing product with a chemical shift of −156.2 ppm in the 31P{1H} NMR spectrum, with 79% spectroscopic yield. 117/119Sn satellite analysis indicated the presence of two Sn atoms attached to phosphorus, confirming the selective monoarylation of (Bu3Sn)3P by PhCl to form (Bu3Sn)2PPh (see Section S3, ESI†).
Following this successful proof of principle, a range of other strong bases were screened as potential alternatives to KHMDS. However, with the sole exception of NaHMDS, which gave similar conversion (76%), all other replacements led to significantly reduced performance, with most resulting in complete loss of the desired reactivity (see Table S1, ESI†). In contrast, screening of other reaction parameters revealed that good conversion to the target product could be achieved without the need for excess base or electrophile (69% after 18 h), although use of small excesses was found to be beneficial, with 1.5 equivalents of both leading to an excellent spectroscopic yield of 91% after 24 h (Scheme 2b; see also Table S2, ESI†).
To complete the transformation of P4 into the parent primary phosphine, the two Bu3Sn moieties in (Bu3Sn)2PPh must be replaced with H atoms. Fortunately, we have previously shown that analogous exchange can be easily achieved by addition of HCl.4c And indeed, addition of excess HCl (4 M in 1,4-dioxane) to the in situ generated (Bu3Sn)2PPh resulted in clean conversion into the parent phenylphosphine (PhPH2) as observed by 31P{1H} and 31P NMR spectroscopy (Scheme 2c; see also Section S4, ESI†).15
With each individual reaction step optimised, the one-pot transformation of P4 into PhPH2 was then targeted. To demonstrate scalability, as well as aid in product isolation, this was pursued at 80 mmol scale (2 × 40 mmol reactions, combined during workup). Thus, after neat reaction of P4 with Bu3SnH and Bu3SnOMe to afford (Bu3Sn)3P, toluene, KHMDS and PhCl were directly added, and the resulting solution heated overnight (to further transform to (Bu3Sn)2PPh). Following removal of volatiles, quenching with HCl in Et2O, and workup via filtration and fractional distillation, the target product PhPH2 could be successfully isolated as a clear, colourless liquid in good yield (20.0 g, 57%; Scheme 3a; see also Section S5, ESI†). This isolated material contains only traces of residual Sn (288 ppm by ICP-OES; Table S4, ESI†), which compares well with guidelines even for sensitive applications such as oral pharmaceuticals (up to 600 ppm).16 Moreover, the final fractional distillation also enabled recovery of the major Sn-containing byproduct, Bu3SnCl, in excellent yield (93%, 292.4 g). Since Bu3SnCl can easily be transformed back into Bu3SnH and Bu3SnOMe using very inexpensive reagents (e.g. NaBH4 or MeOH, respectively),17 this allows the formation of stoichiometric organotin waste to be bypassed, and creates a simple synthetic loop with very cheap terminal reagents (see Fig. S20, ESI†).
 | ||
| Scheme 3 (a) One-pot synthesis and isolation of PhPH2 starting from P4, with recovery Bu3SnCl byproduct; (b) substrate scope of aryl/alkyl halides giving rise to products (Bu3Sn)2PAr or (Bu3Sn)2PAlk, respectively (for full reaction conditions, see Section S6, ESI†); (c) reaction of (Bu3Sn)3P (0.04 mmol), KHMDS, and substituted aryl chlorides and proposed intermediate aryne via dehydrohalogenation by KHMDS (i) or by the potassium phosphide KP(SnBu3)2 (ii); (d) mechanistic probe experiments using 2-chloro-p-xylene (top) and 2-chloro-m-xylene (bottom) (for full details, see Section S6, ESI†). Percentages in part (a) refer to isolated yields and in part (b) to spectroscopic yields determined using an internal standard (Ph3PO; for full details see Section S6, ESI†). Recovered yield in part (a) refers to Bu3Sn moieties used. | ||
Having achieved a successful proof of concept for the parent primary aryl phosphine PhPH2, the generality of this transformation was then investigated by screening the reactivity of isolated (Bu3Sn)3P towards other aryl halides, as well as some alkyl derivatives. Interestingly, replacement of PhCl with the other parent halobenzenes led to inferior results, with conversion to (Bu3Sn)2PPh decreasing in the order Cl (91%) > Br (71%) > I (66%) ≫ F (16%). In contrast, excellent conversions to (Bu3Sn)2PAr could be achieved for certain other model ArCl substrates bearing both electron-donating and electron-withdrawing substituents (m-OMe and o-CF3, 70% and 61% respectively, Scheme 3b; for isomer identification, see Section S8, ESI†). Moreover, the aryl chloride substrates could be directly replaced with alkyl halides, leading to primary alkyl phosphines bearing industrially relevant alkyl motifs (2-ethylhexyl, 2,4,4-trimethylpentyl). In these cases, the bromides were found to slightly outperform the chlorides, although the latter still provided good spectroscopic conversions (64% vs. 49% for 2-ethylhexyl, 67% vs. 46% for 2,4,4-trimethylpentyl; Scheme 3b).
