
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A mini review on photocatalytic lignin conversion into monomeric aromatic compounds†
Shibo
Shao
ab,
Xiangzhou
Wang
c,
Wenbing
Li
*a,
Yiming
Zhang
d,
Shi
Liu
a,
Weisheng
Xiao
a,
Zongyang
Yue
b,
Xu
Lu
*a and
Xianfeng
Fan
 *b
*b
aAdvanced Materials Research Center, Petrochemical Research Institute, PetroChina Company Limited, Beijing 102206, P.R. China
bSchool of Engineering, The University of Edinburgh, Edinburgh, EH9 3JL, UK. E-mail: x.fan@ed.ac.uk
cRefining, Chemical & New Materials Company, PetroChina Company Limited, Beijing, 100007, P.R. China
dSchool of Materials Science and Engineering, Lanzhou University of Technology, Lanzhou 730050, P.R. China
First published on 7th January 2025
Abstract
To meet the growing demand for chemicals and energy while minimizing environmental impact, it is necessary to employ sustainable resources instead of fossil fuel feedstock and develop green processes to upgrade the chemical industry. Lignin, as the only abundant natural aromatic structured biomass, has been identified as a promising feedstock for producing monomeric aromatics through the depolymerisation process. However, the robust structure of lignin, coupled with the limitations of conventional catalytic methods, presents significant challenges for converting lignin into aromatic monomers. Recently, photocatalysis has emerged as a remarkable platform for versatile lignin transformation and has achieved some significant progress. The unique photogenerated reactive species as well as the mild reaction conditions enable the selective cleavage of targeted interunit bonds in lignin while preserving the aromatic structures and functional groups for further processing. In this review, we aimed to provide an overview of the state-of-the-art advances in key photocatalytic processes applied for the conversion of lignin using various photocatalysts. This review focuses on the latest investigations into lignin conversion mechanisms, emphasizing selective C–O and C–C bond cleavage. Additionally, we discuss catalyst modification strategies and key factors influencing the lignin conversion process based on significant recent publications. Furthermore, we elucidate future perspectives and challenges for the photocatalytic conversion of lignin into valuable products. Hopefully, this mini review will inspire future work on the photocatalytic lignin conversion process to achieve lignin valorisation and a sustainable supply of aromatic chemicals.
1. Introduction
1.1. Background
As global awareness of climate change and the depletion of fossil resources intensifies, the transformation of the traditional chemical industry, which remains predominantly reliant on fossil fuel feedstocks, has emerged as a critical societal challenge in the 21st century. In this context, lignocellulosic biomass, the most abundant renewable carbon source, has gained considerable attention as a promising alternative to unsustainable fossil fuel feedstocks for producing chemical products.1–5 Among the three primary components of lignocellulose, lignin serves as both the “structural glue” and “protective barrier,” binding cellulose and hemicellulose together while safeguarding plant tissues from external environmental damage.2,6–9 Lignin, typically comprising over 15 wt% of lignocellulose, represents the only polymer bioresource with aromatic structure available in substantial quantities.7 The current lignocellulose biorefinery industry primarily emphasizes the utilization of carbohydrates (cellulose and hemicellulose) from lignocellulosic biomass as sustainable feedstocks to produce high-value non-aromatic chemicals. Annually, over 70 million tons of lignin are generated from the paper manufacturing and biorefinery sectors, with yields anticipated to increase as an increasing number of biorefineries are being commissioned. Nevertheless, lignin is often regarded as waste or is utilized as a low-quality fuel, which is a significant resource wastage and leading to potential environmental pollution issues.10Although the exact structures of lignin may vary with the origins (e.g., the types of plants) and the environmental factors during plant growth, it is widely recognized that lignin is a highly functionalised, amorphous biopolymer composed of aromatic units with diverse functional groups (methoxyl, hydroxyl and carbonyl groups). As shown in Fig. 1, the aromatic monomer units in lignin are typically interconnected by C–C bonds (referred to as condensed linkages) and C–O bonds (also known as ether linkages). Among these interunit linkages, C–O linkages, particularly Cβ–O linkages, are the predominant type in lignin and typically account for over 50% of all interunit bonds between aromatic monomers.3,6,11–13 Therefore, the conversion of lignin into aromatic monomeric products through the cleavage of Cβ–O linkages has been extensively studied and is regarded as a promising strategy for lignin valorisation. However, it is important to note that the structure of lignin biomass can be altered by various isolation methods, which may lead to the formation of more recalcitrant C–C bonds during lignin degradation and repolymerization processes. For instance, kraft lignin, produced from the papermaking industry under harsh alkaline conditions during the delignification stage, typically contains a significant amount of C–C bonds rather than C–O bonds.14 The fragmentation of C–C bonds should also be systematically investigated to fully utilise lignin bioresources. Due to the complex structure and recalcitrant nature of natural lignin, it is challenging to monitor and understand the reaction chemistry and mechanism of the lignin conversion process. In this case, some aromatic dimers consisting of two aromatic monomers connected by specific C–O or C–C bonds, resembling the interunit chemical bonds in lignin, are widely employed as reactants to mimic the chemical properties and reactive behaviours of the corresponding chemical bonds in lignin. Therefore, the experiments could be simplified and enable more detailed investigations of reaction mechanisms based on the defined structures of lignin model compounds. The typical lignin model compounds employed in the literature are listed in Table S2 in the ESI.† For example, 2-phenoxy-1-phenylethanol, which contains a Cβ–O linkage and a hydroxyl group, is a typical and representative lignin model compound that has been frequently used as the reactant to study the selective cleavage of the Cβ–O bond and Cα–Cβ bond in numerous studies.15–22 In addition to converting lignin into aromatic monomers, the reforming of lignin for hydrogen production also represents another viable strategy for lignin valorisation.23–25
 | ||
| Fig. 1 Representative structure models of lignin and typical linkages (this is a simplified model for illustrating the types of monolignols in lignin structure, it is not the accurate structural formula of lignin). | ||
However, the conversion of lignin into valuable chemicals remains a considerable challenge due to its inherent recalcitrance and complex structure.6,22,26,27 Two conventional thermocatalytic strategies have been developed for converting lignin into chemical products. The first involves the gasification and pyrolysis of lignin biomass to produce syngas (e.g., H2 and CO) or other small molecules. The second strategy focuses on the extensive cleavage of bonds in lignin to produce simple aromatic compounds such as benzene, toluene, and xylene. As shown in Fig. 2, various conventional lignin processing approaches were briefly summarised with the corresponding reaction conditions. In general, lignin processing methods involving acids and bases, as well as oxidative and reductive reagents, can typically be conducted at relatively lower temperatures, whereas processes such as pyrolysis and gasification require significantly higher operating temperatures.28 Among them, lignin oxidation is generally performed at lower temperatures, yielding fine or platform chemicals such as aromatic alcohols, aldehydes, and acids. The reductive hydro-processing is typically carried out at temperatures ranging from 100 to 350 °C and requiring additional hydrogen donors to produce simple aromatic compounds such as toluene, benzene, phenol, etc. Monomeric aromatic compounds can also be obtained through acid/base catalysed depolymerization of lignin, which selectively cleaves C–O or C–C linkages between lignin units. However, conventional thermocatalytic processes often demand excessive chemical reagents (e.g., acids or bases for hydrolysis) and operate under harsh reaction conditions (e.g., high temperature, pressure, and additional hydrogen). These conditions result in significant energy and resource consumption and frequently cause side reactions that generate unwanted by-products, degrading the valuable aromatic moieties and functional groups in lignin (e.g., the simultaneous cracking of aliphatic ether bonds and saturation of arene rings).1,8,29,30
 | ||
| Fig. 2 A brief summary of conventional lignin conversion processes and the corresponding required reaction conditions.28 | ||
Since the aromatic monomeric units in lignin are predominantly linked by C–O and C–C bonds, recent efforts in lignin conversion have concentrated on selectively cleaving these bonds under relatively mild conditions. This strategy allows for the preservation of the natural functionalized aromatic structures, enabling the production of value-added aromatic compounds.3,11,22 Inspired by the photosynthesis of natural plants, photocatalysis, which is also driven by the abundant and green energy from the sun, has been deemed as a promising approach to solve many environmental problems. Compared to traditional energy-intensive thermochemical catalysis, photocatalysis powered by sustainable solar energy can simultaneously facilitate oxidation and reduction processes by generating unique photo-induced charge carriers that interact with reactive species under mild conditions. This capability allows for precise control over reaction selectivity through the cleavage of specific interunit chemical bonds, and in turn leads to a high selectivity for desirable products.20,31 Given these unique features, photocatalysis is regarded as one of the most promising approaches for lignin valorisation, and recently some progress has been achieved regarding the conversion of lignin into value-added outcomes. The photocatalytic conversion of lignin into aromatic monomers, which simultaneously performs oxidation and reduction for selectively cleaving the C–O or C–C interunit linkages in lignin, represents a very promising method that can effectively utilise the unique natural aromatic structures of lignin. Different from the typical products (e.g., defunctionalised aromatics or over-hydrogenated alkanes) generated from the traditional thermocatalytic lignin depolymerisation, photocatalytic lignin conversion can yield a broader range of value-added products, including aromatic ketones, aromatic aldehydes, and phenols. However, akin to numerous other photocatalytic processes, the suboptimal reaction efficiency poses a critical challenge that must be addressed. The limited systematic investigations in this area and the inadequate understanding of photochemical reaction mechanisms represent significant barriers to the design of efficient photocatalysts and the establishment of effective photocatalytic processes for lignin conversion via selective interunit linkage fragmentation.
This review primarily focuses on the literature concerning the photocatalytic conversion of lignin into value-added aromatic monomers via the selective cleavage of interunit C–O and C–C bonds between the aromatic units of lignin. The reaction mechanism for the selective scission of C–O and C–C bonds, the formation of intermediate radicals, the roles of photogenerated charged carriers and radicals are systematically discussed and analysed. In addition, the photocatalysts employed in the lignin conversion process are categorically reviewed. Although the photocatalytic lignin valorisation is still far from being realised at an industrial scale, this study provides some valuable insights to support the development of efficient and reliable photocatalytic systems for lignin valorisation and contribute to expanding the applications of photocatalysis.
