
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cu-catalyzed [1,3]-asymmetric methoxy rearrangement of N-methoxyanilines: mechanistic insight†
Kazuki
Masukawa
b,
Amu
Kojima
b,
Takuma
Sato
b,
Masahiro
Terada
 b and
Itaru
Nakamura
*ab
b and
Itaru
Nakamura
*ab
aInstitute for Excellence in Higher Education, Tohoku University, 980-8576, Japan. E-mail: itaru-n@tohoku.ac.jp
bDepartment of Chemistry, Graduate School of Science, Tohoku University, 980-8578, Japan
First published on 25th February 2025
Abstract
Cu-catalyzed reactions of N-methoxy-2,6-dimethylanilines in the presence of a cationic Cu catalyst ligated to a chiral NHC ligand, which has an (ortho-carbamoyl)phenyl group on the nitrogen atom of (S,S)-diphenylimidazolidinylidene, furnished chiral ortho-quinol imines with good enantioselectivity. In addition, a cascade reaction involving the [1,3]-methoxy rearrangement followed by the Diels–Alder reaction yielded the corresponding three-dimensional molecules in a diastereo- and enantioselective manner.
Asymmetric rearrangements have received much attention for constructing sterically congested structures with a tetrasubstituted carbon as an asymmetric center by changing the connectivity of the starting materials.1 Asymmetric [3,3]-rearrangements, such as the Claisen rearrangement and the Cope rearrangement, have been intensively investigated for the synthesis of enantio-enriched organic molecules (Scheme 1a).2 In contrast, asymmetric [1,3]-rearrangement reactions have received much less attention, because the process inherently proceeds via a strained transition state, making the stereo control of the enantio-determining process difficult (Scheme 1b).3–7 Recent studies have indicated that transition metal catalysts, nucleophilic catalysts, and Brønsted acid catalysts promote asymmetric [1,3]-rearrangements with excellent enantioselectivity. However, these processes are still limited to the migration of the carbon group from the oxygen atom to the carbon atom, such as the aza-Petasis–Ferrier rearrangement3–5 and the Steglich rearrangement,6 involving C–C bond formation (X = O, Y = CR, and x = CR’3).
 | ||
| Scheme 1 Asymmetric [3,3]- and [1,3]-rearrangements. | ||
In this context, we focus on the [1,3]-alkoxy rearrangement of N-alkoxyanilines, which involves N–O bond cleavage and C–O bond formation (Scheme 2a).8–15 We recently found that cationic Cu catalysts ligated to an N-heterocyclic carbene (NHC) ligand effectively promote the [1,3]-alkoxy rearrangement. In particular, in substrates having an electron-donating substituent, such as an alkyl, a p-anisyl, or a methoxy group, at the ortho position, the migration of the alkoxy group preferentially to the substituted ortho position takes place.11–14 Because the resulting ortho-quinol imines function as a versatile intermediate for further transformations, such as the [1,2]-rearrangement,11 the Michael addition,12,13 or the Diels–Alder reaction,14 the [1,3]-alkoxy rearrangements are useful for the synthesis of elaborate organic molecules. Accordingly, we envisioned that properly designed chiral NHC ligands would induce the enantioselective C–O bond formation, realizing a new class of asymmetric [1,3]-rearrangement reactions. Specifically, the reactions of 2,6-disubstituted N-alkoxyanilines would proceed via the differentiation of the two prochiral ortho positions by employing chiral NHC ligands (Scheme 2b). Herein, we report that the Cu-catalyzed reaction of 2,6-disubstituted N-methoxyanilines 1 in the presence of cationic Cu catalysts ligated to a chiral NHC ligand proceeds through an asymmetric [1,3]-methoxy rearrangement to yield chiral ortho-quinol imines 2 with high enantioselectivity. In addition, the cascade reaction of 1 with maleimides 3 produces three-dimensional compounds 4 with good diastereo- and enantioselectivities.
