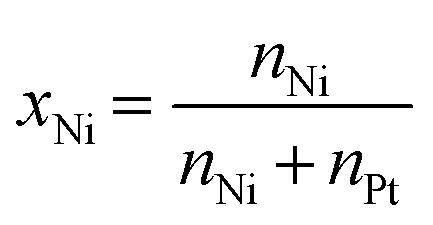NiPt catalysts for the synthesis of iso-butanol: the influence of molar ratio and total metal loading on activity and stability†
Received
21st January 2025
, Accepted 10th February 2025
First published on 11th February 2025
Abstract
If methanol and ethanol are produced regeneratively from green H2 and CO2, a mixture of these two can further react to form longer-chain branched alcohols such as iso-butanol, which can serve as sustainable feedstocks for the transportation and chemical sectors. NiPt catalysts have shown to be promising for this process. This study investigates the influence of the total metal loading Ni + Pt and the molar ratio of Ni to Pt on catalytic behavior. It was observed that the values for the total metal loading and the molar fraction of Pt must be set correctly and precisely during synthesis, as most of all the space–time yield of the catalyst, the conversion of ethanol, and the iso-butanol yield strongly depend on them. Differing mechanisms for the decomposition of acetaldehyde on pure Pt/C and NiPt/C are comparatively discussed. They explain the beneficial effect of Ni in avoiding the formation of carbonaceous deposits on the active centers. In addition, Ni was found to prevent particle growth due to its strong metal–support interaction.
Introduction
The catalytic valorization of blends of green ethanol and green methanol can yield branched higher alcohols. Fig. 1 illustrates how iso-butanol is formed in accordance with the Guerbet reaction, for which acetaldehyde and formaldehyde are initially generated by dehydrogenating the alcohols. The aldehydes then react in the presence of a base and with the abstraction of an H-atom in the α-position to the carbonyl group of the acetaldehyde to form acrolein. Acrolein is then re-hydrogenated on the catalyst, and 1-propanol is formed. This reaction principle is repeated in a further methylation step and iso-butanol is generated. Sustainably-produced iso-butanol can serve as a component in gasoline, thus significantly reducing CO2 emissions in the transport sector.1 In this respect, Häusler et al.2,3 and Pasel et al.4–6 focused on monometallic and, most of all, bimetallic catalysts supported on activated carbon, as the combination of two metals presents the opportunity to optimize the geometric and electronic characteristics of the generated alloy and to create a synergistic interplay between the metals, thus increasing the catalytic activity and stability.7–11 The synthesis of iso-butanol from mixtures of ethanol and methanol has not yet been studied very intensively in the literature. There are two patents based on basic supports, such as MgO, ZrO2 or CeO2, which in turn are doped with various metals or alloys.12,13 For example, its inventors describe a PtNi/MgO sample with 75% selectivity towards iso-butanol and 65% conversion of ethanol. Other patents are based on large-pore mordenites that have been exchanged with precious metals or alkali metals, or on MnZrZn oxides loaded with Pt and Pd.14,15 Moreover, hydrotalcites and hydroxyapatites are used for this reaction in the patent literature.16,17 Carlini et al.18–20 investigated Cu-chromite samples and hydrotalcites with differing MgAl compositions and they are doped with Cu. They worked at temperatures between 200 °C and 300 °C and reached 100% selectivity towards iso-butanol. Siddiki et al.21 not only investigated the methylation of alcohols but also expanded their area of investigation to ketones and indoles using a Pt/C catalyst. Gupta et al.22 used Ir complexes based on a phosphaalkene ligand showing ethanol conversions higher than 60% and selectivities towards iso-butanol higher than 90%. Wingad et al.23 investigated different Ru diphosphine complexes. 99.8% selectivity towards the desired Guerbet product iso-butanol and values of more than 75% ethanol conversion were achieved at a reaction temperature of 180 °C and after approximately 20 hours of time on stream. Sama et al.24 also worked with Ru diphosphine complexes, which were functionalized by various amines. The most active catalyst with diamine ethylene functionalities showed an iso-butanol yield of 74% at 78% ethanol conversion.
 |
| | Fig. 1 Mechanistic course of the formation of iso-butanol via the Guerbet reaction.25 | |
In this study, the influence of the molar ratio of Ni to Pt (xNi/xPt) and the total metal loading of Ni + Pt of activated carbon supported NiPt catalysts on their catalytic behavior during iso-butanol synthesis from mixtures of ethanol and methanol was investigated. For this purpose, fourteen precipitation catalysts were synthesized and divided into four groups. The synthesis of the catalysts in each of these aimed at setting the same molar ratio xNi/xPt. These amounted to 99, 19, 9, and 4, respectively, and were defined by the mass fractions of the metals in the respective precursor solutions. To investigate the influence of loading, summary mass fractions for Ni + Pt of 5%, 10%, 20%, and 30% were applied within each group (or only 10%, 20%, and 30% in some cases). First, the catalysts were characterized using ICP-OES, XRD, and H2-chemisorption. For the experimental evaluation, the parameters measured were iso-butanol concentration, ethanol conversion, selectivity towards iso-butanol, iso-butanol yield, and the space–time yield (STY) of the catalysts.
Experimental
Catalyst synthesis
The synthesis of the precipitation catalysts supported on activated carbon from Table 1 is described in detail in the paper by Pasel et al.,6 using the example of the Ni99Pt1 catalyst with 5 wt% total metal loading. The main steps are the precipitation of Ni(OH)3 on the activated carbon with subsequent impregnation with [Pt(NH3)4](NO3)2 and the calcination of the catalyst precursors with a subsequent reduction with H2.
Table 1 Results from ICP-OES analyses for the different precipitation catalysts
| Group number desired ratio |
Catalyst nominal |
wt(Ni + Pt) [%] |
x
Ni [%] |
x
Pt [%] |
x
Ni/xPt [−] |
Catalyst actual |
| I |
5 wt% Ni99Pt1 |
3.6 |
99.55 |
0.45 |
220 |
3.6 wt% Ni99.5Pt0.5 |
| 10 wt% Ni99Pt1 |
8.5 |
99.43 |
0.57 |
176 |
8.5 wt% Ni99.4Pt0.6 |
|
x
Ni/xPt = 99 |
20 wt% Ni99Pt1 |
18.8 |
99.39 |
0.61 |
164 |
18.8 wt% Ni99.4Pt0.6 |
| 30 wt% Ni99Pt1 |
7.6 |
98.48 |
1.52 |
65 |
7.6 wt% Ni98.5Pt1.5 |
| 5 wt%% Ni95Pt5 |
3.3 |
97.75 |
2.25 |
43 |
3.3 wt% Ni97.8Pt2.2 |
| II |
10 wt% Ni95Pt5 |
6.0 |
96.13 |
3.87 |
25 |
6.0 wt% Ni96.1Pt3.9 |
|
x
Ni/xPt = 19 |
20 wt% Ni95Pt5 |
12.7 |
94.85 |
5.15 |
18 |
12.7 wt% Ni94.9Pt5.1 |
| 30 wt% Ni95Pt5 |
24.3 |
95.91 |
4.09 |
23 |
24.3 wt% Ni95.9Pt4.1 |
| 10 wt% Ni90Pt10 |
7.4 |
95.14 |
4.86 |
20 |
7.4 wt% Ni95.1Pt4.9 |
| III |
20 wt% Ni90Pt10 |
14.5 |
94.46 |
5.54 |
17 |
14.5 wt% Ni94.5Pt5.5 |
|
x
Ni/xPt = 9 |
30 wt% Ni90Pt10 |
19.5 |
92.97 |
7.03 |
13 |
19.5 wt% Ni93.0Pt7.0 |
| 10 wt% Ni80Pt20 |
6.2 |
88.61 |
11.39 |
8 |
6.2 wt% Ni88.6Pt11.4 |
| IV |
20 wt% Ni80Pt20 |
13.0 |
89.77 |
10.23 |
9 |
13.0 wt% Ni89.8Pt10.2 |
|
x
Ni/xPt = 4 |
30 wt% Ni80Pt20 |
21.9 |
86.63 |
13.37 |
6 |
21.9 wt% Ni86.6Pt13.4 |
The calculation of the molar quantities of Ni and Pt (nNi and nPt, respectively) required for the catalysts from Table 1 was determined using the following equations. According to eqn (1), the total catalyst mass to be synthesized is defined as the sum of the masses of Pt and Ni as active components and C as a support. Eqn (2) and (3) establish a relationship between the desired molar fractions for Ni and Pt (xNi and xPt, respectively) and the respective molar quantities of Ni and Pt required for this in the precursor solutions. If eqn (1)–(3) are suitably combined with each other, eqn (4) and (5) can be used to calculate the required molar quantities of Ni and Pt for a specific mass of the catalyst with the defined total metal loadings and molar fractions of Ni and Pt, respectively. The abbreviation ‘TML’ in these equations stands for the ‘total metal loading’ percentage. Finally, it is necessary to relate these calculated molar quantities to the two precursor solutions, Ni(NO3)2·6H2O and [Pt(NH3)4](NO3)2, in order to calculate the required masses in these two solutions.