Alongside these successful substrates, several other alkyl halides were investigated, including secondary and tertiary substrates, but were found to give significantly inferior results (see Section S6, ESI†). Other aryl chlorides provided excellent conversion to (Bu3Sn)2PAr products; however, close inspection typically revealed two (or more) separate product signals in each of the 31P{1H} NMR reaction spectra, with similar chemical shifts and 117/119Sn satellites. This duplication was retained upon acidification with HCl, with 31P{1H} and 31P NMR spectra suggesting the formation of two isomeric primary aryl phosphines, which was further supported by GC-MS (Scheme 3c; for more detailed discussion, see Section S7.2, ESI†).
The observation of regioisomers was unexpected and suggests that the observed arylation reactions are unlikely to proceed via simple nucleophilic attack of (Bu3Sn)3P (or derivatives) on the electrophile, as was assumed for other electrophiles in our previous reports.4c,14 Indeed, SNAr reactivity is unlikely on more fundamental grounds, since SNAr is generally infeasible for Ar–Cl bonds in the absence of strongly electron-withdrawing substituents (and would be more favourable for Ar–F; cf. reduced reactivity of PhF vs. PhCl). Instead, we speculated that the reaction might proceed via an aryne intermediate. It is well known that aryl halides can form aryne intermediates upon dehydrohalogenation by strong bases, and this has been explicitly reported using various aryl halides and KHMDS.18 The aryne thus generated would feature two adjacent carbon atoms that are both viable sites for nucleophilic attack (Scheme 3c, pathway i).19,20 Unless the aryne is symmetrical, this should then lead to the formation of two regioisomeric products (at least in the absence of strong steric or electronic biases).
To test this proposal, two additional substrates were investigated: 2-chloro-1,3-dimethylbenzene and 2-chloro-1,4-dimethylbenzene. If the reaction does proceed via an aryne intermediate, this should lead to the generation of a single product for the former (as the aryne should be symmetrical) or no product for the latter (as aryne generation is precluded by the absence of an adjacent C–H bond). Both outcomes were confirmed experimentally by NMR spectroscopic analysis, albeit with low conversion in the former case, possibly due to steric hindrance (Scheme 3d; for further discussion see also Sections S6 and S7.2, ESI†).
Finally, the reaction between KHMDS and (Bu3Sn)3P in the absence of an organic substrate was also examined. 31P{1H} NMR monitoring showed the formation of a new, more upfield signal at −376.2 ppm with 117/119Sn satellites corresponding to two Sn atoms attached to the phosphorus. A similar upfield shift is observed during the known reaction of (Me3Si)3P and KOtBu (another reaction between a tetrel-substituted phosphine and a strong, non-nucleophilic base) to generate the related potassium phosphide KP(SiMe3)2. As such, the new signal is assigned to the potassium phosphide KP(SnBu3)2 (see Section S7.1, ESI†).21 This suggests that rather than being deprotonated by KHMDS directly, aryne generation could also occur via initial formation of KP(SnBu3)2, which would also be a strong base (Scheme 3c, pathway ii).