 | ||
| Scheme 2 Cu-catalyzed [1,3]-alkoxy rearrangement. | ||
Prior to performing experiments aimed at designing the chiral NHC ligand, we conducted a theoretical investigation of the [1,3]-methoxy rearrangement using a computational approach to elucidate the C–O bond forming process that leads to an ortho-quinol imine-copper complex (Schemes 3, S1, and S3,†Fig. 1 and 2).‡ First, we calculated an achiral reaction of an ortho-toluidine derivative using the IPrCuBF4 catalyst as a model system (Schemes S1 and S3†). In the first stage of the reaction, we assumed that the N–O bond, which has a relatively low bond dissociation energy,15 is cleaved to form the corresponding methoxycopper species either through ionic cleavage, which we proposed based on the Lewis acid-mediated [1,3]-rearrangement (Scheme S1a†) or concerted oxidative addition calculated by Dang (Scheme S1b†). However, despite conducting an exhaustive search, we were unable to find a structurally reasonable transition state for this rearrangement process via these mechanisms.16 Therefore, given that the N–O bond has a relatively low bond dissociation energy, we hypothesized that the first stage of this reaction proceeds through homolytic cleavage of the N–O bond to generate radical pair IM1 (Scheme S1c†).17–19 In fact, the calculated bond dissociation energy of the N–O bond in ortho-toluidine SM, of which the carbonyl is coordinated with the copper catalyst, is 22.8 kcal mol−1. Then, the generated methoxy radical binds again to the copper atom, forming methoxycopper(III) intermediate IM2 with square planar geometry. Although this intermediate has a character of the nitrenium species IM2′, the NBO analysis suggested that contribution of the methoxycopper(III) IM2 is larger than that of Cu(I) IM2′ with the nitrenium moiety (see the ESI†). In addition, the calculations suggest that the C–O bond-forming process through TS2–3 is the rate-determining process, the activation energy of which is calculated to be 25.6 kcal mol−1. It should be noted that the methoxy group preferentially migrates to the methyl-bound ortho position, where the nucleophilic attack on the ortho carbon with a methyl substituent is energetically favored over that on a non-substituted ortho carbon, as evidenced by previously reported experimental results (see the ESI†).7–9 This is because the methyl-bound carbon atom is more positively charged according to the NBO analysis. Moreover, the calculations suggest that the electron-withdrawing group on the N-methoxyaniline substrate decelerates the rearrangement. According to NBO analysis, the C–O bond forming process involves a decrease of electrons at the ortho position forming the C–O bond to facilitate the interaction with a lone pair of the migrating methoxy group. In other words, contribution of the nitrenium character-like IM2′ in the transition state TS2–3 is larger than that in intermediate IM2 (see the ESI†).