| | | mcat,total = mM + mC ⇔ mcat,total = mPt + mPt + mC | (1) |
| |  | (2) |
| |  | (3) |
| | 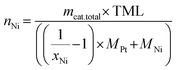 | (4) |
| |  | (5) |
Catalyst characterization
The catalyst characterization experiments were conducted as described by Häusler et al. and Pasel et al.2,5 These utilized X-ray diffraction (XRD) analysis, inductively coupled plasma combined with optical emission spectroscopy (ICP-OES), and the temperature programmed desorption of H2 (H2-TPD).
Carrying out the activity tests
The procedure for carrying out the catalytic experiments is described in detail by Pasel et al.6
Ethanol conversion was calculated using eqn (6). In this equation, cK represents the concentrations of the different products of the Guerbet reaction, while Z stands for their stoichiometric coefficients. Specifically, the stoichiometric coefficient for the formation of iso-butanol was one, whereas those for the generation of 1-butanol and 1-hexanol were two and three, respectively.
| |  | (6) |
For the yield
Yi and the selectivity
Si of a specific component “
i” as well as the space–time yield STY of the respective catalysts, the following equations were formulated:
| |  | (7) |
| |  | (8) |
| |  | (9) |
Results and discussion
Catalyst characterization
Table 1 shows the results of the ICP-OES analyses of the various precipitation catalysts from the four groups mentioned. In the “Catalyst nominal” column, for example, the abbreviation “10% Ni95Pt5@C” means that the synthesis of this catalyst aimed at a total metal loading of 10 wt% Ni + Pt, with a molar ratio of Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt of 95:5, whilst the “Catalyst actual” column summarizes the respective experimentally measured values. It is noticeable in this table that, for each individual catalyst, the total metal loading Ni + Pt measured by ICP-OES was lower than the nominal value targeted during the synthesis. The discrepancies in the metal loadings were further investigated by determining the metal losses for Ni in the individual synthesis steps of the catalyst. For this purpose, aliquots of the precipitation solutions, filtrates, and wash solutions were analyzed using ICP-OES and the Ni losses in the individual test steps were quantified. These analyses showed that a maximum of 7% of the Ni mass was lost in filtration and washing steps. As some of the Ni remained dissolved during precipitation and some of the Ni(OH)3 returned to the solution when the filter cake was washed, a slightly lower loading can be expected. The larger observed discrepancies, however, cannot be explained in this way. It is also apparent that in almost any case, the molar ratios xNi/xPt, which were measured by means of ICP-OES, were higher than the desired values targeted during the synthesis. This means that the Pt particles were deposited on the activated carbon support to a lesser degree than was the case with the Ni particles. It can be concluded that the impregnation step with an aqueous solution of [Pt(NH3)4](NO3)2, followed by calcination and reduction as described in the Experimental section, is highly critical. Nevertheless, the desired trends regarding the total metal loading Ni + Pt and the molar ratio xNi/xPt aimed for in the synthesis of the catalysts from this table could be retrieved in the ICP-OES results, so that the basic approach of this study was confirmed.
:Pt of 95:5, whilst the “Catalyst actual” column summarizes the respective experimentally measured values. It is noticeable in this table that, for each individual catalyst, the total metal loading Ni + Pt measured by ICP-OES was lower than the nominal value targeted during the synthesis. The discrepancies in the metal loadings were further investigated by determining the metal losses for Ni in the individual synthesis steps of the catalyst. For this purpose, aliquots of the precipitation solutions, filtrates, and wash solutions were analyzed using ICP-OES and the Ni losses in the individual test steps were quantified. These analyses showed that a maximum of 7% of the Ni mass was lost in filtration and washing steps. As some of the Ni remained dissolved during precipitation and some of the Ni(OH)3 returned to the solution when the filter cake was washed, a slightly lower loading can be expected. The larger observed discrepancies, however, cannot be explained in this way. It is also apparent that in almost any case, the molar ratios xNi/xPt, which were measured by means of ICP-OES, were higher than the desired values targeted during the synthesis. This means that the Pt particles were deposited on the activated carbon support to a lesser degree than was the case with the Ni particles. It can be concluded that the impregnation step with an aqueous solution of [Pt(NH3)4](NO3)2, followed by calcination and reduction as described in the Experimental section, is highly critical. Nevertheless, the desired trends regarding the total metal loading Ni + Pt and the molar ratio xNi/xPt aimed for in the synthesis of the catalysts from this table could be retrieved in the ICP-OES results, so that the basic approach of this study was confirmed.
