
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cationic Zr catalysts for the sequential polymerisation of alkenes and cyclic oxygenated monomers†
Eszter
Fazekas
 ,
Carole A.
Morrison
and
Jennifer A.
Garden
*
,
Carole A.
Morrison
and
Jennifer A.
Garden
*
EaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: j.garden@ed.ac.uk
First published on 8th April 2025
Abstract
Block copolymers (BCPs) featuring an apolar polyalkene and a polar polyester segment are attractive materials that can be synthesised through one-pot procedures with rare-earth metal catalysts, including those based on Sc. However, examples remain limited to the copolymerisation of conjugated olefins, such as isoprene and myrcene, with cyclic esters. While Zr is diagonally related to Sc in the periodic table, and Zr-based catalysts excel at non-conjugated olefin polymerisations, cationic Zr complexes remain unreported for apolar polyolefin/polar polyester BCPs. Here, we show that cationic Zr amine bisphenolate and zirconocene complexes are effective catalysts for the sequential polymerisation of various alkene and cyclic ester/ether monomers, yet deliver two separate homopolymers instead of copolymers. Mechanistic studies combined with DFT calculations suggest that the alkene monomer is polymerised via a coordination–insertion pathway, whereas ε-caprolactone polymerisation follows a cationic ring-opening mechanism under these conditions.
Introduction
Block copolymers (BCPs) can combine the advantages of two or more homopolymers, opening up access to materials with tunable properties and versatile applications spanning drug delivery, membranes, compatibilisers and lubricants.1,2 Polar/apolar BCPs are particularly attractive, as the synergy of hydrophobicity and hydrophilicity enables self-assembly into tailored nanostructures, and can also deliver enhanced tensile modulus, flexibility and dyeability compared to the respective homopolymers or their blends.3–5 However, BCPs that combine polyolefins with polyesters remain scarce, due to the different monomer classes and thus the disparate mechanisms required to generate the polar and apolar blocks.6–8 Polar/apolar BCPs are generally synthesised via multi-step procedures involving end-group functionalisation of the first block, so that it can be employed as a macroinitiator. Alternative one-pot methods utilising bifunctional catalysts and sequential monomer addition are beneficial, as these can avoid time-consuming and costly purification steps. Relatively recently, catalyst systems have been reported that are capable of copolymerising alkenes with cyclic esters to generate poly(alkene-co-ester) BCPs. These catalysts are predominantly based on M3+ rare earth metals, including Sc, Y (Fig. 1a–c) and lanthanides such as Sm, Er, Yb and Lu.9–17 So far, studies have been almost exclusively limited to the copolymerisation of conjugated alkenes such as 1,3-dienes or styrene with ε-caprolactone (CL). The ability to incorporate industrially more relevant monoalkenes (e.g. ethylene or 1-hexene) remains a challenge.14 | ||
| Fig. 1 Structures of cationic Y-9 (a) and Sc-based10,11 catalysts (b and c) reported for olefin/cyclic ester block copolymer synthesis and the structures of two Zr-based catalysts investigated in this work (counter anions omitted for clarity). | ||
Zirconium catalysts have a strong track record in the homopolymerisation of commodity olefins and cyclic esters.18–27 A range of Zr complexes, including cationic zirconocenes and Zr-phenolates, have been used to prepare apolar/apolar BCPs from olefin monomers such as ethylene, 1-hexene and styrene.28–31 Polar/polar BCPs have also been produced via Zr-catalysed ring-opening polymerisation (ROP) of cyclic esters, cyclic ethers (epoxides) or cyclic carbonates.32–37 Notably, Hadjichristidis and co-workers copolymerised a polar alkene (methyl methacrylate, MMA) with a polar cyclic ester (CL) using a zirconocene catalyst.38 Some Zr-based catalysts have even been reported for the separate homopolymerisation of apolar olefins and polar cyclic esters.39 However, we are not aware of any reports of Zr catalysts combining these two distinct monomer classes to produce apolar/polar BCPs. This is somewhat surprising, as the diagonal relationship between metals in the periodic table is well-known, with Sc and Zr sharing several key similarities relevant to olefin and cyclic ester polymerisation. Specifically, the two elements have similar electronegativities (χ = 1.33 for Zr vs. χ = 1.36 for Sc), oxophilicities (θ = 0.8 for both Sc and Zr) and ionic radii (88 ppm for Zr4+vs. 86 ppm for Sc3+).40–42 The parallels between Sc and Zr for olefin oligomerisation and polymerisation have been reported,43 and both Sc and Zr metallocene complexes catalyse the random copolymerisation of ethylene with polar olefins.44,45 To understand the opportunities and limitations of using cationic Zr catalysts to prepare poly(olefin-co-esters), we selected two zirconium pre-catalysts (ABPZrBn2 (1) and Cp2ZrMe2 (2), Fig. 1) that have, along with structurally related derivatives, previously shown success for both olefin polymerisation and the ROP of cyclic esters. Here, we investigated the potential of these catalysts to synthesise apolar/polar BCPs.46–54