 | ||
| Scheme 3 Reaction coordinates for the Cu-catalyzed [1,3]-rearrangement of N-methoxy-ortho-toluidine. | ||
 | ||
| Fig. 1 Design of chiral NHC ligand L1 and the conformational isomers of methoxycopper(III) intermediate IM2*. | ||
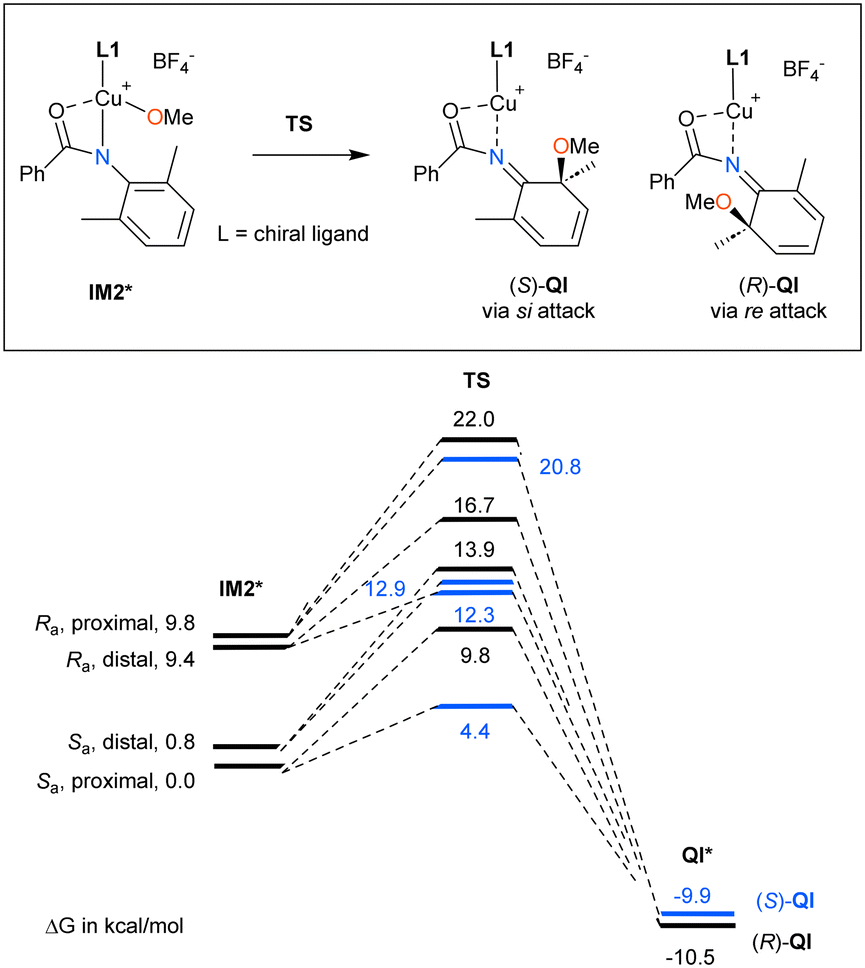 | ||
| Fig. 2 Reaction coordinates for the C–O bond-forming process. | ||
According to these computational results, we envisioned that an NHC ligand with a coordinative group would induce the enantio-recognition of 2,6-dimethylaniline through desymmetrizative C–O bond formation through the methoxycopper(III) intermediate, with square pyramidal geometry. From the perspective of structural simplicity and synthetic accessibility, we selected chiral (S,S)-diphenylimidazolidinylidene ligand L1, which has ortho-(N,N-dimethylcarbamoyl)phenyl and 2,4,6-triisopropylphenyl (Trip) groups as substituents on the nitrogen atoms of NHC. As a result of structural exploration, the transition states for the C–O bond formation were classified into eight states based on the attack of the methoxy group on either the si- or re-face of the four conformational isomers of methoxycopper(III) intermediate IM2*, originated from (1) the C–N axial chirality of the ortho-(N,N-dimethylcarbamoyl)phenyl group on NHC (Ra and Sa) and (2) the relative orientation of the NHC ligand and the substrate molecule (distal and proximal; the conformation in which the methoxy ligand is located close to the ortho-carbamoyl group is named proximal, Fig. 1). From the energy diagram of the eight transition states shown in Fig. 2, we predict that designed NHC ligand L1 has the potential to induce the enantio-differentiation of the two prochiral ortho positions, with the S enantiomer as the main product. It should be noted that interconversion between the conformational isomers occurs through homolytic cleavage of the Cu–O bond in IM2*, generating a radical pair corresponding to IM1, as shown in Scheme 3. In other words, interconversion via rotation of the C–N bond between the nitrogen atom on the imidazolidinylidene ring and the carbon atom on the aryl ring substituted by the ortho-carbamoyl aryl group, as well as the Cu–N bond in IM2* is unlikely due to the bulkiness of the chiral NHC ligand.