In conclusion, we have developed a simple and efficient one-pot reaction for the direct generation of primary phosphines from P4 via stannylation, using inexpensive and readily available terminal reagents. Focusing on the parent primary phosphine PhPH2, we have demonstrated the selectivity and scalability of the reaction, which avoids the chlorination of P4, including the efficient recovery of the key Bu3SnX reagents to minimize the formation of toxic waste. Several aryl chlorides substituted with electronically distinct functional groups also gave very good conversions and selectivity, as did several primary alkyl bromides and chlorides. We have also investigated the underlying reaction mechanism, which for aryl chlorides appears to proceed via dehydrohalogenation to an aryne and can therefore lead to two regioisomeric phosphine products. Further research on this topic is ongoing, particularly concerning the additional functionalisation of the intermediate products (Bu3Sn)2PR (R = aryl, alkyl), which still bear two Bu3Sn moieties. Their further in situ derivatization could therefore potentially yield heteroleptic and even chiral phosphines.
We thank Petra Lugauer for her valuable experimental assistance and Vanessa Tomanek for the ICP-OES measurement. We acknowledge the DFG (RW, WO 1496/12-1, project number 548830090) and EPSRC (DJS, EP/V056069/1) for their financial support, as well as the Alexander von Humboldt Foundation for awarding a postdoctoral fellowship to DJS.
Conflicts of interest
A patent covering all the results described herein has been filed (as of 13 February 2020) by the University of Regensburg (EP 20,157,197.3; inventors, D. J. S. and R. W.). The authors declare no other competing interests.Data availability
The data supporting this article have been included as part of the ESI.†Notes and references
- H. Diskowski and T. Hofmann, Phosphorus, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2020 Search PubMed.
- Some products can instead be accessed via PH3 or P2O5 as the initial intermediates, with these routes suffering similar drawbacks. See: D. E. C. Corbridge, Chemistry Biochemistry and Technology, Elsevier, 2000.
- (a) D. J. Scott, Angew. Chem., Int. Ed., 2022, 61, e202205019 CrossRef CAS PubMed; (b) Y. Liu, X. Chen and B. Yu, Chem. – Eur. J., 2023, 29, e202302142 CrossRef CAS PubMed.
- For selected key reports, see: (a) U. Lennert, P. B. Arockiam, V. Streitferdt, D. J. Scott, C. Rödl, R. M. Gschwind and R. Wolf, Nat. Catal., 2019, 2, 1101–1106 CrossRef CAS PubMed; (b) Y. Wang, T. Szilvasi, S. Yao and M. Driess, Nat. Chem., 2020, 12, 801–807 CrossRef CAS PubMed; (c) D. J. Scott, J. Cammarata, M. Schimpf and R. Wolf, Nat. Chem., 2021, 13, 458–464 CrossRef CAS PubMed; (d) S. Reichl, E. Mädl, F. Riedlberger, M. Piesch, G. Balázs, M. Seidl and M. Scheer, Nat. Commun., 2021, 12, 5774 CrossRef CAS PubMed; (e) M. Donath, K. Schwedtmann, T. Schneider, F. Hennersdorf, A. Bauzá, A. Frontera and J. J. Weigand, Nat. Chem., 2022, 14, 384–391 CrossRef CAS PubMed; (f) T. M. Horsley Downie, A. Velić, L. A. Coelho, R. Wolf and D. J. Scott, ChemSusChem, 2025, 18, e202401895 CrossRef CAS PubMed.
- (a) P. C. J. Kamer and P. W. N. M. van Leeuwen, Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, Wiley, 2012 CrossRef; (b) D. W. Allen, J. C. Tebby, P. Balczewski, D. Loakes, G. Keglevich and M. Migaud, Organophosphorus Chemistry, Royal Society of Chemistry, 2011 Search PubMed; (c) A. Börner, Phosphorus ligands in asymmetric catalysis; synthesis and applications, John Wiley & Sons Incorporated, 2008 Search PubMed.
- A. J. Kendall and D. R. Tyler, Dalton Trans., 2015, 44, 12473 RSC.
