
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Accelerating the identification of the rate controlling steps by conducting microkinetic modeling on surrogate networks†
Hongyu
Li
a,
Jia
Zhang
 *b,
Zihao
Yao
c and
P.
Hu
*ade
*b,
Zihao
Yao
c and
P.
Hu
*ade
aCentre for Computational Chemistry and Research Institute of Industrial Catalysis, East China University of Science and Technology, Shanghai 200237, China
bInstitute of High Performance Computing (IHPC), Agency for Science, Technology and Research (A*STAR), 1 Fusionopolis Way, #16–16 Connexis, Singapore 138632, Republic of Singapore. E-mail: zhangj@ihpc.a-star.edu.sg
cInstitute of Industrial Catalysis, College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310032, China
dSchool of Physical Science and Technology, ShanghaiTech University, Shanghai, 201210, China. E-mail: hupj@shanghaitech.edu.cn
eSchool of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, BT9 5AZ, UK
First published on 17th March 2025
Abstract
Identifying the rate-controlling steps in an unknown reaction network can be time-consuming due to its inherent complexity. Here we present a strategy to simplify this process by focusing expensive barrier calculations on significant elementary steps. The strategy is constructed by determining significant elementary steps using the degree of rate control data, which is derived from microkinetic modeling calculations performed on surrogate networks, which are a series of networks generated by assigning fictitious values to unknown barriers while all the reaction energies are computed using density functional theory. The barriers for significant elementary steps are then calculated iteratively to refine the network. We demonstrate this strategy for the reaction of Fischer–Tropsch synthesis, which has already been extensively studied in our previous work. Applying the strategy, we identified the most rate-controlling step, achieving a 77% reduction in the number of transition state calculations compared to traditional methods. Additionally, a detailed analysis of the strategy reveals the correlation between the parameters in the strategy and its performance. We validate the practicability of the strategy by applying it onto testing networks and the potential limitations of the strategy are also discussed.
Introduction
A catalytic reaction network is typically complex; a system involving hundreds of intermediates can result in a network with numerous reactions and pathways. To extract useful information from such a network, a large number of calculations must be performed. Traditional calculations for mechanistic studies can be divided into three parts, as illustrated in Fig. 1: reaction energy calculations (structural optimizations of initial and final states of each elementary step), transition state (TS) calculations (TS search using NEB or constrained optimization or other methods), and microkinetic modeling (MKM). Conducting high-fidelity calculations for the entire reaction network is both costly and time-consuming. Typically, TS calculations are the most time-consuming, often being several orders of magnitude more computationally expensive than other calculations. Therefore, reducing the number of TS calculations is the key to improving the efficiency of reaction kinetics studies. | ||
| Fig. 1 The traditional method (upper) and our strategy (lower) of determining rate-controlling steps. Our strategy efficiently identifies the rate-controlling steps in the reaction network by minimizing the number of TS calculations. First, surrogate networks with accurate reaction energies and fictitious energy barriers are constructed. MKM on the surrogate networks provides kinetic analysis to identify significant elementary steps. The activation energies of the highest-priority steps are calculated iteratively using DFT, with the network being updated to improve accuracy. This process continues until convergence is achieved. | ||
This process can be significantly simplified by recognizing that only a small portion of reactions controls most of the properties of the entire network. The majority of the network properties, except for the kinetic parameters of the rate-controlling steps, have small impacts on the overall reaction network. The rate-controlling step can be simply interpreted as the slowest step of the favored pathway and it can strongly influence the overall reaction rate of the network. In order to identify rate-controlling steps, one popular method is the degree of rate control (DRC). This approach for analyzing multistep reaction mechanisms was introduced by Campbell,1–4 in which the “degree of rate control for elementary step i”, DRCi, is defined as
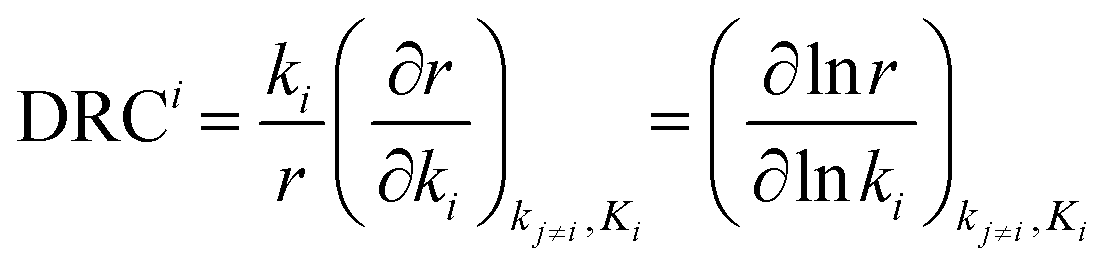 | (1) |
To address this, researchers have proposed various solutions,5–14 primarily relying on approximations like the BEP relation15–20 or machine learning models to minimize computational resources and time. As a representative example, by using the prediction of machine-learning and approximations, such as scaling relations, Ulissi et al. developed an approach to determine the significant reactions on the favored pathway: by focusing only on these reactions with full density functional theory (DFT) calculations at each iteration, they achieved remarkable reduction of time and resource cost on studying the network of syngas.9 Despite all the progress in this field, two main improvements could still be made. One is to achieve a more optimal trade-off between accuracy and efficiency. We need to determine when to use an approximate method that saves time but is less accurate, and when to choose a high-fidelity DFT calculation, which is more precise but also more time-consuming. The second improvement is to develop a better strategy to identify significant elementary steps.
