
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailored support reduction of Cu/SrTiO3 catalysts for enhanced methanol production†
Chiara
Pischetola
 *a,
Luca
Artiglia
a,
Frank
Krumeich
b and
Jeroen A.
van Bokhoven
*ab
*a,
Luca
Artiglia
a,
Frank
Krumeich
b and
Jeroen A.
van Bokhoven
*ab
aCenter for Energy and Environmental Sciences, Paul Scherrer Institute, Villigen 5232, Switzerland. E-mail: chiara.pischetola@psi.ch
bInstitute for Chemical and Bioengineering, ETH Zurich, Zurich 8093, Switzerland. E-mail: jeroen.vanbokhoven@chem.ethz.ch
First published on 10th March 2025
Abstract
Copper supported on strontium titanate is explored as a catalyst in the hydrogenation of carbon dioxide to methanol. We used combined H2-TPR, O2-TPO, XPS, and STEM-EDX to identify the support defects, tailored by the activation procedure. Strontium titanate forms oxygen vacancies under high-temperature reductive treatments. The extent of its reduction is a function of the copper content and of the pressure; the highest extent is achieved at 2 wt% copper and 20 bar hydrogen. The catalytic data agree with a direct relationship between the methanol selectivity and the concentration of the oxygen vacancies, with the best results being: 90% (10% towards carbon monoxide) and an associated methanol space time yield of 0.49 gMeOH gcat−1 h−1. The selectivity is higher than that achieved on a typical copper catalyst on zinc oxide alumina, while keeping the competitive productivity value, despite having thirty times lower copper content. Post-reaction characterisation suggests that these sites are stable under reaction conditions. We propose a dual-site surface mechanism based on oxygen vacancies formed at the copper–support interface and via long-distance hydrogen spillover.
Introduction
The current climate crisis calls for the implementation of mitigation strategies based on carbon dioxide (CO2) emission reduction, capture and utilization. The methanol (CH3OH) economy, postulated by Prof. George A. Olah,1 is a model of reference for the development of sustainable routes in the chemical and energy sectors. Renewable methanol can be produced via (sequestered) carbon dioxide valorisation with green hydrogen (H2) according to the equilibrium equation (eqn (1)): CO2 + 3H2 ↔ CH3OH + H2O. The thermodynamic yield of methanol following eqn (1) under energy-efficient (i.e. T < 250 °C, P < 50 bar) operations is currently not achievable by the industrial catalyst based on CuO/ZnO/Al2O3 (CZA).2 This exothermic reaction is hindered by the activation of stable carbon dioxide and its selective conversion into target methanol. In contrast, the competitive formation of carbon monoxide (CO) via the endothermic reverse water gas shift (RWGS) reaction (CO2 + H2 ↔ CO + H2O; eqn (2)) is thermodynamically favoured under industrial-relevant conditions.An understanding of the structure–performance relationship common to copper (Cu)-based catalysts can lead to improved catalysts and process optimisation. It is widely accepted3–5 that the activation of hydrogen and carbon dioxide occurs following a dual-site mechanism. The copper phase accounts for the hydrogen splitting.6 Regarding the carbon dioxide activation, despite decades-long research in the field,2,7 the topic is still under debate. Multiple types of active sites have been proposed, notably the copper surface and the Cu–Me alloy8–10vs. the MeOx−1 oxygen vacancies (OVs)11–14 at a Cu/MeOx interface.15,16 Given the relevance of the CZA catalyst,2 the Cu/ZnO system has been extensively investigated.17–19 The CuZn alloy sites, identified as prominent both experimentally20–22 and from calculations,23,24 are intertwined with the formation of an oxygen deficient ZnO phase.25–28 To attempt a rational catalyst design, strategies based on controlled 3D morphologies with adjustable structural features29,30 have been proposed, highlighting a direct role of ZnOx−1 oxygen vacancies (OVs) in the surface mechanism. A valid approach to confirm, in a systematic manner, the role of oxygen vacancies in the title reaction is to select a reducible support whose metal(s) cannot alloy with copper. Such a material may potentially be a catalyst of interest.
Perovskite SrTiO3 (STO) well fits this requirement.31,32 STO has an ABO3 structure where SrO layers are alternated to those of TiO2. The perovskite reducibility is linked to the formation of oxygen vacancies at the B sites.33 Metal doping,34–36 electrochemical reduction,37,38 plasma treatment34 and high temperature annealing39 are common strategies to modulate the OV concentration on ABO3 perovskites. The presence of a transition metal (e.g. copper nanoparticles) on the STO surface could help the support reduction, modulating the required annealing temperature. To date, the only example of carbon dioxide hydrogenation over Cu/SrTiO3 is the computational work by Hus et al.,40 where kinetic Monte Carlo (KMC) simulations and density functional theory (DFT) calculations showed that methanol production (90% selectivity) is possible in the 200–240 °C temperature range and below 50 bar pressure. A ternary system (Cu–ZnO–SrTiO3) has been recently tested in the title reaction and the experimental results suggest a beneficial role of SrTiO3 in the Cu–ZnO activity in methanol production.41
The main goal of this work is to test and characterize a novel system based on copper supported on STO, with controlled properties. The laboratory-synthesised Cu/SrTiO3 materials are tested in carbon dioxide hydrogenation targeting methanol. We design a strategy to control the STO reduction and study its effect on the methanol productivity. Annealing under a hydrogen flow of catalysts containing different copper loadings and by increasing the hydrogen pressure is demonstrated to be effective in tailoring the extent of STO reduction. Temperature-programmed measurements under reductive (TPR) and/or oxidative (TPO) conditions combined with X-ray photoelectron spectroscopy and electron microscopy analysis shed light on the structure–activity relationships of Cu/STO. Small copper nanoparticles and homogeneously dispersed copper domains formed at high hydrogen pressure allow hydrogen spill-over with the formation of oxygen vacancies beneficial to methanol production. Cu/SrTiO3 is a catalyst of interest given the high selectivity reached (90%) together with a competitive production level (STYCu/SrTiO3/STYCZA = 0.83).
Results
Catalysis
Fig. 1 and S1† show the time-on-stream response of 1_STO, 2_STO, 5_STO, HP2_STO and 2_STO_C catalysts (see Tables 1 and S1† for specifics) in the continuous gas flow hydrogenation reaction of carbon dioxide at 30 bar and 200 °C. Before the reaction, the catalysts were reduced in hydrogen at 1 bar (20 bar in the case of HP2_STO). All the catalysts were active towards the production of methanol with carbon monoxide as the only by-product formed from the competitive RWGS reaction. At comparable carbon dioxide conversion levels (4 ± 2%) and under steady state conditions, reached within 30 minutes from the start of the reaction, the methanol selectivity spanned from 23 to 90% following the order: 2_STO_C < 1_STO < 5_STO < 2_STO < HP2_STO.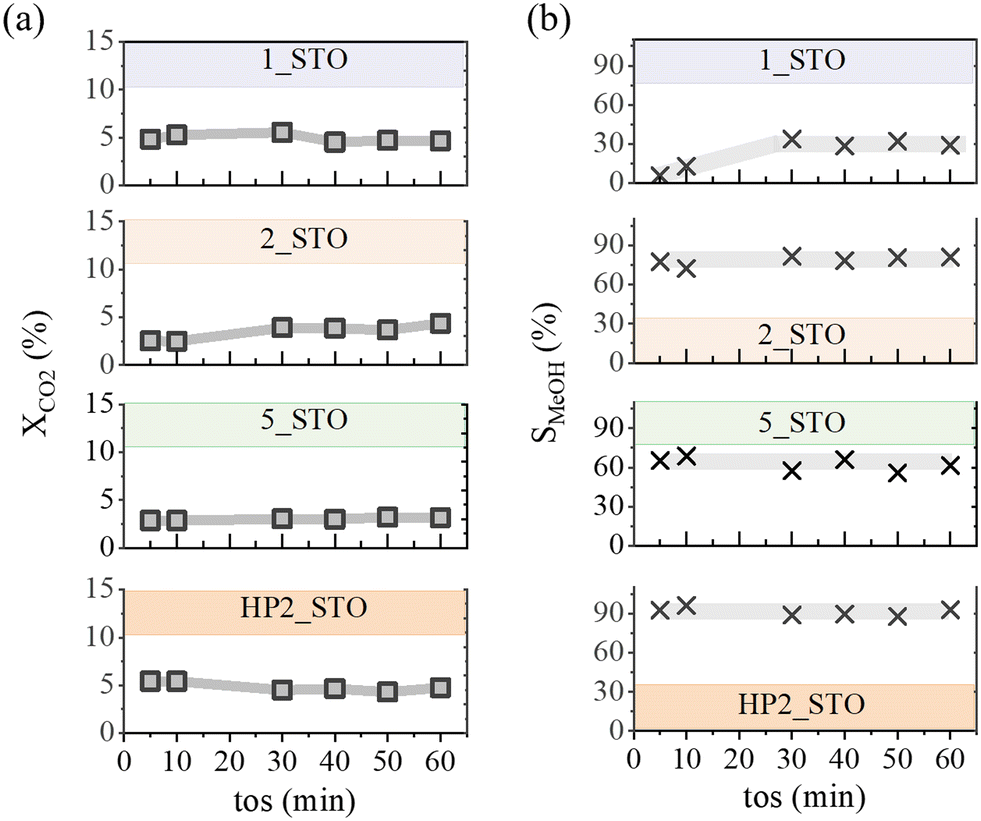 | ||
Fig. 1 Flow test response. Time on stream (tos, min) profiles of (a) carbon dioxide conversion (XCO2, %) and (b) methanol selectivity (SMeOH, %) over (from top to bottom) 1_STO, 2_STO, 5_STO and HP2_STO catalysts, respectively. Reaction conditions: P = 30 bar, T = 200 °C, GHSV = 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. 000 h−1. | ||