1.2. The principle of photocatalysis and its advantages in lignin conversion
The concept of ‘photocatalysis’ was initially proposed based on the experiments involving dye degradation and reduction of metal ions catalysed by metal oxides under external illumination.32–34 Before Fujishima and Honda's groundbreaking report in 1972 on photocatalytic hydrogen production from water splitting, driven by titanium dioxide (TiO2) under UV irradiation, the research on photocatalysis remained in its embryonic stage and attracted limited attention.33–35 Subsequently, heterogeneous photocatalysis garnered widespread interest, and its capabilities in CO2 reduction, pollution abatement, fine chemical synthesis, fuel production, and other applications were gradually demonstrated and explored.36,37 Furthermore, numerous semiconductor materials beyond metal oxides, such as metal sulphides and carbon nitrides, have been extensively investigated for their photocatalytic properties. In recent decades, the development of photocatalysis has been significantly accelerated by intensive efforts from researchers worldwide, demonstrating its immense potential in the fields of environmental and energy science.Unlike metal crystal conductors, semiconductors have discontinuous energy bands. Specifically, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are separated by a region of forbidden electronic states, commonly referred to as the bandgap of semiconductors. Herein, the highest electron-occupied band is known as the valence band (VB), while the lowest unoccupied band is known as the conduction band (CB). Typically, in a photocatalytic system, the light-harvesting centres and active catalytic sites are the key components. In heterogeneous photocatalytic systems, semiconductor materials generally serve as the light-harvesting centres, while the active catalytic sites are typically located on the surface of the semiconductor or on other components (e.g., metal nanoparticles) integrated with semiconductor materials. As shown in Fig. 3, the mechanism of semiconductor photocatalysis can generally be described in the following stages:
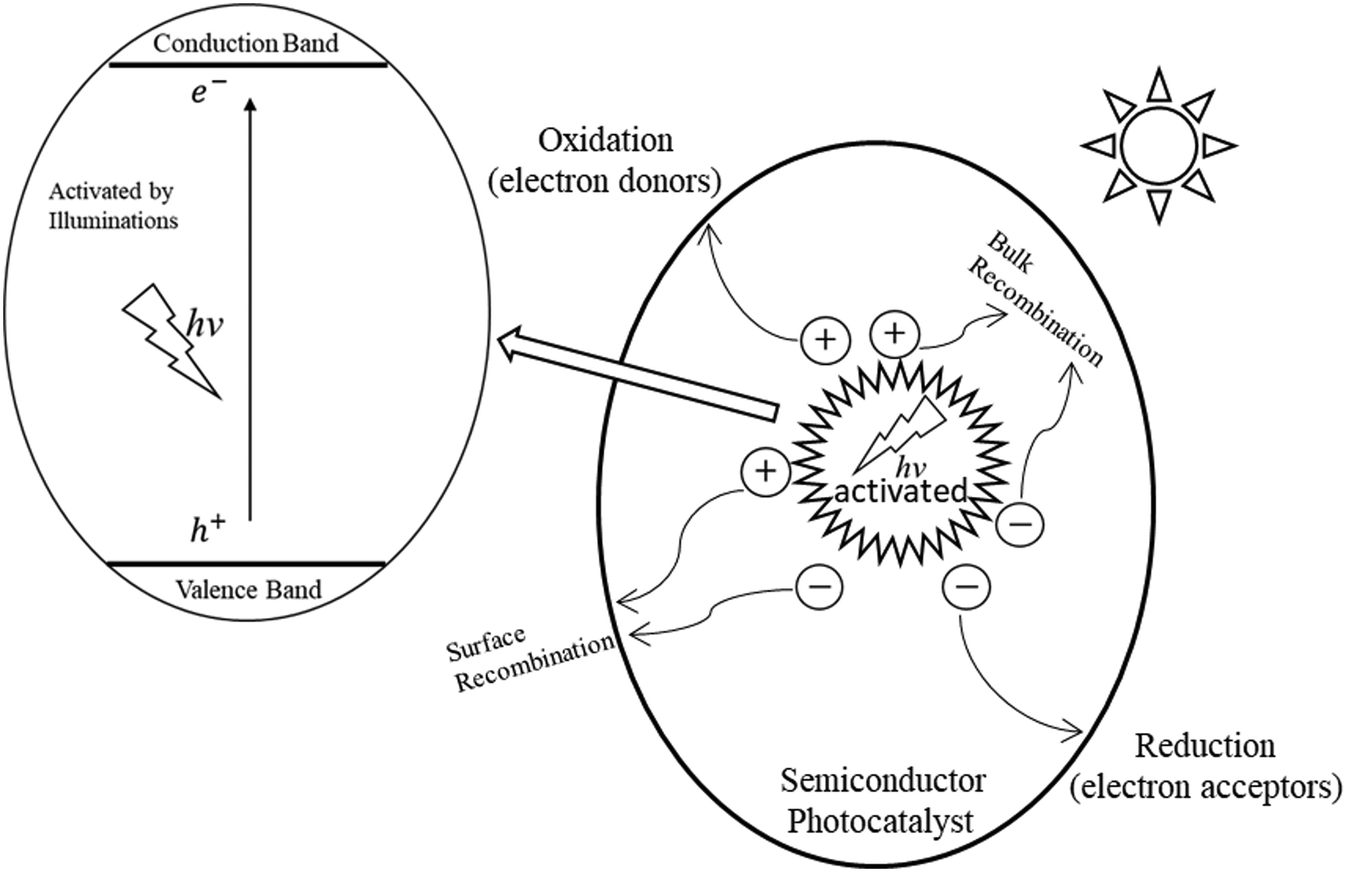 | ||
| Fig. 3 The mechanism of the photocatalytic process on the surface of the semiconductor under the irradiation of light. | ||
i) When photons from light irradiation with energy (hv) equal to or greater than the bandgap of the semiconductor photocatalyst are absorbed by the light-harvesting centres, electrons in the valence band (VB) are excited and transferred to the conduction band (CB), resulting in the formation of electron–hole pairs, with electrons in the CB and holes remaining in the VB.
ii) The photogenerated electrons and holes, once separated from the light-harvesting centres, may recombine either within the bulk or on the surface of the catalysts, resulting in energy dissipation. It is important to note that the recombination of these photogenerated charges reduces catalytic activity, ultimately leading to lower quantum efficiency of the photocatalysts.
iii) The photoexcited electrons and holes migrate to the catalytic sites on the surface of the catalysts, where they can react with the adsorbed species (electron acceptors and donors), thereby initiating the corresponding reduction and oxidation reactions.
Based on the bandgap excitation mechanism of photocatalysis, the photocatalytic process can be divided into three key stages: (1) the generation of photoexcited electron–hole pairs at the light-harvesting centres; (2) the separation and transfer of photogenerated charges from the light-harvesting centres to the active catalytic sites; and (3) the adsorption and activation of reactant species at the active catalytic sites.38–41 In general, the overall photocatalytic performance is primarily determined by the influential factors arising from the processes in the above stages.
At first, the light absorption capability of the light-harvesting centres and the bandgap structure of semiconductors are fundamental aspects of photocatalysis. Given that the natural sunlight spectrum contains only about 5% ultraviolet (UV, 300–400 nm) light, it is crucial to develop catalysts with optimal bandgaps that can be excited by visible light, enabling efficient solar energy utilisation and the generation of photoinduced electron–hole pairs.42 Herein, the bandgap energy could be determined by the equation of electronic absorption spectra:
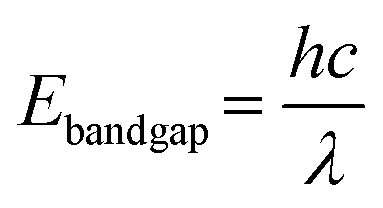
The subsequent separation and transfer of photoexcited charges must overcome the challenge of recombination of the photoinduced electron–hole pairs, which is considered as one of the primary factors limiting the overall photocatalytic performance. In fact, the majority of photogenerated charges are lost due to recombination during the photocatalytic process.39 From a timescale perspective, the recombination of electron–hole pairs is a rapid process occurring within the picosecond (ps) range, while the transfer of photogenerated charges to the catalytic sites typically takes hundreds of picoseconds. Moreover, the reactions between photoinduced charge carriers and adsorbed reactants, however, generally require durations ranging from nanoseconds (ns) to microseconds (μs).43,44 Therefore, the key to achieving high quantum efficiency lies in suppressing recombination while promoting the separation and transport of photogenerated charge carriers throughout the photocatalytic process.
From the perspective of reaction thermodynamics and energy, as depicted in Fig. 4, the activation energy (Ea1) of reactants in uncatalysed systems can be reduced through interactions between the catalysts and reactants. However, high energy inputs are still required to overcome the activation barriers (Ea2 and Ea3) in thermo-driven catalytic reactions. In the case of photocatalysis, the energy barriers (Ea4) can be significantly lowered due to the activation of reactants by highly reactive photoinduced charge carriers from the excited photocatalysts. Consequently, the thermodynamically unfavourable reactions can proceed under mild conditions, and unwanted side reactions, which typically arise under harsh conditions, can be avoided. All these features of photocatalysis allow it to overcome the challenges associated with conventional thermocatalytic lignin processes, particularly in terms of high energy consumption, low selectivity, and the need for additional chemical reagents. The natural aromatic ring structures with functional groups in lignin can be preserved during the lignin decomposition process, yielding monomeric aromatic compounds that can be further upgraded into fine chemicals. In addition to thermocatalysis and photocatalysis, biological methods have also been studied for lignin conversion.45–47 These methods utilise enzymatic or microbial systems to selectively degrade lignin and produce aromatic compounds under mild conditions. Similar to photocatalysis, biological methods also offer advantages including the environmentally friendly nature and the ability to selectively cleave specific lignin bonds without the need for high temperatures or additional chemicals. However, due to the complex and heterogeneous structure of lignin, biological methods face significant challenges, including relatively low efficiency, prolonged reaction times, and the inability to achieve complete lignin depolymerisation. Moreover, in order to sustain microbial activity, biological methods generally require more restricted conditions in comparison to photocatalysis, such as pH condition, temperature, and nutrient availability, which further complicates the scalability and industrial application of the biological lignin conversion approach.45
 | ||
| Fig. 4 The energy diagram illustrates the transformation of reactants to products, showing the activation energies required for each process (Ea1, Ea2, Ea3 and Ea4 is the activation energy for the uncatalysed reaction, step reactions of a thermocatalytic reaction and photocatalytic reaction). | ||
2. Photocatalytic conversion of lignin into aromatics via selective bond scission
In recent years, various photocatalytic systems have been developed to convert lignin into valuable chemicals, positioning photocatalytic approaches as a promising strategy for lignin valorisation. Most studies focused on the field of photocatalytic transformation of lignin to related platform compounds, in particular to precisely cleave targeted C–O or C–C bonds in lignin and obtaining aromatic monomers. The photocatalytic performance of representative studies focusing on C–O or C–C bond cleavage is summarised in Tables S3 and S4 in the ESI.†2.1. Selective C–O bond fragmentation
Since the Cβ–O bond is the predominant interunit linkage in natural lignin structures, it has been widely targeted for cleavage in the conversion of lignin into monomeric aromatics. The reaction pathways for selective Cβ–O bond fragmentation can generally be classified into two categories: stepwise conversion and direct photocatalytic redox-neutral conversion (Fig. 5). Additionally, a few studies have also explored the photocatalytic selective cleavage of other C–O bonds in lignin, such as Cα–O bonds and 4-O-5 bonds. | ||
| Fig. 5 Two different reaction strategies of photocatalytic Cβ–O bond cleavage. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, thereby facilitating the subsequent cleavage of the Cβ–O bond. For instance, in one of the most commonly used dimeric lignin model compounds, the pre-oxidation of 2-phenoxy-1-phenylethanol to 2-phenoxyacetophenone decreases the BDE of the Cβ–O bond from 289.53 kJ mol−1 to 233.89 kJ mol−1 (Scheme 1b).50
O, thereby facilitating the subsequent cleavage of the Cβ–O bond. For instance, in one of the most commonly used dimeric lignin model compounds, the pre-oxidation of 2-phenoxy-1-phenylethanol to 2-phenoxyacetophenone decreases the BDE of the Cβ–O bond from 289.53 kJ mol−1 to 233.89 kJ mol−1 (Scheme 1b).50
 | ||
| Scheme 1 (a) A stepwise oxidation–reduction strategy and the calculated BDE of the C–C and C–O bonds in the β-O-4 linkage before and after the oxidation; (b) a two-step conversion strategy of 2-phenoxy-1-phenylethanol (this scheme has been reproduced from ref. 7 and 51 with permission from Elsevier & American Chemical Society, copyright 2021 & copyright 2016). | ||
Stephenson's group was the first to demonstrate that this stepwise reaction strategy can also be applied to the photocatalytic cleavage of Cβ–O bonds in lignin model compounds. In their early studies, the initial pre-oxidation stage was carried out using non-photocatalytic methods, including stoichiometric oxidants (e.g., [4-AcNH-TEMPO]BF4), a palladium-catalysed aerobic oxidation system (Pd(OAc)2/DMSO catalyst system) or electrocatalytic oxidation via hydrogen atom transfer (HAT). After the pre-oxidation, the subsequent Cβ–O bond cleavage in oxidised β-O-4 lignin model compounds (ketones) was catalysed by various Ir-based molecular photocatalysts (e.g., [Ir(ppy)2(dtbbpy)]PF6) in the presence of organic bases (e.g., diisopropylethylamine) as the electron donor and hydrogen donors (e.g., formic acid).52–54 The mechanism studies indicated that the reductive fragmentation of Cβ–O bonds was driven by the reductive quenching cycle of Ir-based molecular photocatalysts. Specifically, the light-excited photocatalysts can initiate the metal-to-ligand charge transfer process, generating the excited state [Ir]3+*. Subsequently, [Ir]3+* can be converted to [Ir]2+ with strong reductive capability after accepting an electron from the electron donor. The benzylic ketone or aliphatic aldehyde generated from the pre-oxidation stage can react with [Ir]2+via a single electron transfer (SET) process and generate radical anions, which could be further decomposed to the corresponding intermediate radicals through the C–O bond fragmentation. And the target monomeric aromatic products could be obtained following the protonation and hydrogen atom transfer process.52–54 In addition to designing the catalyst system, Stephenson and co-workers developed a series of flow reaction systems to facilitate the pre-oxidation and Cβ–O bond fragmentation. These systems could enhance light absorption and mass transfer efficiency, significantly improving the catalytic performance in the depolymerization of dimeric β-O-4 lignin model compounds.53
The two-step Cβ–O bond cleavage processes developed by Stephenson and co-workers were proven to be able to break the Cβ–O linkages from a wide range of dimeric lignin model compounds. However, the iridium complexes are homogeneous photocatalysts, which have the inherent shortages in recycling for long-term use. To overcome this problem, the iridium complexes were immobilised onto a mesoporous cellular silica foam (MCF) via a facile thiolene click reaction between prefabricated thiol-functionalised mesoporous cellular silica foams and vinyl-tagged iridium complexes.55 Compared with the corresponding homogeneous Ir-based complex, this elaborate Ir-MCF catalyst can be easily recycled for reuse and exhibited similar excellent catalytic performance in the fragmentation of Cβ–O bonds in dimeric β-O-4 ketones under visible-light irradiation due to the aerogel-like 3D porous structure and high visible-light transparency.55
In addition to iridium based photocatalysts in this field, π-conjugated carbazolic copolymers (CzCP) with a finely tuned ratio of carbazolic electron donor (D) and electron acceptor (A) were found to efficiently cleave Cβ–O bonds in lignin model compounds.56 The redox potentials of CzCP materials can be modulated by the tuneable D/A ratios. For example, the pre-oxidation of Cα–OH to CαO is driven by CzCP100 (D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :A = 0:100), which possesses the strongest oxidative capability and operates via a direct photoinduced single-electron transfer (SET) process. In the subsequent step, Cβ–O bond cleavage is facilitated by photoexcited CzCP33 (D:A = 66:33), which reacts with a sacrificial electron donor to generate highly reductive CzCP33˙−, enabling the reduction of ketones formed during the pre-oxidation stage.56 Moreover, this heterogeneous catalyst could be easily separated from the reaction system through filtration or centrifugation and has demonstrated stable catalytic activity during reuse, increasing the potential for employing porous organic frameworks (POFs) as heterogeneous photocatalysts in lignin conversion via a stepwise reaction pathway.