Results and discussion
Exploring monomer scope with cationic ABPZrBn+ and Cp2ZrMe+ complexes
The ABPZrBn2 pre-catalyst was synthesised via literature procedures (Fig. S1 and S2†),55,56 whilst commercially available Cp2ZrMe2 was used as received. Both pre-catalysts were activated towards olefin polymerisation via the in situ addition of one equivalent of tris(pentafluorophenyl)borane (BCF) or trityl tetrakis(pentafluorophenyl)borate (TBCF), to generate cationic ABPZrBn+ or Cp2ZrMe+ (1 and 2, Fig. 1) along with a weakly coordinating BnB(C6F5)3−/MeB(C6F5)3− or B(C6F5)4− counter anion (Fig. S3†).57 To establish the monomer scope and the potential for block copolymer formation, catalysts 1–2 were screened for the homopolymerisation of a diverse range of monomers (Table 1), including olefins and conjugated dienes (1-hexene, styrene, isoprene and myrcene), cyclic esters (CL, rac-lactide and β-butyrolactone) and cyclic ethers (propylene oxide, cyclohexene oxide and limonene oxide).| Entry | Monomer | Pre-cat. | Co-cat. | Mon. equiv. | T (°C) | t (h) | Conv. (%) | M n,th (kg mol−1) | M n,SEC (kg mol−1) | Đ |
|---|---|---|---|---|---|---|---|---|---|---|
| Conditions: 0.0125 mmol of ABPZrBn2 or Cp2ZrMe2 and BCF or TBCF pre-stirred in toluene for 10 minutes prior to the addition of monomer to afford 1 M concentration of monomer in toluene. SEC – uncorrected values against polystyrene standards, except for PCL samples, which were corrected with a correction factor of 0.56.58 | ||||||||||
| 1 | 1-Hexene | ABPZrBn2 | BCF | 100 | r.t. | 0.33 | 100 | 8.4 | 13.9 | 1.64 |
| 2 | 1-Hexene | ABPZrBn2 | TBCF | 100 | r.t. | 0.33 | 100 | 8.4 | 10.5 | 1.59 |
| 3 | 1-Hexene | Cp2ZrMe2 | BCF | 100 | r.t. | 0.33 | 100 | 8.4 | <1 | — |
| 4 | Isoprene | ABPZrBn2 | BCF | 50 | 75 | 18 | 0 | — | — | — |
| 5 | Isoprene | ABPZrBn2 | TBCF | 50 | r.t. | 24 | 100 | 3.4 | <1 | — |
| 6 | Myrcene | ABPZrBn2 | BCF | 50 | 75 | 48 | 0 | — | — | — |
| 7 | Myrcene | ABPZrBn2 | TBCF | 50 | r.t. | 24 | 47 | 3.2 | <1 | — |
| 8 | Styrene | ABPZrBn2 | BCF | 50 | r.t. | 24 | 21 | 1.0 | <1 | — |
| 9 | Styrene | Cp2ZrMe2 | BCF | 50 | r.t. | 0.5 | 43 | 2.2 | 2.0 | 1.5 |
| 10 | ε-CL | ABPZrBn2 | BCF | 100 | r.t. | 2 | 49 | 5.6 | 12.4 | 1.07 |
| 11 | ε-CL | ABPZrBn2 | TBCF | 100 | r.t. | 2 | 47 | 5.3 | 13.3 | 1.04 |
| 12 | ε-CL | ABPZrBn2 | BCF | 100 | 75 | 2 | 100 | 11.4 | 12.4 | 1.19 |
| 13 | PO | ABPZrBn2 | BCF | 200 | r.t. | 2 | 100 | 11.6 | <1.0 | — |
| 14 | PO | ABPZrBn2 | TBCF | 200 | r.t. | 1 | 67 | 7.7 | <1.0 | — |
| 15 | PO | — | BCF | 100 | r.t. | 0.33 | 100 | 5.8 | <1.0 | — |
| 16 | PO | — | TBCF | 100 | r.t. | 0.33 | 100 | 5.8 | <1.0 | — |
While 1/BnB(C6F5)3− was previously shown to polymerise 1-hexene without solvent or in heptane,47,56 here the reactions were performed in toluene solvent at a 1 M monomer concentration. This aligns with reaction conditions often used for cyclic ester ROP, which were selected to facilitate subsequent investigations into the sequential addition of polar monomers for BCP synthesis (vide infra).59–64 Under these adapted conditions, 100 equiv. of 1-hexene was quantitatively converted at room temperature in <20 minutes using ABPZrBn2 with both the BCF and TBCF co-catalysts (Table 1, entries 1–2). The Mn values of the resultant polyhexene (PH) were higher than expected, which is commonly observed with cationic catalysts and is attributed to the inefficient generation of the active catalyst species.59 Control reactions revealed that the neutral ABPZrBn2 pre-catalyst and BCF co-catalyst were individually inactive towards 1-hexene polymerisation (Table S1,† entries 2–3), confirming the cationic polymerisation mechanism. Notably, under identical conditions, 2/MeB(C6F5)3− afforded low-molecular-weight oligomers with a high prevalence of olefinic (vinylene or vinylidene) end groups observed in the 1H NMR spectrum (Table 1, entry 3, Fig. S5†). This corresponds to significant β-hydride elimination reactions,65 which limits the applicability of 2 for the in situ synthesis of BCPs, where a living polymer chain end is required. Notably, the occurrence of β-H elimination using catalyst 1 cannot be fully excluded, however, as the obtained PH showed relatively well-controlled Mn and relatively low quantities of olefinic end groups (<1% vs. the PH resonances), 1/BnB(C6F5)3− was investigated for block copolymerisation studies.