- K. V. Katti, H. Gali, C. J. Smith and D. E. Berning, Acc. Chem. Res., 1998, 32, 9–17 CrossRef.
- H. Dorn, R. A. Singh, J. A. Massey, A. J. Lough and I. Manners, Angew. Chem., Int. Ed., 1999, 38, 3321–3323 CrossRef CAS PubMed.
- T. Hanaya and H. Yamamoto, Bull. Chem. Soc. Jpn., 1989, 62, 2320–2327 CrossRef CAS.
- E. P. Kyba and S.-T. Liu, Inorg. Chem., 1985, 24, 1613–1616 CrossRef CAS.
- J. Cammarata, F. F. Westermair, P. Coburger, D. Duvinage, M. Janssen, M. K. Uttendorfer, J. Beckmann, R. M. Gschwind, R. Wolf and D. J. Scott, Angew. Chem., Int. Ed., 2024, 63, e202408423 CAS.
- Y. Zabolotna, D. M. Volochnyuk, S. V. Ryabukhin, D. Horvath, K. S. Gavrilenko, G. Marcou, Y. S. Moroz, O. Oksiuta and A. Varnek, J. Chem. Inf. Model., 2021, 62, 2171–2185 CrossRef PubMed.
- (a) L. Pause, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 1999, 121, 7158–7159 CrossRef CAS; (b) M. Cybularczyk-Cecotka, J. Szczepanik and M. Giedyk, Nat. Catal., 2020, 3, 872–886 CrossRef CAS; (c) S. Wu, J. Kaur, T. A. Karl, X. Tian and J. P. Barham, Angew. Chem., Int. Ed., 2022, 61, e202107811 CrossRef CAS PubMed; (d) L.-L. Liao, L. Song, S.-S. Yan, J.-H. Ye and D.-G. Yu, Trends Chem., 2022, 4, 512–527 CrossRef CAS.
- (a) M. Till, J. Cammarata, R. Wolf and D. J. Scott, Chem. Commun., 2022, 58, 8986–8989 RSC; (b) J. Cammarata, D. J. Scott and R. Wolf, Chem. – Eur. J., 2022, 28, e202202456 CrossRef CAS PubMed.
- I. Kalinina and F. Mathey, Organometallics, 2006, 25, 5031–5034 CrossRef CAS.
- EMA scientific guidelines on elemental impurities can be found here: https://www.ema.europa.eu/en/ich-q3d-elemental-impurities-scientific-guideline (accessed July 2025).
- E. R. Birnbaum and P. H. Javora, J. Organomet. Chem., 1967, 9, 379–382 CrossRef CAS.
- (a) A. V. Lis, I. P. Tsyrendorzhieva, A. I. Albanov, V. I. Rakhlin and M. G. Voronkov, Russ. J. Org. Chem., 2013, 49, 1451–1453 CrossRef CAS; (b) A. V. Lis, I. P. Tsyrendorzhieva, A. I. Albanov, B. A. Gostevskii and V. I. Rakhlin, Russ. Chem. Bull., Int. Ed., 2015, 64, 2090–2094 CrossRef CAS; (c) A. V. Lis, I. P. Tsyrendorzhieva, A. I. Albanov, B. A. Shainyan and V. I. Rakhlin, Russ. J. Org. Chem., 2015, 51, 335–340 CrossRef CAS; (d) A. V. Lis, I. P. Tsyrendorzhieva, A. I. Albanov and V. I. Rakhlin, Russ. Chem. Bull., Int. Ed., 2016, 65, 592–594 CrossRef CAS.
- For an overview see: T. L. Gilchrist, Science of Synthesis, Thieme, 2008, vol. 43, pp. 151–224 Search PubMed.
- For a recent overview on aryne reactivity towards organophosphorus compounds, see: J. Chen, R. Fan, Z. Liu and J. Tan, Adv. Synth. Catal., 2021, 363, 657–667 CrossRef CAS.
- C. A. Russel and N. S. Townsend, Des. Synth., 2012, 11, 343–354 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc03444b |
| This journal is © The Royal Society of Chemistry 2025 |