:A = 0:100), which possesses the strongest oxidative capability and operates via a direct photoinduced single-electron transfer (SET) process. In the subsequent step, Cβ–O bond cleavage is facilitated by photoexcited CzCP33 (D:A = 66:33), which reacts with a sacrificial electron donor to generate highly reductive CzCP33˙−, enabling the reduction of ketones formed during the pre-oxidation stage.56 Moreover, this heterogeneous catalyst could be easily separated from the reaction system through filtration or centrifugation and has demonstrated stable catalytic activity during reuse, increasing the potential for employing porous organic frameworks (POFs) as heterogeneous photocatalysts in lignin conversion via a stepwise reaction pathway.
As the pre-oxidation of Cα–OH to CαO is a crucial step for initiating the stepwise Cβ–O bond fragmentation process, several studies have focused on this pre-oxidation stage and achieved significant progress. In later work by Stephenson's group, they developed a hybrid photocatalytic redox system that combined an Ir-based molecular photocatalyst with Pd(OAc)2 to effectively drive the pre-oxidation of lignin model compounds.57 Pd(OAc)2 serves as the mediator in this system, interacting with the excited Ir photocatalyst to produce high-valent Pd(IV) species, which then oxidize the target reactants.57 In addition, the 4,4′-bis(diphenylamino)-benzophenone (DPA-BP)/N-hydroxyphthalimide (NHPI)/O2 photocatalytic system was developed and demonstrated high efficiency in the aerobic oxidation of β-O-4 lignin models.58 The mechanistic investigation revealed that this aerobic oxidation process followed a photoinduced proton-coupled electron transfer (PCET) reaction pathway. Specifically, phthalimide-N-oxyl (PINO) radicals that were converted from NHPI by the DPA-BP photocatalyst can abstract hydrogen from Cα–H in benzylic alcohol and generate the Cα-hydroxy benzylic radical. This radical then reacts with O2 to produce a peroxyl radical, which ultimately converts to a ketone after further reaction with NHPI.58 Moreover, based on a similar oxidation mechanism involving reactive oxidative species (ROS), Cao et al. employed various persulfates (e.g., (NH4)2S2O8) as the radical initiator and CuBr2 as the photocatalyst, which can effectively facilitate the selective oxidation of C–OH in dimeric lignin model compounds under visible light irradiation.59 The semiconductor δ-MnO2 can also generate active oxygen species (O2− or O22−) from molecular oxygen and oxidise Cα–OH in various dimeric β-O-4 alcohols under blue light (470 nm), but it is of interest that γ-CH2OH in lignin model compounds could be retained in this reaction process.60 Otherwise, the oxidation of three technical lignin sources including organosolv lignin, kraft lignin, and alkali lignin was conducted. The IR and 2D-NMR characterisation results indicated that the δ-MnO2 photocatalytic system successfully oxidised these lignin samples and the results from the subsequent depolymerization of the pre-oxidized lignin further demonstrated the effectiveness of δ-MnO2.60 In addition to the investigations that focused on photocatalytic oxidation for the first step, Fang et al. found that Pd/TiO2 can efficiently catalyse the fragmentation of Cβ–O bonds in dimeric β-O-4 ketones, generating the corresponding aromatic monomers through hydrogenolysis and hydrogenation processes.61
However, the reaction conditions for the individual oxidation and reduction processes are typically different; for instance, distinct additional chemical reagents are required for single-electron transfer (SET) and hydrogen atom transfer (HAT) processes. As a result, the complex separation after the first oxidation step and the subsequent transfer of reactants to the reduction step pose significant challenges in this two-step strategy. To address this issue, Luo et al. developed a tandem dual-light driven photocatalytic system capable of cleaving β-O-4 bonds in lignin model compounds (e.g., 2-phenoxy-1-phenylethanol) and producing the corresponding aromatic monomers in a single pot.50 In this case, two catalysts (Pd–ZnIn2S4 and TiO2) with different bandgap structures co-exist in the same reaction system, therefore, this system can consecutively carry out the oxidation and hydrogenolysis reactions in a single pot under different wavelengths of light irradiation. Specifically, H–Cα–OH is firstly oxidised to CαO by the photogenerated holes from Pd–ZnIn2S4 under blue LED light (455 nm). Subsequently, after switching to UV LED light irradiation, the cleavage and reduction of Cβ–O bonds are driven by the photogenerated electrons from TiO2, with the assistance of NaOAc, producing the corresponding aromatic monomeric products.50 In the hydrogenolysis process of Cβ–O bonds, it is worth pointing out the (101) crystal facet of TiO2 was identified as the most active crystal facet, and the photoexcited Ti3+ on the surface of this facet can weaken the Cβ–O bonds of the adsorbed ketone, thereby facilitating their cleavage.50 This dual-wavelength light driven stepwise C–O bond cleavage strategy was also realised using TiO2 with varying NiO loadings.62 In this system, the photo-redox capability of TiO2 could be rationally tuned by the NiO loading, allowing the catalysts to respectively facilitate the dehydrogenation and hydrogenolysis processes in the stepwise C–O bond cleavage reaction.
Overall, the use of the stepwise photocatalytic reaction strategy in this field has shown some potential, but some certain inherent limitations of the stepwise processes remain unsolved. Specifically, the pre-oxidation and subsequent reduction leading to C–O bond cleavage are challenging to carry out simultaneously, necessitating the complicated separation and purification of pre-oxidized reactants in most stepwise reaction pathways. Although the use of flow reactors and the combination of different catalysts can to some extent address this issue at the laboratory scale, these approaches may present challenges for scale-up. Additionally, stepwise photocatalytic processes often require additives, such as radical initiators, scavengers for photogenerated charges, or stoichiometric chemical reagents, further complicating the reaction systems.
Heterogeneous photocatalysts have also been employed to depolymerize β-O-4 lignin model compounds and organosolv lignin. Luo et al. followed a similar reaction route and developed a heterogenous zinc indium sulphide photocatalyst. In this photocatalytic system, the reaction begins with the oxidation of Cα–OH to form ketone intermediates, and then further cleavage of the Cβ–O bond in lignin model compounds via a self-hydrogen transfer hydrogenolysis mechanism.15 It should be noted that no additional chemical reagents or co-catalysts were required in this process. The oxidation of the CαH–OH moiety and the reductive cleavage of Cβ–O bonds can be respectively driven by photogenerated holes and electrons on the same catalyst under blue LED irradiation. In addition, it is interesting that the hydrogen atoms transferred to the final aromatic monomer during the Cβ–O bond fragmentation originate from the alcoholic group (Cα–H–OH) of the lignin model compounds. More specifically, the photogenerated holes can abstract hydrogen atoms from the Cα–H–OH moiety, generating ketone intermediates and protons. These protons can be adsorbed onto the catalyst surface to form a “hydrogen pool” after reacting with the photogenerated electrons. This hydrogen pool subsequently reacts with the ketone intermediates to produce the corresponding aromatic monomers. The results from deuterium-labelled experiments and kinetic isotopic effect studies indicate that the adsorbed hydrogen species are critical for the fragmentation of the Cβ–O bond. The hydrogen from the hydroxyl group ultimately transfers to acetophenone while the hydrogen from Cα–H can be transferred to phenol. This hydrogen transfer hydrogenolysis process was further expanded and investigated, revealing that external hydrogen donors (e.g., isopropanol) can also provide hydrogen for the fragmentation of Cβ–O bonds in ketone lignin model compounds lacking the alcoholic group. Additionally, the depolymerization of organosolv poplar lignin was conducted in this ZnIn2S4 photocatalytic system with isopropanol as the external hydrogen donor. The generation of a variety of monomers and p-hydroxyacetophenone derivatives after the reaction further demonstrated the effectiveness of this photoinduced self-hydrogen transfer hydrogenolysis strategy for lignin conversion.
Based on a similar reaction mechanism, the fragmentation of Cβ–O bonds in dimeric Cβ–O lignin model compounds was also achieved employing ultrathin Ni-layer decorated CdS nanosheets as the photocatalyst via this reaction route.18 In this study, the solvent environment was found to be a crucial factor in determining the overall reaction performance. When pure acetonitrile was used as the solvent, ketone (2-phenoxy-1-phenylethanone) was the only aromatic product obtained from the corresponding alcohol lignin model compound (2-phenoxy-1-phenylethanol). This is because the abstracted hydrogen from the alcoholic group (CαH–OH) was primarily reduced to H2 due to the strong reductive capability of Ni/CdS, rather than forming a “hydrogen pool” on the catalyst surface, which is essential for the C–O hydrogenolysis process. Interestingly, the reaction selectivity was altered by mixing acetonitrile with water, decreasing the selectivity for the dimeric aromatic ketone product and yielding around 50% aromatic monomers. The selectivity for aromatic monomers was further enhanced by altering the pH of the solvent to an alkaline condition using KOH, where over 90% of 2-phenoxy-1-phenylethanol was converted into the corresponding aromatic monomers (acetophenone and phenol), with a significant improvement in reaction rate. The improved performance was attributed to the decreased solubility of 2-phenoxy-1-phenylethanone in the water–acetonitrile mixture, which increased its adsorption on the catalyst surface. Simultaneously, the evolution of H2 from the abstracted hydrogen (CαH–OH) was suppressed by the formation of oxidized nickel species on the catalyst surface under alkaline conditions. This allowed the extracted protons to be accumulated, and thus facilitate the cleavage of Cβ–O bonds.