In contrast to the aforementioned Sc- and Y-based catalysts (Fig. 1a–c), which successfully homopolymerised and copolymerised conjugated dienes with cyclic esters, 1/BnB(C6F5)3− was inactive for the polymerisation of 1,3-dienes (myrcene and isoprene), even under relatively harsh reaction conditions and extended reaction times (Table 1, entries 4 and 6). In contrast, using TBCF as the co-catalyst led to oligomerisation at room temperature (entries 5 and 7). Both 1/BnB(C6F5)3− and 2/MeB(C6F5)3− showed activity towards styrene, however, only oligomeric product mixtures were obtained (entries 8–9). Therefore, 1-hexene was selected as the most promising olefinic monomer for further investigations into BCP synthesis.
The homopolymerisation of CL occurred smoothly with catalyst 1 using either BCF or TBCF cocatalysts at room temperature (Table 1, entries 10–11). Increasing the reaction temperature to 75 °C 1/BnB(C6F5)3− gave complete monomer conversion in 2 hours, with well-controlled molecular weights and narrow dispersity (entry 12, Mn,SEC = 12.4 kg mol−1vs. Mn,theo = 11.4 kg mol−1, Đ = 1.19). Catalyst 1/BnB(C6F5)3− also polymerised rac-lactide and β-butyrolactone, albeit with lower conversions and poorer polymerisation control than CL (Table S1,† entries 15–16). This is perhaps unsurprising, as most literature on olefin/cyclic ester block copolymers features poly(ε-caprolactone) (PCL) as the polar block although the reason for this is currently unclear. Specifically, size exclusion chromatography (SEC) analysis revealed a multimodal distribution for poly(lactic acid) and low-molecular-weight oligomers for poly(β-butyrolactone). Therefore, CL was selected as the cyclic ester of choice for copolymerisation studies.
Along with cyclic esters, cyclic ethers (epoxides) were also investigated. Using 1/BnB(C6F5)3− in toluene solvent, propylene oxide (PO), cyclohexene oxide (CHO) and limonene oxide (LO) were all ring-opened with essentially quantitative monomer conversion, although SEC analysis revealed that only oligomers were produced (<1.2 kg mol−1, Table 1, entries 13–14 and Table S1,† entries 21–22). The reactions with CHO and LO were highly exothermic, increasing the reaction temperature above the boiling point of the toluene solvent, and so these monomers were not studied further. Inspection of the 1H NMR spectra for the crude PO polymerisation mixture revealed a diagnostic triplet at 9.72 ppm in CDCl3, attributed to the C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)H unit of propionaldehyde.66 Control reactions using solely the BCF or TBCF co-catalysts also gave quantitative conversion of PO into oligomers and propionaldehyde (Table 1, entries 15–16; Table S1,† entries 19–20), which suggests that organoborane-catalysed isomerisation and oligomerisation of propylene oxide occurs.67,68 Whilst the Zr pre-catalysts and co-catalysts were stirred together for 10 minutes to generate the active cationic species prior to monomer addition, traces of unreacted co-catalyst may be present.9 In contrast, the neutral ABPZrBn2 pre-catalyst showed no activity (Table S1,† entry 17).
O)H unit of propionaldehyde.66 Control reactions using solely the BCF or TBCF co-catalysts also gave quantitative conversion of PO into oligomers and propionaldehyde (Table 1, entries 15–16; Table S1,† entries 19–20), which suggests that organoborane-catalysed isomerisation and oligomerisation of propylene oxide occurs.67,68 Whilst the Zr pre-catalysts and co-catalysts were stirred together for 10 minutes to generate the active cationic species prior to monomer addition, traces of unreacted co-catalyst may be present.9 In contrast, the neutral ABPZrBn2 pre-catalyst showed no activity (Table S1,† entry 17).
Overall, the homopolymerisation studies revealed that catalyst 1 with BCF or TBCF gave high activities and good control over the polymerisation of 1-hexene and CL, and also generated oligomeric poly(propylene oxide) (PPO). On the basis of the potential affinity to form apolar polymers (1-hexene) and polar polymers/oligomers (CL/PO), these three monomers were selected for further studies on the synthesis of poly(olefin-block-ester)s and poly(olefin-block-ether)s.