| Material | Name | Cu contenta (wt%) | H2-TPR experiments | O2-TPO experimentsb | ||
|---|---|---|---|---|---|---|
| Reduction T (°C) | Hydrogen consumed (mmol gcat−1) | Oxidation T (°C) | Oxygen consumed (mmolO2 mmolX−1) | |||
| a From ICP measurements. b Post-reaction. c Experiment at 5 bar. d Experiment at 10 bar. e Experiment at 20 bar. f X is STO. g X is Cu. | ||||||
| SrTiO3 | STO | 0 | — | 0 | 250 | 1.60f |
| Cu/SrTiO3 | 1_STO | 1.1 | 151 | 0.16 | — | — |
| 157 | 0.01 | |||||
| 2_STO | 2.0 | 133 | 0.06 | — | — | |
| 140 | 0.28 | |||||
| 146 | 0.03 | |||||
| 146c | 0.37 | |||||
| 129d | 0.39 | |||||
| 123e | 0.41 | |||||
| HP2_STO | —b | 0 | 130 | 0.49g | ||
| 155 | ||||||
| 250 | 2.18f | |||||
| 5_STO | 5.1 | 148 | 0.8 | — | — | |
| 158 | 0.02 | |||||
The trend highlights the fair performance of the Cu catalyst based on commercial SrTiO3. A direct comparison among the tested catalysts is drawn in Fig. 2 by using the methanol space time yield as a metric: in (a) “STYCu” is normalised per copper content and in (b) “STY” is normalised with respect to the total catalyst mass. Commercial CZA is used as a reference. Fig. S2† reports the performance correlation plots with the comparison of our data vs. the literature. Over the range of temperature (150–250 °C) and pressure (20–60 bar) scanned, the maximum methanol selectivity reached is 80%, at the expense of the corresponding STY. The striking feature of Fig. 2(a) is that, at comparable conversion levels (see Fig. 1 and S2(c)†), STO-based catalysts outperform commercial CZA. The methanol productivity is non-linearly dependent on copper loading. A volcano-trend is in evidence with the highest value obtained from the catalysts with 2 wt% copper: 2_STO (by comparing catalysts activated under 1 bar hydrogen) and HP2_STO (overall). The materials have been engineered to present diversified content of support defects, tailored as oxygen vacancies. Fig. 2(b) shows a positive correlation of the productivity values with the support partial reduction calculated from hydrogen temperature programmed reduction (H2-TPR) measurements. Specifically, the extent of support reduction (calculation reported in the Materials and methods section) spans from 0% (2_STO_C) to 0.62% (2_STO) after high-temperature hydrogen activation at 1 bar and increases to 1.63% in the case of the HP2_STO sample. The dashed line refers to the STY calculated for commercial CZA showing a value of 0.59 h−1 with an associated methanol selectivity of 60%. The STY from SrTiO3-based catalysts increases following the order 2_CML (0.04 gMeOH gcat−1 h−1) < 1_STO (0.07 gMeOH gcat−1 h−1) < 5_STO (0.19 gMeOH gcat−1 h−1) < 2_STO (0.34 gMeOH gcat−1 h−1). The activation of 2_STO in hydrogen at 20 bar (sample HP2_STO, dark orange square) boosts the methanol productivity to the highest recorded value of 0.49 h−1 with an associated selectivity of 90%. It is important to stress that the STY metric from HP2_STO is obtained with ca. 30 times less Cu content vs. CZA.
 | ||
| Fig. 2 Catalytic performances. (a) Commercial CZA, 1_STO, 2_STO, 5_STO and HP2_STO comparison in terms of the methanol time space yield with respect to the copper content (STYCu, molMeOH molCu−1 h−1). (b) Methanol time space yield (STY, gMeOH gcat−1 h−1) dependence on support partial reduction (TPRSTOreduction, %); solid (grey (2_STO_C), violet (1_STO), light orange (2_STO), green (5_STO)) and open (dark orange, HP2_STO) symbols indicate catalyst activation at 1 and 20 bar, respectively. The dashed line refers to the STY from CZA (63.5% CuO, 24.7% ZnO, 10.1% Al2O3 and 1.3% MgO). Reaction conditions: P = 30 bar, T = 200 °C, GHSV = 26000 h−1. | ||
Catalyst reducibility
The extent of support reduction is tuned during catalyst activation following high-temperature treatment in hydrogen. H2-TPR measurements were performed over fresh bare STO, 1_STO, 2_STO, 5_STO and 2_STO_C. The experimental profiles are plotted in Fig. 3, S1 and S3,† while the results from data fitting are listed in Tables 1 and S1.† In the absence of copper, no variation in the TCD response is detected for STO over the 50–250 °C temperature range and at 1 bar overall pressure. A positive peak appears for the Cu-containing materials. The maxima are centred at 145 ± 5 °C for the samples based on lab synthesised STO, whereas a shift towards higher temperature (190 °C) is observed from the STO_C support. The increase in copper loading is reflected by larger peak areas (see Fig. S3† and Table 1). However, the calculated hydrogen consumption is not proportional to the mole content of copper; it exceeds the stoichiometric requirement for the complete copper reduction from Cu(II) to metallic Cu(0) with the only exemption in the case of the 2_STO_C sample. | ||
| Fig. 3 Catalytic reducibility. (a) H2-TPR peak deconvolution (from top to bottom) of 1_STO, 2_STO and 5_STO. The envelope is given by the dashed black line; the blue areas refer to hydrogen overconsumption with respect to the Cu content. (b) XRD patterns of fresh (light) and post-H2-TPR (dark) STO (grey), 1_STO (violet), 2_STO (orange) and 5_STO (green) with associated Miller indices from standard SrTiO3; the yellow area and the inset highlight the (1 1 1) reflection of metallic Cu. (c) H2-TPR profiles of 2_STO at (from bottom to top): 1 (70 mgcat), 5 (46 mgcat), 10 (65 mgcat) and 20 (45 mgcat) bar. (d) Hydrogen overconsumption (%) dependence on total pressure (bar) during H2-TPR experiments of the 2_STO catalyst. The dashed black line is included for visual help. | ||
The TCD profiles of 1_STO and 5_STO can be fitted with two Gaussian contributions (Fig. 3(a)). The red curves at lower temperature (151 °C and 148 °C, respectively) have an area equivalent to the theoretical hydrogen moles required for complete copper reduction, while the blue curves at higher temperature (157 °C and 158 °C, respectively) close the mass balance. A three-curve fitting is required for 2_STO where two asymmetrical contributions at low temperature (red curves at 133 °C and 140 °C) have a combined area consistent with the hydrogen requirement for the Cu(II) → Cu(0) transition; such a shape is indicative of non-homogeneous particle size distribution (as further confirmed by microscopy, see infra). The blue area at 146 °C represents the excess of hydrogen consumed. The calculated values of support partial reduction from data fitting follow the trend: 2_STO_C ≪ 1_STO < 5_STO < 2_STO.
The structural integrity of the materials after the high-temperature treatment is confirmed by comparing the X-ray diffractograms (Fig. 3(b)) of the samples recovered after H2-TPR measurements with those collected from the fresh materials. The characteristic reflections of SrTiO3 (JCPDS No. 35-734) at 2θ = 32.5°, 40.0°, 46.6° and 57.9°, corresponding to the (1 1 0), (1 1 1), (2 2 0) and (2 1 1) planes, respectively, are present in all the patterns; 2_STO and 5_STO post-H2-TPR have an extra peak at 43.3° assigned to the Cu (1 1 1) reflection (JCPDS No. 04-0836). The extent of support reduction in 2_STO, which showed the highest value from the experiments at 1 bar, was further tuned by increasing the pressure during H2-TPR measurements up to 20 bar. The profiles in Fig. 3(c) gradually shift towards lower reduction temperatures (from 140 to 123 °C) with increasing pressure. The area of the peaks correspond to the hydrogen overconsumption values of Fig. 3(d) which follow a linear positive correlation with respect to the cell pressure.