The oxidation of the alcohol group to a ketone group is crucial for the fragmentation of Cβ–O bonds in all the aforementioned studies, whether following stepwise or direct conversion routes. The fundamental principle behind these reaction pathways is that the oxidative transformation step reduces the bond dissociation energy (BDE) of the Cβ–O bond. In addition to this pre-oxidation approach for lowering the BDE, another pathway involving the formation of Cα radical intermediates has also been investigated. This mechanism was first proposed by Wang's group and was successfully demonstrated to be feasible in the depolymerization of lignin model compounds, organosolv lignin, and even natural lignin biomass.22 In the early stage, CdS nanoparticles (NPs) in the form of cubic zinc blende structure exhibited potential in the decomposition of 2-phenoxy-1-phenylethanol (PP-ol) through the cleavage of Cβ–O bonds, yielding acetophenone and phenol as the major products.31 CdS NPs were further optimised by reducing their particle size to below the Bohr exciton diameter to enhance the photocatalytic performance, and CdS quantum dots (QDs) with average size ranging from 3.8 nm to 6.1 nm were obtained.22 Among these, the CdS QDs with a size of 4.4 nm demonstrated the highest catalytic performance, which can be attributed to the increased surface areas and enhanced redox capability. The mechanistic studies supported by both experimental and computational results revealed that the formation of the Cα radical intermediate is crucial to the subsequent fragmentation of Cβ–O bonds as illustrated in Scheme 2(a). This intermediate drastically lowers the bond dissociation energy (BDE) of the Cβ–O bond in 2-phenoxy-1-phenylethanol (PP-ol) from 55 to 7.8 kcal mol−1, even lower than the reduced BDE of the Cβ–O bond in the ketone intermediate (2-phenoxy-1-phenylethanone, PP-one).22 The controlled experiments (Scheme 2(b)) using different quenching reagents indicated that both photogenerated holes and electrons are involved in the reaction, thus a so-called electron–hole coupled photocatalytic reaction mechanism was proposed. Specifically, the Cα–H is firstly oxidised and dehydrogenated by photogenerated holes, generating a Cα radical intermediate. Upon reacting with the photogenerated electrons, the Cα radical intermediate undergoes the cleavage of the weakened Cβ–O bond and ultimately yields acetophenone and phenol. Herein, the formation of Cα radical intermediates was confirmed through the radical trapping experiment, where 5-diisopropoxy-phosphoryl-5-methyl-1-pyrroline-N-oxide (DIPPMPO) was employed as the trapping reagent to capture radicals. This led to the formation of stable hydroxylamine and nitrone products, which could be analysed using positive-ion electrospray ionization mass spectrometry and 31P nuclear magnetic resonance (NMR). Notably, the results unambiguously confirmed the presence of the Cα radical intermediate in the reaction.22 Various dimeric lignin models with various functionalised groups (e.g., γ-hydroxyl group) were successfully converted into corresponding aromatic monomers without unwanted degradation of these functional groups using this photocatalytic system, further demonstrating the high selectivity of the catalyst. Furthermore, this study found that the yield of monomeric aromatics increased with the Cβ–O linkage content during the depolymerisation of various technical lignin and natural lignin in birch woodmeal and nearly approached the theoretical maximum yields of aromatic monomers. Otherwise, the CdS QDs can be easily recycled from the reaction system through a reversible aggregation–colloidisation process for reuse. Afterwards, the dissolved aromatic monomer products in the solvent, along with the residual cellulose and hemicellulose solids, could be readily separated. This study, for the first time, demonstrated a “lignin-first” strategy that converts lignin into aromatic monomers via photocatalysis without sacrificing cellulose and hemicellulose, further confirming the immense potential of photocatalysis for producing monomeric aromatics from native lignocellulosic biomass.20,22,31
 | ||
| Scheme 2 (a) Proposed electron–hole coupled (EHCO) mechanism for CdS-catalysed Cβ–O bond cleavage of a dimeric β-O-4 lignin model compound through a Cα radical intermediate; (b) the control experiments with various reactants or additives (this scheme has been reproduced from ref. 22 with permission from Nature Publication Group, copyright 2018). | ||
Subsequently, CdS quantum dots (QDs) were further modified and systematically investigated in terms of the effects of organic ligands attached to their surface.64 Given that the colloidal stability of CdS QDs is highly dependent on the hydrophilic or hydrophobic properties of the surface organic ligands, the dispersibility of CdS QDs in the reaction solvent (CH3OH/H2O solution) could be enhanced by loading hydrophilic organic ligands (e.g., mercaptoalkanoic acid and HS(CH2)nCOOH) onto their surface. This modification can facilitate intimate contact between CdS QDs and solid native lignin. In this manner, the CdS QD catalysts could function as a pseudo homogeneous catalyst and significantly improve the efficiency of solid–solid contact between the heterogeneous catalyst and solid reactants, thereby significantly improving the catalytic performance. Regarding the impact of ligands on the surface of the photocatalysts, the characterization results from femtosecond transient absorption spectroscopy revealed that the rate constant for electron transfer decreased exponentially with increasing ligand length (HS(CH2)nCOOH, n = 2, 5, 10). And the photocatalyst with shorter alkyl chains of ligands exhibited better catalytic activity due to the lower energy barrier for charge transfer. Thus, the surface ligands are considered to act as the electron-tunnelling mediator, facilitating the transfer of interfacial charges from CdS QDs to the substrate.
The photocatalytic performance of CdS could be further improved through a simple Ag+ cation exchange approach, which had been commonly applied in the field of photocatalysis.17 Ag+ can substitute Cd2+ during the cation exchange process, forming Ag2S on the surface of CdS. This modification can significantly suppress the recombination and promote the transfer of photogenerated electrons. Consequently, the conversion rate of PP-ol could be increased from 35% to nearly 100% under the same reaction conditions. The improved photocatalytic activity is attributed to the reduced of recombination of photogenerated electron–hole pairs and the accelerated transfer of photogenerated electrons in Ag2S@CdS. The characterisation results indicated that the intrinsic band structure of CdS was not significantly altered, however, the Fermi level of Ag2S@CdS was downshifted due to the lower Fermi level of Ag2S compared to CdS. This allows photogenerated electrons to transfer from the conduction band of CdS to Ag2S, promoting the separation of photogenerated charges and enhancing the proton affinity of the catalyst. As illustrated in Scheme 3, Ag2S@CdS can effectively reduce recombination while promoting the separation and migration of photogenerated electrons and holes, thereby providing more photogenerated charges at the surface to trigger the redox cleavage of Cβ–O bonds via a similar electron–hole coupled oxidation (EHCO) mechanism.65
 | ||
| Scheme 3 Schematic representation comparing the fragmentation of the β-O-4 bond in Ag2S@CdS (yellow) with CdS (blue) (this figure has been reproduced from ref. 17 with permission from American Chemical Society, copyright 2020). | ||
In addition to enhancing the contact and interfacial charge transfer between the photocatalyst and lignin substrates, energy band engineering was employed to optimise the photocatalysts and deepen the understanding of the mechanisms involved in the photocatalytic conversion of lignin model compounds and lignin itself. A series of ZnmIn2Sm+3 (m = 1–6) in the form of aggregated multi-layered nanosheets were synthesised using a simple hydrothermal method, and the energy band structures of the materials were modified by adjusting the atomic ratio of Zn to In, as illustrated in Scheme 4(a).16 The controlled experiments conducted under UV irradiation or visible light irradiation indicated that the enlarged valence-band maximum (VBM) and conduction-band minimum (CBM) can enhance photo-redox capabilities and promote the conversion of PP-ol. More specifically, a lower (more positive) VB edge can facilitate hydrogen atom abstraction (HAA) of the Cα–H bond of PP-ol and generate the Cα radical intermediate while a higher (more negative) CB edge can promote the reductive cleavage of the Cβ–O bonds. However, an excessively wide energy band gap can result in poor responses to visible light, adversely affecting the photocatalytic performance of Zn5In2S8 and Zn6In2S9.16,20 As illustrated in Scheme 4(b), the mechanistic studies suggested that both photogenerated electrons and holes are involved in the fragmentation of Cβ–O bonds. And the reaction essentially follows the same mechanism involving the Cα radical intermediate that was proposed in previous studies.16,20,22 However, a minor pathway for Cβ–O cleavage via a CO intermediate reaction also occurs during the photocatalytic process.16 The thiol group (–SH) was found to be critical to catalytic activity, as it can trap photogenerated holes and form thiyl radicals that selectively activate the Cα–H bond, leading to the formation of the Cα radical intermediate. Further experiments on the depolymerization of native lignin were conducted, achieving a high yield of functionalized monomeric aromatic products (18.4%) from the photocatalytic conversion of dioxasolvs birch lignin.
 | ||
| Scheme 4 (a) Band structure diagram of ZnmIn2Sm+3 (m = 1–6); (b) proposed photocatalytic mechanism for β-O-4 bond cleavage in 2-phenoxy-1-phenylethanol over the Zn4In2S7 catalyst (this scheme has been reproduced from ref. 16 with permission from Wiley-VCH, copyright 2019). | ||
Most of the previous studies on the photocatalytic C–O bond cleavage in lignin model compounds have primarily focused on modifying photocatalysts by optimizing material properties, such as the light absorption edge, energy band structure, recombination rate, and transfer efficiency of photoexcited electrons. Additionally, material surface engineering has been explored to enhance the intimate contact between catalysts and the substrate.17,18,66 Otherwise, certain critical factors that can considerably influence the photocatalytic performance have not been systematically investigated, which greatly impeded the rational design of photocatalysts and the development of efficient photocatalytic systems for C–O bond fragmentation in lignin. For example, it has been confirmed that the use of an external hydrogen donor is an effective strategy for reaching great catalytic performance in the thermo-catalytic lignin depolymerisation process,1,67 but this mechanism had not been studied in the photocatalytic lignin conversion process. In fact, in previously reported studies, photocatalytic C–O bond fragmentation reactions were typically carried out in solvents without hydrogen-donating properties. The lignin model compound itself often served as the hydrogen donor, providing the necessary hydrogen for the hydrogenolysis cleavage of C–O bonds.15,18,20 It is plausible that the reaction rate and selectivity could be constrained by insufficient supply of hydrogen abstracted from the lignin substrate itself. Furthermore, previous research also found that hydrogen gas could be generated during the cleavage of lignin model compounds, which competes with the essential demand for hydrogen in the hydrogenolysis cleavage of C–O bonds, thereby reducing lignin conversion efficiency and the selectivity for monomeric aromatics.15,18 Herein, inspired by the fact that certain catalysts can abstract and activate hydrogen from hydrogen donors to promote the hydrogenolysis process, Shao and his co-workers successfully developed a series of metal sulphide photocatalysts capable of accelerating hydrogen atom transfer (HAT) during the hydrogenolysis of Cβ–O bonds.68–71 This was accomplished by supplying adequate hydrogen from the hydrogen-evolving half-reaction of the water dissociation process, resulting in a significant improvement in both the reaction rate and selectivity for the target monomeric aromatic products. The isotopic labelled experiments confirmed that the hydrogen transferred from water to the monomeric aromatics during the photocatalytic process. In addition, KIE measurements indicated that the protons generated from water decomposition are crucial to the overall reaction, further highlighting the beneficial role of water in promoting the reaction. Moreover, Shao and his co-workers confirmed the promoting effect of adsorbed reactive radical species generated from photocatalytic water dissociation on the cleavage of target C–O bonds in lignin by DFT calculations. As shown in Scheme 5, H* and *OH generated from water dissociation and absorbed on the surface of the photocatalyst can remarkably increase the energy barrier for the oxy-dehydrogenation of key reactive intermediates, which is energetically favourable in the absence of these adsorbed reactive radical species. Specifically, although the energy required for the generation of the radical intermediate via C–H activation and hydrogen atom abstraction (HAA) increases from 1.23 eV to 1.65 eV with the presence of adsorbed radical species on the surface of photocatalyst, the side-reaction of further dehydrogenation (Scheme 5(b) R1) of the key radical intermediate on the bare catalyst surface remains a kinetically favourable reaction. In contrast, this dehydrogenation process requires an activation energy (Ea) of 0.44 eV on the catalyst surface when H* and *OH are adsorbed (Scheme 5(b) R2). Therefore, the reaction can proceed in the direction of the target C–O bond cleavage while inhibiting side reactions of oxy-dehydrogenation. The difference between R3 and R4 lies in the origin of hydrogen in the final monomeric aromatic products. Reaction pathway R3 has the lowest activation energy (Ea) of 0.09 eV, making it the most favourable from a kinetic perspective. In this pathway, H* adsorbed on the surface of photocatalyst transfers to acetophenone, while the hydrogen from the C–OH group of the radical intermediate is transferred to the hydroxy group of phenol. In contrast, pathway R4, which has the highest Ea of 1.03 eV, involves phenol receiving H* from the catalyst surface, with the hydrogen from the C–OH group of the radical intermediate transferring to acetophenone. These DFT calculated results align well with the findings from isotope-labelled experiments, providing further insight into the hydrogen transfer behaviours in this reaction process.
 | ||
| Scheme 5 (a) Proposed reaction mechanism for photocatalytic fragmentation of aryl ether C–O bonds in lignin model compounds over quaternary cadmium zinc indium sulphide photocatalysts; (b) the calculated reaction profiles of four different PP-ol conversion pathways in two different catalytic microenvironments (this scheme has been reproduced from ref. 68 with permission from Elsevier, copyright 2024). | ||
In summary, compared to the stepwise reaction routes, the direct photocatalytic cleavage of Cβ–O bonds via a redox-neutral reaction not only can decrease or avoid the use of additive stoichiometric chemical reagents but also can simplify the complicated separation and purification process for the pre-oxidised reactants after the first step. Moreover, as both photogenerated holes and electrons could be utilised for driving the oxidation and reduction simultaneously, the theoretical reaction efficiency could also be improved in this reaction route.
The commercial TiO2 (Degussa P25) loaded with Pd nanoparticles as the co-catalyst was found capable of cleaving Cα–O bonds under UV light irradiation and mild conditions. When benzyl phenyl ether (BPE) was used as the reactant in a solvent of 2-propanol containing 20 vol% water, the reaction selectively produced toluene and phenol as the only products.72 In this reaction process, the reduction of H+ at the Pd sites (H–Pd) generates active hydrogen species with strong reductive capabilities. These active hydrogen species, analogous to those reactive species formed via the dissociative adsorption of H2 in thermocatalytic processes, were identified as crucial participants in accomplishing the hydrogenolysis of Cα–O bonds.
Inspired by the use of the Ni/TiN nanocomposite as the catalyst in the thermolysis and hydrogenolysis of commercial kraft lignin, Liu et al. discovered that TiN could also efficiently cleave C–O bonds in phenethyl phenyl ether (PPE) under relatively mild conditions (100 °C, KOH, isopropanol) in a visible-light-driven photocatalytic process, yielding styrene and phenol as the products.73,74 The action spectrum study indicated that this reaction is driven by direct photon energy, with the strong localized surface plasmon resonance (LSPR) effect of TiN resulting in exceptionally high light absorption properties. To further investigate the active sites in this reaction, the chemical states of titanium (TiO2 (+4), Ti–N–O (+1), and TiN (+3)) in TiN were modified through calcination. The results revealed that Ti–N–O species are the active sites for this photocatalytic reaction. A possible mechanism involving radical intermediates is proposed in this work, as illustrated in Scheme 6. The reaction initiates with the abstraction of Cβ–H, generating Cβ-radical intermediates, followed by C–C shift and Cβ–O cleavage, ultimately producing styrene and phenol. Notably, this photocatalytic system demonstrates high selectivity for styrene, avoiding the formation of over-reduced products (e.g., ethylbenzene) typically seen in thermal reactions. The unsaturated CC bonds in the product can be further processed to yield various value-added products (e.g., through C–N coupling hydroamination reactions), thereby enhancing the applicability of products derived from lignin conversion.
 | ||
| Scheme 6 Proposed reaction mechanism of the photocatalytic fragmentation of PPE by TiN NPs through Cβ-radical intermediates (this scheme has been reproduced from ref. 74 with permission from American Chemical Society, copyright 2021). | ||
The localized surface plasmon resonance (LSPR) effect of noble metals has also been explored in this field and was found to enhance the inherent bond-breaking ability of transition-metal complexes in the cleavage of Cα–O linkages in α-O-4 lignin model compounds. Noble metal nanoparticles, such as Au or Ag, were co-immobilised with Ni(II) complexes on Al2O3 nanofibers grafted with a silane containing an amino group, forming Au-ASN-Ni2+ and Ag-ASN-Ni2+photocatalysts.75 As illustrated in Scheme 7, the plasmonic metal nanoparticles function as antennas (step i), harvesting light energy and transferring it to the active sites. It should be noted that the intense electromagnetic near-fields of the plasmonic metal nanoparticles can remarkably enhance the chemisorption of the lignin mocel substrates to the Ni2+ active sites (step ii). The light-excited hot electrons can migrate from the plasmonic metal nanoparticles to Ni2+ sites through a molecular bridge of the aromatic ring of the reactants (step iii and iv). This mechanism also eliminates the need for intense thermal heating, which is typically required to activate the Ni complex and overcome the reaction energy barriers. Eventually, the lignin model compound benzyl phenyl ether could be converted to phenol and toluene with high selectivity using Au-ASN-Ni2+ or Ag-ASN-Ni2+ photocatalysts under visible light irradiation at 80–90 °C in isopropanol with KOH (step v). Notably, no by-products from the over-reduction of aromatic rings were detected, which was likely attributed to the relatively mild reaction conditions. Further mechanistic studies indicated that the interband excitation of Au nanoparticles and the intraband excitation of Ag (LSPR effect) generate photoinduced hot electrons, which migrate to the Ni2+ complex via the unoccupied anti-bonding π* orbitals of the aromatic ring of the ether, reducing Ni2+ to catalytically active Ni0 species. The active Ni0 species then drive the fragmentation of Cα–O linkages, yielding phenol and toluene, with hydrogen released from the oxidation of isopropanol on the plasmonic metal nanoparticles.
 | ||
| Scheme 7 Proposed reaction mechanism of the plasmonic-antenna-promoted catalysts in benzyl phenyl ether decomposition (this scheme has been reproduced from ref. 75 with permission from Elsevier, copyright 2019). | ||
Nickel, as a relatively inexpensive non-plasmonic metal from the platinum group, is a well-known hydrogenation catalyst employed for the reductive cleavage of aryl ether C–O bonds via thermocatalysis. Recently, Li et al. developed Ni nanoparticles that can selectively catalyse the hydrogenolysis of various aryl ether C–O bonds in lignin under visible light irradiation and relatively mild conditions.9 Mechanistic studies revealed that the Ni nanoparticles on the catalyst surface serve as active sites, absorbing visible light through interband electronic transitions and generating hot electrons to drive the cleavage of C–O bonds in various lignin model compounds. In this reaction system, additional reductive agents (e.g., hydrazine hydrate or NaBH4) are required to maintain the metallic Ni0 on the surface, thereby providing more active sites for the reaction. As expected, the reaction temperatures required for the fragmentation of different C–O bonds corresponded to the bond dissociation energies (BDE) of the C–O bonds in diphenyl ether (DPE, 4-O-5 structure), 2-phenylethyl phenyl ether (PPE, β-O-4 structure), and benzyl phenyl ether (BPE, α-O-4 structure). The photocatalytic conversion of dealkaline lignin was also conducted to assess the system's feasibility, resulting in the generation of 14 monomeric aromatic products with an overall yield of 9.84 wt% after 35 hours of reaction at 80 °C.
The photocatalytic system developed by Shao and his co-workers can also selectively cleave Cα–O bonds in benzyl phenyl ether68 with the assistance of radicals from water dissociation. Notably, as shown in Scheme 5(a), the simultaneous fragmentation of Cα–O and Cβ–O bonds was accomplished without additional chemical reagents. The α-O-4 lignin model compound (BPE) could be completely converted at room temperature with nearly 90% selectivity for monomeric aromatic products (benzaldehyde and phenol) after 90 min of visible light irradiation. Moreover, it should be noted that the generation of benzaldehyde from this study is different from that presented in the previous literature, which usually produced toluene rather than benzaldehyde.1,29,72,75 Typically, the generation of benzaldehyde is more favourable than toluene, as toluene is more challenging to be further processed due to the highly stability of its (sp3) C–H bonds. And the isotopic labelled experiments with D2O and H2O18 directly revealed that the radicals generated from water dissociation participate in the C–O bond cleavage and the extra oxygen atom in the generated benzaldehyde originates from water.
Due to the higher bond dissociation energy (77.74 kcal mol−1) of 4-O-5 bonds compared to other major C–O bonds, such as Cα–O bonds (57.28 kcal mol−1) and Cβ–O bonds (69.35 kcal mol−1), the 4-O-5 linkages in lignin are the most challenging C–O bonds to cleave.65,76,77 Tan et al. developed an aryl carboxylic radical-mediated acidolysis approach to break the aryl ether bond of the 4-O-5 lignin linkage under blue LED irradiation.76 In this reaction, the aryl carboxylic radicals were generated from acridinium photocatalysts with strong oxidative capability, subsequently, these electrophilic radical species would attack the electron-rich aromatic rings of the reactants, yielding metastable intermediates. Cu(TMHD)2 was employed as a Lewis acid catalyst to coordinate and further stabilise these intermediates. Notably, Cu(TMHD)2 can also facilitate the subsequent C–O fragmentation through a single electron transfer (SET) with the reduced photocatalyst, ultimately producing esters and phenols.65,76
Unlike Tan's work, which introduced external active radical species to activate the robust 4-O-5 bonds and initiate the reaction, Zhou et al. utilised uranyl species as the photocatalysts, leveraging their ligand-to-metal charge transfer (LMCT) properties and superior oxidative capability (Eox = +2.60 V vs. SCE) to directly activate the robust C–O bonds in 4-O-5 lignin model compounds.77 As depicted in Scheme 8(a), an interesting outcome of this reaction is that two phenol molecules can be generated from one molecule of diaryl ether. The results from O18 isotopic labelling experiments revealed that the additional oxygen in the produced phenol originate from uranyl photolysis of water through the oxygen atom transfer (OAT) process. It should be noted that these findings also demonstrated that both C–O bonds in the 4-O-5 structure can be cleaved, as all phenol derivatives, whether substituted with tert-butyl or cyano groups labelled with O18. The possible reaction mechanism was further investigated and is illustrated in Scheme 8(b), the process begins with a single electron transfer (SET) between photo-excited *UO22+ and the diary ether via the ligand-to-metal charge transfer (LMCT) process, generating UO2+ and radical cation A. Subsequently radical cation A reacts with the uranyl peroxide complexes generated from the uranyl photolysis of water, which further decomposed to the radical cation of phenol B and phenol oxygen anion C through the cleavage of C–O bonds. The final products of phenol were generated from another SET process between B and UO2+, followed by the hydrogenation of C.
 | ||
| Scheme 8 (a) The results from O18 labelling experiments and investigation of the dual cut-off mechanism; (b) proposed mechanism for uranyl-photocatalysed hydrolysis of the 4-O-5 linkage in lignin model compounds (this scheme has been reproduced from ref. 77 with permission from American Chemical Society, copyright 2021). | ||
In addition, the cleavage of Caryl–O bonds in β-O-4 structures was also investigated to obtain various non-phenolic monomeric aromatic products. However, this fragmentation faces significant challenges due to the high bond dissociation energy (BDE) of Caryl–O bonds and low selectivity, which is affected by the presence of weaker aliphatic ether bonds.20,65 To address these challenges, Li et al. developed an intramolecular substitution approach to cleave the Caryl–O bond in β-O-4 lignin model compounds as displayed in Scheme 9.78 The radical precursor, acyl oxime, was firstly introduced into the β-O-4 structures through the stepwise oximation and acylation reactions. Then, the iminyl radicals were generated by the photoexcited organic molecular photocatalyst (phenothiazine, PTH) via a single electron transfer (SET) mechanism. These iminyl radicals could attack the Caryl–O bonds, forming the spiro intermediates through aryl migration in a proposed five-membered ring. The spiro intermediates subsequently decomposed through C–O bond cleavage, yielding arylamine and ketone products.78

2.2. Selective C–C bond fragmentation
Due to the significant presence of C–C linkages in lignin, the yield of monomeric aromatic products from lignin depolymerisation can be limited if only C–O linkages are targeted for fragmentation. Furthermore, lignin isolated from conventional industrial processes, such as the kraft, sulphite, and organosolv processes, as well as from commercial biorefining operations, typically contains higher fractions of C–C bonds. This is because additional C–C bonds form through condensation reactions, often at the expense of Cβ–O bonds.6,79 Therefore, it is crucial to target the cleavage of C–C interunit linkages to further increase the yields of monomeric aromatic products from lignin depolymerisation. However, the fragmentation of C–C bonds poses greater challenges than that of C–O bonds due to their non-polar and robust characteristics. Currently, C–C bond scission primarily focuses on β-O-4 and β-1 linkages (specifically the cleavage of the Cα–Cβ bond), which are the most abundant and second most abundant linkages in most lignin biomass.Photocatalytic lignin depolymerisation through C–C bond scission has achieved some progress. It has been found that the generation of various radical species, which can activate reactants or weaken the bond dissociation energy of C–C bonds, is crucial in these reaction processes, generally resulting in the production of aldehydes and phenols as the primary products. Based on the specific radical species formed during the C–C bond cleavage, most approaches aimed at selectively cleaving Cα–Cβ bonds can be classified into three main categories: alkoxy radical approach, carbon radical mediated oxidation approach, and cation radical mediated approach.
 | ||
| Scheme 10 Proposed reaction mechanism of Cα–Cβ bond cleavage by the Ir complex via a PCET process (this scheme has been reproduced from ref. 82 with permission from Elsevier, copyright 2019). | ||
Due to the chelation property of the benzyl Cα–OH structure, the active metal sites can coordinate with the benzyl Cα–OH group in lignin substates and be excited by light irradiation. This interaction can generate the alkoxy radical intermediate, facilitating the subsequent scission of C–C bonds through a light-driven ligand-to-metal charge transfer (LMCT) process.84–86 The chelating interaction with the targeted hydroxyl group along with the oxidative capability was found to be crucial for the cleavage of C–C bonds via the LMCT process. Various vanadium complexes, synthesised with rational ligand modification, were developed as the catalysts to facilitate C–C bond fragmentation in β-O-4 and β-1 linkages using this LMCT approach, yielding the corresponding aromatic monomer products.84–86 Wang et al. further advanced this LMCT strategy for C–C bond cleavage by employing CeCl3-based photocatalysts and introducing additional reagents of di-tert-butyl azodicarboxylate (DBAD).87 As shown in Scheme 11, the mechanistic studies indicated that this process begins with the coordination of cerium (CeIII) with the benzyl Cα–OH group, which is then oxidised by DBAD* to form a CeIV/lignin complex and DBAD−. Next, the key alkoxy radical intermediate, featuring a weakened C–C bond, is generated through the photoinduced LMCT homolysis of the CeIV/lignin complex and subsequently decomposed into an aldehyde product and an alkyl radical. Unlike the previous studies where the generated alkyl radical typically reacts with a hydrogen donor or oxygen species to produce formic acid or ether, in this case, hydrazinium compounds were formed by coupling the alky radical with DBAD.65,87
 | ||
| Scheme 11 The plausible reaction mechanism for CeCl3/DBAD photocatalytic fragmentation and amination of the Cα–Cβ bond in lignin model compounds (this scheme has been reproduced from ref. 65 and 87 with permission from Nature Publication Group & Chinese Chemical Society, copyright 2022 & copyright 2020). | ||
Herein, Hou et al. firstly developed a hybrid CuOx/ceria/anatase nanotube photocatalyst capable of catalysing the fragmentation of Cα–Cβ bonds in various β-1 lignin model compounds with high selectivity under visible light illumination.88 As shown in Scheme 12(a), the reaction is initiated by the preferential activation of the Cβ–H bond, leading to the generation of a Cβ-radical intermediate induced by photogenerated holes and protons. Subsequently, the Cβ-radical intermediate reacts with protons and the peroxide radical anion derived from the photo-reduced O2, forming an unstable peroxide intermediate. This intermediate undergoes Cα–Cβ bond fragmentation, producing monomeric aromatics through a six-membered-ring transition state. This study further confirmed that the activation and transformation of the CαH–OH group represent a competing side reaction, resulting in the formation of 1,2-diphenylethanone as a by-product alongside minor Cα–Cβ bond fragmentation. Moreover, the role of each catalyst component was systematically investigated. Specifically, the competing side reaction of Cα–H activation primarily occurred on anatase TiO2, while ceria was found to narrow the band gap and enhance the visible light absorption of the catalyst. The highly dispersed CuOx nanoclusters on the surface served as hole trapping centres, suppressing the side reaction leading to ketone formation due to their inactivity in Cα–H activation. Additionally, the presence of CuOx nanoclusters on ceria increased the concentration of surface defects, including oxygen vacancies (Vo) and Ce3+ ions, which are the catalytically active sites of ceria.
 | ||
| Scheme 12 The proposed reaction mechanism for photocatalytic cleavage of the Cα–Cβ bond in the (a) β-1 lignin model compounds over the CuOx/CeO2/TiO2 catalyst and (b) in the β-O-4 lignin model compounds over mpg-C3N4 catalysts (this scheme is reproduced from ref. 66 and 88 with permission from American Chemical Society, copyright 2017 & copyright 2018). | ||
Based on a similar reaction mechanism involving the Cβ radical intermediate, Liu et al. developed a mesoporous graphitic carbon nitride (mpg-C3N4) that can efficiently catalyse the fragmentation of the Cα–Cβ bond in both β-O-4 and β-1 lignin model compounds under visible light irradiation and aerobic conditions (Scheme 12(b)).66 Notably, the results of solid-state NMR characterisation and theoretical studies confirmed that the presence of π–π stacking interactions between the corrugated mpg-C3N4 surface and lignin model compounds may promote the Cβ–H bond activation, and the photogenerated holes are essential to the formation of Cβ radical intermediates. In addition, various carbon nitride based semiconductor photocatalysts were further investigated based on different material modification strategies, including the construction of a Z-scheme heterojunction system and the introduction of sulphur dopants and cyano (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) groups on the surface, all aimed at enhancing photocatalytic activity for breaking Cα–Cβ bonds in lignin.14,89
N) groups on the surface, all aimed at enhancing photocatalytic activity for breaking Cα–Cβ bonds in lignin.14,89
Among them, Shao and his co-workers found that the cleavage of lignin C–C bonds can be promoted by creating multiple defects on g-C3N4 as the active sites driving the key step reactions.90 Compared to the pristine g-C3N4, the modified g-C3N4 photocatalyst with defective sites showed a superior catalytic activity (improved 102%) and selectivity (∼90%) for monomeric aromatics. This study proved that the construction of defects and the ultrathin structure optimise the electronic properties of g-C3N4, facilitating better separation and transfer of photoinduced charges. And as shown in Scheme 13, the control experiments and DFT calculations further suggested that the constructed defect sites can facilitate the generation of essential reactive radicals (e.g., activation of O2) and radical intermediates (C–H activation).90 Moreover, an urchin-like hierarchical Nb2O5 hollow microsphere was found to catalyse this bond dissociation reaction through the same reaction mechanism, with the (001) facet as the active site for the reaction.91
 | ||
| Scheme 13 Proposed mechanism of multi-defective g-C3N4 photocatalysed cleavage of C–C bonds in β-O-4 and β-1 structures; the marked area 1, 2 and 3 represent the –CN, N vacancy and C vacancy, respectively (this scheme has been reproduced from ref. 90 with permission from Elsevier, copyright 2024). | ||

3. Photocatalysts and key factors in the photocatalytic lignin conversion process
To efficiently convert lignin into value-added monomeric aromatic products, it is essential to meticulously design and control the photocatalysts and the key factors that influence the photocatalytic performance. This section discusses the development of photocatalysts, the construction of effective reactor systems, and the optimization of reaction conditions.Surface reactions that occur at the catalytic sites, which involve the effective adsorption and activation of reactant species, are critical to the performance of photocatalytic reactions. Typically, the catalytic sites are located on the surface, either at positions where photoexcitation occurs or within components integrated with the semiconductors.39 However, the photocatalytic processes generally involve multiple reaction pathways and are frequently accompanied by competitive side reactions.40 In this context, the catalytic sites are crucial for regulating reaction performance by enhancing the adsorption and activation of reactive species, thereby achieving high catalytic activity and selectivity. Additionally, the overall performance of the photocatalytic process can be influenced by common issues encountered in catalytic reactions, including reaction temperature, pH of the reaction system, external energy inputs (such as electric fields), and the dispersion of catalysts, among other factors.94–96
3.1. Photocatalysts for lignin conversion
The catalyst plays a pivotal role in photocatalytic lignin conversion systems. In early studies, particularly those focusing on the photocatalytic mineralisation of lignin in pulping black liquor, photocatalysis was often considered an unselective process. However, more recent studies have demonstrated that through careful screening and material modifications—such as optimising catalyst composition, morphology, and electronic structure—the catalytic activity and product selectivity can be finely tuned, thereby significantly improving the overall reaction performance.Both homogeneous and heterogeneous photocatalysts have been developed and applied in photocatalytic lignin conversion. Homogeneous photocatalysts, typically organometallic complexes formed by metal ions (e.g., vanadium, iridium) and organic ligands, have exhibited superior catalytic performance, particularly in the selective cleavage of C–O or C–C linkages. These photocatalysts offer tuneable catalytic activity and intimate contact with the substrates, enabling high photocatalytic efficiency. However, the inherent challenges of product–catalyst separation and catalyst recyclability remain unresolved.
In contrast, heterogeneous photocatalysts, widely used in lignin conversion, can be easily separated from the reaction mixture for reuse. However, they are often limited by insufficient light-harvesting capabilities, relatively low reactivity, and a lack of understanding of the active sites. Further research is needed to address these issues. To date, photocatalysts developed for lignin conversion can be broadly categorized into metal oxides, metal sulphides, carbon-based materials, and immobilized homogeneous catalysts, based on their primary active components.
Among them, binary metal sulphide based photocatalysts (e.g., CdS, ZnS) and multinary metal sulphides (e.g., zinc indium sulphide, ZnxCd1−xS) have been used as photocatalysts and demonstrated considerable potential in various studies. Mechanistic investigations revealed that metal sulphide photocatalysts can facilitate the fragmentation of C–O bonds in a redox-neutral manner. Specifically, the lignin substrates are oxidised by photogenerated holes, leading to the formation of radical intermediates or intermediates with weakened targeted C–O bonds. The photogenerated electrons can then drive the cleavage of these C–O bonds.15,18 Various material modification methods that could enhance the activity of photocatalysts in other applications are also applicable in improving the photocatalytic performance of metal sulphide catalysts in the lignin conversion process. For instance, introducing an appropriate amount of AgS into CdS and employing graphene oxide as the supporting material can facilitate the separation and migration of photogenerated charge carriers, demonstrating their effectiveness.17,98 Additionally, as insufficient contact between heterogeneous catalysts and the substrates poses a great challenge to achieving high catalytic performance, various approaches have been explored to address this issue. For example, Wu et al. synthesised CdS quantum dots (QDs) with surface organic ligand surfactants, enabling them to function as pseudo-homogeneous catalysts in the reaction system due to their high colloidal stability.99,100
Additionally, carbazolic porous organic frameworks (POFs) composed of cross-linked carbazoles, featuring tuneable photo-redox properties, can also oxidatively cleave C–O bonds in lignin model compounds through a stepwise reaction pathway.56 However, additional chemical reagents are often required as co-catalysts to assist the POF photocatalysts in driving the reactions, which represents a notable drawback compared to the carbon nitride photocatalytic system that operates effectively without additives.
Recently, covalent organic frameworks (COFs) have garnered increasing attention and demonstrated significant potential in photocatalytic applications. Compared to traditional semiconductor photocatalysts, COFs can offer flexible tunability in their structure and composition, which is highly advantageous for photocatalysis, including superior visible light absorption and efficient separation and transfer of photogenerated charges.109 Moreover, the tailorable pore structures of COFs allow for precise control over the reaction directions, particularly in shape-selective catalytic reactions.13 However, the use of COFs as photocatalysts in lignin conversion remains in its preliminary stage, and further investigation is needed to realise the potential of COFs in this field.