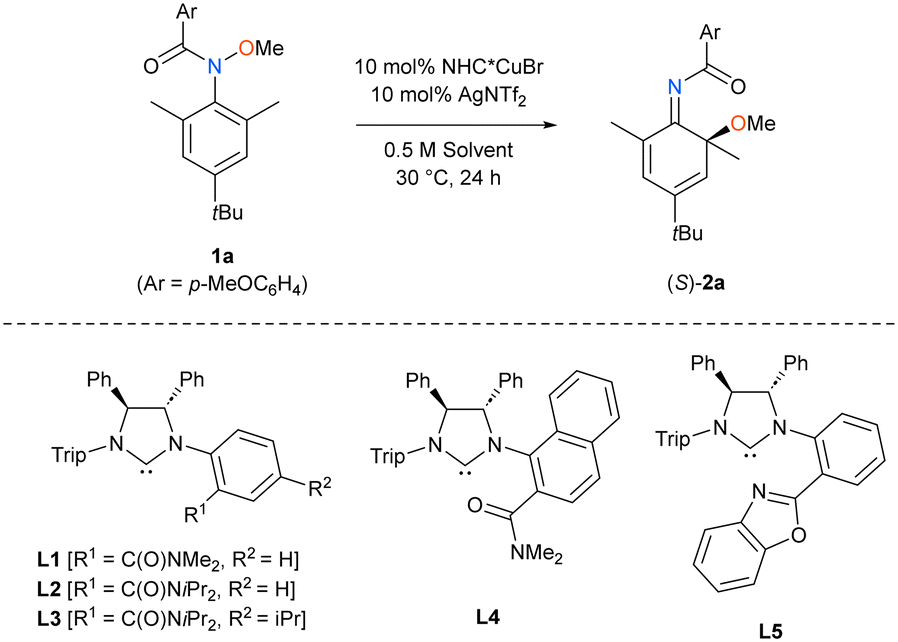
Based on these computational results, we examined the applicability of chiral NHC (NHC*) ligands L1 to L5 to the Cu-catalyzed reactions of 2,6-dimethyl-N-methoxyaniline 1a. The results are summarized in Table 1. Chiral ligands L1 and L2, which have an N,N-dimethylcarbamoyl group and an N,N-diisopropylcarbamoyl group, respectively, at the ortho position of the phenyl ring attached to the nitrogen atom, induced the enantio-differentiation of the ortho position of the aniline ring, yielding S product (S)-2a as a major stereoisomer, as predicted (Table 1, entries 1 and 2).§ In contrast, chiral ligand L4, the N,N-dimethylcarbamoyl group of which is located at the 2 position of the 1-naphthyl group yielded (R)-2a as a major stereoisomer with low enantioselectivity (entry 4). In chiral ligand L5, the 2-benzoxazolyl group was an effective substituent in place of the carbamoyl group (entry 5), whereas other functional groups, such as benzoyl and methoxy groups, were much less efficient (see the ESI†).¶ Among the reaction conditions tested, the combination of chiral ligand L3, which has an isopropyl group at the para position of the benzene ring with the ortho-carbamoyl group, and ethyl acetate as a solvent led to good enantioselectivity, albeit a low chemical yield (entry 6).|| Because a considerable amount of starting material 1a was recovered, we examined the effect of increasing the loading amount of the chiral NHC*–Cu catalyst (entry 7). However, we found that the use of 30 mol% of L3CuBr and AgNTf2 resulted in a decrease in enantioselectivity, presumably owing to the partial racemization of obtained product 2a (see the ESI†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | NHC* | Solvent | Yield/%a | Erb | Recovery/%a |
| a Yields were determined by 1H NMR analysis using CH2Br2 as an internal standard. b Enantiomeric ratio was determined by chiral SFC analysis. c 30 mol% of each of L3CuBr and AgNTf2 was used. | |||||
| 1 | L1 | DCE | 27 | 82![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :18 :18 |
70 |
| 2 | L2 | DCE | 33 | 80:20 |
65 |
| 3 | L3 | DCE | 31 | 74:26 |
60 |
| 4 | L4 | DCE | 49 | 34:66 |
50 |
| 5 | L5 | DCE | 13 | 82:18 |
85 |
| 6 | L3 | EtOAc | 24 | 89:11 |
51 |
| 7c | L3 | EtOAc | 58 | 83:17 |
18 |
To solve the racemization problem of ortho-quinol imine 2a, we carried out a cascade reaction involving the [1,3]-alkoxy rearrangement followed by the Diels–Alder reaction with maleimides 3 (Scheme 4). To our delight, excellent enantioselectivity was maintained even in the reaction of 1a with N-methylmaleimide 3a (5 equiv.) using 30 mol% of the chiral NHC catalyst, affording desired product 4aa in 63% yield with 91:9 er. It should be noted that the reactions yielded a single diastereomer derived from the approach of maleimides 3 to ortho-quinol imine 2 from the side of the methoxy group in an endo manner.14 As expected, substrate 1c having an electron-withdrawing bromo group in the aroyl group on the nitrogen atom exhibited lower reactivity than 1a, which has a methoxy group, although the enantioselectivity was maintained. Similarly, the substrates 1e and 1f, which have a phenyl group and a vinyl group, respectively, at the para position of the aniline ring, underwent the cascade reaction to yield products 4ea and 4fa, respectively, with good enantioselectivity but in low chemical yields. The low chemical yields are presumably due to the inhibition of the catalyst turnover through the formation of a homoleptic [(L3)2Cu]+ complex, which is inactive in the [1,3]-rearrangement reactions.