The following experimental protocol allows us to confirm whether the modifications occurred on the catalysts during activation are preserved after the reaction, thus being relevant to the discussion of the structure–activity relationship of the material. It consists of four in situ experiments performed in sequence: (i) H2-TPR at 20 bar over 2_STO to produce HP2_STO, (ii) carbon dioxide hydrogenation reaction at 30 bar and 200 °C over HP2_STO, followed by (iii) H2-TPR and (iv) oxygen-temperature programmed oxidation (O2-TPO) at 1 bar. The results from the post-reaction characterisation of HP2_STO are reported in Fig. 4(a) and Table 1.
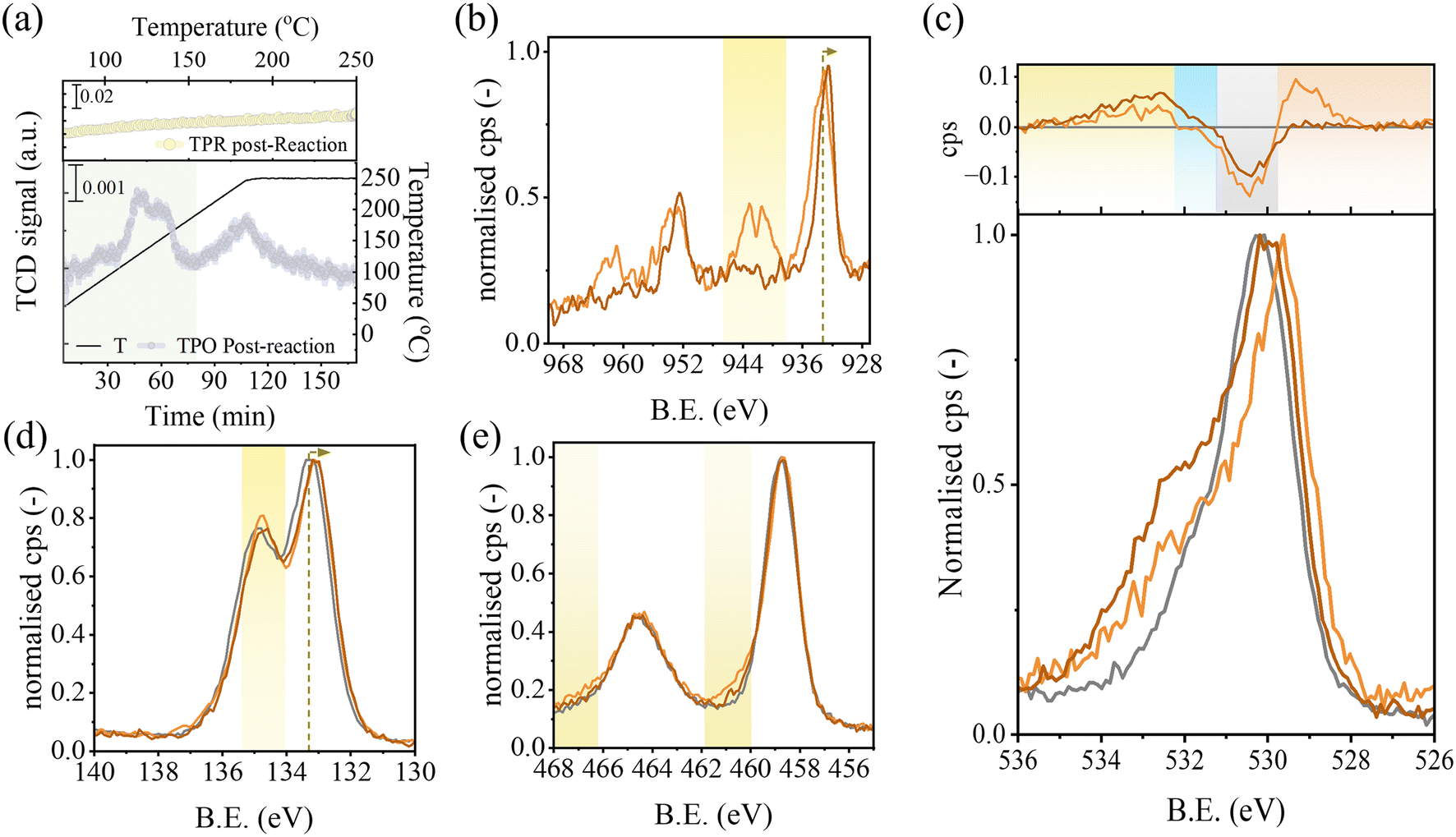 | ||
| Fig. 4 Post-reaction characterisation. (a) In situ O2-TPO (bottom panel) and H2-TPR (top panel) profiles of HP2_STO post-reaction at 30 bar and 200 °C. (b)–(e) High-resolution photoemission spectra of bare STO (grey), fresh (light orange) 2_STO and post-reaction (dark orange) HP2_STO samples over the Cu 2p, O 1s, Ti 2p and Sr 3d regions, respectively. The inset in (c) is the area-normalized O 1s profiles of STO, fresh 2_STO and post-reaction HP2_STO subtracted from the O 1s spectrum of STO. | ||
A flat TCD response is recorded during H2-TPR over the 50–250 °C temperature range. On the other hand, two positive peaks are in evidence in the O2-TPO profile. The low-temperature peak appears as a doublet with maxima centred at 130 °C and 155 °C, respectively, whereas the second peak is observed at 250 °C. Peak assignment is done on the basis of the results from O2-TPO control experiments on bulk CuO and bare STO (Fig. S4(a)†), which delivered a positive peak at 170 and 250 °C, respectively. Therefore, the first peak in Fig. 4(a) can be attributed to the re-oxidation of the copper phase occurring in a step-wise Cu(0) → Cu(I) → Cu(II) fashion given the profile shape and total peak area consistent with the reaction stoichiometry (see Table 1). The high-temperature peak can be attributed to the re-oxidation of the support with an oxygen consumption exceeding by 25% the partial reduction extent of HP2_STO calculated from H2-TPR experiments. The oxygen consumption extracted from the high-temperature peak is further analysed in Fig. S4(b),† where the values are normalised with respect to that from bare STO. A linear trend with respect to the support partial reduction from H2-TPR experiments is in evidence.