In this work, we aim to address these issues. We developed a workflow to identify the rate-controlling steps of a reaction network, significantly reducing computational effort by performing a limited number of TS calculations. The strategy used DFT calculations to obtain reaction energies, and the MKM was conducted using CATKINAS, a software developed by our group and widely used.21–30 We saved time and computational resources by reducing barrier calculations, as shown in Fig. 1. Our workflow uses DRC data as an indicator to find significant elementary steps and calculate the corresponding TS energy at each iteration. The characteristics of the reaction network are refined at each iteration, and the rate-controlling steps of the network are determined at the end of the refinement. We demonstrated this workflow for the Fischer–Tropsch (FT) reaction,31–46 a process that utilizes synthesis gas to convert into hydrocarbons and oxygenated hydrocarbons. Using the strategy, we identified the most rate-controlling step with reducing the number of TS calculations by 77%.
Result
General workflow
As mentioned before, traditional reaction mechanism calculations consist of three parts: calculating reaction energies, determining the TSs, and performing the microkinetic modeling (MKM). Comparing the computational cost of each calculation, it is well known that the TS calculations are much more expensive than the other calculations. This leads to the core of our strategy: minimizing the number of TS calculations while maintaining a high level of accuracy. We managed to achieve this goal by constructing a workflow to find the significant elementary steps and calculate their TS energies iteratively. As illustrated in Fig. 2a, the entire workflow is divided into four parts: surrogate network construction, surrogate network MKM, evaluation and surrogate network refinement.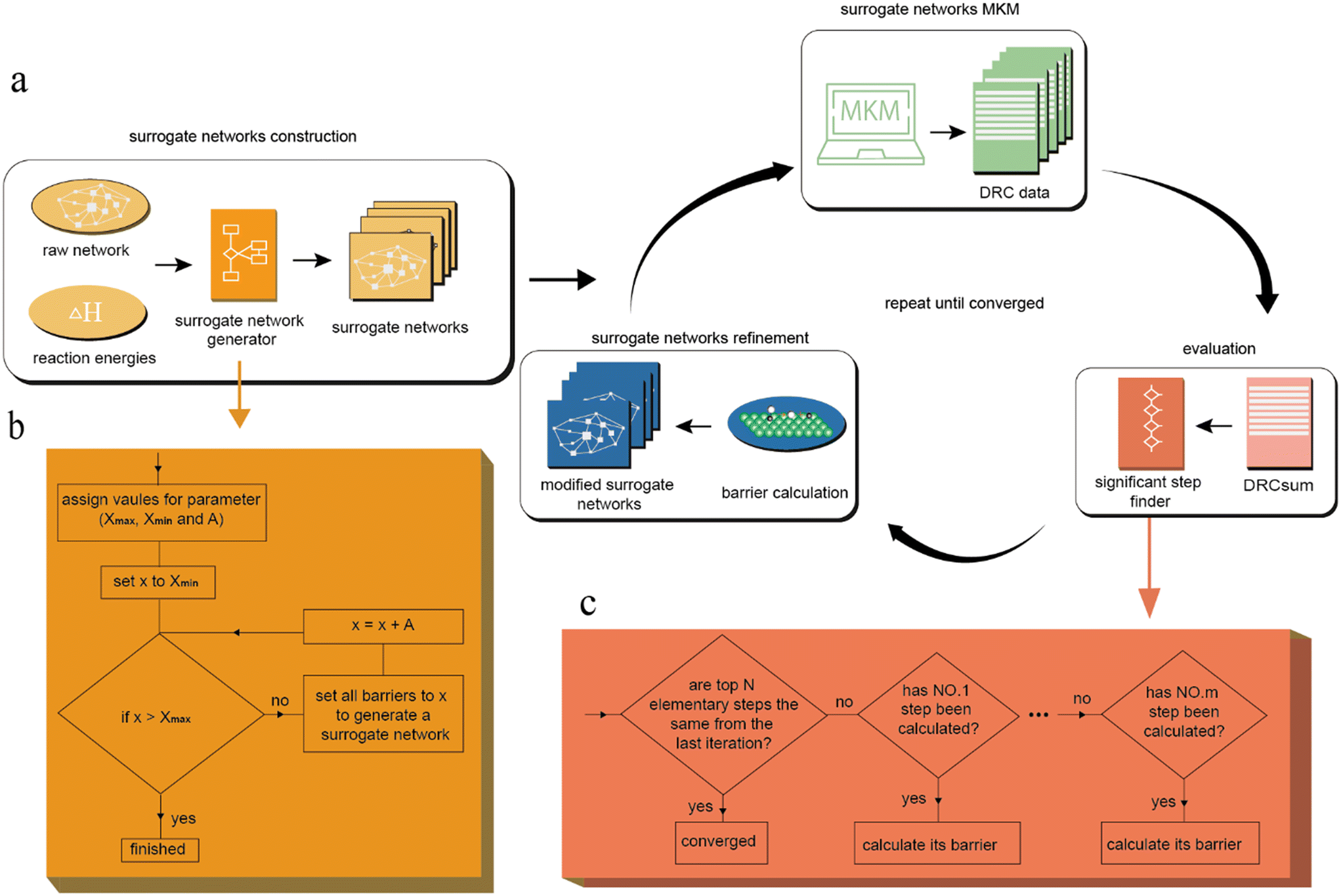 | ||
| Fig. 2 Workflow used to generate surrogate networks to iteratively determine significant elementary steps. (a) Network refinement methodology. In each iteration, the significant elementary step is determined based on kinetic information obtained from MKM of surrogate networks and TS calculation will be conducted on the significant elementary step using DFT to refine surrogate networks. (b) Flow chart of surrogate network generator in the surrogate networks construction phase. (c) Flow chart of the significant step finder in the evaluation phase. | ||
In constructing the surrogate networks, the first step is to define all possible chemical species and elementary steps to be considered in the network. Then the adsorption energies of all intermediates are calculated to obtain the reaction energy for each elementary step. In this way, a raw reaction network with only the thermodynamic information is built. Next, a surrogate network is generated by assigning fictitious values to energy barriers of all the elementary steps in the network. To construct a series of surrogate networks, as shown in Fig. 2b, we began with assigning values to Xmax (the biggest fictitious barrier value), Xmin (the smallest fictitious barrier value) and A (the interval of fictitious barrier between surrogate networks) in our strategy. As the fictitious barrier x changes from Xmax to Xmin, a series of surrogate networks were constructed. For clarity, when we built the surrogate networks, the fictitious energy barrier x is only added to the exothermic forward/backward reactions in each step. For example, assuming the energies of the initial state and the final state for a specific elementary step are E1 and E2, respectively, the energy of TS will be E1 + x when E1 < E2; otherwise, it will be E2 + x if E2 < E1.