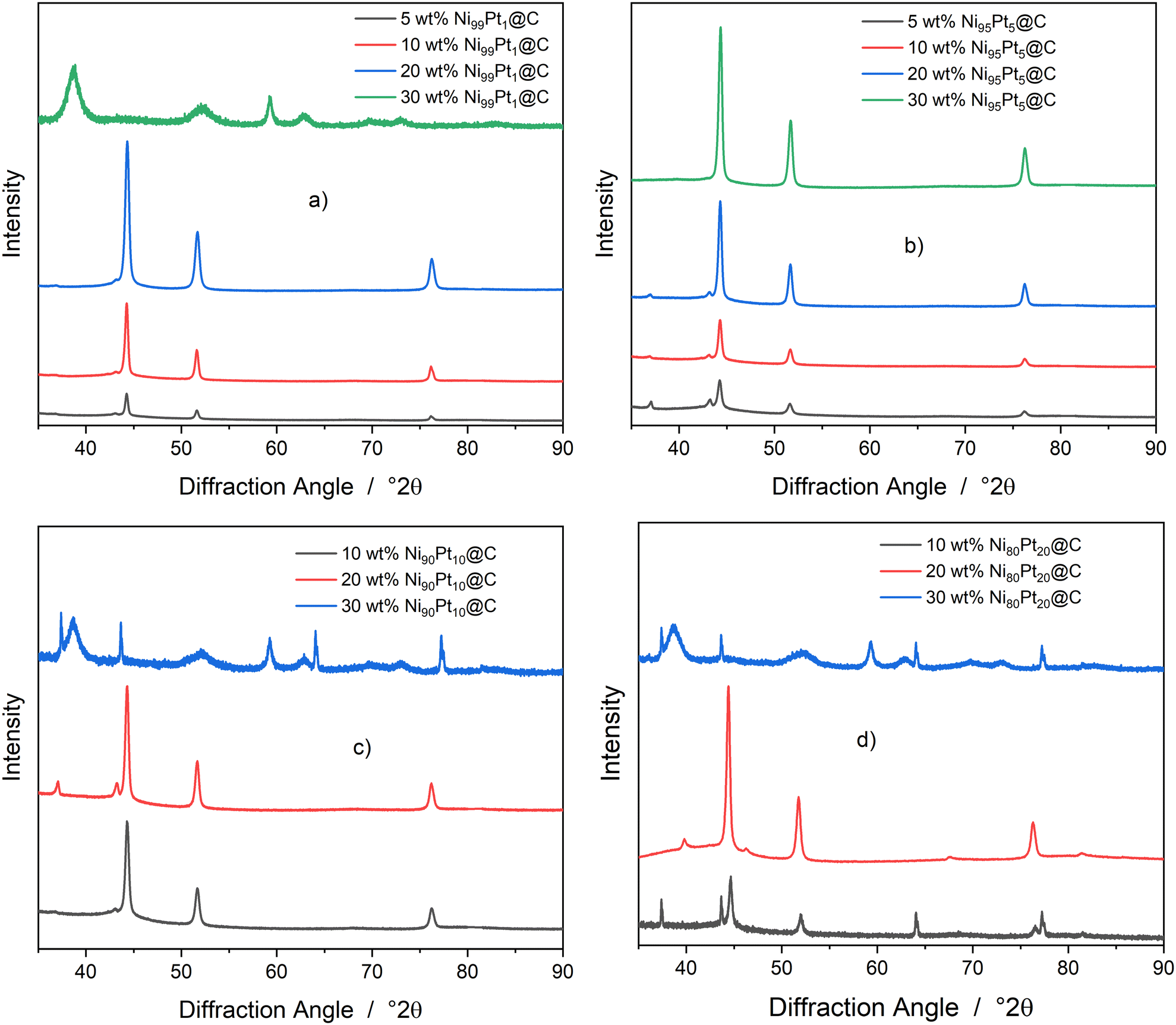
Fig. 2 shows the diffractograms of the calcined and reduced catalysts with differing total metal loadings Ni + Pt and various molar ratios xNi/xPt. To simplify and standardize the descriptions and discussions in this section, the nominal catalyst designations are used. Thereby, Fig. 2a displays the diffractograms for the catalysts of group I with a molar ratio of xNi/xPt 99:1. For the catalysts with 5 wt%, 10 wt%, and 20 wt%, reflections for NiPt occurred at 44°, 51°, and 76°, whereas at 43° a weak reflection for a cubic NiO phase can be seen. For the 30 wt% Ni99Pt1@C catalyst, a strongly deviating diffractogram was obtained. The broad reflexes can be assigned to the theophrastite phase and thus be identified as Ni(OH)2. The Rietveld analysis resulted in a pure phase for Ni(OH)2 with isotropic crystalline domains. They are platelet-shaped crystals with dimensions of 60.6 nm × 60.6 nm × 3.425 nm in the directions (100), (010), and (001). It can be assumed that for the 30 wt% Ni99Pt1@C catalyst, the calcination process was defective. For the other catalyst materials in this group, the intensities for the reflexes increased with increasing total metal loading. This increase was valid for the signal intensity but not for the half-width or position of the reflexes, which remained constant within group I. This qualitative observation is in good agreement with the quantitative measurement of the crystallite sizes obtained from the phase analysis being displayed in Table 2. For the catalysts of group I, only the NiPt and NiO phases were fitted. Due to the small signal intensity, however, the phase fraction of the NiO phase can only be specified with great uncertainty and is, therefore, not listed in Table 2 as it represented less than one weight percent of the crystalline portion. The crystallite sizes for the NiPt phase were in the range from 34 nm to 21 nm, whereby the largest crystalline domains were found for the 5 wt% Ni99Pt1@C catalyst. The sizes of these domains decreased with increasing total metal loading. As the NiO phase only had very low signal intensities, the resulting crystallite size could not be reliably determined. The possible reflections for a Pt phase at positions 39.79°, 46.28°, 67.53°, 81.34°, and 85.80° did not occur.
 |
| | Fig. 2 Display of the X-ray diffraction patterns for the NiPt@C catalyst with differing total metal loadings and various molar ratios xNi/xPt, 2θ = 10° to 90°, and Cu Kα-radiation (λ = 1.5419 Å), step width = 0.02 deg s−1. a) Ni99Pt1. b) Ni95Pt5. c) Ni90Pt10. d) Ni80Pt20. | |
Table 2 Results of the Rietveld analysis of the differing precipitation catalysts, phase fractions with less than half a percent were not listed. w(phase) refers to the mass fraction of the crystalline material
| Catalyst nominal |
wt(NiPt) [%] |
d(NiPt) [nm] |
w(Pt) [%] |
d(Pt) [nm] |
w(NiO) [%] |
d(NiO) [nm] |
| 5% Ni99Pt1 |
95.0 ± 3.3 |
33.88 ± 0.13 |
— |
— |
— |
— |
| 10% Ni99Pt1 |
100.0 ± 7.1 |
30.53 ± 0.10 |
— |
— |
— |
— |
| 20% Ni99Pt1 |
99.23 ± 0.13 |
21.35 ± 0.07 |
— |
— |
— |
— |
| 30% Ni99Pt1 |
— |
— |
— |
— |
— |
— |
| 5% Ni95Pt5 |
99.0 ± 2.5 |
26.7 ± 1.0 |
— |
— |
1.0 ± 2.5 |
26.25 ± 0.48 |
| 10% Ni95Pt5 |
98.5 ± 6.4 |
23.88 ± 0.42 |
— |
— |
1.5 ± 6.4 |
29.42 ± 0.61 |
| 20% Ni95Pt5 |
98.2 ± 1.1 |
26.93 ± 0.07 |
1.3 |
3.98 |
0.5 ± 1.1 |
16.64 ± 0.42 |
| 30% Ni95Pt5 |
98.96 ± 0.03 |
25.48 ± 0.05 |
1.04 ± 0.03 |
3.97 |
— |
— |
| 10% Ni90Pt10 |
98.78 |
23.78 ± 0.07 |
— |
— |
1.21 ± 0.01 |
39.7 ± 5.7 |
| 20% Ni90Pt10 |
99.30 ± 0.40 |
25.90 ± 0.09 |
— |
— |
— |
— |
| 30% Ni90Pt10 |
— |
— |
— |
— |
— |
— |
| 10% Ni80Pt20 |
98.06 ± 0.87 |
19.32 |
— |
— |
10.17 ± 0.87 |
134 ± 10 |
| 20% Ni80Pt20 |
92.6 ± 0.4 |
30.79 |
1.96 ± 0.06 |
31.83 |
5.4 ± 0.4 |
125 ± 10 |
| 30% Ni80Pt20 |
— |
— |
— |
— |
— |
— |
Fig. 2b shows the diffractograms for the catalysts of group II with a molar ratio xNi/xPt of 95:5. They differed from the group I diffractograms already described in such a way that now the reflections of the NiO phase were clearly visible at 37° and 43° for the catalysts with total metal loadings of 5 wt%, 10 wt%, and 20 wt%, respectively. The Rietveld analyses showed that the corresponding phase fractions amounted to approximately one percent by weight. The crystallite sizes for the NiO phase were in the range from 17 nm to 26 nm. NiO was not found for the catalyst with the highest metal loading of 30 wt%. The fraction for the NiPt phase was almost 100% for all four catalysts, and their crystallite sizes were closely distributed around 26 nm. Reflections for the Pt phase could not be found. Rietveld refinement, however, resulted in a Pt phase with a mass fraction of approximately one percent of the crystalline material in the case of the catalysts with 20 wt% and 30 wt% total metal loading. Due to the low proportion and the amorphous activated carbon background, the latter results are uncertain. In group II, the catalyst with the targeted 30 wt% total metal loading also revealed the NiPt reflexes. The theophrasite phase was fully converted into NiO, which in turn was changed to Ni in the subsequent reduction with H2.