Ex situ X-ray photoelectron spectroscopy (XPS) is used to confirm the nature of the reduced species on the catalyst generated by the (designed) hydrogen (over)consumption, namely metallic copper and oxygen vacancies (OVs). The STO (grey), fresh 2_STO (light orange) and post-reaction HP2_STO (dark orange) materials are examined in Fig. 4(b–e) by comparing the high-resolution photoemission spectra over the Cu 2p (b), O 1s (c), Ti 2p (d) and Sr 3d (e) regions. Peak component data analysis derived from curve fitting is reported in Tables S2 and S3† and in the XPS data analysis section of the ESI.† The Cu 2p signal of 2_STO reproduces the spectral features of CuO, used as a reference for Cu2+ (see Fig. S5(a)†). The copper Auger (L3M4.5M4.5) spectrum (Fig. S5(b)†) superimposes well to that of the reference CuO, further proving the presence of Cu2+. Moreover, the value of the modified Auger parameter  matches well with that reported for CuO.42 The Cu 2p spectrum of HP2_STO shifts towards lower binding energy (BE) values. The main difference between the HP2_STO and 2_STO spectra is the quenching of shake-up satellites (see Fig. S5(c)†), suggesting the quantitative reduction of Cu2+. The calculated Auger parameter
matches well with that reported for CuO.42 The Cu 2p spectrum of HP2_STO shifts towards lower binding energy (BE) values. The main difference between the HP2_STO and 2_STO spectra is the quenching of shake-up satellites (see Fig. S5(c)†), suggesting the quantitative reduction of Cu2+. The calculated Auger parameter  of 1851.5 eV (vs. literature 1849.2 eV of Cu2O and 1851.2 eV of Cu0 (ref. 42)) confirms the presence of metallic copper. However, the line shape of Cu LMM differs from that of bulk metallic copper, taken as a reference in Fig. S5(b).† The main reasons to explain such a difference are as follows: (i) the reference Cu LMM spectrum for Cu0 was for foil and not powder, which would better reproduce the large amount of coordinatively unsaturated sites of NPs deposited on the sample; (ii) the broader feature of the spectrum of HP2_STO reveals the presence of a small fraction of Cu(I) (probably due to partial reoxidation of copper nanoparticles during the sample transfer from the reactor to the XPS setup) in agreement with ref. 43. A direct comparison between the O 1s spectra of STO, 2_STO and HP2_STO is provided in the inset of Fig. 4(c), where the area-normalised profile of STO is subtracted from the area-normalised profiles of the other two catalysts; the deconvolution of the O 1s spectrum of HP2_STO is reported in Fig. S6† as exemplary fitting. The positive feature of 2_STO at low BE can be attributed to lattice oxygen from CuO supported on the fresh catalyst,44,45 in agreement with the interpretation of the Cu 2p profiles. The copper nanoparticles on 2_STO and HP2_STO attenuate the photoemission signal generated by the lattice oxygen of the STO support resulting in the negative feature of the inset. Unsaturated oxygen atoms adjacent to surface defects like oxygen vacancies46,47 are in evidence in the blue spectral region only for HP2_STO. These defects can form by the overconsumption of hydrogen observed during the H2-TPR experiment. Chemisorbed CO32−,48,49 OH− groups50,51 and water52,53 can generate the positive contributions observed at high BE; their relative abundance is: HP2_STO > 2_STO > STO. This is consistent with reports correlating the higher content of surface hydroxyls with the higher density of oxygen vacancies via reversible repopulation.54 The Ti 2p and Sr 3d spectra collected from the three materials respectively overlap, with the only differences being as follows. The Ti 2p1/2 peak broadens towards higher BE (Δ = +1.7 eV) for 2_STO, suggesting the presence of a new component (highlighted in yellow in Fig. 4d and shown in Fig. S7(a),† 13.7%). Other Ti 2p1/2 spectra collected from other CuO/TiO2 systems55–57 present as well this feature, which seems to be an effect of doping Cu(II)-oxo species, like on 2_STO. The presence of mixed Cu–Ti oxides has been invoked as an explanation.56Post-reduction, copper segregates onto the surface in HP2_STO; i.e. the component at 460.4 eV disappears. A slight negative binding energy shift (Δ = −0.2 eV) of Sr 3d upon copper deposition is observed. Given the complexity of the materials under investigation, we cannot exclude that such a small shift can arise from energy scale alignment effects. The Sr 3d spectra of 2_STO and HP2_STO differ in terms of shape (highlighted in yellow in Fig. 4(e)), where the new feature can be attributed to strontium carbonate (SrCO3) at the surface.58,59 Maximum-normalised XRD patterns of STO, 2_STO and HP2_STO in Fig. S8† confirm the presence of strontium carbonate (reflections at 2θ = 25.2 and 25.9°, JCPDS No. 005-0418) in the copper-containing materials.
of 1851.5 eV (vs. literature 1849.2 eV of Cu2O and 1851.2 eV of Cu0 (ref. 42)) confirms the presence of metallic copper. However, the line shape of Cu LMM differs from that of bulk metallic copper, taken as a reference in Fig. S5(b).† The main reasons to explain such a difference are as follows: (i) the reference Cu LMM spectrum for Cu0 was for foil and not powder, which would better reproduce the large amount of coordinatively unsaturated sites of NPs deposited on the sample; (ii) the broader feature of the spectrum of HP2_STO reveals the presence of a small fraction of Cu(I) (probably due to partial reoxidation of copper nanoparticles during the sample transfer from the reactor to the XPS setup) in agreement with ref. 43. A direct comparison between the O 1s spectra of STO, 2_STO and HP2_STO is provided in the inset of Fig. 4(c), where the area-normalised profile of STO is subtracted from the area-normalised profiles of the other two catalysts; the deconvolution of the O 1s spectrum of HP2_STO is reported in Fig. S6† as exemplary fitting. The positive feature of 2_STO at low BE can be attributed to lattice oxygen from CuO supported on the fresh catalyst,44,45 in agreement with the interpretation of the Cu 2p profiles. The copper nanoparticles on 2_STO and HP2_STO attenuate the photoemission signal generated by the lattice oxygen of the STO support resulting in the negative feature of the inset. Unsaturated oxygen atoms adjacent to surface defects like oxygen vacancies46,47 are in evidence in the blue spectral region only for HP2_STO. These defects can form by the overconsumption of hydrogen observed during the H2-TPR experiment. Chemisorbed CO32−,48,49 OH− groups50,51 and water52,53 can generate the positive contributions observed at high BE; their relative abundance is: HP2_STO > 2_STO > STO. This is consistent with reports correlating the higher content of surface hydroxyls with the higher density of oxygen vacancies via reversible repopulation.54 The Ti 2p and Sr 3d spectra collected from the three materials respectively overlap, with the only differences being as follows. The Ti 2p1/2 peak broadens towards higher BE (Δ = +1.7 eV) for 2_STO, suggesting the presence of a new component (highlighted in yellow in Fig. 4d and shown in Fig. S7(a),† 13.7%). Other Ti 2p1/2 spectra collected from other CuO/TiO2 systems55–57 present as well this feature, which seems to be an effect of doping Cu(II)-oxo species, like on 2_STO. The presence of mixed Cu–Ti oxides has been invoked as an explanation.56Post-reduction, copper segregates onto the surface in HP2_STO; i.e. the component at 460.4 eV disappears. A slight negative binding energy shift (Δ = −0.2 eV) of Sr 3d upon copper deposition is observed. Given the complexity of the materials under investigation, we cannot exclude that such a small shift can arise from energy scale alignment effects. The Sr 3d spectra of 2_STO and HP2_STO differ in terms of shape (highlighted in yellow in Fig. 4(e)), where the new feature can be attributed to strontium carbonate (SrCO3) at the surface.58,59 Maximum-normalised XRD patterns of STO, 2_STO and HP2_STO in Fig. S8† confirm the presence of strontium carbonate (reflections at 2θ = 25.2 and 25.9°, JCPDS No. 005-0418) in the copper-containing materials.
To summarize, treatment of 2_STO at high temperature and high pressure in hydrogen (activation step) and hydrogen + carbon dioxide (reaction step) resulted in the post-reaction HP2_STO sample which differs from 2_STO by: (i) quantitative reduction of Cu2+ and formation of metallic copper reflected in (ii) the loss of Cu–Ti mixed oxides with copper surface segregation, (iii) formation of OV sites and (iv) lower content of strontium carbonate.
Catalyst morphology
Conclusive considerations on the structure–activity relationship of the Cu/STO catalysts can be drawn by complementing the description of their electronic properties with structural/morphological characterisation. Adherence of the laboratory synthesised STO materials to the crystallographic structure of SrTiO3 perovskite is confirmed by XRD (Fig. 3(b)). N2-physisorption measurements (Fig. S9(a)†) show a type IV isotherm with an associated H1 hysteresis loop, typical of mesoporous materials arranged as agglomerates. The computed surface area is one order of magnitude higher than that of STO_C (see Fig. S1(c)†). The representative scanning electron microscopy (SEM) image of STO in Fig. S9(b)† agrees with an agglomerate conformation for the material, revealing hierarchically structured mesoporous aggregates with a multi-plated morphology (vs. the bulk and smooth rod-like structures reported in Fig. S1(c)†). Inductive-coupled plasma (ICP) analysis confirms the nominal copper content on the supported catalysts (see Tables 1 and S1†). The associated BET surface areas differ by a maximum of 5% from that of the bare support.A morphology study based on transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM) with energy dispersive X-ray (EDX) spectroscopy was performed on 1_STO, 2_STO, and 5_STO post H2-TPR. High-angle annular dark field (HAADF) representative STEM pictures are reported in Fig. S10(a–c).† The EDX elemental maps of selected areas in Fig. 5 show in green the contributions coming from copper species. The increment of copper loading from 1 to 5 wt% (from (a) to (c)) is reflected by an increase of the counts of spherical-like Cu nanoparticles on top of the support but does not result in a size growth (dCu = 27 ± 2 nm), as shown by the associated particle size distribution in (d–f), thus resulting in a comparable dispersion.60 Both 1_STO and 5_STO follow a standard Gaussian distribution, whereas a bimodal trend is observed for 2_STO. The bimodal trend in Fig. 5(e) mimics well the TCD profile of Fig. 2(a); a correlation between small copper nanoparticles (7 nm average) and the low-temperature reduction event (133 °C) can be drawn from the collected data. From statistical analysis, at least 85% of copper in 2_STO is arranged as ill-defined round-shaped nanoparticles (see STEM-EDX and HRTEM images in Fig. S10(d–f)†), but evidence of homogeneous copper distribution with a low concentration over STO is provided in Fig. S11.† In particular, the EDX spectrum in (b), referring to the area in (a), shows two distinct signals with similar intensity at 0.93 and 8.04 keV, which are assigned to the characteristic CuLα and Kα lines, respectively.61 The elemental mapping in (c–f) is characterised by comparable dispersion of the O, Sr, Ti and Cu elements, respectively. The OKα, SrLα, TiKα and Kβ lines appear in Fig. S11(b)† at 0.53, 1.81, 4.51 and 4.95 keV, respectively, in agreement with tabulated values.61