Sequential monomer addition: homopolymerisation or copolymerisation?
To investigate the potential synthesis of poly(hexene-block-caprolactone), 1-hexene and CL were sequentially added to a solution of 1/BnB(C6F5)3− using optimised conditions from the homopolymerisation studies. 1-Hexene was quantitatively converted (10 minutes, r.t., toluene), then CL was added and the temperature was raised to 75 °C for 1 hour. A variety of 1-hexene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CL ratios were tested (50:50, 50:100 and 100:100), and in all cases, complete conversion of 1-hexene and >75% conversion of CL was observed (Table 2, entries 1a–3b).
:CL ratios were tested (50:50, 50:100 and 100:100), and in all cases, complete conversion of 1-hexene and >75% conversion of CL was observed (Table 2, entries 1a–3b).
| Entry | Equiv. 1-hexene | Conv. 1-hexene (%) | Equiv. M2 | Conv. M2 (%) | M n,th (kg mol−1) | M n, SEC (kg mol−1) | Đ |
|---|---|---|---|---|---|---|---|
| Conditions: 0.0125 mmol of ABPZrBn2 and BCF pre-stirred in toluene for 10 minutes prior to the addition of monomer to afford 1 M concentration of monomer in toluene. SEC – uncorrected values against polystyrene standards.a Bimodal distribution in SEC analysis, the peaks were analysed separately. | |||||||
| 1a | 50 | >99 | — | — | 4.2 | 7.7 | 1.34 |
| 1b | 50 | >99 | 50 CL | 97 | 9.7 | 13.6 | 1.70 |
| 2a | 100 | >99 | — | — | 8.4 | 13.7 | 1.70 |
| 2b | 100 | >99 | 100 CL | 78 | 17.3 | 30.2 | 1.46 |
| 3a | 50 | >99 | — | — | 4.2 | 8.6 | 1.49 |
| 3ba | 50 | >99 | 100 CL | 87 | 14.1 | 47.3/9.6 | 1.12/1.31 |
| 4a | 50 | >99 | — | — | 4.2 | 6.4 | 1.75 |
| 4b | 50 | >99 | 50 PO | >99 | 7.1 | 7.9 | 1.51 |
| 5a | 100 | >99 | — | — | 8.4 | 8.8 | 1.91 |
| 5b | 100 | >99 | 50 PO | >99 | 11.3 | 10.7 | 1.61 |
When using a 100:100 ratio of 1-hexene:CL, the SEC analysis of the resultant polymers indicated an increase in molecular weight compared to the PH samples taken prior to the addition of CL, with the Mn increasing from 13.7 kg mol−1 to 30.2 kg mol−1 (Table 2, entry 2a–b) and with a seemingly monomodal distribution (Fig. 2, top). However, other monomer feed ratios showed bimodal SEC traces (e.g. using a 50:100 ratio of 1-hexene:CL, Fig. 2, bottom, Table 2). This indicates the presence of homopolymers, which was subsequently corroborated by DOSY NMR analysis. The PH100–PCL100 polymerisation (Table 2, entry 3b) clearly shows two distinct diffusion coefficients for the separate PH and PCL polymers (Fig. 3), emphasising the importance of not relying solely on 1H NMR and SEC analysis to characterise BCPs. Indeed, closer inspection of the SEC trace (Fig. 2, top, orange) reveals that the “monomodal” peak overlaps with both the higher Mn PCL100 fraction (Fig. 2, bottom, orange trace, left peak) and the PH50 precursor (Fig. 2, top, blue). Furthermore, no ketone resonance was present in the 13C NMR or the HMBC spectrum (Fig. S9†); a ketone unit would be formed from the insertion of CL into a Zr–C(polyhexene) bond.69 The two homopolymers could be mostly separated via precipitation of the PCL component (in methanol or hexane), or hot hexane washes/Soxhlet extraction (in refluxing hexane for 5 days) to remove the PH component. While pure PH was obtained, 1H NMR analysis of the PCL fraction showed the presence of some residual PH.