** In addition, the result that the opposite enantiomer was obtained as a major product when L4 was used indicates that the transition state for the C–O bond-forming process is very sensitive to trivial changes in the chemical structure. Further development of chiral NHC ligands for the catalytic [1,3]-alkoxy rearrangement to solve these problems is underway in our laboratory. Nevertheless, the present cascade reaction involving the asymmetric [1,3]-methoxy rearrangement is efficient for the synthesis of three-dimensional molecules in a highly stereoselective and enantioselective manner. In addition, it should be noted that stereocontrol of alkoxy groups is still limited to oxy-Michael addition reactions of alcohols, due to low nucleophilicity or high pKa.20 In this context, we realized enantio-control of the alkoxy group by a totally different approach based on the rearrangement in this investigation.
 | ||
| Scheme 4 Cu-catalyzed asymmetric [1,3]-rearrangement and Diels–Alder reaction of 1 with 3. a The reactions of 1 (0.1 mmol) with 3 (0.5 mmol) were carried out in the presence of L3CuBr (30 mol%) and AgNTf2 (30 mol%) in EtOAc (0.2 mL) at 30 °C for 24 h. Isolated yield. The enantiomeric ratio was determined by chiral SFC analysis. b 10 mol% of each of L3CuBr and AgNTf2 was used. c At 50 °C. | ||
In conclusion, we have developed a chiral NHC ligand for the catalytic asymmetric [1,3]-methoxy rearrangement of N-methoxyaniline derivatives. Because ortho-quinol imines can be further manipulated to furnish favourable compounds, the present method is useful for the synthesis of chiral building blocks in a unique manner.
Data availability
The exploratory investigation, experimental procedures, computational data, and characterization data are available. The data supporting this article have been included as part of the ESI.† Crystallographic data for 2c have been deposited at the CCDC under 2415308.Author contributions
Conceptualization: IN; methodology: KM and IN; investigation: KM and AK; formal analysis: TS and IN; writing: TS, MT and IN; supervision: MT. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The computation was performed using the Research Centre for Computational Science, Okazaki, Japan (Project: 22-IMS-C127, 23-IMS-C116, and 24-IMS-C110). This work was supported by JSPS KAKENHI Grant Number JP20H02731, Grant-in-Aid for Scientific Research (B) from MEXT, Japan.Notes and references
- X.-L. Liu, M.-L. Gong, X. Yang, P. Tian and Q.-H. Li, Org. Chem. Front., 2024, 11, 4934 RSC.
- H. Ren and W. D. Wulff, J. Am. Chem. Soc., 2011, 133, 5656 CrossRef CAS PubMed.
- Y. Zhang and Y. Chen, Org. Chem. Front., 2022, 9, 4744 RSC.
- M. Terada and Y. Toda, J. Am. Chem. Soc., 2009, 131, 6354 CrossRef CAS.