In the MKM phase of surrogate networks, we conducted MKM on all the surrogate networks to obtain their kinetic information, which contains DRC data. Since our goal in this work was to identify the key elementary steps within a highly complex reaction network, coverage dependence was not explicitly considered to streamline the calculation process. In the evaluation phase, we introduced an indicator, DRC(sum), to assess the significance of each elementary step. For a specific elementary step i, DRCi(sum) is defined as the sum of the absolute value of the corresponding DRC in each surrogate network as follows:
 | (2) |
In the refinement phase of surrogate networks, we used DFT calculations to obtain the barrier of the chosen elementary step and updated all the surrogate networks by replacing the fictitious barrier of the selected step with its actual barrier. This process is repeated until the convergence criterion is met.
Identification of rate-controlling steps
In this work, we demonstrated this workflow for the FT reaction network on Co(0001), which has been thoroughly studied in our previous work.47 Since the structure of the reaction network and the reaction energies and activation energies have already been calculated,47 we skipped the network determination, reaction energy and barrier calculations in our current workflow, and focused on demonstrating how our workflow determines the significant elementary step iteratively and refines the network. We set parameters Xmax, Xmin and A to 1.5 eV, 0.0 eV and 0.1 eV, respectively, for the construction of surrogate networks. Detailed explanation about parameters setting will be included in the next section. For simplicity, the weight wz in the definition formula of DRC(sum) (eqn (1)) was uniformly set to 1 for every surrogate network.The refinement procedure is illustrated in Fig. 3a, which shows the entire reaction network with nodes as reaction species and lines as elementary steps. For visual simplicity, hydrogen is omitted. Refinements were performed under various settings of parameter N. As N increased, a greater number of TSs were required to be calculated for the iteration to terminate. The additional elementary steps that needed to be calculated by DFT, as N was increased to N + 1, are indicated by colored lines in Fig. 3a. Since parameter N defines the convergence criterion in the evaluation phase, increasing N requires more rate-controlling steps to converge before the iteration stops. This would result in more TS calculations and potentially more accurate outcomes. The accuracy of the final result and the number of iterations before convergence are both plotted against the value of N in Fig. 3b. In this work, ‘accuracy’ is defined as the ratio of the sum of DRC values of the calculated M elementary steps to the total DRC sum across all elementary steps, with all DRC values derived from a reference set based on the full reaction network from prior work,47 which conducted calculations of all the TS energies through DFT calculations. This metric is designed specifically for internal assessment of the strategy's predictive accuracy within this study and is not intended for direct application in real-world determinations of rate-controlling steps.