 | ||
| Fig. 2 SEC traces of the polymers produced from the sequential polymerisation of 1-hexene and caprolactone using 1/BnB(C6F5)3− catalyst, with 1-hexene:CL ratios of 100:100 (top) and 50:100 eq. (bottom). | ||
 | ||
| Fig. 3 DOSY NMR spectrum of polymer products from the sequential polymerisation of 100 eq. hexene and 100 eq. CL using 1/BnB(C6F5)3−. | ||
Overall, catalyst 1/BnB(C6F5)3− is clearly capable of the one-pot polymerisation of both olefin and cyclic ester monomers through sequential addition. Yet under these conditions, the reaction delivers homopolymers. Cationic 1/BnB(C6F5)3− has been reported to initiate the living polymerisation of 1-hexene through a coordination–insertion mechanism.47,56,61 Here, a similar mechanism occurs in spite of the different reaction conditions, as evidenced by observation of benzyl units in the 1H NMR spectrum. Notably, these resonances have the same diffusion coefficient as PH in the DOSY NMR spectra of the purified homopolymer, providing further support for benzyl-end-capped PH. Following the 1-hexene polymerisation, the organometallic species features a Zr+–C(polyhexene) bond as the active chain end (Scheme 1, centre), assuming that no significant β-hydride elimination occurs. This could either initiate the ROP of CL via nucleophilic attack of the Zr+–C(polyhexene) bond upon a coordinated CL monomer (Scheme 1, left), and/or could initiate the cationic ROP (cROP) of cyclic esters via an activated chain end mechanism (Scheme 1, right).54
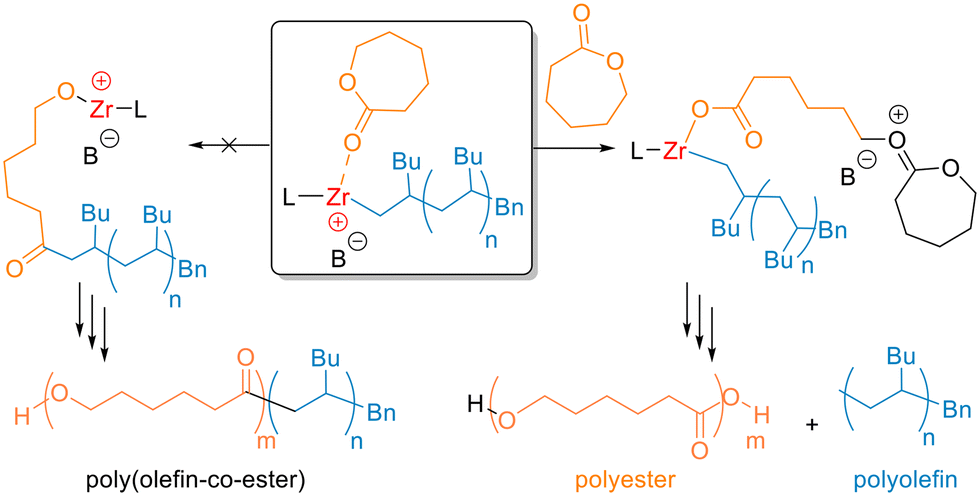 | ||
| Scheme 1 Possible mechanisms for the coordination–insertion (left) and cationic (right) ring-opening of CL following 1-hexene polymerisation (L = amine bisphenolate ligand, B− = BnB(C6F5)3−). | ||
To gain mechanistic insight into the formation of PCL using 1/BnB(C6F5)3−, MALDI-ToF analysis was performed on both the polymer products produced via CL homopolymerisation (Table 1, entry 12) and sequential 1-hexene/CL polymerisations (Table 2, entry 2b). In both cases, two dominant series were detected corresponding to OH/H-capped PCL with Na+ or K+ ions (Fig. S10 and S11†), suggesting a cationic ROP mechanism.54,70 Moreover, no Bn end-groups were observed in the PCL obtained from CL homopolymerisation, which suggests that the Bn group on catalyst 1 does not participate in significant initiation under these reaction conditions (75 °C, toluene). Intriguingly, upon addition of CL to cationic 1, an immediate colour change from yellow to colourless was observed. In contrast, 1 remains yellow upon 1-hexene addition. While the exact reason for this difference remains unclear, this may be due to the migration of the cationic charge from the Zr centre to the propagating end of the PCL chain, with concomitant formation of a Zr–alkoxide species (Scheme 1, right). In 1-hexene polymerisation, the cationic charge remains localised on Zr. Overall, the data suggests that the cationic ROP mechanism occurs instead of initiation from a Zr+–C(polyalkene) bond under these reaction conditions. Notably, experiments performed with the reverse order of monomer addition gave no 1-hexene polymerisation after the PCL was formed. This potentially indicates that the Zr centre is no longer cationic, although 1-hexene coordination may also be blocked by polar functional groups in the PCL structure.