- M. Terada, T. Komuro, Y. Toda and T. Korenaga, J. Am. Chem. Soc., 2014, 136, 7044 CrossRef CAS PubMed.
- J. Zhang, X. Han, X. Wu, Y. Liu and Y. Cui, ACS Sustainable Chem. Eng., 2019, 7, 5065 CrossRef CAS.
- L. Yao and K. Ishihara, Chem. Sci., 2019, 10, 2259 RSC.
- I. Nakamura, T. Jo, Y. Ishida, H. Tashiro and M. Terada, Org. Lett., 2017, 19, 3059 CrossRef CAS PubMed.
- I. Nakamura, S. Nozawa, Y. Ishida, I. Muranushi, A. Mayerweg and M. Terada, Org. Chem. Front., 2021, 8, 6390 RSC.
- I. Nakamura, R. Konta, Y. Ishida, M. Tachibana and M. Terada, Adv. Synth. Catal., 2023, 365, 4677 CrossRef CAS.
- Y. Ishida, I. Nakamura and M. Terada, J. Am. Chem. Soc., 2018, 140, 8629 CrossRef CAS PubMed.
- I. Nakamura, H. Tashiro, Y. Ishida and M. Terada, Org. Lett., 2020, 22, 3794 CrossRef CAS PubMed.
- I. Nakamura, M. Tachibana, R. Konta, H. Tashiro and M. Terada, Molecules, 2023, 28, 4251 CrossRef CAS PubMed.
- I. Nakamura, K. Masukawa, Y. Ishida and M. Terada, Org. Lett., 2021, 23, 4127 CrossRef CAS PubMed.
- O. Mó, M. Yáñez, M. Eckert-Maksić, Z. B. Maksić, I. Alkorta and J. Elguero, J. Phys. Chem. A, 2005, 109, 4359 CrossRef PubMed.
- Y.-Q. Fang and L. Dang, Org. Lett., 2020, 22, 9178 CrossRef CAS PubMed.
- Y.-N. Zheng, H. Zheng, T. Li and W.-T. Wei, ChemSusChem, 2021, 14, 5340 CrossRef CAS PubMed.
- H.-M. Jiang, Y.-L. Zhao, Q. Sun, X.-H. Ouyang and J.-H. Li, Molecules, 2023, 28, 1775 CrossRef CAS PubMed.
- X. Tang, Z. Zhu, C. Qi, W. Wu and H. Jiang, Org. Lett., 2016, 18, 180 CrossRef CAS PubMed.
- G. Su, M. Formica, K. Yamazaki, T. A. Hamlin and D. J. Dixon, J. Am. Chem. Soc., 2023, 145(23), 12771 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2415308. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cy00106d |
| ‡ Quantum chemical calculations were performed using the Gaussian 16 program package. Structural optimization based on density functional theory (DFT) were carried out using the B3LYP functional including Grimme's D3 dispersion correction with Becke–Johnson damping and the def2-SVP basis set. Intrinsic reaction coordinate (IRC) calculations were performed to verify the connectivity between the transition state and the corresponding intermediates. All structures of the intermediates (no imaginary frequency) and the transition states (a single imaginary frequency) were verified by vibrational analysis. |
| § The absolute configuration of the product was deduced from the reaction of 1c using L2 as a chiral ligand. See the ESI.† |
| ¶ The use of previously reported chiral NHC ligands was not effective for the present reaction. See the ESI.† |
| || We found that the use of ethyl acetate provided the best enantioselectivity for the cascade reaction between 1 and maleimide 2a (Table S2†). Moreover, it was observed that L3 performed better than L2 in ethyl acetate (Table S2,† entry 4 versus entry 7). |
| ** A molecular ion peak, which corresponded to [(L2)2Cu]+ was observed by HRMS. Additionally, [(IMes)2Cu]NTf2 did not promote the [1,3]-rearrangement reaction. See the ESI.† |
| This journal is © The Royal Society of Chemistry 2025 |