 | ||
| Fig. 5 Morphological characterisation. (a)–(c) EDX maps of selected areas from 1_STO, 2_STO, and 5_STO post H2-TPR with associated (d)–(f) particle size distribution. Note: green areas in (a–c) represent counts from copper species on the Ti + Sr + O overlay given in purple. | ||
Discussion
The Cu/STO catalysts developed in this study are synergistic systems able to yield methanol. The suboptimal catalytic results of 2_STO_C originate from the morphology of the commercial perovskite (see Fig. S1†), unsuitable to be used as an efficient support for Cu nanoparticles. On the other hand, the microstructure of the lab-synthesised STO (Fig. S9†), obtained from confinement effects induced by hydrophobic interactions between the solvents used during synthesis,32 is key to control the surface properties required for competitive methanol productivity. Given the volcano-type trend of STYCu, one can conclude that the copper phase is not the sole involved in the surface mechanism. Indeed, Fig. 2 suggests that supports with different reduction extents, as a key property, deliver different responses from the activity tests. Recent literature reports making use of Cu–ZnO/SrTiO3,41 Cu/CeO2 (ref. 62 and 63) and In2O3 (ref. 64 and 65) support the beneficial role of oxygen vacancies (OVs) in methanol synthesis. Nonetheless, the published efforts29,30,66 have failed so far in providing a system with engineered OVs able to deliver methanol STYs (0.03–0.45 gMeOH gcat−1 h−1, at 220–250 °C and 30–60 bar) matching the productivity from the CZA catalyst (used as a standard for general reference, Fig. S2†). In this work, we succeeded in achieving a competitive STY value vs. CZA, despite having 30 times lower Cu loading, by exploiting the reducibility property of STO. H2-TPR analysis combined with surface-sensitive XPS measurements demonstrated that STO redox features can be tailored during the high-temperature hydrogen pre-treatment by changing the (i) copper content and (ii) hydrogen pressure. The in situ formed metallic copper nanoparticles act as active sites for hydrogen dissociation,67 which can migrate from the Cu/support interface via spill-over and extract lattice oxygen generating vacancies.68 For a given reducible support, the extent of the spill-over is temperature- and pressure-dependent.69 Generally speaking, at low hydrogen partial pressure (e.g. <10−1 bar) and high temperature (e.g. >150 °C), the support reduction can be limited at the metal/support interface and linked to oxygen consumption via water release.69 As a consequence, samples with a higher Cu/support interface are expected to deliver a greater partial reduction of the support. The values reported in Fig. 2(b), obtained from data fitting (Fig. 3(a), (c), S1(b),†Tables 1 and S1†) agree with the results from the structural and morphological characterisation. No support reduction is calculated for 2_STO_C, affected by the lowest Cu dispersion, given its BET surface area. An increasing STO reduction extent is observed from 1_STO to 5_STO, where a higher loading (see Table 1), but comparable copper particle size (extracted by microscopy, Fig. 5(d)–(f)), results in more support being in direct contact with copper; thus greater consumption of hydrogen can occur. The 2_STO sample is an outlier from the trend, being affected by a larger hydrogen overconsumption than expected. This can be attributed to the presence of copper nanoparticles with an average mean diameter equal to 7 nm (ca. 20%) which can act as extra (Cu/support) interface sites for STO reduction. The increase in the activation pressure results in higher hydrogen uptake (Table 1) and allows for the hydrogen migration on the surface to occur over a longer distance, thus promoting further support reduction (Fig. 2(b) and 3(d)). The data collected for the Cu/STO system agree with the pressure-dependence study of hydrogen coverage on Pt/TiO2 by Beck et al.70 and provide experimental proof to the postulated effect of pressure and the support on the distance covered by hydrogen spill-over.69,71H2-TPR experiments combined with operando XAS on CZA showed that the Cu and Zn speciation is strongly pressure-dependent with reduction events enhanced at increasing pressures.17 Here, high-temperature (250 °C) and high-pressure (20 bar) hydrogen treatment is used as a successful strategy to form metallic copper and maximise the STO partial reduction. The  of HP2_STO is consistent with that of Cu(0), whereas the accounted fraction of Cu(I) from the visual inspection of the Auger line shape in Fig. S5(b)† can originate from the exposure to air of the sample before the ex situ analysis.72 The good correlation between the hydrogen overconsumption (from H2-TPR measurements) and the data analysis of the O 1s spectra of STO, 2_STO and HP2_STO (Fig. 4(c)) confirms that surface defects, here identified as oxygen vacancies (OVs), are clearly present only in HP2_STO. Such OVs are not repopulated during the reaction. Indeed, no further hydrogen is consumed to regenerate them during the post-reaction H2-TPR experiment, whereas a high temperature oxidizing atmosphere is required to uptake oxygen on the support (Fig. 4(a)). The trend shown in Fig. S4(b)† confirms the dependence of the TCD profile solely on the material structure, excluding any contribution from the (possible still) adsorbed surface intermediates. Reduction at 20 bar of 2_STO delivered a hydrogen consumption (with respect to the STO phase) that did not match the oxygen uptake during TPO by reduced HP2_STO (see Table 1). This can be linked to the oxygen uptake ability of STO (Fig. S4(a)† and Table 1), as previously reported in other studies focused on perovskites.73,74 The excess electrons per OV are transferred to the neighbouring titanium atoms,75,76 inducing the Ti4+ → Ti3+ transition.39 However, the deconvolution of Ti 2p photoemission spectra in Fig. 4(d) does not allow for the fitting of an extra feature at lower BE than that of Ti4+, to account for any Ti3+.36 The exposure to air during sample transfer for the ex situ XPS analysis can compromise the stability of Ti3+ species. Moreover, evidence that oxygen vacancies are formed without the Ti3+ detection from XPS analysis has been previously reported77,78 for TiO2 materials. It was proposed that Ti3+ species were formed at a sublayer deeper than the probing depth of the XPS analysis.70,78 DFT computational calculations on Fe-doped SrTiO3 showed that the energetically favoured configuration when oxygen vacancies are formed is obtained by the sinking of the neighbouring titanium atom into the bulk and the migration of the vacancies towards the surface.79 This is consistent with the XPS results for O 1s and Ti 2p regions. More experiments with operando and bulk-sensitive techniques are required to pinpoint the exact electronic configuration and structure of STO partially reduced materials and exclude any artefacts caused by the ex situ characterisation.
of HP2_STO is consistent with that of Cu(0), whereas the accounted fraction of Cu(I) from the visual inspection of the Auger line shape in Fig. S5(b)† can originate from the exposure to air of the sample before the ex situ analysis.72 The good correlation between the hydrogen overconsumption (from H2-TPR measurements) and the data analysis of the O 1s spectra of STO, 2_STO and HP2_STO (Fig. 4(c)) confirms that surface defects, here identified as oxygen vacancies (OVs), are clearly present only in HP2_STO. Such OVs are not repopulated during the reaction. Indeed, no further hydrogen is consumed to regenerate them during the post-reaction H2-TPR experiment, whereas a high temperature oxidizing atmosphere is required to uptake oxygen on the support (Fig. 4(a)). The trend shown in Fig. S4(b)† confirms the dependence of the TCD profile solely on the material structure, excluding any contribution from the (possible still) adsorbed surface intermediates. Reduction at 20 bar of 2_STO delivered a hydrogen consumption (with respect to the STO phase) that did not match the oxygen uptake during TPO by reduced HP2_STO (see Table 1). This can be linked to the oxygen uptake ability of STO (Fig. S4(a)† and Table 1), as previously reported in other studies focused on perovskites.73,74 The excess electrons per OV are transferred to the neighbouring titanium atoms,75,76 inducing the Ti4+ → Ti3+ transition.39 However, the deconvolution of Ti 2p photoemission spectra in Fig. 4(d) does not allow for the fitting of an extra feature at lower BE than that of Ti4+, to account for any Ti3+.36 The exposure to air during sample transfer for the ex situ XPS analysis can compromise the stability of Ti3+ species. Moreover, evidence that oxygen vacancies are formed without the Ti3+ detection from XPS analysis has been previously reported77,78 for TiO2 materials. It was proposed that Ti3+ species were formed at a sublayer deeper than the probing depth of the XPS analysis.70,78 DFT computational calculations on Fe-doped SrTiO3 showed that the energetically favoured configuration when oxygen vacancies are formed is obtained by the sinking of the neighbouring titanium atom into the bulk and the migration of the vacancies towards the surface.79 This is consistent with the XPS results for O 1s and Ti 2p regions. More experiments with operando and bulk-sensitive techniques are required to pinpoint the exact electronic configuration and structure of STO partially reduced materials and exclude any artefacts caused by the ex situ characterisation.
The complex system of active sites (i.e. small and big copper nanoparticles, OVs at the interface and ‘long-distance’ OVs) on hydrogen-treated Cu/STO materials is summarised in Scheme 1. An educated guess of the structure–activity relationship is proposed. The oxygen vacancies formed at the interface with big copper nanoparticles (common to 1_STO, 2_STO and 5_STO) are adsorption sites for the activation of carbon dioxide.63 The trend of methanol production in Fig. 1 and 2 suggests that these sites are selective towards the formation of key intermediates involved in the methanol synthesis, as proposed for a Cu/TiO2−x defective catalyst.80 A theoretical study on Cu/STO identified a geometric effect on the carbon monoxide formation rate (via RWGS), shown to be enhanced in the presence of small copper nanoparticles.81 However, the higher SMeOH of 2_STO vs. 1_STO and 5_STO and of HP2_STO vs. 2_STO indicates that the long-distance oxygen vacancies (OVs as isolated sites or clustered79) can act as sites for direct carbon dioxide conversion to methanol. Moreover, combined KMC simulations with DFT calculations on Cu/TiO2 agreed with the carbon monoxide-mediated pathway as a possible route contributing to methanol production from carbon dioxide hydrogenation.82 Our system delivers unmatched methanol selectivity (90%) while keeping a productivity value 5 times higher than that of the closest formulation (i.e. Cu–ZnO/SrTiO3 by Liu et al.) reported in the literature.41 In an independent computational study, Hus et al.40 have reported consistent results to our findings.
 | ||
| Scheme 1 Structure–activity relationship. (a) Effect of pressure (blue coded) and metal loading (purple coded) on the type of oxygen vacancy (open square; OVs) and the size of copper nanoparticles (semi-sphere), respectively. Low pressure allows for OV sites to form only at the interface, whereas higher pressures result in longer-distance OV sites. Intermediate loading (2 wt%) results in the simultaneous presence of small and big nanoparticles. (b) Proposed structure–activity relationship between product formation (carbon monoxide and methanol) and the type of active site (small copper nanoparticles, copper nanoparticles + interface OVs and long-distance OVs). The key feature is the conversion at the long-distance OVs of carbon dioxide. | ||
Conclusions
Cu/STO catalysts show exceptionally high methanol productivity from carbon dioxide, paving the road for the application of a novel class of methanol catalysts. For the first time, a different formulation than CuO/ZnO/Al2O3 can deliver methanol yields up to industrial standards. High-surface area Cu/STO materials can be synthesised with a tailored number of surface defects, which are identified by combined H2-TPR, O2-TPO and XPS measurements as oxygen vacancies (OVs). The partial STO reduction can occur at the Cu/support interface or at longer distances via hydrogen spill-over. The extent of reduction is a function of the dispersion of the copper nanoparticles and of the hydrogen reduction pressure. The OV sites are suggested to play a role in carbon dioxide and carbon monoxide activation. This work shows that such OVs are selective sites towards the formation of methanol. The results from our investigation highlight how critical the activation conditions are to tune the desired properties of a given catalyst. Cu/STOs are novel catalysts that can not only be easily engineered but also deliver excellent performances in selectively reducing carbon dioxide to methanol.Materials and methods
All the gasses employed in this work (argon, helium, nitrogen, hydrogen, oxygen, hydrogen + carbon dioxide mix) are of extra-high purity (≥5.0) from Messer and PanGas. The reagents (strontium carbonate, titanium(IV) butoxide, copper(II) nitrate trihydrate, purity ≥99%; sodium hydroxide solution, >97%) and solvent (ethylene glycol, ≥99%) involved in the catalyst(s) synthesis were purchased from Merck. The commercial bulk CuO (Sigma Aldrich, 99%), SrTiO3 (Sigma Aldrich, 99%) and methanol catalyst CZA (Alfa Aesar, catalogue no. 45776, lot no. I06Z036) were used without further purification.Catalyst synthesis
The hydrothermal synthesis of STO was based on the protocol developed by Zhou et al.32 Briefly, strontium carbonate (1.088 g, Sr/Ti = 1.0) was dissolved in 10 mL of deionized water and dropwise added to ethylene glycol (EG) solution containing titanium(IV) butoxide (1.75 g). A sodium hydroxide solution (5 M, 5 mL) was added (total EG:H2O = 2 vol/vol) and the mixture was stirred (700 rpm) for 1 h at room temperature. The solution was transferred into an autoclave and thermally treated at 180 °C, 10 °C min−1 for 24 h. The white powder was collected by centrifugation, washed 3 times with H2O + EtOH (50/50 vol) and dried in a vacuum at 70 °C for ca. 12 h.
Copper-supported catalysts (2_STO_C, 1_STO, 2_STO and 5_STO) were synthesised by a deposition–precipitation method. A copper(II) nitrate aqueous solution was added to the support (300–500 mg), under constant (700 rpm) stirring. The copper concentration was kept constant at 0.10 g L−1 (for loading of 2 and 5 wt%) and 0.20 g L−1 (for loading of 1 wt%), as a result of fixed STO/water at 2.8 ± 0.2 g L−1. The pH was increased to 7.8 ± 3 by adding dropwise a sodium hydroxide solution (0.1 M) at 80 °C, under reflux. The mixture was aged for 4 h. The precipitate was recovered by centrifugation, washed with H2O + EtOH (50/50 vol) until pH = 7 and dried in a vacuum oven at 70 °C for ca. 12 h.
Catalyst characterisation
ICP measurements were performed on an Ultima2 HORIBA Jobin Yvon from the extract in nitric acid/chloric acid (1:3 vol/vol) solution of digested 60–70 mg of sample following a 1:60 dilution. Instrument calibration was performed before each analysis with standard copper solutions over the 0.5–10 ppm range.
H2-TPR and O2-TPO analyses were carried out using a Micrometrics Autochem 2950 HP unit equipped with a TCD. Samples (ca. 40–100 mg) were loaded between two layers of quartz wool inside a quartz (for experiments at P = 1 bar) or stainless steel (P > 1 bar) U-shaped cell. In a standard experiment, a continuous flow of 20 cm3 min−1 of 10% vol/vol hydrogen (oxygen) in Ar (He) was fed to the cell while increasing the temperature (2 °C min−1) to 250 °C (dwell 1 h); the output passed through a cold trap (i.e. isopropanol + liquid nitrogen; −80 °C) before reaching the detector. The profile curve fitting was performed in Origin 2024 employing Gaussian curves after subtracting a non-linear background; positive peaks are conventionally attributed to (hydrogen or oxygen) gas consumption. The areas from peak deconvolution were converted into hydrogen and oxygen consumption (mmol) on the basis of calibration plots. The extent of the support reduction is defined as the ratio between the moles of hydrogen extracted from the high-temperature peak of H2-TPR profiles and the moles of SrTiO3 used in the experiments.
XRD patterns were collected with a Bruker D8 ADVANCE diffractometer (Bragg–Brentano geometry) under CuKα radiation (λ = 1.54 Å, 40 kV and 40 mA) at a scan rate of 0.01° step−1 over the 4–80° 2θ range.
Ex situ XPS measurements were carried out in a laboratory XPS setup (base pressure 9 × 10−9 mbar) equipped with a Specs Phoibos 150-EP electron analyzer, using a Mg Kα non-monochromatic source (excitation energy of 1253.6 eV, pass energy of 20 eV). Powder samples were obtained by pressing 20–30 mg of catalyst powder onto an Ag grid, to obtain thin and homogeneous pellets. The analysis performed on the used catalyst was carried out directly after the reaction, with a transfer time in air (from the cold reactor to the vacuum chamber) within a ten minute range. The full range survey (0–1050 eV BE range), high-resolution spectra of the Sr 3d, Ti 2p, O 1s and Cu 2p core levels and Cu LMM Auger region were collected. The energy scale alignment was obtained using the Ti 2p3/2 peak (BE = 458.7 eV) as a reference to correct for charging effects. The CasaXPS software (Casa Software Ltd., UK) was used for data treatment. The spectra were fitted using Gaussian–Lorentzian curves after subtracting a Shirley background and by fixing the spin–orbit splitting and the full width at half maximum (FWHM). The modified Auger parameter α′ was calculated as the difference between the KE of the most intense Auger and photoelectron lines of copper plus the excitation energy.
N2-physisorption experiments were performed on 3Flex Micromeritics Inc. equipment at 77 K. Prior to analysis, samples were evacuated at 250 °C in a Micromeritics VacPrep 061 system. The specific surface area was calculated according to the Brunauer–Emmett–Teller (BET) method by fitting the isotherms in the 0.02 to 0.1 p/p0 range. The pore volume was determined according to the BLH method.
SEM pictures were taken with a Zeiss Supra VP5 microscope equipped with an FEG electron detector. The materials in the form of fine powder were directly deposited on carbon tape mounted on the sample holder. TEM and STEM-EDX investigation was performed on a JEOL JEM F200 microscope equipped with a high-angle annular dark field detector (HAADF-STEM, 200 kV) and two SDD energy-dispersive X-ray (EDX) detectors. High-resolution TEM (HRTEM) images were recorded with a JEM-ARM300F microscope (GrandARM, JEOL, 300 kV). The sample was prepared by depositing the material dispersed in ethanol on carbon foil supported on the Mo TEM grid. Data analysis was done by using ImageJ software; the distribution of the copper nanoparticles was described by Gaussian fittings based on the counting of 130–260 individuals. The copper nanoparticle size (dCu) is given as a surface-weighted mean diameter defined as:  , where ni is the number of particles of diameter di. Data reproducibility from the characterisation analyses was better than ±10%.
, where ni is the number of particles of diameter di. Data reproducibility from the characterisation analyses was better than ±10%.
Catalytic tests
The Phoenix reactor system unit (ThalesNano) was used for the catalytic tests. The gas phase carbon dioxide hydrogenation reactions were carried out in continuous mode inside a stainless-steel reactor (0.6 cm internal diameter) with a fixed-bed catalyst (250–500 μm fraction) layer reduced in situ (20 mL min−1 hydrogen, 2 °C min−1 up to 250 °C) prior to each reaction. The catalyst layer consisted of a homogeneous physical mixture of the active catalyst and inert SiC up to a total mass of 0.1 g; this allowed the conversion level to be controlled while keeping the contact time fixed (τ = 0.14 s). The reactants (pre-mixed 24.86 mol% carbon dioxide + 75.14 mol% hydrogen) were delivered with a calibrated mass flow controller at a fixed flow of 50 mL min−1. The reactions were performed at 200 °C and 30 bar; blank tests over the empty reactor, SiC fixed-bed and under pure hydrogen flow resulted in undetected carbon dioxide conversion and/or methanol production. The gaseous effluent was analyzed using an on-line micro-GC (Inficon 3000 Micro GC) equipped with a TCD. Isothermal conditions were maintained over molsieve (80 °C, argon carrier) and PlotU (120 °C, helium carrier) columns for the detection of non-polar and polar compounds, respectively. The catalysts performances were evaluated under steady-state conditions (i.e. fluctuations of carbon dioxide molar flow of ± 5% with respect to the average value) on the basis of (i) carbon dioxide conversion (XCO2, %), (ii) methanol selectivity (SMeOH, %) and (iii) methanol space time yield (STY, gMeOH gcat−1 h−1), i.e. catalyst performance indicators, see the ESI† for more details. Carbon mass balance was better than ±7%.Data availability
All the data relevant to the study have been included in the manuscript and as part of the ESI.† The raw data will be provided by the corresponding authors upon request.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge funding from the Paul Scherrer Institut.References
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CAS.
- A. Beck, M. A. Newton, L. G. A. van de Water and J. A. van Bokhoven, Chem. Rev., 2024, 124, 4543–4678 CrossRef CAS PubMed.
- D. Bianchi, T. Chafik, M. Khalfallah and S. J. Teichner, Appl. Catal., A, 1995, 123, 89–110 CAS.
- I. A. Fisher and A. T. Bell, J. Catal., 1998, 178, 153–173 CAS.
- M. D. Rhodes and A. T. Bell, J. Catal., 2005, 233, 198–209 CrossRef CAS.
- S. Dang, H. Yang, P. Gao, H. Wang, X. Li, W. Wei and Y. Sun, Catal. Today, 2019, 330, 61–75 CAS.
- J. Niu, H. Liu, Y. Jin, B. Fan, W. Qi and J. Ran, Int. J. Hydrogen Energy, 2022, 47(15), 9183–9200 CAS.
- S. Kuld, C. Conradsen, P. G. Moses, I. Chorkendorff and J. Sehested, Angew. Chem., 2014, 126, 6051–6055 Search PubMed.
- M. Ren, Y. Zhang, X. Wang and H. Qiu, Catalysts, 2022, 12, 403–436 CAS.
- M. M.-J. Li, Z. Zeng, F. Liao, X. Hong and S. C. E. Tsang, J. Catal., 2016, 343, 157–167 CAS.
- B. Ouyang, W. Tan and B. Liu, Catal. Commun., 2017, 95, 36–39 CAS.
- D. Ferrah, A. R. Haines, R. P. Galhenage, J. P. Bruce, A. D. Babore, A. Hunt, I. Waluyo and J. C. Hemminger, ACS Catal., 2019, 9, 6783–6802 CrossRef CAS.
- S. Tada, S. Kayamori, T. Honma, H. Kamei, A. Nariyuki, K. Kon, T. Toyao, K. Shimizu and S. Satokawa, ACS Catal., 2018, 8, 7809–7819 CAS.
- H. Chen, H. Cui, Y. Lv, P. Liu, F. Hao and W. Xiong, Fuel, 2022, 314, 123035 CAS.
- J. C. Frost, Nature, 1988, 334, 577–580 CAS.
- C. Tisseraud, C. Comminges, T. Belin, H. Ahouari, A. Soualah, Y. Pouilloux and A. Le Valant, J. Catal., 2015, 330, 533–544 CrossRef CAS.
- A. Beck, M. Zabilskiy, M. A. Newton, O. Safonova, M. G. Willinger and J. A. van Bokhoven, Nat. Catal., 2021, 4, 488–497 CrossRef CAS.
- A. Urakawa, Nat. Catal., 2021, 4, 447–448 Search PubMed.
- N. J. Divins, D. Kordus, J. Timoshenko, I. Sinev, I. Zegkinoglou, A. Bergmann, S. W. Chee, S. Widrinna, O. Karslıoğlu and H. Mistry, Nat. Commun., 2021, 12, 1435 CrossRef CAS PubMed.
- P. Amann, B. Klötzer, D. Degerman, N. Köpfle, T. Götsch, P. Lömker, C. Rameshan, K. Ploner, D. Bikaljevic and H.-Y. Wang, Science, 2022, 376, 603–608 CrossRef CAS PubMed.
- J. Nakamura, Y. Choi and T. Fujitani, Top. Catal., 2003, 22, 277–285 CrossRef CAS.
- T. Fujitani, J. Jpn. Pet. Inst., 2020, 63, 43–51 CrossRef CAS.
- S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjær, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2016, 352, 969–974 CrossRef CAS PubMed.
- Y.-F. Shi, P.-L. Kang, C. Shang and Z.-P. Liu, J. Am. Chem. Soc., 2022, 144, 13401–13414 CrossRef CAS PubMed.
- X. Liu, J. Luo, H. Wang, L. Huang, S. Wang, S. Li, Z. Sun, F. Sun, Z. Jiang and S. Wei, Angew. Chem., Int. Ed., 2022, 61, e202202330 (1–10) Search PubMed.
- Z. Li, T. Du, Y. Li, H. Jia, Y. Wang, Y. Song and X. Fang, Fuel, 2022, 322, 124264 CrossRef CAS.
- J. Guo, Z. Luo, G. Hu and Z. Wang, Greenhouse Gases:Sci. Technol., 2021, 11, 1171–1179 CrossRef CAS.
- J. Wang, G. Li, Z. Li, C. Tang, Z. Feng, H. An, H. Liu, T. Liu and C. Li, Sci. Adv., 2017, 3, e1701290 (1–10) Search PubMed.
- D. Kordus, S. Widrinna, J. Timoshenko, M. Lopez Luna, C. Rettenmaier, S. W. Chee, E. Ortega, O. Karslioglu, S. Kühl and B. Roldan Cuenya, J. Am. Chem. Soc., 2024, 146, 8677–8687 CrossRef CAS PubMed.
- C. Zhang, L. Wang, Y. Chen, X. He, Y. Song, O. M. Gazit and Z. Zhong, Chem. Eng. J., 2023, 475, 146102 CrossRef CAS.
- J. Wang, T. Wang, Z. Zhao, R. Wang, C. Wang, F. Zhou, S. Li, L. Zhao and M. Feng, J. Alloys Compd., 2022, 902, 163865 CrossRef CAS.
- M. Zhou, J. Chen, Y. Zhang, M. Jiang, S. Xu, Q. Liang and Z. Li, J. Alloys Compd., 2020, 817, 152796 CrossRef CAS.
- S. Carlotto, Appl. Surf. Sci., 2022, 602, 154376 CrossRef CAS.
- Y. Lu, A. Ma, Y. Yu, R. Tan, C. Liu, P. Zhang, D. Liu and J. Gui, ACS Sustainable Chem. Eng., 2018, 7, 2906–2910 CrossRef.
- E. A. Rüdiger, Phys. Chem. Chem. Phys., 2021, 13, 368–384 Search PubMed.
- Z. Su, F. Fang, X. Li, W. Han, X. Liu and K. Chang, J. Colloid Interface Sci., 2022, 626, 662–673 CrossRef CAS PubMed.
- Y. Zhu, L. Zhang, B. Zhao, H. Chen, X. Liu, R. Zhao, X. Wang, J. Liu, Y. Chen and M. Liu, Adv. Funct. Mater., 2019, 29, 1901783 (1–12) Search PubMed.
- X. Liu, H. Xie, X. Qu, K. Yu, H. Yin, Q. Song and Z. Ning, J. Solid State Chem., 2021, 302, 122387 CrossRef CAS.
- H. Trabelsi, M. Bejar, E. Dhahri, M. Sajieddine, K. Khirouni, P. R. Prezas, B. M. G. Melo, M. A. Valente and M. P. F. Graça, J. Alloys Compd., 2017, 723, 894–903 CrossRef CAS.
- M. Hus, D. Kopac and B. Likozar, ACS Catal., 2018, 9, 105–116 Search PubMed.
- Y. Liu, X. Wang, Z. Wang, C. Chen, J. Song, L. Zhang, W. Bao, B. Sun, L. Wang and D. Liu, ACS Catal., 2024, 14, 12610–12622 CAS.
- C. D. Wagner, A. V. Naumkin, A. Kraut-Vass, J. W. Allison, C. J. Powell and J. R. Rumble Jr, The NIST X-Ray Photoelectron Spectroscopy (XPS) Database, US Department of Commerce, 1991 Search PubMed.
- L. Martínez, K. Lauwaet, G. Santoro, J. M. Sobrado, R. J. Peláez, V. J. Herrero, I. Tanarro, G. J. Ellis, J. Cernicharo, C. Joblin, Y. Huttel and J. A. Martín-Gago, Sci. Rep., 2018, 8, 7250 Search PubMed.
- Y. Wang, Y. Lü, W. Zhan, Z. Xie, Q. Kuang and L. Zheng, J. Mater. Chem. A, 2015, 3, 12796–12803 RSC.
- K. L. Chavez and D. W. Hess, J. Electrochem. Soc., 2001, 148, G640 (1–5) Search PubMed.
- Y. Tang, H. Shen, J. Cheng, Z. Liang, C. Qu, H. Tabassum and R. Zou, Adv. Funct. Mater., 2020, 30, 1908223 (1–9) Search PubMed.
- J. Yang, S. Hu, Y. Fang, S. Hoang, L. Li, W. Yang, Z. Liang, J. Wu, J. Hu and W. Xiao, ACS Catal., 2019, 9, 9751–9763 CAS.
- J. Stoch and J. Gablankowska-Kukucz, Surf. Interface Anal., 1991, 17, 165–167 CAS.
- N. A. Merino, B. P. Barbero, P. Eloy and L. E. Cadús, Appl. Surf. Sci., 2006, 253, 1489–1493 CAS.
- K. Aravinthkumar, E. Praveen, A. J. R. Mary and C. R. Mohan, Inorg. Chem. Commun., 2022, 140, 109451 CAS.
- M. C. Biesinger, L. W. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CAS.
- X. Peng, J. Zheng, Y. Zhang and Z. Wang, Mater. Lett., 2022, 309, 131317 (1–4) Search PubMed.
- W. Y. Hernández, M. N. Tsampas, C. Zhao, A. Boreave, F. Bosselet and P. Vernoux, Catal. Today, 2015, 258, 525–534 Search PubMed.
- T. J. Frankcombe and Y. Liu, Chem. Mater., 2023, 35, 5468–5474 CAS.
- H. Hou, M. Shang, F. Gao, L. Wang, Q. Liu, J. Zheng, Z. Yang and W. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 20128–20137 CAS.
- E. Mańkowska, M. Mazur, J. Domaradzki, P. Mazur, M. Kot and J. I. Flege, Sensors, 2023, 23, 3822–3835 CrossRef PubMed.
- M. S. P. Francisco, V. R. Mastelaro, P. A. P. Nascente and A. O. Florentino, J. Phys. Chem. B, 2001, 105, 10515–10522 CrossRef CAS.
- P. A. W. Van der Heide, Q. D. Jiang, Y. S. Kim and J. W. Rabalais, Surf. Sci., 2001, 473, 59–70 CrossRef CAS.
- B. Ura, J. Trawczyński, A. Kotarba, W. Bieniasz, M. J. Illán-Gómez, A. Bueno-López and F. E. Lopez-Suarez, Appl. Catal., B, 2011, 101, 169–175 CrossRef CAS.
- G. Bergeret and P. Gallezot, Handbook of Heretogeneous Catalysis, Wiley-VCH, 2008, vol. 2, pp. 738–765 Search PubMed.
- Lawrence Berkeley National Laboratory Database, https://xdb.lbl.gov/Section1/Table_1-2.pdf, (accessed July 5, 2024).
- R. Singh, K. Tripathi and K. K. Pant, Fuel, 2021, 303, 121289 (1–10) Search PubMed.
- W. Wang, Z. Qu, L. Song and Q. Fu, J. Energy Chem., 2020, 40, 22–30 CrossRef.
- A. Cao, Z. Wang, H. Li and J. K. Nørskov, ACS Catal., 2021, 11, 1780–1786 CrossRef CAS.
- W. Wang, K. Huo, Y. Wang, J. Xie, X. Sun, Y. He, M. Li, J. Liang, X. Gao, G. Yang, S. Lin, F. Cao, H. Jiang, M. Wu and N. Tsubaki, ACS Catal., 2024, 14, 9887–9900 CrossRef CAS.
- X. Chang, X. Han, Y. Pan, Z. Hao, J. Chen, M. Li, J. Lv and X. Ma, Ind. Eng. Chem. Res., 2022, 61, 6872–6883 CrossRef CAS.
- R. Prins, Chem. Rev., 2012, 112, 2714–2738 CrossRef CAS PubMed.
- K. Shun, K. Mori, S. Masuda, N. Hashimoto, Y. Hinuma, H. Kobayashi and H. Yamashita, Chem. Sci., 2022, 13, 8137–8147 RSC.
- A. Beck, D. Kazazis, Y. Ekinci, X. Li, E. A. Müller Gubler, A. Kleibert, M.-G. Willinger, L. Artiglia and J. A. van Bokhoven, ACS Nano, 2023, 17, 1091–1099 CrossRef CAS PubMed.
- A. Beck, H. Frey, M. Becker, L. Artiglia, M. G. Willinger and J. A. van Bokhoven, J. Phys. Chem. C, 2021, 125, 22531–22538 CrossRef CAS.
- W. Karim, C. Spreafico, A. Kleibert, J. Gobrecht, J. VandeVondele, Y. Ekinci and J. A. van Bokhoven, Nature, 2017, 541, 68–71 CrossRef CAS PubMed.
- N. Jardón-Maximino, M. Pérez-Alvarez, R. Sierra-Ávila, C. A. Ávila-Orta, E. Jiménez-Regalado, A. M. Bello, P. González-Morones and G. Cadenas-Pliego, J. Nanomater., 2018, 2018, 9512768 Search PubMed.
- A. Mroziński, S. Molin, P. Błaszczak, T. Miruszewski, K. Górnicka, J. Karczewski and P. Jasiński, Int. J. Hydrogen Energy, 2023, 48, 35250–35266 CrossRef.
- W. Si, Y. Wang, S. Zhao, F. Hu and J. Li, Environ. Sci. Technol., 2016, 50, 4572–4578 CAS.
- D. A. Muller, N. Nakagawa, A. Ohtomo, J. L. Grazul and H. Y. Hwang, Nature, 2004, 430, 657–661 CAS.
- R. Astala and P. D. Bristowe, Modell. Simul. Mater. Sci. Eng., 2001, 9, 415–422 CAS.
- L. Liu, F. Gao, H. Zhao and Y. Li, Appl. Catal., B, 2013, 134, 349–358 Search PubMed.
- S. Hoang, S. P. Berglund, N. T. Hahn, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2012, 134, 3659–3662 CAS.
- A. Staykov, H. Téllez, T. Akbay, J. Druce, T. Ishihara and J. Kilner, Chem. Mater., 2015, 27, 8273–8281 CAS.
- C. Zhang, L. Wang, U. J. Etim, Y. Song, O. M. Gazit and Z. Zhong, J. Catal., 2022, 413, 284–296 CrossRef CAS.
- D. Kopač, B. Likozar and M. Huš, ACS Catal., 2020, 10, 4092–4102 Search PubMed.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional details on the catalyst performance indicators (i.e. definition and calculations), experimental details and materials characterisation data (H2-TPR, O2-TPO, XPS, XRD, STEM), including tables with XPS fitting parameters and data analysis (DOC). See DOI: https://doi.org/10.1039/d4cy01487a |
| This journal is © The Royal Society of Chemistry 2025 |