The sequential addition of 1-hexene followed by PO revealed essentially quantitative conversion of both monomers at room temperature (Table 2, entry 4a–b), yet the evidence shows that no BCPs were formed. SEC studies of the product mixtures showed multimodal distributions with the PO component in the oligomeric range (Mn < 1 kg mol−1, Fig. S15†), and MALDI-ToF analysis of the PO oligomers showed H/OH end groups consistent with a cROP mechanism (Fig. S12†). Traces of unreacted BCF may oligomerise/rearrange PO, preventing the formation of BCPs as well as limiting the cROP of PO to the formation of oligomers.67 This observation is significant, as it shows that the co-catalyst is unlikely to be an innocent spectator in the group IV metal-catalysed cROP of epoxides.71
Computational studies
Given the aforementioned diagonal relationship between Sc and Zr, combined with the literature precedence for cationic Sc complexes to produce olefin/cyclic ester BCPs (Fig. 1b and c), we were curious to understand why Zr+–C(polyolefin) complexes appear to undergo cROP under these conditions (Scheme 1, right), while structurally similar Sc+–C(polyolefin) complexes can follow a coordination–insertion pathway (Scheme 1, left). To probe the difference in metal–O and metal–C bonds between structurally similar Zr+ and Sc+ catalysts, DFT studies were performed on simplified metallocene structures (see ESI† for more details). An isobutyl unit was selected to represent the polyolefin chain and the counteranion was omitted to decrease the computational cost (Scheme 2). Moreover, the Me and SiMe3 Cp substituents from the Cp*Sc catalyst reported in the literature (Fig. 1c) were omitted to allow better structural comparison with catalyst 2. The thermodynamic feasibility of the coordination–insertion mechanism for Sc+vs. Zr+ was compared, with two units of CL inserted into the M–C bond (Scheme 2). For the Sc complex an exothermic reaction (ΔG = −54.0 kJ mol−1) was observed, while the identical insertion for the Zr+ analogue was endothermic (ΔG = +11.8 kJ mol−1, see Table S5†). This falls in line with recently reported experimental and DFT studies on the cROP of CL via zirconocene complexes.72,73 The preferential formation of the coordination–insertion Sc product (Scheme 2, top right) may be due to the stronger Sc–O bond (local force constant = 4.33 mDyn Å−1) compared to the analogous Zr–O bond (3.91 mDyn Å−1) (Table S3†).74 Notably, some Zr catalysts are known to be poisoned by polar monomers, which has been attributed to the formation of a strong Zr–O bond hindering any further propagation.75 Here, the DFT calculations show that this may not always be the case. Intriguingly, with these metallocene complexes the Sc–O bond is stronger, which may in fact increase the thermodynamic stability of the product enabling a coordination–insertion ROP mechanism. While our studies focused solely on thermodynamics, a clear difference between the two metals was shown, with Sc being exothermic for coordination–insertion of CL, whereas Zr is endothermic. | ||
| Scheme 2 Calculated free energy differences for the insertion of a ε-caprolactone unit into the Sc and Zr metallocene complexes shown. | ||
Conclusions
In conclusion, two cationic Zr–alkyl complexes based on aminobisphenolate or Cp ligands have been tested for the polymerisation of olefins, cyclic ethers and cyclic esters, including sequential addition studies to investigate the potential one-pot synthesis of polar/apolar block copolymers. While the homopolymerisation of 1-hexene and ε-caprolactone occurred efficiently, SEC and DOSY analyses of polymers with various monomer ratios showed that separate homopolymers were formed. Mechanistic studies revealed that block copolymer formation did not occur, and that instead, cationic ring-opening polymerisation of the cyclic ester takes place under these reaction conditions. DFT calculations show that the ring-opening and insertion of CL into the cationic zirconocene Zr+–C(polyolefin) bond is an endothermic process. In contrast, CL ring-opening and insertion into the analogous Sc+–C(polyolefin) bond is exothermic, which aligns with previous reports for cationic scandium complexes successfully producing diblock copolymers from olefins and cyclic esters. In spite of the diagonal relationship and similarities between Sc and Zr, these differences in bond strengths may hamper the effectiveness of the zirconocene catalyst for BCP synthesis, although the impact of side-reactions such as β-hydride elimination cannot be unequivocally ruled out and this is currently being studied in our laboratory. Overall, this work highlights additional challenges to overcome in synthesising polyolefin/polyester diblock copolymers with cationic Zr+ catalysts.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge Prof. Michael Shaver, Prof. Kyoko Nozaki and Dr Raju Chambenahalli for useful discussions. We also thank the UKRI Future Leaders Fellowship (MR\T042710\1, J. A. G. and E. F.) and the Royal Society (RSG\R1\180101, J. A. G.) for funding, and the Edinburgh Compute and Data Facility (ECDF) for high performance computing access.References
- H. Dau, G. R. Jones, E. Tsogtgerel, D. Nguyen, A. Keyes, Y.-S. Liu, H. Rauf, E. Ordonez, V. Puchelle, H. Basbug Alhan, C. Zhao and E. Harth, Chem. Rev., 2022, 122(18), 14471–14553 CrossRef CAS PubMed.
- A. Dorigato, Adv. Ind. Eng. Polym. Res., 2021, 4(2), 53–69 CAS.
- Y.-Y. Li, X.-Z. Zhang, H. Cheng, J.-L. Zhu, S.-X. Cheng and R.-X. Zhuo, Macromol. Rapid Commun., 2006, 27(22), 1913–1919 CrossRef CAS.
- U. Capasso Palmiero, M. Sponchioni, N. Manfredini, M. Maraldi and D. Moscatelli, Polym. Chem., 2018, 9(30), 4084–4099 RSC.
- G. Desurmont, M. Tanaka, Y. Li, H. Yasuda, T. Tokimitsu, S. Tone and A. J. Yanagase, J. Polym. Sci., Part A: Polym. Chem., 2000, 38(22), 4095–4109 CrossRef CAS.
- A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Chem. Rev., 2016, 116(4), 1969–1992 CrossRef CAS PubMed.
- D. J. Walsh, M. G. Hyatt, S. A. Miller and D. Guironnet, ACS Catal., 2019, 9(12), 11153–11188 CrossRef CAS.
- C. Diaz and P. Mehrkhodavandi, Polym. Chem., 2021, 12(6), 783–806 RSC.
- S. C. Kosloski-Oh, Y. Manjarrez, T. J. Boghossian and M. E. Fieser, Chem. Sci., 2022, 13, 9515–9524 RSC.
- J. Wang, S. Xu, X. Hu, Y. Huo and X. Shi, Organometallics, 2022, 41(2), 115–123 CrossRef CAS.
- L. Pan, K. Zhang, M. Nishiura and Z. Hou, Macromolecules, 2010, 43(23), 9591–9593 CrossRef CAS.
- H.-J. Jung, I. Yu, K. Nyamayaro and P. Mehrkhodavandi, ACS Catal., 2020, 10(11), 6488–6496 CrossRef CAS.
- H. Yasuda, M. Furo, H. Yamamoto, A. Nakamura, S. Miyake and N. Kibino, Macromolecules, 1992, 25(19), 5115–5116 CrossRef CAS.
- G. Desurmont, T. Tokimitsu and H. Yasuda, Macromolecules, 2000, 33(21), 7679–7681 CrossRef CAS.
- L. Wang, D. Cui, Z. Hou, W. Li and Y. Li, Organometallics, 2011, 30(4), 760–767 CrossRef CAS.
- C. Yao, D. Liu, P. Li, C. Wu, S. Li, B. Liu and D. Cui, Organometallics, 2014, 33(3), 684–691 CrossRef CAS.
- Y. Wang, C. Zhou and J. Cheng, Macromolecules, 2020, 53(9), 3332–3338 CrossRef CAS.
- R. A. Collins, A. F. Russell and P. Mountford, Appl. Petrochem. Res., 2015, 5(3), 153–171 CrossRef CAS.
- C. Chen, Nat. Rev. Chem., 2018, 2(5), 6–14 CrossRef CAS.
- H. G. Alt and A. Köppl, Chem. Rev., 2000, 100(4), 1205–1222 CrossRef CAS PubMed.
- A. Buchard, C. J. Chuck, M. G. Davidson, G. Gobius du Sart, M. D. Jones, S. N. McCormick and A. D. Russell, ACS Catal., 2023, 13(4), 2681–2695 CrossRef CAS PubMed.
- A. Stopper, J. Okuda and M. Kol, Macromolecules, 2012, 45(2), 698–704 CrossRef CAS.
- L.-C. Liang, S.-T. Lin, C.-C. Chien and M.-T. Chen, Dalton Trans., 2013, 42(25), 9286–9293 RSC.
- D. T. Jenkins, E. Fazekas, S. B. H. Patterson, G. M. Rosair, F. Vilela and R. D. McIntosh, Catalysts, 2021, 11(5), 551 CrossRef CAS.
- A. J. Chmura, M. G. Davidson, M. D. Jones, M. D. Lunn and M. F. Mahon, Dalton Trans., 2006, 887–889 RSC.
- X. Wang, A. Thevenon, J. L. Brosmer, I. Yu, S. I. Khan, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136(32), 11264–11267 CrossRef CAS PubMed.
- A. L. Zelikoff, J. Kopilov, I. Goldberg, G. W. Coates and M. Kol, Chem. Commun., 2009, 6804–6806 RSC.
- M. Białek and J. Fryga, Polymers, 2021, 13(24), 4456 CrossRef PubMed.
- S. E. Reybuck, A. L. Lincoln, S. Ma and R. M. Waymouth, Macromolecules, 2005, 38(7), 2552–2558 CrossRef CAS.
- C. Hohberger, T. P. Spaniol and J. Okuda, Macromol. Chem. Phys., 2014, 215(20), 2001–2006 CrossRef CAS.
- D. J. Arriola, E. M. Carnahan, P. D. Hustad, R. L. Kuhlman and T. T. Wenzel, Science, 2006, 312(5774), 714–719 CrossRef CAS PubMed.
- S. M. Quan, X. Wang, R. Zhang and P. L. Diaconescu, Macromolecules, 2016, 49(18), 6768–6778 CrossRef CAS.
- R. Dai and P. L. Diaconescu, Dalton Trans., 2019, 48(9), 2996–3002 RSC.
- Z. C. Hern, S. M. Quan, R. Dai, A. Lai, Y. Wang, C. Liu and P. L. Diaconescu, J. Am. Chem. Soc., 2021, 143(47), 19802–19808 CrossRef CAS PubMed.
- S. Pappuru, D. Chakraborty, V. Ramkumar and D. K. Chand, Polymer, 2017, 123, 267–281 CrossRef CAS.
- P. Dobrzynski and J. J. Kasperczyk, J. Polym. Sci., Part A: Polym. Chem., 2006, 44(10), 3184–3201 CrossRef CAS.
- P. J. Dobrzynski, J. Polym. Sci., Part A: Polym. Chem., 2002, 40(10), 1379–1394 CrossRef CAS.
- K. Kostakis, S. Mourmouris, G. Karanikolopoulos, M. Pitsikalis and N. J. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2007, 45(16), 3524–3537 CrossRef CAS.
- V. V. Burlakov, P. Arndt, W. Baumann, A. Spannenberg and U. Rosenthal, Organometallics, 2006, 25(2), 519–522 CrossRef CAS.
- K. P. Kepp, Inorg. Chem., 2016, 55(18), 9461–9470 CrossRef CAS PubMed.
- A. F. Wells, Structural Inorganic Chemistry, Clarendon Press, 2012 Search PubMed.
- L. Pauling, The Nature of the Chemical Bond, 1960, pp. 97–101 Search PubMed.
- K. Mashima, Organometallics, 2021, 40(21), 3497–3505 CrossRef CAS.
- J. Chen, A. Motta, B. Wang, Y. Gao and T. J. Marks, Angew. Chem., Int. Ed., 2019, 58(21), 7030–7034 CrossRef CAS PubMed.
- B. Liu, K. Qiao, J. Fang, T. Wang, Z. Wang, D. Liu, Z. Xie, L. Maron and D. Cui, Angew. Chem., Int. Ed., 2018, 57(45), 14896–14901 CrossRef CAS PubMed.
- S. Gendler, S. Segal, I. Goldberg, Z. Goldschmidt and M. Kol, Inorg. Chem., 2006, 45(12), 4783–4790 CrossRef CAS PubMed.
- E. Y. Tshuva, I. Goldberg, M. Kol, H. Weitman and Z. Goldschmidt, Chem. Commun., 2000, 379–380 RSC.
- E. Y. Tshuva, I. Goldberg, M. Kol and Z. Goldschmidt, Organometallics, 2001, 20(14), 3017–3028 CrossRef CAS.
- E. Villaseñor, R. Gutierrez-Gonzalez, F. Carrillo-Hermosilla, R. Fernández-Galán, I. López-Solera, A. R. Fernández-Pacheco and A. Antiñolo, Eur. J. Inorg. Chem., 2013, 2013(7), 1184–1196 CrossRef.
- J. Jitonnom and W. J. Meelua, Theor. Comput. Chem., 2017, 16(01), 1750003 CrossRef.
- H. Sinn and W. Kaminsky, Ziegler-Natta Catalysis, in Advances in Organometallic Chemistry, ed. F. G. A. Stone and R. West, Academic Press, 1980, vol. 18, pp. 99–149 Search PubMed.
- Y. Ning, Y. Zhang, A. Rodriguez-Delgado and E. Y. X. Chen, Organometallics, 2008, 27(21), 5632–5640 CrossRef CAS.
- M. Teruaki, H. Michiya, O. Kunihiro, M. Makoto and Y. Tohru, Chem. Lett., 1995, 24(8), 737–738 CrossRef.
- M. Hayakawa, M. Mitani, T. Yamada and T. Mukaiyama, Macromol. Chem. Phys., 1997, 198(5), 1305–1317 CrossRef CAS.
- E. Y. Tshuva, M. Versano, I. Goldberg, M. Kol, H. Weitman and Z. Goldschmidt, Inorg. Chem. Commun., 1999, 2(8), 371–373 CrossRef CAS.
- E. Y. Tshuva, I. Goldberg and M. Kol, J. Am. Chem. Soc., 2000, 122(43), 10706–10707 CrossRef CAS.
- Y. Zhang, L. Qu, Y. Wang, D. Yuan, Y. Yao and Q. Shen, Inorg. Chem., 2018, 57(1), 139–149 CrossRef CAS PubMed.
- M. Save and A. Soum, Macromol. Chem. Phys., 2002, 203(18), 2591–2603 CrossRef CAS.
- J. M. Switzer, N. E. Travia, D. K. Steelman, G. A. Medvedev, K. T. Thomson, W. N. Delgass, M. M. Abu-Omar and J. M. Caruthers, Macromolecules, 2012, 45(12), 4978–4988 CrossRef CAS.
- D. K. Steelman, P. D. Pletcher, J. M. Switzer, S. Xiong, G. A. Medvedev, W. N. Delgass, J. M. Caruthers and M. M. Abu-Omar, Organometallics, 2013, 32(17), 4862–4867 CrossRef CAS.
- D. K. Steelman, S. Xiong, P. D. Pletcher, E. Smith, J. M. Switzer, G. A. Medvedev, W. N. Delgass, J. M. Caruthers and M. M. Abu-Omar, J. Am. Chem. Soc., 2013, 135(16), 6280–6288 Search PubMed.
- P. D. Pletcher, J. M. Switzer, D. K. Steelman, G. A. Medvedev, W. N. Delgass, J. M. Caruthers and M. M. Abu-Omar, ACS Catal., 2016, 6(8), 5138–5145 CrossRef CAS.
- J. M. Switzer, P. D. Pletcher, D. K. Steelman, J. Kim, G. A. