
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective isomerization of β-pinene: a sustainable method for total utilization of turpentine as a biomass resource†
Robert E.
Munday‡
 a,
Ryan Z.
Whitehead‡
a,
Elsa
Iacono‡
a,
Philippa L.
Jacob
a,
Steven M.
Howdle
a,
Gary M.
Walker
b,
Vincenzo
Taresco
a and
Anabel E.
Lanterna
*a
a,
Ryan Z.
Whitehead‡
a,
Elsa
Iacono‡
a,
Philippa L.
Jacob
a,
Steven M.
Howdle
a,
Gary M.
Walker
b,
Vincenzo
Taresco
a and
Anabel E.
Lanterna
*a
aSchool of Chemistry, University of Nottingham, University Park, NG7 2RD, UK. E-mail: anabel.lanterna@nottingham.ac.uk
bThe Knowle, Nether Lane, Hazelwood, Lubrizol Limited, Derbyshire, DE56 4AN, UK
First published on 5th March 2025
Abstract
Abundant and naturally available terpenes, such as α-pinene, are gaining attention as polymer building blocks, providing a biogenic source to replace fossil carbon feedstocks. However, α-pinene extraction from the natural source, turpentine, requires energy demanding and wasteful distillation processes. Here, we used a facile and green photocatalytic approach to obtain highly pure α-pinene, allowing total utilization of turpentine as a biomass resource.
Introduction
The use of nature-derived precursors in the polymer industry has gained momentum in the last two decades. In particular, paper manufacture provides two main products that are of high interest: lignin and turpentine. Turpentine is composed of several terpenes and has been extensively used in fragrances, flavours, paint thinners, adhesives, disinfectants and pharmaceutical drugs amid other applications,1–6 mainly exploiting its chief components, α- and β-pinenes. Other terpenes are also present in lesser amounts, such as carene, camphene, limonene, and terpinolene (Scheme 1), depending on the turpentine's origin.7 The natural abundance of pinenes, their molecular structure and the fact that their extraction is a well-established industrial process makes them the ideal compounds to be used in the bio-polymer industry as a raw material.8,9 Turpentine is used as a solvent in paints, but the use of turpentine as a chemical requires energy demanding processes including separation and purification of the main components via distillation.10 α-Pinene is particularly interesting because polymers formed from α-pinene acrylate (71 °C) and methacrylate (142 °C) show higher glass transition temperatures (Tg) than those achieved from analogous β-pinene acrylate (41 °C) and methacrylate (115 °C) structures, demonstrating the versatile and useful nature of the α-pinene isomer.8 The high Tg of these polymers has facilitated the synthesis of bio-based pressure sensitive adhesives11 as well as a bio-based poly high internal phase emulsion (HIPE).12 Therefore, finding a low energy process to separate the two major components of turpentine (i.e., α- and β-pinenes) could lead to better availability of pure α-pinenes and provide significant opportunities for several industries including pharmaceutical, cosmetic13 and also the polymer industry. | ||
| Scheme 1 Turpentine natural components and their variable compositions.7 | ||
Currently, α-pinene is obtained via distillation,10 but beyond the energy cost of this separation methodology, the process is also wasteful as the remaining β-pinene isomer is not used, making inefficient mass utilisation of this biogenic feedstock. Alternatively, α- and β-pinenes could be interconverted via isomerisation into one another. Work by Periasamy et al.14 showed the isomerisation of β-pinene can be carried out at room temperature in a mixture of iron and copper salts. Nevertheless, the reaction required 12 h for completion and only yielded 80% of the product, requiring chromatographic separation from the homogeneous catalyst (Scheme 2a). More recently, König et al. have shown exocyclic alkenes can be fully isomerised using visible light.15 The reaction requires the presence of homogeneous photocatalysts, a base, dimethyl formamide as solvent and 24 h of irradiation to proceed. Albeit with excellent yields (93%), the process still requires chromatographic separation (Scheme 2b). Work published by Gómez et al. demonstrated isomerisation and hydrogenation of alkenes via ionic liquid-stabilised palladium nanoparticles.16 However, a hydrogen gas flow was required, and using β-pinene as the starting material resulted in a mixture of α/β-pinene and pinane (Scheme 2c). In contrast, our earlier work17 showed that palladium-decorated TiO2 (Pd@TiO2) could be used for the isomerisation of allylbenzenes in a simple reaction setup that required only methanol as solvent and blue light. Building on this, we have now found that the same catalyst can be used for the isomerisation of exocyclic alkenes, namely β-pinene, into the more stable endocyclic alkene (α-pinene) in an unprecedented clean and specific manner (Scheme 2d). The methodology requires a heterogeneous photocatalyst based on Pd@TiO2, UVA excitation (via an LED centred at 365 nm), inert atmosphere (N2) and the presence of an alcohol, such as isopropanol (IPA). The reaction proceeds to completion in as short as five minutes and can be scaled to grammes in batch. This isomerisation process can transform β-pinene into α-pinene in a single step, thereby reducing waste generation and avoiding chromatographic separation and energy-demanding distillation methods.
 | ||
| Scheme 2 Different approaches for isomerisation of β-pinene into α-pinene: a) thermal isomerisation using iron and copper salts, b) homogeneous photocatalytic isomerisation using cobalt, c) isomerisation and hydrogenation of alkenes via ionic liquid-stabilised palladium nanoparticles, and d) heterogeneous photocatalytic isomerisation (this work). | ||
Results and discussion
Considering the ability of Pd@TiO2 to isomerise allylbenzenes17 under blue light irradiation, we decided to carry out the isomerisation of β-pinene into α-pinene under similar conditions. First, we synthesised Pd@TiO2 following a reported protocol,17 the presence of Pd nanoparticles was confirmed by electron microscopy (Fig. S1†), the metal loading (1.3 wt% Pd) determined by ICP-OES (see ESI†) and the oxidation state studied via X-ray photoelectron spectroscopy (XPS, Fig. S5†). Then, the reaction was performed by dispersing β-pinene and Pd@TiO2 in IPA and irradiating under an inert atmosphere (N2). As shown in Table 1, the reaction works under visible light irradiation (Table 1, entry i), yielding 83% of α-pinene as the only product. The difference between conversion and yield are attributed to pinene adsorption onto the catalyst surface, which we determined to be of at least 9% when using the highest irradiance. Thermogravimetric analysis (TGA) indicates small amounts, around 2%, of organic material is present on the catalyst (Fig. S6 and Table S1†) after exposure in the dark, which is in agreement with the ca. 4% of pinene loss found for Table 1, entry v. Increasing irradiation energy by using UVA light (entries ii–iv) drastically reduced the reaction time to 30 min and 5 min respectively. The reaction does not proceed in the dark at room temperature (Table 1, entry v) or at 80 °C (Table 1, entry vi), supporting the photocatalytic nature of the reaction. Similarly, the reaction does not proceed in the absence of catalyst (Table 1, entry vii), or when employing bare P25 TiO2 as the catalyst (Table 1, entry viii). What is more, the reaction supernatant (from Table 1, entry iv) showed an almost negligible amount of Pd (0.08% of the total Pd mass used in the reaction), suggesting the catalyst is stable under reaction conditions. Interestingly, the reaction works in air, albeit with longer reaction times (Table 1, entry ix and Fig. S7†). The presence of IPA is crucial for the reaction to take place. As shown in Table 1 (entry x) the absence of IPA yielded negligible conversion, suggesting the solvent plays a key role in the isomerisation process. Screening of different solvents (Table S2†) supports this, with only IPA, a polar protic solvent, showing any activity. Analysis of the material after reaction in polar solvents like IPA and MeCN shows Pd(II) is reduced to Pd(0), whereas the reaction in less polar/non-polar solvents (i.e., toluene and heptane) have a lesser effect in the oxidation state of the material. Despite the differences observed in the material oxidation state before and after reaction, the reaction only works in IPA, which suggests the oxidation state of the metal is important but cannot be the only parameter driving this process. TEM analysis show particle size remains fairly constant, however, we note that changes at atomic level cannot be distinguished by the spatial resolution of this technique. Finally, the reaction did not proceed in the presence of the radical trapping agent TEMPO (Table 1, entry xi and xii), suggesting a radical mediated mechanism. To explore the reaction mechanism further, we assayed isotopic experiments using deuterated IPA. The analyses of the 1H and 2H NMR spectra (Fig. S10–S19†) indicate that the reaction proceeds in deuterated solvent and that no deuterium incorporation is found in the α-pinene product. Thus, the isomerisation process can be rationalised as intramolecular H transfer, as opposed to intermolecular H transfer suggested in other systems.17 Nevertheless, the reaction seems to slow down in the deuterated solvent, indicating the solvent is involved in the reaction (Fig. S8†). Given that the reaction does not proceed in the absence of palladium nanoparticles, it is likely that these serve as the active sites for the isomerisation. A plausible mechanism (Scheme 3) could involve the association of β-pinene to the Pd surface followed by an oxidative addition to form an allyl-Pd hydride intermediate. This facilitates the intramolecular H transfer through a reductive elimination forming the thermodynamically favoured α-pinene. Upon excitation of TiO2, the alcohol acts as a sacrificial electron donor reducing surface PdO to Pd, as seen by post-reaction XPS analysis. As mentioned before, the reaction also works under blue light irradiation (Table 1, entry i). As shown in Fig. S3,† the emission of the blue lamp has some contribution below 410 nm, which could be sufficient to excite TiO2 (Fig. S4†). As the intensity of these higher energy photons is much lower with the blue lamp, the reaction takes longer times when compared to the reaction under UVA irradiation.| Entry | λ (nm) | Irradiance (W cm−2) | Time (h) | Conversion (%) | Yield (%) |
|---|---|---|---|---|---|
| a Reaction conditions: β-pinene (0.25 mmol, 34 mg), Pd@TiO2 catalyst (8 mg, 0.4 mol%), 4 ml IPA, N2 atmosphere. Yields calculated via GC-FID using tridecane as external standard. Relative errors: 2–4%. b Reaction carried out at 80 °C. c No catalyst. d Bare P25 TiO2 was used as the catalyst. e Reaction carried out under air. f Neat reaction, no IPA. g TEMPO (1.1 eq.) added to the reaction. | |||||
| i | 455 | 9 | 6 | 96 | 83 |
| ii | 365 | 0.07 | 0.5 | >99 | 90 |
| iii | 365 | 0.14 | 0.5 | 97 | 75 |
| iv | 365 | 1.3 | 5 min | >99 | 84 |
| v | 365 | 0 (dark) | 72 | 4 | 0 |
| vib | 365 | 0 (dark) | 1 | 5 | 0 |
| viic | 365 | 1.3 | 24 | 0 | 0 |
| viiid | 365 | 1.3 | 0.5 | 3 | 0 |
| ixe | 365 | 1.3 | 1 | 72 | 72 |
| xf | 365 | 0.07 | 22 | 3 | 1 |
| xig | 365 | 1.3 | 5 min | 2 | 0 |
| xiig | 455 | 9 | 6 | 1 | 0 |
 | ||
| Scheme 3 Plausible reaction mechanism for isomerisation of β-pinene involving an allyl-Pd hydride intermediate and a sacrificial electron donor (IPA) to promote the reduction of Pd surface enabling the oxidative addition step.17 | ||
Furthermore, the selectivity of the reaction is remarkable with >99% of β-pinene converted into α-pinene. Alternatively, when the same reaction is run using α-pinene as starting material no isomerisation to β-pinene is detected. In fact, when an artificial mixture of α-pinene and β-pinene (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40) is used, the reaction yields only α-pinene (Table 2, entry ii). What is more, irradiation of commercial turpentine in IPA showed that β-pinene had a conversion into α-pinene of >99% which resulted in an α-pinene yield of 95% (Table 2, entry iii), bringing the total content of β-pinene in turpentine to <0.1% and the α-pinene content to 97% from 79%.
:40) is used, the reaction yields only α-pinene (Table 2, entry ii). What is more, irradiation of commercial turpentine in IPA showed that β-pinene had a conversion into α-pinene of >99% which resulted in an α-pinene yield of 95% (Table 2, entry iii), bringing the total content of β-pinene in turpentine to <0.1% and the α-pinene content to 97% from 79%.
| Entry | Terpene | Time | Conversion (%) | Yield (%) |
|---|---|---|---|---|
|
a Reaction conditions: terpene (0.25 mmol), Pd@TiO2 catalyst (8 mg, 0.4 mol%), 4 ml IPA, N2 atmosphere, LED irradiation centred at 365 nm, 0.07 W cm−2. Yields calculated via GC-FID using tridecane as external standard.
b 60:40 α/β artificial mixture.
c Commercial turpentine: 18% β-pinene, 79% α-pinene.
|
||||
| i | β-Pinene | 50 min | >99 | 92 |
| ii | α-/β-Pineneb | 4 h | >99 | >99 |
| iii | Turpentinec | 2 h | >99 | 95 |
The reusability of the catalyst was evaluated at lower production rates (around 75%) by lowering the light intensity four times (from 1.3 to 0.3 W cm−2). The catalyst works efficiently for the first three cycles, with activity dropping to ca. 50% during the fourth cycle. Notably, reusing the catalyst under the reaction conditions (i.e., 1.3 W cm−2) shows no significant loss in activity (Fig. 1, Table S3†), with yield remaining above 95% after 4 cycles. The catalyst required no additional regeneration or cleaning steps between cycles and was simply removed by centrifugation and dried via vacuum desiccation. This represents a key advancement upon the current state-of-the-art, as the photocatalyst may be easily retrieved and re-used.
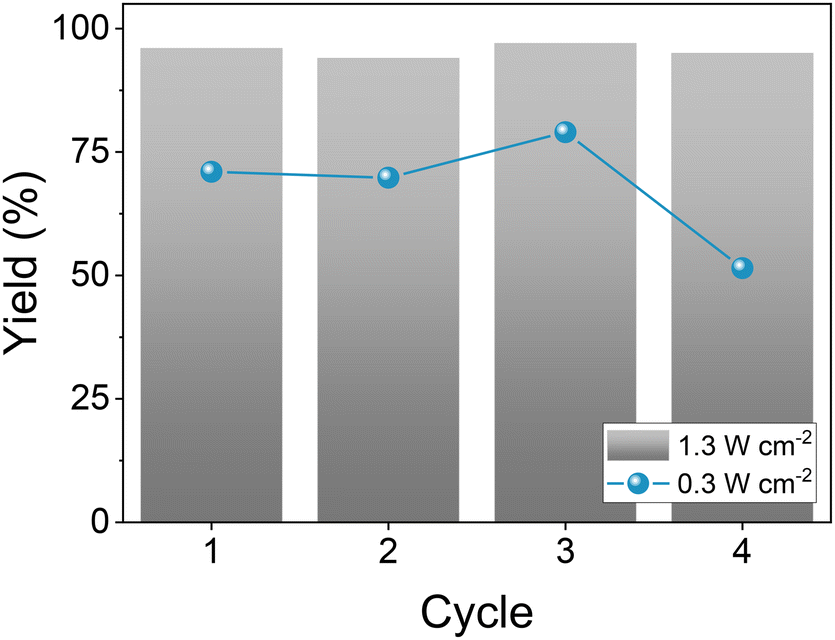 | ||
| Fig. 1 Successive cycles of the isomerisation of β-pinene. Reaction conditions: β-pinene (0.25 mmol), 8 mg Pd@TiO2 catalyst, 4 ml IPA, N2 atmosphere, LED irradiation centred at 365 nm and working at 1.3 W cm−2 (grey bars) or at 0.3 W cm−2 (blue dots) for 5 minutes. Pd@TiO2 was removed via centrifugation and dried in a vacuum desiccator before use in the next cycle. Yields calculated via GC-FID using tridecane as external standard. | ||
It is important to highlight that all these reactions were performed in batch conditions, yet we were able to scale the reaction up to grammes (Table 3), indicating this approach has the potential for large scale applications. Table 3 shows different attempts to scale the reaction, first to hundreds of milligrams (Table 3, entry ii) and then to grams (Table 3, entry iii). As Fig. S9† shows, the 10-fold increase in concentration did not significantly affect the reaction kinetics. A 50-fold increase, i.e., 1.7 g of pinene, slow down the kinetics reaching ca. 60% conversion in two hours and 90% conversion in four hours. This could be caused by mass and heat transfer issues faced when scaling up batch reactions.
| Entry | β-Pinene (g) | Catalyst loading (mol%) | Conversion (%) | Yield (%) |
|---|---|---|---|---|
| a Reaction conditions: β-pinene, 8 mg Pd@TiO2 catalyst, 4 ml IPA, N2 atmosphere, LED irradiation centred at 365 nm and working at 0.3 W cm−2 for 60 min. Yields calculated via GC-FID using tridecane as external standard. b Total reaction volume 6.7 ml, value between brackets indicates yield after 4 hours. | ||||
| i | 0.034 | 0.4 | 98 | 89 |
| ii | 0.340 | 0.04 | 99 | 92 |
| iiib | 1.727 | 0.04 | 3(89) | 0(90) |
To demonstrate the selectivity of the process towards β-pinene isomerisation, the reactivity of other terpenes was tested under the same reaction conditions (Table 4, entries i–vi). Interestingly, the terpenes analysed did not show reaction (or very low conversions) under these conditions. Extending the irradiation times to one or sixteen hours showed higher conversions as described in the ESI† (Scheme S1). Isomerisation reactions were additionally carried out with both cyclic and linear aliphatic alkenes (Table 4, entries vii–x). The reaction with exo-cyclic methylene cyclohexane predominantly yielded the corresponding endo-cyclic alkene, with a minor component of the hydrogenated methylcyclohexane product (Table 4, entry vii). Similar results were obtained when starting from the endo-cyclic alkene (Table 4, entry viii). Subjecting α- or β-diisobutylene to the reaction conditions resulted in a mixture containing predominately α-diisobutylene dimer, with a small component of the hydrogenated product (Table 1, entry ix and x). The mixtures of isomers obtained in these reactions clearly demonstrates that the photoisomerisation of β-pinene to α-pinene is unique in its selectivity. Additionally, the formation of hydrogenated products in the reactions of methylene cyclohexane/methyl cyclohexene and α/β-diisobutylene indicates, in these cases, the involvement of IPA solvent as a H donor, similar to a previous report.17
| Entry | Alkene | Conversion (%) | Isomer yield (%) | Alkane yield (%) |
|---|---|---|---|---|
| a Reaction conditions: alkene (0.25 mmol), 8 mg Pd@TiO2 catalyst, 4 ml IPA, N2 atmosphere, LED irradiation centred at 365 nm and working at 1.3 W cm−2 for 5 minutes. Values in brackets correspond to 1-hour reaction time for entries i and ii and v–x and 16-hour reaction time for entries iii and iv. b See Scheme S1† for full description of product distribution. N/A: not applicable. | ||||
| i |
 β-Pinene β-Pinene |
>99 (>99) |
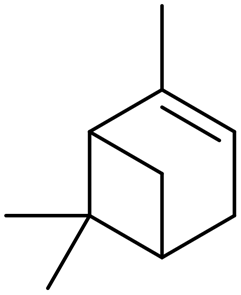 84 (98) 84 (98) |
 0 (0) 0 (0) |
| ii |
 α-Pinene α-Pinene |
0 (0) |
 0 (0) 0 (0) |
 0 (0) 0 (0) |
| iii |
 δ3-Carene δ3-Carene |
0 (20) |
 0 (25) 0 (25) |
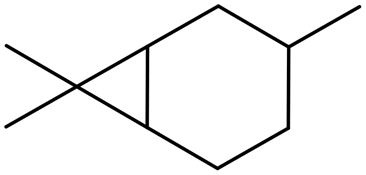 0 (0) 0 (0) |
| iv |
 δ2-Carene δ2-Carene |
1 (9) |
 2 (8) 2 (8) |
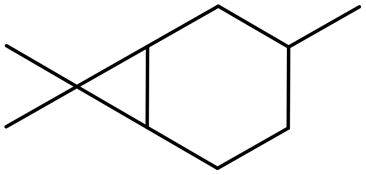 0 (0) 0 (0) |
| v |
 Camphene Camphene |
0 (35) | N/A |
 0 (39) 0 (39) |
| vib |
 D-Limonene
D-Limonene |
11 (80) | 3, 4 (23, 4) | 4, 2 (22, 11) |
| vii |
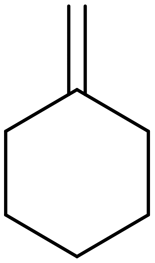 Methylene cyclohexane Methylene cyclohexane |
99 (>99) |
 85 (17) 85 (17) |
 13 (65) 13 (65) |
| viii |
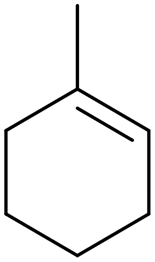
 Methyl cyclohexene Methyl cyclohexene |
20 (97) |
 <1 (0) <1 (0) |
 7 (65) 7 (65) |
| ix |
 α-Diisobutylene α-Diisobutylene |
25 (39) |
 15 (13) 15 (13) |
 2 (13) 2 (13) |
| x |
 β-Diisobutylene β-Diisobutylene |
83 (86) |
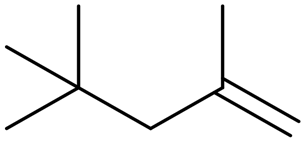 74 (65) 74 (65) |
 2 (11) 2 (11) |
Our approach provides a simple and quantitative method to selectively deliver α-pinene from turpentine by transforming the less useful β-pinene isomer whilst avoiding the use of wasteful chromatographic separation and energy-intensive distillation processes. To better understand the advantages of our approach, we calculated the process mass intensification (PMI) and compared it to previous methodologies for isomerisation of β-pinene to α-pinene (Scheme 2 and ESI†). We found that the heterogeneous catalysis process has PMI of 223 for a 1 M scale and the homogeneous catalysis and photocatalysis approaches have PMIs of 178 and 664 for scales of 167 mM and 100 mM respectively. In contrast, when working at ca. 1.8 M scale our approach lowers the PMI to 3.5, showing the huge benefit of our process from a sustainability standpoint. As this mass-based metric alone cannot account for several other environmental effects, we collected information available for a qualitative comparison18 (Tables S5 and S6†). Overall, our approach uses lower amounts of harmful and toxic materials, while generating less mass waste.
Conclusion
In conclusion, we have successfully developed a rapid and efficient methodology for the isomerisation of β-pinene into α-pinene under mild conditions. This approach operates at room temperature and atmospheric pressure, making it highly convenient. Utilizing a heterogeneous photocatalytic system, we can achieve the conversion of common α- and β-pinene mixtures and feedstocks (such as turpentine) into highly pure α-pinene within less than an hour. Furthermore, this process demonstrates good air tolerance. Our approach adds to a series of strategies dedicated to modifying biomass derivates using heterogeneous photocatalysis.19–21 Our methodology has been evaluated in batch conditions at various scales, ranging from milligrammes to grammes. This showcases the potential of our approach for large-scale isolation of α-pinene, holding great promise for commercial application and broader industrial implementation. By providing a rapid, environmentally friendly, and cost-effective route to obtain high-purity α-pinene, our methodology contributes significantly to the field and offers exciting prospects for the synthesis of bio-based polymers.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
E. I., R. Z. W. and R. E. M. carried out the experimental work presented in the study and conducted analysis of acquired data and equally contributed as first authors. P. L. J., S. M. H., G. M. W., and V. T. assisted with supervision and reviewing and editing of the manuscript. A. E. L. conceived the study, led the supervision, wrote the first draft of the manuscript and worked on reviewing and editing it. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council (EPSRC) Programme grant MASI (EP/V000055/1), the University of Nottingham, and Lubrizol Ltd. V. T. would like to thank the University of Nottingham for his Nottingham Research Fellowship. P. L. J. acknowledges funding support from EPSRC Doctoral Prize (EP/W524402/1). P. L. J. also acknowledges funding support from the Engineering and Physical Sciences Research Council and SFI Centre for Doctoral Training in Sustainable Chemistry (EP/S022236/1). The authors thank the Nanoscale and Microscale Research Centre (nmRC) for providing access to instrumentation.Notes and references
-
P. Bajpai, Biermann's Handbook of Pulp and Paper – Raw Material and Pulp Making, Elsevier, 3rd edn, 2018 Search PubMed
.
- B. Salehi, S. Upadhyay, I. E. Orhan, A. K. Jugran, S. L. D. Jayaweera, D. A. Dias, F. Sharopov, Y. Taheri, N. Martins, N. Baghalpour, W. C. Cho and J. Sharifi-Rad, Biomolecules, 2019, 9, 738 CrossRef CAS PubMed
-
A. J. D. Silvestre and A. Gandini, Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2008, ch. 2, pp. 17–38, DOI:10.1016/B978-0-08-045316-3.00002-8
-
K. C. da Silva Rodrigues-Corrêa, J. C. de Lima and A. G. Fett-Neto, in Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes, ed. K. G. Ramawat and J.-M. Mérillon, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 4037–4060, DOI:10.1007/978-3-642-22144-6_175
- K. C. D. S. Rodrigues-Corrêa, J. C. d. Lima and A. G. Fett-Neto, Food Energy Secur., 2012, 1, 81–93 CrossRef
- T. B. Adams, C. L. Gavin, M. M. McGowen, W. J. Waddell, S. M. Cohen, V. J. Feron, L. J. Marnett, I. C. Munro, P. S. Portoghese, I. M. C. M. Rietjens and R. L. Smith, Food Chem. Toxicol., 2011, 49, 2471–2494 CrossRef CAS PubMed
-
M. Gscheidmeier and H. Fleig, Ullmann's Encyclopedia of Industrial Chemistry, 2000, DOI:10.1002/14356007.a27_267
- M. F. Sainz, J. A. Souto, D. Regentova, M. K. G. Johansson, S. T. Timhagen, D. J. Irvine, P. Buijsen, C. E. Koning, R. A. Stockman and S. M. Howdle, Polym. Chem., 2016, 7, 2882–2887 RSC
- M. Winnacker, Angew. Chem., Int. Ed., 2018, 57, 14362–14371 CrossRef CAS PubMed
-
L. Zhang, L. Zeng, L. Zhang, G. Pu and W. Zhang, Method for producing alpha-pinene from turpentine, CN Pat., CN103497084B, 2015 Search PubMed
- M. T. Elsmore, R. L. Atkinson, D. J. Irvine, S. M. Howdle and D. S. A. De Focatiis, Polym. Test., 2022, 109, 107530 CrossRef CAS
- O. R. Monaghan, S. T. Skowron, J. C. Moore, M. Pin-Nó, K. Kortsen, R. L. Atkinson, E. Krumins, J. C. Lentz, F. Machado, Z. Onat, A. Brookfield, D. Collison, A. N. Khlobystov, D. De Focatiis, D. J. Irvine, V. Taresco, R. A. Stockman and S. M. Howdle, Polym. Chem., 2022, 13, 5557–5567 RSC
- T. M. Bennett, J. Portal, V. Jeanne-Rose, S. Taupin, A. Ilchev, D. J. Irvine and S. M. Howdle, Eur. Polym. J., 2021, 157, 110621 CrossRef CAS
- M. R. Reddy and M. Periasamy, J. Organomet. Chem., 1995, 491, 263–266 CrossRef
- Q.-Y. Meng, T. E. Schirmer, K. Katou and B. König, Angew. Chem., Int. Ed., 2019, 58, 5723–5728 CrossRef CAS PubMed
- G. Garg, S. Foltran, I. Favier, D. Pla, Y. Medina-González and M. Gómez, Catal. Today, 2020, 346, 69–75 Search PubMed
- A. Elhage, A. E. Lanterna and J. C. Scaiano, ACS Catal., 2017, 7, 250–255 Search PubMed
- P. Jessop, Green Chem., 2020, 22, 13–15 RSC
- Q. Yan, X. Wu, H. Jiang, H. Wang, F. Xu, H. Li, H. Zhang and S. Yang, Coord. Chem. Rev., 2024, 502, 215622 CrossRef CAS
- H. Zhou, H. Wang, C. Yue, L. He, H. Li, H. Zhang, S. Yang and T. Ma, Appl. Catal., B, 2024, 344, 123605 CrossRef CAS
- L. He, L.-L. Zhang, H. Zhou, Y. Nie, H. Wang, B. Tang, H. Li, T. Ma and H. Zhang, Adv. Energy Mater., 2025, 15, 2403168 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Catalyst characterisation, leaching tests, thermogravimetric analyses, solvent screening, kinetic studies, NMR spectra, reusability studies, extended description of substrate scope, calculations of environmental impact. See DOI: https://doi.org/10.1039/d4cy01247j |
| ‡ Authors equal contribution |
| This journal is © The Royal Society of Chemistry 2025 |