Fig. 2c depicts the diffractograms for the catalysts of group III. Just as in the cases of groups I and II, the NiPt phases again occurred at 44°, 51°, and 76° for the samples with 10 wt% and 20 wt% total metal loading. For the latter catalyst, the reflections for the NiO phase were stronger than before. In the case of the catalyst with 30 wt% metal loading, the theophrastite phase was again the main phase. At 37°, 44°, 64°, and 77°, the reflections of an undetermined NiO phase were observed. The reflections are atypical because they are of low intensity and yet are sharp and symmetrical. It cannot be ruled out that they are caused by the measurement itself due to false scattering. As before, no pure Pt phase was recognizable in these catalysts either. The crystallite sizes for the NiPt phase were in the range between 23.78 nm and 25.90 nm with a phase fraction close to 100%.
Finally, Fig. 2d presents the diffractograms for the catalysts of group IV. Again, reflections for the NiPt and NiO phases, and, in the case of the catalyst with a 30 wt% total metal loading (the theophrastite phase) were observed. For the 20 wt% Ni80Pt20@C catalyst, a Pt phase could be detected in the diffractogram for the first time. The Pt reflections occurred at the positions 40°, 46°, 68°, and 81°. The Pt mass proportion of 1.96% calculated via the Rietveld analysis was made up of the amount of Pt in the NiPt phase and the Pt phase itself. In the literature, segregation into the disordered Ni1−xPtx phase and the Ni3Pt phase is only described for alloys of Ni and Pt with a Pt mass fraction of 23% or more.26 The reason for the detected Pt phase in Fig. 2d may lie in an additional segregating effect of the activated carbon support. In addition, if the homogenization during the synthesis procedure is insufficient, locally high concentrations of the Pt precursor might occur, which in turn form the Pt phase.
In summary, it can be stated that the crystallite sizes of the Ni particles for all catalysts, irrespective of their total metal loading or molar ratio xNi/xPt, lay in the range from 19 nm to 34 nm. The smallest crystallite size was found for the 10 wt% Ni80Pt20@C catalyst with 19.32 nm. In the group of Ni95Pt5@C catalysts, the crystallite sizes were around 26 nm, regardless of the loading. It was assumed that the loading of the catalysts might influence the crystallite size in such a way that increasing metal loading might lead to larger metal particles and thereby also to larger crystal domains. This influence cannot be proven based on the measured data. The catalysts in group I even show the opposite trend. Here, as the loading increased, the crystal sizes decreased. Regardless of the metal loading and molar ratio, the preparation method led to almost quantitatively reduced catalysts with a low proportion of NiO. None of the catalyst materials exhibited considerable proportions of Pt as a differentiated Pt phase, which indicates that in all cases, a NiPt alloy was produced that did not segregate under the conditions used.
To investigate the catalytically active surface, chemisorption experiments were carried out with all catalysts. For this purpose, the catalyst precursors were first calcined and reduced in the chemisorption apparatus under the conditions outlined in the Experimental section. By means of a rinsing phase with Ar at 35 °C, physisorbed H2 from the reduction process was removed to clean the surface. Subsequently, at 35 °C 20 pulses of pure H2 were pulsed onto the catalyst surfaces and the adsorbed quantities of H2 were calculated. Immediately afterwards, temperature-programmed desorption was carried out to determine the quantities of desorbed H2. The results of these experiments are listed in Table 3 and illustrated in Fig. 3. As this figure illustrates the trends for adsorption and desorption based on the actual metal loadings Ni + Pt and the actual molar fractions of Pt, the actual catalyst designations will be used in the following discussion. It is noticeable that in most cases, the quantities of desorbed H2 were higher than those of H2 that had previously been adsorbed on the respective catalysts. This can be explained by the fact that chemically bound H2 was released from the carbon support during the temperature-programmed desorption at high temperatures (up to 800 °C). It is also clear that the adsorbed quantities of H2 increased almost continuously when the total metal loading and the molar fraction of Pt increased. Exceptions were the catalysts 24.3 wt% Ni95.9Pt4.1 and 21.9 wt% Ni86.6Pt13.4, which showed low values for the molar quantities of adsorbed H2. The maximum molar quantity of 308 μm g−1 was found in the case of the 13 wt% Ni89.8Pt10.2 sample. This can be explained by the fact that H2 was preferentially adsorbed on Pt active sites, as the adsorption energies for H2 are higher on Pt than on Ni. Chemisorption with H2 can therefore be used to determine the number of Pt active sites directly. As a result, the 13 wt% Ni89.8Pt10.2 catalyst revealed the highest number of Pt centers on its surface, whereas the Ni99Pt1 catalysts from group 1 only had a comparably small number of Pt active sites. The Ni95Pt5 particles synthesized by Taniguchi et al.27 show that, depending on the synthesis method, Pt and Ni can form either a Pt core with an Ni shell or an Ni cube with Pt enrichment along the cube corners and edges. These results show that Ni and Pt are already subject to segregation and local enrichment, even at low mass fractions. These effects are mass fraction- and size-dependent.28–30 The described trends for the adsorbed and desorbed quantities of H2 are also observed in Fig. 3a and b. The figure once more illustrates the 13 wt% Ni89.8Pt10.2 catalyst as the sample that exhibited the largest quantities of H2 during pulse chemisorption and temperature-programmed desorption, respectively. All experimental data for these figures are summarized in Tables S1 and S2.†
Table 3 Results of the H2 adsorption and desorption experiments for the various precipitation catalysts with differing total metal loadings and molar ratios xNi/xPt
| Catalyst nominal |
Catalyst actual |
n
adsorption (H2) [μmol g−1] |
n
desorption (H2) [μmol g−1] |
| 5 wt% Ni99Pt1 |
3.6 wt% Ni99.5Pt0.5 |
3 |
37 |
| 10 wt% Ni99Pt1 |
8.5 wt% Ni99.4Pt0.6 |
0 |
84 |
| 20 wt% Ni99Pt1 |
18.8 wt% Ni99.4Pt0.6 |
6 |
52 |
| 30 wt% Ni99Pt1 |
7.6 wt% Ni98.5Pt1.5 |
9 |
3 |
| 5 wt% Ni95Pt5 |
3.3 wt% Ni97.8Pt2.2 |
2 |
71 |
| 10 wt% Ni95Pt5 |
6.0 wt% Ni96.1Pt3.9 |
17 |
53 |
| 20 wt% Ni95Pt5 |
12.7 wt% Ni94.9Pt5.1 |
65 |
103 |
| 30 wt% Ni95Pt5 |
24.3 wt% Ni95.9Pt4.1 |
24 |
50 |
| 10 wt% Ni90Pt10 |
7.4 wt% Ni95.1Pt4.9 |
39 |
64 |
| 20 wt% Ni90Pt10 |
14.5 wt% Ni94.5Pt5.5 |
42 |
40 |
| 30 wt% Ni90Pt10 |
19.5 wt% Ni93.0Pt7.0 |
163 |
0 |
| 10 wt% Ni80Pt20 |
6.2 wt% Ni88.6Pt11.4 |
49 |
44 |
| 20 wt% Ni80Pt20 |
13.0 wt% Ni89.8Pt10.2 |
308 |
300 |
| 30 wt% Ni80Pt20 |
21.9 wt% Ni86.6Pt13.4 |
5 |
4 |
 |
| | Fig. 3 Representation of the determined quantities of H2 as a function of the total metal loadings and the molar fraction of Pt from two differing measurement methods and for NixPt1−x@C catalysts. The measuring points are marked with black crosses. The data points in between were linearly interpolated. The values for the total metal loadings and the molar fraction of Pt were taken from the ICP-OES measurements. a) Molar quantity of adsorbed H2 on the precipitation catalysts, which was measured by means of pulse-chemisorption, ![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) Ar = 30 ml min−1, T = 35 °C, Δt = 20 min, 20 pulses of H2, Vpulse = 0.2056 ml, Cf = (1.67 ± 0.02) × 10−8 mmol mV−1 s−1. b) Molar quantity of desorbed H2 from the precipitation catalysts, which was measured by means of temperature-programmed desorption following prior pulse-chemisorption, ΔT from 35 °C to 800 °C, β = 10 k min−1, Ar = 30 ml min−1, Cf = (1.67 ± 0.02) × 10−8 mmol mV−1 s−1. Ar = 30 ml min−1, T = 35 °C, Δt = 20 min, 20 pulses of H2, Vpulse = 0.2056 ml, Cf = (1.67 ± 0.02) × 10−8 mmol mV−1 s−1. b) Molar quantity of desorbed H2 from the precipitation catalysts, which was measured by means of temperature-programmed desorption following prior pulse-chemisorption, ΔT from 35 °C to 800 °C, β = 10 k min−1, Ar = 30 ml min−1, Cf = (1.67 ± 0.02) × 10−8 mmol mV−1 s−1. | |
Catalytic experiments
Fig. 4 depicts the concentration profiles of iso-butanol as a function of time on stream and the total metal loading Ni + Pt for the catalysts from the different groups I to IV. For the representation of this figure, the actual values for the catalyst composition were chosen. The reactions were carried out at 165 °C and an initial ethanol concentration of 600 mmol l−1.
 |
| | Fig. 4 Concentrations of iso-butanol as a function of time on stream and total metal loading Ni + Pt for the catalysts from the different groups: a) group I; b) group II; c) group III; d) group IV; T = 165 °C, m(catalyst) = 250 mg, d(powder) < 75 μm, V(reactor) = 70 ml, c0(EtOH) = 600 mmol l−1, c(NaOH) = 450 mmol l−1, c(n-decane) = 15 mmol l−1, methanolic solution. | |
Fig. 4a depicts the concentration trends of iso-butanol for the catalysts from group I. The sample with an actual metal loading of 3.6 wt% reached a maximum final iso-butanol concentration of 31.01 mmol l−1. The iso-butanol concentration increased with a constant formation rate of (7.16 ± 0.41) mmol h l−1, which can be described very well linearly (R2 = 97.47%). In the case of the catalyst with an actual metal loading of 8.5 wt%, a final iso-butanol concentration after four hours of time on stream of 50.23 mmol l−1 with a constant formation rate of (11.93 ± 0.26) mmol h l−1 was observed, which represents the local maximum for this catalyst group. With further increases of the total metal loading (catalyst with 18.8 wt%), the formation rate decreased to (5.95 ± 0.41) mmol h l−1. It thus fell below the value of the catalyst, with 3.6 wt% total metal loading. The final iso-butanol concentration in this case was only 22.29 mmol l−1 and the concentration trend of iso-butanol can be mapped very well with a straight line with R2 = 96.72%. The catalyst with the actual composition 7.6 wt% Ni98.5Pt1.5@C revealed the lowest final iso-butanol concentration of 7.40 mmol l−1. Here, the formation rate for iso-butanol was also the lowest at (1.92 ± 0.11) mmol h l−1, although the increase in concentration was also linear (R2 = 96.72%). This low activity can be attributed to two points: firstly, the actual total metal loading was only 7.6 wt% and thus deviated strongly from the nominal 30 wt%. The second factor is the finding from the previous chapters that this catalyst was not sufficiently calcined and reduced. This means that less elemental active surface centers were available for the synthesis of iso-butanol.
For the catalysts from group II shown in Fig. 4b, the picture is like that of the catalysts from group I. The maximum iso-butanol concentration in this case was 74.44 mmol l−1 and was reached with the 6.0 wt% Ni96.1Pt3.9 catalyst. The formation rate amounted to (16.90 ± 0.21) mmol h l−1 with another linear increase (R2 = 99.89%). Even with the 3.3 wt% loaded catalyst, the formation rate at the end of the reaction after four hours on stream was (12.40 ± 0.26) mmol h l−1, yielding an iso-butanol concentration of 50.26 mmol l−1 (R2 = 99.70%). The final concentrations for the catalysts with the highest actual total metal loadings in this group of 12.7 wt% and 24.3 wt%, respectively, were lowest amongst the samples from group II. However, with both catalysts the formation of iso-butanol was also linear without deactivation phenomena.
The catalysts from groups III and IV in Fig. 4c and d showed significantly differing trends if compared with those of the samples from groups I and II. The iso-butanol concentrations achieved were not that strongly influenced by the total metal loading. For example, in group III, the 7.4 wt% Ni95.1Pt4.9 catalyst achieved a final concentration of 17.26 mmol l−1, whereas the 14.5 wt% Ni94.5Pt5.5 catalyst revealed an even lower final iso-butanol concentration of 14.00 mmol l−1. A similar behavior was found for the 6.2 wt% Ni88.6Pt11.4 and 13.0 wt% Ni89.8Pt10.2 catalysts from group IV. Although the final iso-butanol concentration increased significantly from 13.83 mmol l−1 to 23.99 mmol l−1 for these two samples, the concentrations achieved were significantly lower than those detected with the catalysts from groups I and II with comparable total metal loadings. The reason for the deviating catalytic behavior of the samples from groups III and IV lies in their rapid deactivation with increasing time on stream. In this respect, Fig. 5 shows exemplarily the time-dependent concentrations of the iso-butanol concentration for the 7.4 wt% Ni95.1Pt4.9, 6.2 wt% Ni88.6Pt11.4, and 13.0 wt% Ni89.8Pt10.2 catalysts from groups III and IV. It becomes obvious that for each catalyst, the values for the concentration of iso-butanol were almost constant after approximately two hours on stream, indicating a strong deactivation of the catalytically active centers. From the findings shown in Fig. 4, it can be concluded that an increasing molar fraction of Pt in the active NiPt phase favors deactivation. Häusler et al.3 reported a comparable deactivation of their pure Pt/C catalyst for the synthesis of iso-butanol from ethanol/methanol blends. All experimental data for these figures are summarized in Tables S3–S6.†
 |
| | Fig. 5 Iso-butanol concentrations as a function of time on stream for the 7.4 wt% Ni95.1Pt4.9, 6.2 wt% Ni88.6Pt11.4, and 13.0 wt% Ni89.8Pt10.2 samples, m(catalyst) = 250 mg, T = 165 °C, d(powder) < 75 μm, V(reactor) = 70 ml, c0(EtOH) = 600 mmol l−1, c(NaOH) = 450 mmol l−1, c(n-decane) = 15 mmol l−1; methanolic solution. | |
Several reasons for this Pt molar fraction-driven deactivation phenomenon are discussed in the literature. One explanation can be found in the formation of carbonaceous deposits on the Pt/C surface, which were observed by Pasel et al.,25 when for this work ethanol molecules were pulsed on the surface of a Pt/C catalyst under ultra-high vacuum conditions. It was concluded from the pulse response experiments that ethanol rapidly reacted to from acetaldehyde and H2, and that the detected carbonaceous deposits originated from the decomposition of intermediate acetaldehyde into H2, CH4, CO, and C. In this case, it was possible to remove the deposits by simply pulsing O2 molecules onto the active centers, yielding CO and CO2. It stands to reason that this aspect of carbonaceous deposits also applies to the deactivations found in Fig. 5. Panchenko et al.31 and Ezzhelenko et al.32 concluded from their experiments with Pt, Pd, Ir, Ru, and Rh on various supports (C, Al2O3, TiO2, CeO2, ZrO2) that strong metal–CO complexes were formed, which blocked the catalytically active sites and thus deactivated them. Theoretical DFT calculations support the results of the pulse response experiments33 and are consistent with the observations of Ezzhelenko et al.32 All experimental data for this figure are summarized in Table S7.† The top part of Fig. 6 summarizes these findings and explanations with a scheme, showing a possible reaction mechanism for the decomposition of acetaldehyde on a pure Pt/C catalyst. An additional explanation for the observed deactivation phenomenon lies in the agglomeration of the Pt particles on the activated carbon support, for which Fig. 7 gives evidence. It shows high-angle annular dark-field (HAADF)-TEM images of a fresh Pt/C catalyst (Fig. 7a) and of a used Pt/C sample after iso-butanol synthesis under standard reaction conditions for four hours at 180 °C (Fig. 7b). It becomes clear from these figures that considerable particle growth took place during the reaction.
 |
| | Fig. 6 Schemes for a possible reaction mechanism for the decomposition of acetaldehyde on a pure Pt/C catalyst (top) and on an NiPt/C sample (bottom). | |
 |
| | Fig. 7 High-angle annular dark-field (HAADF)-TEM images of a fresh Pt/C catalyst (a), a used Pt/C sample after iso-butanol synthesis under standard reaction conditions for four hours at 180 °C (b), a fresh Ni99Pt1/C catalyst (c), and a used Ni99Pt1/C sample after iso-butanol synthesis under standard reaction conditions for four hours at 180 °C (d). | |
As described above, NiPt/C catalysts with low actual molar fractions of Pt in the range between 0.5% and 5.1% from groups I and II did not show signs of deactivation after four hours on stream. In this respect, a higher molar fraction of Ni turned out to be favorable, as Ni is known as an active component in methanation catalysts.34–36 This activity results in precursors for CH4, such as methyl groups, on the Ni surface being hydrogenated to methane rather than decomposing into H2 and carbonaceous deposits. In addition, it is conceivable that carbonaceous deposits that have already formed on the Ni active centers are converted into CO and CO2 through oxidation by means of the adsorbed OH-groups. It is further assumed that the increase in the Pt molar fraction leads to the formation of localized Pt islands that are too large to allow the migration of adsorbed carbon precursors, so that they cannot be methanized or oxidized quickly enough on the Ni particles. The bottom portion of Fig. 6 illustrates these conclusions with respect to the special role of Ni in the reaction mechanism of acetaldehyde decomposition. In addition, Fig. 7c and d show that the NiPt particles were not subject to any significant sintering process. The HAADF image for the fresh Ni99Pt1/C catalyst indicates that the particles are statistically distributed and exhibit only slight signs of agglomeration. The particle size was approximately six nanometers, and the particles were separated from each other.6 The corresponding image for the catalyst being used in iso-butanol synthesis for four hours at 180 °C displays a very similar appearance of the nanoparticles. There were no signs of particle growth. Ni prevented the particles from sintering due to stronger metal–support interactions compared to those of pure Pt.37–39
As a complement to the previous figures, Fig. 8 depicts the space–time yield STY of the catalyst (Fig. 8a), the conversion of ethanol (Fig. 8b), the iso-butanol yield (Fig. 8c), and the selectivity towards iso-butanol (Fig. 8d) as a function of the total metal loading Ni + Pt and the molar fraction of Pt. Thereby, the partial images show very similar trends for STY, ethanol conversion, and iso-butanol yield. The 6.0 wt% Ni96.1Pt3.9 catalyst achieved the highest value for each of these calculated variables. The space–time yield amounted to 4.72 mmol h−1 g−1, the ethanol conversion was 12.7%, and the iso-butanol yield reached a total of 12.4%. Higher or lower values for the actual total metal loading and the actual molar fraction of Pt turned out to be detrimental. In the case of the selectivities towards iso-butanol, no clear trend emerged. They were the highest and very close to 100% in two separate areas. The first was spanned at low total metal loadings between 3.6 wt% and 8.5 wt% and low molar fractions of Pt between 0.3% and 3.9%, respectively. The second area was characterized by total metal loadings close to 20 wt% and was almost independent of the molar fraction of Pt. The results in the partial images of Fig. 8 show that the values for the total metal loading and the molar fraction of Pt must be set correctly and precisely during synthesis, as most of all the STY, the conversion of ethanol, and the iso-butanol yield strongly depend on them. All experimental data for this figure are summarized in Table S8.†
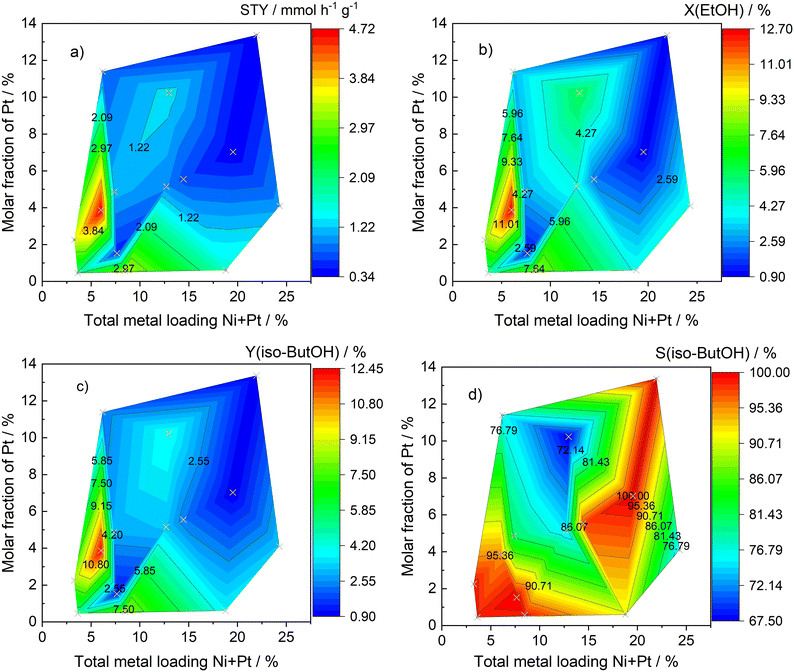 |
| | Fig. 8 STY of the catalyst (a), conversion of ethanol (b), iso-butanol yield (c), and the selectivity towards iso-butanol (d) as a function of the total metal loading Ni + Pt and the molar fraction of Pt; m(cat.) = 250 mg, d(powder) < 75 μm, V(reactor) = 70 ml, c0(EtOH) = 600 mmol l−1, c(NaOH) = 450 mmol l−1, c(n-decane) = 15 mmol l−1; time on stream: 4 h, methanolic solution; the measuring points are shown as crosses. | |
Conclusions
The scientific approach of this study was to investigate the effect of the molar ratio of Ni to Pt and the total metal loading Ni + Pt of activated carbon-supported NiPt catalysts on their catalytic behavior during iso-butanol synthesis from green mixtures of ethanol and methanol. It was found using ICP-OES that the actual values for these two parameters differed significantly from the theoretical values based on the synthesis. The discrepancies observed can be partially explained by the loss of Ni ions because of synthesis-related washing and filtration steps. Nevertheless, the desired trends regarding the total metal loading Ni + Pt and the molar ratio of Ni to Pt aimed for in the synthesis could be retrieved in the ICP-OES results, and so the basic approach of this study was confirmed. XRD measurements showed that catalysts with 30 wt% total metal loading contained Ni(OH)2 phases (theophrastite), indicating an inadequate calcination procedure. For all other catalysts, the typical reflections of Ni and, in some cases, also of NiO, were present. The XRD measurements additionally showed average crystallite sizes in the range of 34 nm to 19 nm. Contrary to the assumption that crystallite sizes increase with increasing total metal loading of the catalyst, there was a slight decrease in crystallite sizes with increasing metal loading. For the nominal 20 wt% Ni80Pt20 (actual 13.0 wt% Ni89.8Pt10.2) catalyst, a Pt phase was formed in addition to the Ni one. H2 adsorption measurements show that the quantity of adsorbed H2 reached a maximum for the nominal 20 wt% Ni80Pt20 (actual 13.0 wt% Ni89.8Pt10.2) catalyst, which also revealed the Pt phase in the XRD spectrum. In particular, the mass fraction of Pt had a strong influence on the quantity of adsorbed H2. The total metal loading correlated positively with the adsorbed quantity of H2, with some exceptions at high total metal loadings. Against this background, it can be concluded that the synthesized catalysts also reveal an increasing number of Pt atoms on the surface with increasing molar fractions of Pt. Considering the different adsorption energies for H2 on Ni (−0.166 eV to 0.464 eV) and Pt (−0.841 eV to −0.126 eV), the observed adsorption behavior of H2 on the catalysts can be rationalized. The catalytic experiments showed that the highest concentrations of iso-butanol could be achieved with low molar fractions of Pt between 0.5% and 5.1% and correspondingly high molar fractions of Ni. In contrast, higher molar fractions of Pt led to deactivation phenomena, which were caused by carbonaceous deposits or strong metal–CO complexes on the Pt particles, which were no longer accessible for iso-butanol synthesis. It can be concluded from the experiments and data from the literature that high molar fractions of Ni promoted the hydrogenation of surface adsorbates, such as methyl groups, which would otherwise have decomposed into H2 and carbonaceous deposits. Furthermore, it is conceivable that carbonaceous deposits that had already formed on the Ni active centers were oxidized into CO and CO2 by means of adsorbed OH-groups. Accordingly, the 6.0 wt% Ni96.1Pt3.9 catalyst with a comparatively low molar fraction of Pt showed the highest values for the space–time yield STY, the conversion of ethanol, and the iso-butanol yield.
Data availability
The data supporting this article have been included as part of the ESI.† This applies to all figures whose data points have been listed in additional tables. The contents of the tables already shown in the main text have not been added again.
Author contributions
Conceptualization – Johannes Häusler and Joachim Pasel; funding acquisition – Ralf Peters; investigation – Johannes Häusler; methodology – Johannes Häusler and Joachim Pasel; supervision – Detlef Stolten; writing original draft – Joachim Pasel; writing review & editing – Joachim Pasel, Johannes Häusler, and Ralf Peters.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
Special thanks are due to the fuel synthesis team at Jülich and all project and cooperation partners. This study was funded by the Deutsche Forschungsgemeinschaft, Germany, (DFG, German Research Foundation) – 491111487. In addition, this work was funded by the German Federal Ministry of Economic Affairs and Energy, Germany, (BMWi), funding number: 19I18006P.
References
- S. Schemme, J. L. Breuer, R. C. Samsun, R. Peters and D. Stolten, Promising catalytic synthesis pathways towards higher alcohols as suitable transport fuels based on H2 and CO2, J. CO2 Util., 2018, 27, 223–237 CrossRef CAS.
- J. Häusler, J. Pasel, C. Wöllhaf, R. Peters and D. Stolten, Dilute Alloy Catalysts for the Synthesis of Isobutanol via the Guerbet Route: A Comprehensive Study, Catalysts, 2024, 14, 215 CrossRef.
- J. Häusler, J. Pasel, F. Woltmann, A. Everwand, M. Meledina, H. Valencia, M. Lipińska-Chwałek, J. Mayer and R. Peters, Elucidating the Influence of the d-Band Center on the Synthesis of Isobutanol, Catalysts, 2021, 11(3), 406 CrossRef.
- J. Pasel, J. Häusler, D. Schmitt, H. Valencia, J. Mayer and R. Peters, Aldol condensation of acetaldehyde for butanol synthesis: A temporal analysis of products study, Appl. Catal., A, 2023, 324, 122286 CrossRef CAS.
- J. Pasel, F. Woltmann, J. Häusler and R. Peters, Surface Redox Reaction for the Synthesis of NiPt Catalysts for the Upgrading of Renewable Ethanol/Methanol Mixtures, Catalysts, 2024, 14, 77 CrossRef CAS.
- J. Pasel, J. Häusler, R. Peters and D. Stolten, Catalytic activity and stability of NiPt/C catalysts for the synthesis of iso-butanol from methanol/ethanol mixtures, Catal. Sci. Technol., 2024, 7048 RSC.
- B. Iqbal, M. Saleem, S. N. Arshad, J. Rashid, N. Hussain and M. Zaheer, One-Pot Synthesis of Heterobimetallic Metal–Organic Frameworks (MOFs) for Multifunctional Catalysis, Chem. – Eur. J., 2019, 25, 10490–10498 CrossRef CAS PubMed.
- L. Nguyen, S. Zhang, L. Wang, Y. Li, H. Yoshida, A. Patlolla, S. Takeda, A. I. Frenkel and F. Tao, Reduction of Nitric Oxide with Hydrogen on Catalysts of Singly Dispersed Bimetallic Sites Pt1Com and Pd1Con, ACS Catal., 2016, 6, 840–850 CrossRef CAS.
- M. Ouyang, K. G. Papanikolaou, A. Boubnov, A. S. Hoffman, G. Giannakakis, S. R. Bare, M. Stamatakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Directing reaction pathways via in situ control of active site geometries in PdAu single-atom alloy c atalysts, Nat. Commun., 2021, 12, 1549 CrossRef CAS PubMed.
- H. Yu, W. Tang, K. Li, S. Zhao, H. Yin and S. Zhou, Enhanced Catalytic Performance for Hydrogenation of Substituted Nitroaromatics over Ir-Based Bimetallic Nanocatalysts, ACS Appl. Mater. Interfaces, 2019, 11, 6958–6969 CrossRef CAS PubMed.
- W. Zhong, Y. Liu and D. Zhang, Theoretical Study of Methanol Oxidation on the PtAu(111) Bimetallic Surface: CO Pathway vs Non-CO Pathway, J. Phys. Chem. C, 2012, 116, 2994–3000 CrossRef CAS.
-
W. Feng and L. Shushuang, CN Pat., CN108117480A, 2016 Search PubMed.
-
O. Fuchs and W. Querfurth, US Pat., US2050788A, 1932 Search PubMed.
-
T. Vanderspurt and J.-L. Kao, US Pat., US5493064, 1994 Search PubMed.
-
T. Vanderspurt, J. K. Russel and K. R. Miller, US Pat., US5811602A, 1995 Search PubMed.
-
K. Kourtakis, M. B. d'Amore and L. E. Manzer, US Pat., US7700813B, 2008 Search PubMed.
-
T. Tsuchida, S. Sakuma and T. Yoshioka, US Pat., US20090205246A1, 2009 Search PubMed.
- C. Carlini, C. Flego, M. Marchionna, M. Noviello, A. M. Raspolli Galletti, G. Sbrana, F. Basile and A. Vaccari, Guerbet condensation of methanol with n-propanol to isobutyl alcohol over heterogeneous copper chromite/Mg–Al mixed oxides catalysts, J. Mol. Catal. A: Chem., 2004, 220, 215–220 CrossRef CAS.
- C. Carlini, M. Marchionna, M. Noviello, A. M. R. Galletti, G. Sbrana, F. Basile and A. Vaccari, Guerbet condensation of methanol with n-propanol to isobutyl alcohol over heterogeneous bifunctional catalysts based on Mg–Al mixed oxides partially substituted by different metal components, J. Mol. Catal. A: Chem., 2005, 232, 13–20 CrossRef CAS.
- C. Carlini, M. Di Girolamo, A. Macinai, M. Marchionna, M. Noviello, A. M. Raspolli Galletti and G. Sbrana, Selective synthesis of isobutanol by means of the Guerbet reaction: Part 2. Reaction of methanol/ethanol and methanol/ethanol/n-propanol mixtures over copper based/MeONa catalytic systems, J. Mol. Catal. A: Chem., 2003, 200, 137–146 CrossRef CAS.
- S. M. A. H. Siddiki, A. S. Touchy, M. A. R. Jamil, T. Toyao and K.-I. Shimizu, C-Methylation of Alcohols, Ketones, and Indoles with Methanol Using Heterogeneous Platinum Catalysts, ACS Catal., 2018, 8, 3091–3103 CrossRef CAS.
- P. Gupta, H. J. Drexler, R. Wingad, D. Wass, E. Baráth, T. Beweries and C. Hering-Junghans, P,N-type phosphaalkene-based Ir(i) complexes: synthesis, coordination chemistry, and catalytic applications, Inorg. Chem. Front., 2023, 10, 2285–2293 RSC.
- R. L. Wingad, E. J. E. Bergström, M. Everett, K. J. Pellow and D. F. Wass, Catalytic conversion of methanol/ethanol to isobutanol – a highly selective route to an advanced biofuel, Chem. Commun., 2016, 52, 5202–5204 RSC.
- F. J. Sama, R. A. Doyle, B. M. Kariuki, N. E. Pridmore, H. A. Sparkes, R. L. Wingad and D. F. Wass, Backbone-functionalised ruthenium diphosphine complexes for catalytic upgrading of ethanol and methanol to iso-butanol, Dalton Trans., 2024, 53, 8005–8010 RSC.
- J. Pasel, J. Häusler, D. Schmitt, H. Valencia, M. Meledina, J. Mayer and R. Peters, Ethanol Dehydrogenation: A Reaction Path Study by Means of Temporal Analysis of Products, Catalysts, 2020, 10, 1151 CrossRef CAS.
- A. A. Popov, A. D. Varygin, P. E. Plyusnin, M. R. Sharafutdinov, S. V. Korenev, A. N. Serkova and Y. V. Shubin, X-ray diffraction reinvestigation of the Ni-Pt phase diagram, J. Alloys Compd., 2022, 891, 161974 CrossRef CAS.
- K. Taniguchi, K. Shinoda, J. L. C. Huaman, S. Yokoyama, M. Uchikoshi, T. Matsumoto, K. Suzuki, H. Miyamura and B. Jeyadevan, Designed synthesis of highly catalytic Ni–Pt nanoparticles for fuel cell applications, SN Appl. Sci., 2018, 1, 124 CrossRef.
- G. Wang, M. A. Van Hove, P. N. Ross and M. I. Baskes, Monte Carlo simulations of segregation in Pt-Ni catalyst nanoparticles, J. Chem. Phys., 2005, 122, 24706 CrossRef PubMed.
- G. Wang, M. A. Van Hove, P. N. Ross and M. I. Baskes, Quantitative prediction of surface segregation in bimetallic Pt–M alloy nanoparticles (M=Ni,Re,Mo), Prog. Surf. Sci., 2005, 79, 28–45 CAS.
- B. S. Mun, M. Watanabe, M. Rossi, V. Stamenkovic, N. M. Markovic and P. N. Ross, The Study of Surface Segregation, Structure, and Valence Band Density of States of Pt3Ni(100), (110), and (111) crystals, Surf. Rev. Lett., 2006, 13, 697–702 CrossRef CAS.
- V. N. Panchenko, E. A. Paukshtis, D. Y. Murzin and I. L. Simakova, Solid Base Assisted n-pentanol Coupling over VIII Group Metals: Elucidation of the Guerbet Reaction Mechanism by DRIFTS, Ind. Eng. Chem. Res., 2017, 56, 13310–13321 CrossRef CAS.
- D. I. Ezzhelenko, S. A. Nikolaev, A. V. Chistyakov, P. A. Chistyakova and M. V. Tsodikov, Deactivation Mechanism of Palladium Catalysts for Ethanol Conversion to Butanol, Pet. Chem., 2021, 61, 504–515 CrossRef CAS.
- O. Cheong, J. Pasel, J. Häusler, R. Peters, M. H. Eikerling and P. M. Kowalski, Rationalizing the mechanism of ethanol dehydrogenation on Pt/C, Surf. Sci., 2024, 739, 122396 CrossRef CAS.
- X. Chen, Y. Zhang, C. Sun, Y. Wang, G. Song, C. Li, K. Hui Lim, R. Ye, Y. Peng, H. Arandiyan, Z. H. Lu, G. Feng, R. Zhang and S. Kawi, Lanthanum-mediated enhancement of nickel nanoparticles for efficient CO2 methanation, Fuel, 2024, 371(Part B), 131998 CrossRef CAS.
- T. Nguyen, B. L. Do, P. A. Nguyen, T. T. V. Nguyen, C. A. Ha, T. C. Hoang and C. L. Luu, Nickel/ceria nanorod catalysts for the synthesis of substitute natural gas from CO2: Effect of active phase loading and synthesis condition, J. Sci.: Adv. Mater. Devices, 2024, 9(3), 100752 CAS.
- X. Tang, C. Song, H. Li, W. Liu, X. Hu, Q. Chen, H. Lu, S. Yao, X. N. Li and L. Lin, Thermally stable Ni foam-supported inverse CeAlOx/Ni ensemble as an active structured catalyst for CO2 hydrogenation to methane, Nat. Commun., 2024, 15, 3115 CrossRef CAS PubMed.
- Q. Sheng, Y. Wang, P. Zhang, Y. Jiang, J. Zhang, Q. Liu, Q. Chang, Y. Yao, X. Liao, Z. Li, S. Lyu and Y. Qin, Highly dispersed Ni on defective carbon with metal-support interaction for efficient and selective cinnamaldehyde hydrogenation, Appl. Surf. Sci., 2024, 666, 160369 CrossRef CAS.
- P. Wang, D. Li, L. Wang, S. Guo, Y. Zhao, H. Shang, D. Wang and B. Zhang, Ultrafine CoNi alloy nanoparticles anchored on surface-roughened halloysite nanotubes for highly efficient catalytic hydrogenation of 4-nitrophenol, Chem. Eng. J., 2024, 495, 153631 CrossRef CAS.
- M. Zhang, T. Yang, K. Jiang, Y. Gao, J. Yang, Z. Liu and Y. Han, Rationally constructing metastable ZrO2 supported Ni catalysts for highly efficient and stable dry reforming of methane, Appl. Catal., B, 2024, 353, 124102 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Johannes
Häusler
ab,
Ralf
Peters
*a,
Johannes
Häusler
ab,
Ralf
Peters