 | (3) |
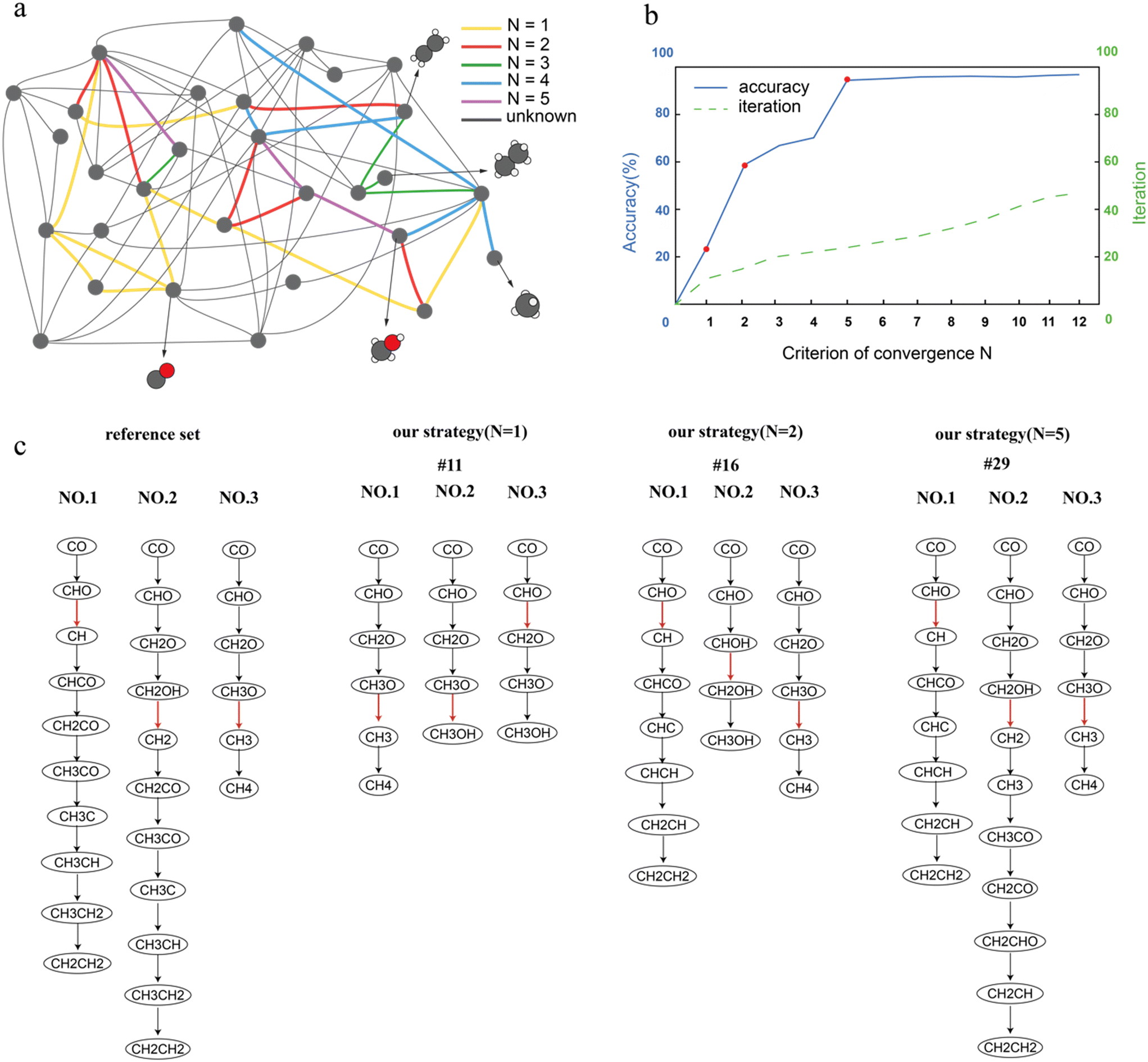 | ||
| Fig. 3 Rate-controlling step determination under different convergence criteria. (a) The FT reaction network on Co(0001) calculated in our previous work.47 In this work, the reaction network with the barriers we previously calculated is used as the reference set. The nodes represent reaction intermediates and each line represents an elementary step between the two nodes. (b) Illustration of accuracy of rate-controlling step determination (solid line) and iterations before convergence (dashed line) as functions of the criterion of convergency N. As N increases, the accuracy of the final result and number of iterations increase. There are three significant increases on the accuracy curve when N is set to 1, 2 and 5. (c) The top 3 most rate-controlling steps and the corresponding pathways determined by traditional calculations and our strategy. To identify the favored pathways using our strategy, the surrogate network in which a specific rate-controlling step reaches its highest DRC was determined firstly, and then the favored pathway for the corresponding rate-controlling step was identified based on MKM data of this surrogate network. | ||
The first accuracy significant increase occurs when N was set to 1; with 11 iterations in total, the calculation converged. The most rate-controlling step was determined as CH3O ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) CH3 + O, which is the third most rate-controlling step according to the reference set. The second significant accuracy increase occurs when N was set to 2, with the convergence achieved after 16 iterations. In this case, the elementary step of CHO CH + O was identified as the most rate-controlling step, which aligns with the result from the reference set. This means that using the method developed in this work, we only conducted 15 TS calculations to correctly predict the most rate-controlling step for the FT reaction network on Co(0001), reducing 77% of TS calculations. The third accuracy significant increase occurs when N was set to 5, with the convergence achieved after 29 iterations. This time we correctly identified the top 3 most rate-controlling steps and reached a value of accuracy of 0.97.
CH3 + O, which is the third most rate-controlling step according to the reference set. The second significant accuracy increase occurs when N was set to 2, with the convergence achieved after 16 iterations. In this case, the elementary step of CHO CH + O was identified as the most rate-controlling step, which aligns with the result from the reference set. This means that using the method developed in this work, we only conducted 15 TS calculations to correctly predict the most rate-controlling step for the FT reaction network on Co(0001), reducing 77% of TS calculations. The third accuracy significant increase occurs when N was set to 5, with the convergence achieved after 29 iterations. This time we correctly identified the top 3 most rate-controlling steps and reached a value of accuracy of 0.97.
Surrogate network assessment
We investigated the most important and innovative aspects of our workflow. Namely, we quantitatively assessed the construction and utilization of the surrogate networks. It is worth mentioning that the general benefit for incorporating surrogate networks into our workflow is that it prevents the refining process of the reaction network from stopping prematurely. For simplicity, we assume that the network consists of three pathways, shown in Fig. 4a, illustrating how the rate-controlling steps can vary as the fictitious barrier levels change. The three schematic diagrams represent the reaction pathways created solely for explanatory purposes and do not carry any intrinsic chemical significance. In these diagrams, solid lines represent elementary steps with barriers previously calculated in the network refining process using our strategy, while dashed lines denote elementary steps with unknown barriers, which are assigned to various fictitious barriers across different surrogate networks. According to the energetic-span model,48 the elementary step that contains the TS with the highest energy on the favored pathway is considered as the rate-controlling step when intermediate energies are all larger than the initial state. As illustrated in Fig. 4a, which represents one iteration of determining the rate-controlling step using our strategy, the unknown barriers in different surrogate networks are assigned to different fictitious values. For example, in a surrogate network with high fictitious barriers, all undetermined barriers are assigned to high values. In this case, pathway 3 becomes the favored pathway because the TS with the highest energy level in pathway 3 is still lower than the TSs with highest energy levels in the other two pathways. As a result, the elementary step containing this TS on pathway 3 is considered the rate-controlling step for the network. A similar process occurs in the networks with low and median fictitious barriers, leading to variations in the rate-controlling steps across different surrogate networks. This variation may result in additional TS calculations for potentially important steps, thereby increasing the likelihood of identifying the actual rate-controlling step. | ||
| Fig. 4 The construction of surrogate networks plays an important role in the identification of rate-controlling steps. (a) Schematic representation of rate-controlling step variations across different surrogate networks, assuming that the reaction network consists of three pathways. Solid lines indicate elementary steps with a calculated barrier, while dashed lines represent elementary steps with undetermined barriers. Different surrogate networks assign various fictitious barriers to these undetermined steps, leading to shifts in the rate-controlling step across networks. The figure demonstrates how varying fictitious barrier levels can shift the rate-controlling step within the reaction network, leading to calculations of more potentially significant elementary steps. (b) Illustration of accuracy and number of iterations before convergence as functions of Xmax. As Xmax increases, the number of iterations required for the calculation to converge increases. The highest accuracy was reached when Xmax was set to 1.5 eV. | ||
To investigate how the choice of surrogate networks influences the efficiency and accuracy of our strategy, we applied our workflow on the FT reaction network on Co(0001) with different range of fictitious barriers. We constructed 5 calculation groups, which sets Xmax to 0.0 eV, 0.5 eV, 1.0 eV, 1.5 eV and 2.0 eV, respectively, while Xmin, A and N were set to 0.0 eV, 0.1 eV and 5 uniformly. As shown in Fig. 2b, as Xmax increases, the range of fictitious barriers become wider and more surrogate networks with higher fictitious barriers will be generated for the corresponding group, leading to different results of refining processes. The accuracy of the final result and the number of iterations needed before convergence for each calculation are shown in Fig. 4b. It can be seen that as Xmax increases, the number of iterations increases. The calculation group with Xmax set to 1.5 eV reached the highest accuracy. The reason for this is that most of the barriers in the reaction network we studied lies in the range of 0.0–1.5 eV. Hence, the calculation group with Xmin and Xmax set to 0.0 eV and 1.5 eV is suitable for probing the rate-controlling steps of the network because its range of fictitious barriers matches the range of real barriers, ensuring that there are enough surrogate networks to investigate potentially significant elementary steps. In contrast, the groups with the range of fictitious barriers too narrow will not have enough surrogate networks to detect some rate-controlling steps and might miss some significant elementary steps. In addition, for the groups where the range of fictitious barriers is too wide, such as the calculation group with Xmax set to 2.0 eV, the surrogate networks with unusually high fictitious barriers will disproportionately influence the identification of the rate-controlling steps, thereby reducing the accuracy of the results.
Examination with testing networks
We generated testing networks to demonstrate the generality of our rate-controlling step identification strategy and to assess its potential limitations. As shown in Fig. 5a, these testing networks were constructed based on the FT reaction network on Co(0001) previously studied.47 By assigning random values to the barriers of all elementary steps while keeping the network structure and reaction energies unchanged, the testing networks were generated. Then we used the same strategy demonstrated above to determine the rate-controlling steps of those new testing networks. The parameters Xmax, Xmin, A and N were set to 1.5 eV, 0 eV, 0.1 eV and 5, respectively, as they achieved relatively high accuracy and efficiency for the original network, as shown in Fig. 4b. | ||
| Fig. 5 Constructing new testing networks to further examine our current strategy. (a) Workflow of generating the testing networks. The testing network are generated by assigning random values to the barriers of all elementary steps while keeping the network structure and reaction energies of previously studied FT reaction network unchanged. All the random barriers we generated were between 0.0 eV and 1.5 eV. (b) Two types of scenarios where our strategy may encounter difficulties discussed in the main text. (c) Favored pathways determined by the reference set and our strategy with rate-controlling steps highlighted in red. The reason for the disaccord is that there was an elementary step with an extremely low barrier in the favored pathway according to our generated reference set, which is highlighted in blue. | ||
Four testing networks were generated in total as the testing reference sets, and our strategy successfully predicted the most rate-controlling step for 3 of them as shown in Table 1. After analyzing the failed case, we find that there are mainly two types of scenarios where our strategy may encounter difficulties on the task of identifying the rate-controlling steps. As shown in Fig. 5b, the first scenario is that the rate-controlling step exists on a thermodynamically unfavorable but kinetic favorable pathway, which means that the energies of intermediates are relatively high but energies of TSs are relatively low on this pathway. There is a possibility that our strategy would identify this pathway as an unfavored pathway and start searching for a possible rate-controlling step elsewhere. In the second scenario, if there is a pathway that is thermodynamically favorable but kinetically unfavorable and does not include the rate-controlling step, our strategy might mistakenly identify this pathway as favored and converge on an incorrect rate-controlling step. Thus, our strategy may encounter difficulties when certain types of elementary steps have unexpected high or low barriers.
| Testing networks | Amount of iterations | Top 3 reactions with biggest DRC(sum) | |
|---|---|---|---|
| Our strategy (reaction #) | Reference set (reaction #) | ||
| T1 | 21 | 59, 3, 58 | 59, 3, 58 |
| T2 | 15 | 43, 4, 6 | 3, 25, 58 |
| T3 | 20 | 59, 58, 6 | 59, 6, 30 |
| T4 | 20 | 3, 58, 59 | 3, 58, 59 |
In the failed case of testing network T2, when the iteration stopped, our strategy identified the most rate-controlling step as C + C CC as shown in Fig. 5c. However, the actual most rate-controlling step, according to the testing reference set, is C + O CO. After thorough inspection, we found that the reason network T2 converged to an incorrect elementary step is that the actual favored pathway, according to the reference set, is a thermodynamically unfavorable pathway. This means that the energies of the species involved in this pathway are relatively high. However, the barrier of an elementary step (CH2CH3 CH2 + CH3) on this pathway, highlighted in blue in Fig. 5c, is unrealistically low (0.037 eV), which unexpectedly makes this pathway favorable. To identify the most rate-controlling step for the testing network T2, we increased the value of N to calculate more potential significant steps. After we increased N to 22, our strategy converged at the correct rate-controlling steps after 47 iterations.
Discussion
The strategy presented in this paper could efficiently reduce the amount of TS calculations; it successfully identified the most rate-controlling step of the FT reaction network on Co(0001), which was calculated in our previous work,47 with only 23% of TS calculations compared to the traditional method. The challenging aspect of this strategy lies in determining its parameters, including those for constructing surrogate networks and setting the convergence criterion. Based on our findings, for an unknown reaction network, we recommend setting the fictitious barrier range to 0.0–2.0 eV and N to above 5% of the total number of the elementary steps of the reaction network. This setting achieved a great trade-off between accuracy and efficiency as shown in Fig. 3b and 4b.For the reaction network tested in this work, which includes 26 species and 67 reactions, the good parameters Xmax, Xmin, A, and N were found to be 1.5 eV, 0.0 eV, 0.1 eV, and 5, respectively. This set choice has accurately predicted the top 3 rate-controlling steps while reducing the number of TS calculations by 55%, as shown in Fig. 4b. Compared to traditional calculations, our strategy focuses on identifying the rate-controlling steps as quickly as possible and performing barrier calculations only on those steps identified as significant. This approach saves substantial time and computational resources. Additionally, unlike methods that rely on machine learning or other approximations, the strategy developed in this work has greater generality. However, it is important to note that our strategy does not guarantee 100% accuracy in predicting rate-controlling steps if the parameters are not properly selected.
Conclusion
In the current work, we developed a strategy to efficiently identify the rate-controlling steps of a given reaction network by leveraging DRC data to highlight significant elementary steps and allocate computational resources accordingly. By applying our strategy to the FT reaction network on Co(0001), we achieved a 77% reduction on TS calculations compared to the traditional method. This approach allows for significant reductions in time and computational costs when exploring unknown reaction networks.The advancements of our method compared to previous approaches are summarized as follows:
1. Efficient rate-control predictions: by conducting MKM calculations on surrogate networks, our method enables the prediction of rate-controlling steps using DRC without the extensive barrier calculations.
2. Balanced accuracy and efficiency: by reducing the number of TS calculations while using high accuracy methods to conduct the calculations of reaction energies and MKM, our strategy reached excellent balance between accuracy and efficiency.
3. General applicability: unlike methods that rely on approximations such as BEP relations, our strategy does not depend on specific approximations, making it applicable to the study of any reaction network.
Finally, our method could be further advanced by integrating experimental data or machine learning models to predict transition state energies or adsorption energies. Combining these approaches has the potential to reduce computational costs and improve accuracy when exploring new reaction networks. Future work may focus on refining strategies to efficiently identify critical elementary steps and incorporating additional catalytic effects, such as coverage effects, to enhance the practicality and effectiveness of the current method.
Methods
All microkinetic modeling calculations are carried out via the CATKINAS package, which is a microkinetic simulation package developed by our group and widely used.21–30Data availability
The data that support the findings of this study are available from the corresponding author on reasonable request.Author contributions
P. H. and J. Z. conceived the project. H. Y. L. carried out all the calculations. H. Y. L. wrote the first draft of the manuscript and prepared figures and all the authors contributed to the revision of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The National Key Research and Development Program of China (2021YFA1500700) and the National Natural Foundation of China (22433004, 92045303) are acknowledged. This work is supported by the ShanghaiTech AI4S Initiative SHTAI4S202404. J. Z. would like to acknowledge the support from the A*STAR AME IAF-PP (Grant No. A19E9a0103) and National Research Foundation Singapore under its Low-Carbon Energy Research (Grant No. U2305D4001).References
- C. Stegelmann, A. Andreasen and C. T. Campbell, J. Am. Chem. Soc., 2009, 131, 13563 CrossRef CAS.
- C. T. Campbell, ACS Catal., 2017, 7, 2770–2779 CAS.
- C. T. Campbell, Top. Catal., 1994, 1, 353–366 CAS.
- C. T. Campbell, J. Catal., 2001, 204, 520–524 CAS.
- J. X. Liu, S. Lu, S. B. Ann and S. Linic, ACS Catal., 2023, 13, 8955–8962 CrossRef CAS.
- T. Lan and Q. An, J. Am. Chem. Soc., 2021, 143, 16804–16812 CrossRef CAS PubMed.
- Y. Kim, J. W. Kim, Z. Kim and W. Y. Kim, Chem. Sci., 2018, 9, 825–835 RSC.
- E. M. Sunshine, M. Shuaibi, Z. W. Ulissi and J. R. Kitchin, J. Phys. Chem. C, 2023, 127, 23459–23466 CrossRef CAS.
- Z. Ulissi, A. Medford, T. Bligaard and J. K. Nørskov, Nat. Commun., 2017, 8, 14621 CrossRef PubMed.
- F. A. Döppel and M. Votsmeier, Chem. Eng. Sci., 2022, 262, 117964 CrossRef.
- A. G. Garrison, J. Heras-Domingo, J. R. Kitchin, G. dos Passos Gomes, Z. W. Ulissi and S. M. Blau, J. Chem. Inf. Model., 2023, 63, 7642–7654 CrossRef CAS PubMed.
- X. Wang, J. Musielewicz, R. Tran, S. Kumar Ethirajan, X. Fu, H. Mera, J. R. Kitchin, R. C. Kurchin and Z. W. Ulissi, Mach. Learn.: Sci. Technol., 2024, 5, 025018 Search PubMed.
- J. Lan, A. Palizhati, M. Shuaibi, B. M. Wood, B. Wander, A. Das, M. Uyttendaele, C. L. Zitnick and Z. W. Ulissi, npj Comput. Mater., 2023, 9, 172 CrossRef CAS.
- B. Wander, M. Shuaibi, J. R. Kitchin, Z. W. Ulissi and C. L. Zitnick, ACS Catal., 2025, 15, 5283–5294 CrossRef.
- W. Yang, K. E. Abdelfatah, S. K. Kundu, B. Rajbanshi, G. A. Terejanu and A. Heyden, ACS Catal., 2024, 10148–10163 CrossRef CAS.
- B. Wang, S. Chen, J. Zhang, S. Li and B. Yang, J. Phys. Chem. C, 2019, 123, 30389–30397 CrossRef CAS.
- J. K. Nørskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943 CrossRef PubMed.
- M. G. Evans and M. Polanyi, Trans. Faraday Soc., 1935, 31, 875–894 RSC.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- J. Jin, J. Chen, H. Wang and P. Hu, Chin. Chem. Lett., 2019, 30, 618–623 CrossRef CAS.
- H. Yuan, J. Chen, H. Wang and P. Hu, ACS Catal., 2018, 8, 10864–10870 CrossRef CAS.
- Z. Chen, Y. Mao, J. Chen, H. Wang, Y. Li and P. Hu, ACS Catal., 2017, 7, 4281–4290 CrossRef CAS.
- H. Yuan, N. Sun, J. Chen, J. Jin, H. Wang and P. Hu, ACS Catal., 2018, 8, 9269–9279 CrossRef CAS.
- J. F. Chen, Y. Mao, H. F. Wang and P. Hu, ACS Catal., 2016, 6, 7078–7087 CrossRef CAS.
- H. Yuan, J. Chen, Y. Guo, H. Wang and P. Hu, J. Phys. Chem. C, 2018, 122, 25365–25373 CrossRef CAS.
- M. Yang, H. Yuan, H. Wang and P. Hu, Sci. China:Chem., 2018, 61, 457–467 CrossRef CAS.
- C. Guo, Y. Mao, Z. Yao, J. Chen and P. Hu, J. Catal., 2019, 379, 52–59 CrossRef CAS.
- J. F. Chen, Y. Mao, H. F. Wang and P. Hu, ACS Catal., 2019, 9, 2633–2638 CrossRef CAS.
- D. Wang, T. Sheng, J. Chen, H. F. Wang and P. Hu, Nat. Catal., 2018, 1, 291–299 CrossRef CAS.
- P. Biloen and W. M. H. Sachtler, Adv. Catal., 1981, 30, 165–216 CAS.
- M. E. Dry, Appl. Catal., A, 1996, 138, 319–344 CrossRef CAS.
- M. E. Dry, Catal. Today, 2002, 71, 227–241 CrossRef CAS.
- J. J. C. Geerlings, J. H. Wilson, G. J. Kramer, H. P. C. E. Kuipers, A. Hoek and H. M. Huisman, Appl. Catal., A, 1999, 186, 27–40 CrossRef CAS.
- E. Iglesia, Appl. Catal., A, 1997, 161, 59–78 CrossRef CAS.
- C. K. ROFER-DePOORTER, Chem. Rev., 1981, 81, 447–474 CrossRef CAS.
- H. Schulz, Appl. Catal., A, 1999, 186, 3–12 CrossRef CAS.
- A. T. Bell, Catal. Rev.:Sci. Eng., 1981, 23, 203–232 CrossRef CAS.
- W. Chen, I. A. W. Filot, R. Pestman and E. J. M. Hensen, ACS Catal., 2017, 7, 8061–8071 CrossRef CAS PubMed.
- R. A. Dictor and A. T. Bell, J. Catal., 1986, 97, 121–136 CrossRef CAS.
- J. G. Ekerdt and A. T. Bell, J. Catal., 1980, 62, 19–25 CrossRef CAS.
- A. Kiennemann and J. P. Hindermann, Catal. Rev.:Sci. Eng., 1993, 35, 1–127 CrossRef.
- H. C. Long, M. L. Turner, P. Fornasiero, J. Kašpar, M. Graziani and P. M. Maitlis, J. Catal., 1997, 167, 172–179 CrossRef CAS.
- P. M. Maitlis, R. Quyoum, H. C. Long and M. L. Turner, Appl. Catal., A, 1999, 186, 363–374 CrossRef CAS.
- S. B. Ndlovu, N. S. Phala, M. Hearshaw-Timme, P. Beagly, J. R. Moss, M. Claeys and E. Van Steen, Catal. Today, 2002, 71, 343–349 CrossRef CAS.
- G. P. Van Der Laan and A. A. C. M. Beenackers, Catal. Rev.:Sci. Eng., 1999, 41, 255–318 CrossRef CAS.
- Z. Yao, C. Guo, Y. Mao and P. Hu, ACS Catal., 2019, 9, 5957–5973 CrossRef CAS.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy01336k |
| This journal is © The Royal Society of Chemistry 2025 |