3.2. Reactor system
The construction of the reactor system is essential for achieving high photocatalytic performance in lignin conversion. Since photocatalytic lignin conversion reactions are typically conducted under moderate conditions (e.g., atmospheric pressure and room temperature), the illumination serves as a critical driving force for the reaction. Specifically, the efficiency of the light sources and the transparency of the reaction mixture must be carefully considered to ensure optimal photocatalytic performance. Traditional batch reactors have been commonly employed in previous studies using heterogeneous photocatalysts due to their simple structures and ease of operation.30 However, it is necessary to investigate and develop a continuous flow reactor system as the fundamental work for future scale-up development.3.3. Additional reagents and atmosphere
Various additional reagents have been employed in previous studies to facilitate or enhance photocatalytic lignin conversion reactions. For instance, Ir-based photocatalysts typically require scavengers, organic bases, and formic acid to effectively perform photocatalytic C–O bond fragmentation via a stepwise reaction pathway.7,20,54,55 And KOH, NaBH4, and hydrazine hydrate were employed to control the chemical states of the active sites on the photocatalysts and maintain the photocatalytic activity during the reactions.9,18 However, it is important to note that the use of additives can increase the cost of the lignin conversion process and pose challenges in product separation and purification. Therefore, it is necessary to develop advanced photocatalysts that can drive the reaction without the need for additives. For instance, various metal sulphide photocatalysts have been developed to cleave C–O bonds in lignin without the need for additional reagents as mediators or scavengers via a photo-redox-neutral reaction mechanism. Otherwise, traditional expensive additives could be replaced with more economically sustainable alternatives. Specifically, since the fragmentation of C–O bonds in lignin typically involves hydrogenolysis and hydrogenation processes, various alcohols and organic acids (e.g., 2-butanol, ethanol, formic acid) have been employed as the external hydrogen donors in previous studies for promoting the reaction.50,110 In this case, it is meaningful to identify the alternative sustainable hydrogen source that can provide hydrogen and promote the photocatalytic C–O bond cleavage.Otherwise, the atmosphere has been confirmed as a significant factor influencing the overall catalytic performance in lignin conversion. Specifically, oxygen acts as an oxidant and plays a crucial role in the oxidative photocatalytic conversion of lignin by generating various oxidative radicals through the photoinduced oxygen process.14 However, the presence of oxygen should be avoided in certain photocatalytic reductive conversions that rely on the radical intermediate mechanism. In these cases, oxygen can significantly promote the oxidative transformation of C–OH to CO, thereby inhibiting the formation of radical intermediates that are essential for C–O bond cleavage.15,68
4. Key challenges in the development of industrial-scale photocatalytic lignin conversion technology
Although photocatalysis has shown exceptional potential in upgrading biomass to produce valuable aromatics, its application has predominantly been confined to laboratory-scale experiments, typically on the millimole scale. The scalability of photocatalytic lignin conversion and its potential industrial applications remain as significant challenges, and no pilot-scale or commercial implementations has been reported to date. Specifically, the following challenges must be addressed to achieve the scale-up and practical implementation of the photocatalytic lignin conversion process.Firstly, the challenge of scaling up photocatalytic lignin conversion and improving process efficiency is primarily reliant on the photocatalyst material itself. Despite advancements in material modification and reaction system intensification leading to significant improvements in catalytic performance compared to the early-stage studies in this field, the overall photocatalytic efficiency is still low, typically remaining in the micromolar range after a few hours of irradiation. Generally, the current catalytic activity reported in the literature is considerably below the minimum requirements for catalysts intended for use in a prototype system. Otherwise, the photocatalysts reported in this field are not currently available commercially and generally synthesised at a laboratory scale. However, for any process to be practical on a large scale, photocatalyst materials must be available in quantities of at least kg-scale quantities. In addition to the photocatalyst, insufficient mass transfer and light distribution are key challenges in the photocatalytic lignin conversion process, due to the recalcitrant and insoluble nature of lignin. As a result, the reaction performance could be significantly worsened by the presence of the suspended lignin substrate in either batch or flow reactors. In this case, the interaction between photocatalysts (or ROSs reported in some previous studies) and substrates could be inhibited. Otherwise, the effectiveness of light irradiation and photon distribution within the reactor is difficult to maintain at a high level during the reaction process, because lignin substrates can also absorb and scatter light. It has been reported that the light penetration could be reduced by the biomass suspension and consequently decrease the photon absorption at the surface of the photocatalyst, leading to lower catalytic activity.111
5. Conclusion and future perspective
In this review, the theoretical principle of semiconductor photocatalysis and recent advances in various approaches for photocatalytic lignin conversion into monomeric aromatics are described and reviewed. Although photocatalytic lignin valorisation has shown great potential in substituting fossil fuel feedstock for producing functional aromatic chemicals, the research of photocatalytic lignin conversion is still at the early stage. Some critical challenges are still unsolved and limit the future industrial implementation in this field.The primary challenge for photocatalytic lignin valorisation is still the undesirable reaction performance including the slow reaction rate and poor selectivity towards valuable products. Firstly, the photocatalytic reaction performance could be significantly restrained by the inherent limitations of photocatalysis, for example, the insufficient utilisation of light caused by the wide band gap and fast recombination rate of photogenerated charges. More importantly, the reaction mechanisms need to be further studied to give guidance on the design of the photocatalytic system for lignin valorisation. In addition, the interaction and contact between the photocatalysts and the lignin substrates could also affect the overall reaction performance.
Many studies in the field of photocatalysis employed theoretical calculations to investigate the reaction mechanism and the electronic structure of the photocatalysts. However, the mechanism studies on lignin conversion through photocatalytic interunit bond cleavage are still mostly based on the experimental results, and only a few studies performed DFT calculation but with simplified lignin model compounds owing to the complex structure of lignin substrates. Since the activation and generation of reactive radicals or intermediates are very critical to the overall reaction performance, it is necessary to perform theorical calculation to simulate the step of the activation and formation of reactive radicals and radical intermediates as the short-term future work. Therefore, the reaction mechanism could be further clarified, and the understanding of the promoting effect of the material modification could also be advanced.
Additionally, from the perspective of long-term future work, the following issues should be investigated and addressed to further advance the photocatalytic utilization of lignin.
(1) The standard reaction substrate must be established for scaled lignin transformation; specifically, the pre-treatment or lignin extraction processes should be further refined to produce lignin substrates with an increased proportion of β-O-4 linkages or other specific structures conducive to catalytic depolymerisation.
(2) The profitability of lignin conversion into monomeric aromatic compounds as well as the life cycle assessment (LCA) of the photocatalytic system should be carefully considered and evaluated. Beyond managing the costs of system construction and catalysts, the target products from lignin depolymerization should prioritize high-demand aromatic monomers for synthetic polymerisation or focus on low-demand but high-value chemical compounds and highly functionalized motifs for specific applications (e.g., medicine).
(3) The reaction mechanism should be further investigated from both width and depth perspectives. From the perspective of width, the range of reactants, including lignin model compounds and actual lignin, should be expanded to ensure a more accurate representation of lignin depolymerization, which remains a fundamental issue not yet fully resolved. For instance, the effects of various functional groups within complex lignin structures are still unclear. Currently, the lignin model compounds commonly used in many studies do not fully capture the intricacies of the lignin depolymerization process. Therefore, it is essential to examine the differences between lignin model compounds and actual lignin arising from simplified molecular structures. As for the depth perspective, the understanding of the reaction mechanism in photocatalytic lignin conversion is still in its early stages. This requires a deeper investigation into the mechanisms involved. In this regard, certain in situ experimental techniques (e.g., EPR, ATR-FTIR, Raman) need to be modified and employed to study surface reactions on the catalyst and the formation of radical intermediates during the reactions. Additionally, theoretical calculations could provide valuable insights into the chemical reaction details at the atomic scale.
(4) The innovative integration of photocatalysis with thermo-catalysis, electro-catalysis, and even enzymatic catalysis and biotechnology may provide an effective approach to enhance the efficiency of lignin conversion. Additionally, the downstream processes, including the separation, collection, and purification of target products from lignin depolymerization, should also be carefully considered.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Shibo Shao: writing – original, review and editing, revision; Xiangzhou Wang: writing, review and editing; Wenbing Li: review and editing; revision; Yiming Zhang: review and editing, revision; Shi Liu: review and editing; Weisheng Xiao: review and editing; Zongyang Yue: review and editing; Xu Lu: supervision; revision; Xianfeng Fan: revision; supervision; project administration.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the research grants from PetroChina (CNPC). This work was also funded by the Engineering and Physical Sciences Research Council (Tender No. EP/W027593/1, EP/V041665/1). The authors gratefully acknowledge the Royal Society of Chemistry, ELSEVIER, American Chemical Society, Chinese Chemical Society, Wiley-VCH and the owners of figures reproduced and used in this paper for their permissions to use the figures.References
- L. Jiang, H. Guo, C. Li, P. Zhou and Z. Zhang, Chem. Sci., 2019, 10, 4458–4468 RSC.
- J. C. Colmenares and R. Luque, Chem. Soc. Rev., 2014, 43, 765–778 RSC.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. M. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- S. Gazi, Appl. Catal., B, 2019, 257, 117936 CrossRef CAS.
- H. Chen, K. Wan, F. Zheng, Z. Zhang, Y. Zhang and D. Long, Renewable Sustainable Energy Rev., 2021, 147, 111217 CrossRef CAS.
- S. H. Li, S. Liu, J. C. Colmenares and Y. J. Xu, Green Chem., 2016, 18, 594–607 RSC.
- P. Li, Y. Ouyang, G. Xiao, Y. Zhao, S. Sarina, J. Baeyens, H. Su and H. Y. Zhu, Green Chem., 2021, 23, 7780–7789 RSC.
- L. I. Granone, F. Sieland, N. Zheng, R. Dillert and D. W. Bahnemann, Green Chem., 2018, 20, 1169–1192 RSC.
- M. Zaheer and R. Kempe, ACS Catal., 2015, 5, 1675–1684 CrossRef CAS.
- C. Zhang and F. Wang, Acc. Chem. Res., 2020, 53, 470–484 CrossRef CAS PubMed.
- Z. Xiang, W. Han, J. Deng, W. Zhu, Y. Zhang and H. Wang, ChemSusChem, 2020, 13, 4199–4213 CrossRef CAS PubMed.
- X. Wu, J. Lin, H. Zhang, S. Xie, Q. Zhang, B. F. Sels and Y. Wang, Green Chem., 2021, 23, 10071–10078 RSC.
- N. Luo, M. Wang, H. Li, J. Zhang, T. Hou, H. Chen, X. Zhang, J. Lu and F. Wang, ACS Catal., 2017, 7, 4571–4580 CrossRef CAS.
- J. Lin, X. Wu, S. Xie, L. Chen, Q. Zhang, W. Deng and Y. Wang, ChemSusChem, 2019, 12, 5023–5031 CrossRef CAS PubMed.
- H. Yoo, M.-W. Lee, S. Lee, J. Lee, S. Cho, H. Lee, H. G. Cha and H. S. Kim, ACS Catal., 2020, 10, 8465–8475 CrossRef CAS.
- G. Han, T. Yan, W. Zhang, Y. C. Zhang, D. Y. Lee, Z. Cao and Y. Sun, ACS Catal., 2019, 9, 11341–11349 CrossRef CAS.
- X. Liu, X. Duan, W. Wei, S. Wang and B. J. Ni, Green Chem., 2019, 21, 4266–4289 RSC.
- X. Wu, N. Luo, S. Xie, H. Zhang, Q. Zhang, F. Wang and Y. Wang, Chem. Soc. Rev., 2020, 49, 6115–6516 RSC.
- J. C. Colmenares, R. S. Varma and V. Nair, Chem. Soc. Rev., 2017, 46, 6675–6686 RSC.
- X. Wu, X. Fan, S. Xie, J. Lin, J. Cheng, Q. Zhang, L. Chen and Y. Wang, Nat. Catal., 2018, 1, 772–780 CrossRef CAS.
- H. Kasap, D. S. Achilleos, A. Huang and E. Reisner, J. Am. Chem. Soc., 2018, 140, 11604–11607 CrossRef CAS PubMed.
- M. F. Kuehnel and E. Reisner, Angew. Chem., Int. Ed., 2018, 57, 3290–3296 CrossRef CAS PubMed.
- K. L. Orchard, K. H. Ly, E. Reisner, T. E. Rosser, M. F. Kuehnel and D. W. Wakerley, Nat. Energy, 2017, 2, 1–9 Search PubMed.
- C. Zhang, H. Li, J. Lu, X. Zhang, K. E. Macarthur, M. Heggen and F. Wang, ACS Catal., 2017, 7, 3419–3429 CrossRef CAS.
- S. Li, S. Kim, A. H. Davis, J. Zhuang, E. W. Shuler, D. Willinger, J. Lee, W. Zheng, B. D. Sherman, C. G. Yoo and G. Leem, ACS Catal., 2021, 11(7), 3771–3781 CrossRef CAS.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- A. Shivhare, D. Jampaiah, S. K. Bhargava, A. F. Lee, R. Srivastava and K. Wilson, ACS Sustainable Chem. Eng., 2021, 9, 3379–3407 CrossRef CAS.
- J. Xu, P. Zhou, C. Zhang, L. Yuan, X. Xiao, L. Dai and K. Huo, Green Chem., 2022, 24, 5351–5378 RSC.
- X. Wu, S. Xie, H. Zhang, Q. Zhang, B. F. Sels and Y. Wang, Adv. Mater., 2021, 2007129, 1–20 Search PubMed.
- A. Albini and M. Fagnoni, ChemSusChem, 2008, 1(1–2), 63–66 CrossRef CAS PubMed.
- K. H. A. Fujishima, Nature, 1972, 238, 37–38 CrossRef PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- K. Hashimoto, H. Irie and A. Fujishima, Jpn. J. Appl. Phys., Part 1, 2005, 44, 8269–8285 CrossRef CAS.
- K. H. T. Inoue, A. Fujishima and S. Konishi, Nature, 1979, 277, 637 CrossRef.
- S. N. Frank and A. J. Bard, J. Phys. Chem., 1977, 81, 1484–1488 CrossRef CAS.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- C. Gao, J. Wang, H. Xu and Y. Xiong, Chem. Soc. Rev., 2017, 46, 2799–2823 RSC.
- Y. Bo, C. Gao and Y. Xiong, Nanoscale, 2020, 12, 12196–12209 RSC.
- S. Meng, H. Wu, Y. Cui, X. Zheng, H. Wang, S. Chen, Y. Wang and X. Fu, Appl. Catal., B, 2020, 266, 118617 CrossRef CAS.
- X. Chen and C. Burda, J. Am. Chem. Soc., 2008, 130, 5018–5019 CrossRef CAS PubMed.
- A. Hussain, J. Hou, M. Tahir, S. Ali, Z. U. Rehman, M. Bilal, T. Zhang, Q. Dou and X. Wang, Catal. Rev.: Sci. Eng., 2022, 1–55 CAS.
- H. H. Mohamed and D. W. Bahnemann, Appl. Catal., B, 2012, 128, 91–104 CrossRef CAS.
- Z. Chen and C. Wan, Renewable Sustainable Energy Rev., 2017, 73, 610–621 CrossRef CAS.
- T. D. H. Bugg and R. Rahmanpour, Curr. Opin. Chem. Biol., 2015, 29, 10–17 CrossRef CAS PubMed.
- T. D. H. Bugg, M. Ahmad, E. M. Hardiman and R. Singh, Curr. Opin. Biotechnol., 2011, 22, 394–400 CrossRef CAS PubMed.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS PubMed.
- N. Luo, M. Wang, H. Li, J. Zhang, H. Liu and F. Wang, ACS Catal., 2016, 6, 7716–7721 CrossRef CAS.
- M. Wang, J. Lu, X. Zhang, L. Li, H. Li, N. Luo and F. Wang, ACS Catal., 2016, 6, 6086–6090 CrossRef CAS.
- I. Bosque, G. Magallanes, M. Rigoulet, M. D. Kärkäs and C. R. J. Stephenson, ACS Cent. Sci., 2017, 3, 621–628 CrossRef CAS PubMed.
- G. Magallanes, M. D. Kärkäs, I. Bosque, S. Lee, S. Maldonado and C. R. J. Stephenson, ACS Catal., 2019, 9, 2252–2260 CrossRef CAS.
- J. D. Nguyen, B. S. Matsuura and C. R. J. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed.
- Z. Hao, S. Li, J. Sun, S. Li and F. Zhang, Appl. Catal., B, 2018, 237, 366–372 CrossRef CAS.
- J. Luo, X. Zhang, J. Lu and J. Zhang, ACS Catal., 2017, 7, 5062–5070 CrossRef CAS.
- M. D. Kärkäs, I. Bosque, B. S. Matsuura and C. R. J. Stephenson, Org. Lett., 2016, 18, 5166–5169 CrossRef PubMed.
- J. Luo and J. Zhang, J. Org. Chem., 2016, 81, 9131–9137 CrossRef CAS PubMed.
- Y. Cao, N. Wang, X. He, H. R. Li and L. N. He, ACS Sustainable Chem. Eng., 2018, 6, 15032–15039 CrossRef CAS.
- J. Dai, A. F. Patti, G. N. Styles, S. Nanayakkara, L. Spiccia, F. Arena, C. Italiano and K. Saito, Green Chem., 2019, 21, 2005–2014 RSC.
- J. Fang, P. Ye, M. Wang, D. Wu, A. Xu and X. Li, Catal. Commun., 2018, 107, 18–23 CrossRef CAS.
- Z. Song, Y. Hu, S. Li, J. Liu, X. Zhang, L. Ma, L. Chen and Q. Zhang, ACS Sustainable Chem. Eng., 2023, 11, 8562–8572 CrossRef CAS.
- K. Chen, J. Schwarz, T. A. Karl, A. Chatterjee and B. König, Chem. Commun., 2019, 55, 13144–13147 RSC.
- X. Wu, S. Xie, C. Liu, C. Zhou, J. Lin, J. Kang, Q. Zhang, Z. Wang and Y. Wang, ACS Catal., 2019, acscatal.9b02171 Search PubMed.
- Z. Huang, N. Luo, C. Zhang and F. Wang, Nat. Rev. Chem., 2022, 6, 197–214 CrossRef PubMed.
- H. Liu, H. Li, J. Lu, S. Zeng, M. Wang, N. Luo, S. Xu and F. Wang, ACS Catal., 2018, 8, 4761–4771 CrossRef CAS.
- H. Li and G. Song, ACS Catal., 2019, 9, 4054–4064 CrossRef CAS.
- S. Shao, K. Wang, Y. Zhang, J. B. Love, Z. Yue, W. Li, L. Huang, Y. Wang, X. Yao, T. Chang and X. Fan, Chem Catal., 2024, 4, 101066 CrossRef CAS.
- S. Shao, K. Wang, J. B. Love, J. Yu, S. Du, Z. Yue and X. Fan, Chem. Eng. J., 2022, 435, 134980 CrossRef CAS.
- Z. Yue, S. Shao, J. Yu, G. Lu, W. Wei, Y. Huang, K. Zhang, K. Wang and X. Fan, ACS Appl. Mater. Interfaces, 2024, 16(23), 29991–30009 CrossRef CAS PubMed.
- Z. Yue, G. Lu, W. Wei, Y. Huang, Z. Chen, F. Dingwall, S. Shao and X. Fan, ACS Appl. Mater. Interfaces, 2024, 16, 47724–47740 CrossRef CAS PubMed.
- K. Imamura, H. Kato, Y. Wada, K. Makabe, A. Onda, A. Tanaka, H. Kominami, K. Sato and K. Nagaoka, Chem. Commun., 2018, 54, 7298–7301 RSC.
- V. Molinari, G. Clavel, M. Graglia, M. Antonietti and D. Esposito, ACS Catal., 2016, 6, 1663–1670 CrossRef CAS.
- Z. Liu, Y. Huang, G. Xiao, P. Li, H. Su, S. Sarina and H. Zhu, Energy Fuels, 2021, 35, 13315–13324 CrossRef CAS.
- P. Han, T. Tana, Q. Xiao, S. Sarina, E. R. Waclawik, D. E. Gómez and H. Zhu, Chem, 2019, 5, 2879–2899 CAS.
- F. F. Tan, X. Y. He, W. F. Tian and Y. Li, Nat. Commun., 2020, 11, 1–11 CrossRef PubMed.
- Y. Zhou, D. Hu, D. Li and X. Jiang, JACS Au, 2021, 1, 1141–1146 CrossRef CAS PubMed.
- H. Li, A. Bunrit, J. Lu, Z. Gao, N. Luo and H. Liu, ACS Catal., 2019, 9, 8843–8851 CrossRef CAS.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- H. G. Yayla, H. Wang, K. T. Tarantino, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2016, 138, 10794–10797 CrossRef CAS PubMed.
- E. Ota, H. Wang, N. L. Frye and R. R. Knowles, J. Am. Chem. Soc., 2019, 141, 1457–1462 CrossRef CAS PubMed.
- Y. Wang, Y. Liu, J. He and Y. Zhang, Sci. Bull., 2019, 64, 1658–1666 CrossRef CAS PubMed.
- S. T. Nguyen, P. R. D. Murray and R. R. Knowles, ACS Catal., 2020, 10, 800–805 CrossRef CAS.
- S. Gazi, W. K. Hung Ng, R. Ganguly, A. M. Putra Moeljadi, H. Hirao and H. Sen Soo, Chem. Sci., 2015, 6, 7130–7142 RSC.
- H. Liu, H. Li, N. Luo and F. Wang, ACS Catal., 2020, 10, 632–643 CrossRef CAS.
- S. Gazi, M. Dokić, A. M. P. Moeljadi, R. Ganguly, H. Hirao and H. Sen Soo, ACS Catal., 2017, 7, 4682–4691 CrossRef CAS.
- Y. Wang, J. He and Y. Zhang, CCS Chem., 2020, 2, 107–117 CrossRef CAS.
- T. Hou, N. Luo, H. Li, M. Heggen, J. Lu, Y. Wang and F. Wang, ACS Catal., 2017, 7, 3850–3859 CrossRef CAS.
- C. Ku, K. Li, H. Guo, Q. Wu and L. Yan, Appl. Surf. Sci., 2022, 592, 153266 CrossRef CAS.
- M. Cao, S. Shao, W. Wei, J. B. Love, Z. Yue, Y. Zhang, X. Zhang, Y. Xue, J. Yu and X. Fan, Appl. Surf. Sci., 2024, 643, 158653 CrossRef CAS.
- H. Chen, D. Hong, K. Wan, J. Wang, B. Niu, Y. Zhang and D. Long, Chin. Chem. Lett., 2022, 33, 4357–4362 CrossRef CAS.
- D. W. Cho, R. Parthasarathi, A. S. Pimentel, G. D. Maestas, H. J. Park, U. C. Yoon, D. Dunaway-Mariano, S. Gnanakaran, P. Langan and P. S. Mariano, J. Org. Chem., 2010, 75, 6549–6562 CrossRef CAS PubMed.
- D. W. Cho, J. A. Latham, H. J. Park, U. C. Yoon, P. Langan, D. Dunaway-Mariano and P. S. Mariano, J. Org. Chem., 2011, 76, 2840–2852 CrossRef CAS PubMed.
- J. Fernández, J. Kiwi, J. Baeza, J. Freer, C. Lizama and H. D. Mansilla, Appl. Catal., B, 2004, 48, 205–211 CrossRef.
- Z. Zhao, H. Zhou, L. Zheng, P. Niu, G. Yang, W. Hu, J. Ran, S. Qiao, J. Wang and H. Zheng, Nano Energy, 2017, 42, 90–97 CrossRef CAS.
- J. Li, Q. Pei, R. Wang, Y. Zhou, Z. Zhang, Q. Cao, D. Wang, W. Mi and Y. Du, ACS Nano, 2018, 12, 3351–3359 CrossRef CAS PubMed.
- V. Etacheri, C. Di Valentin, J. Schneider, D. Bahnemann and S. C. Pillai, J. Photochem. Photobiol., C, 2015, 25, 1–29 CrossRef CAS.
- J. Xu, M. Li, J. Qiu, X. F. Zhang and J. Yao, Int. J. Biol. Macromol., 2021, 185, 297–305 CrossRef CAS PubMed.
- X. Wu, S. Xie, C. Liu, C. Zhou, J. Lin, J. Kang, Q. Zhang, Z. Wang and Y. Wang, ACS Catal., 2019, 9, 8443–8451 CrossRef CAS.
- Y. Wu, H. Wang, W. Tu, Y. Liu, S. Wu, Y. Z. Tan and J. W. Chew, Appl. Catal., B, 2018, 233, 58–69 CrossRef CAS.
- R. Fang, A. Dhakshinamoorthy, Y. Li and H. Garcia, Chem. Soc. Rev., 2020, 49, 3638–3687 RSC.
- Q. Li, X. Luo, F. Liu, J. Cai, W. Han, X. Cao, X. Zhang and Y. Lan, Int. J. Biol. Macromol., 2024, 279, 135492 CrossRef PubMed.
- Y. Niu, C. Guo, X. Cao, J. Li, S. Yang and J. Wang, J. Colloid Interface Sci., 2025, 677, 342–351 CrossRef CAS PubMed.
- M. Zhou, C. Tang, H. Xia, J. Li, J. Liu, J. Jiang, J. Zhao, X. Yang and C. Chen, Fuel, 2022, 320, 123993 CrossRef CAS.
- D. Wang, H. Yang, J. Yang, B. Wang, P. Wasnik, B. Bin Xu and Z. Shi, Adv. Compos. Hybrid Mater., 2023, 6, 1–14 CrossRef.
- C. Li, Y. Xu, W. Tu, G. Chen and R. Xu, Green Chem., 2017, 19, 882–899 RSC.
- D. Masih, Y. Ma and S. Rohani, Appl. Catal., B, 2017, 206, 556–588 CrossRef CAS.
- C. Ku, H. Guo, K. Li, Q. Wu and L. Yan, Chin. Chem. Lett., 2023, 34(1), 107298 CrossRef CAS.
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou, X. Li, D. Huang, Y. Zhu, Z. Wang and J. Tang, Chem. Soc. Rev., 2020, 49, 4135–4165 RSC.
- S. Li, Z. Hao, K. Wang, M. Tong, Y. Yang, H. Jiang, Y. Xiao and F. Zhang, Chem. Commun., 2020, 56, 11243–11246 RSC.
- N. Skillen, H. Daly, L. Lan, M. Aljohani, C. W. J. Murnaghan, X. Fan, C. Hardacre, G. N. Sheldrake and P. K. J. Robertson, Photocatalytic Reforming of Biomass: What Role Will the Technology Play in Future Energy Systems, Springer International Publishing, 2022, vol. 380 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: http://doi.org/10.1039/d4cy01187b |
| This journal is © The Royal Society of Chemistry 2025 |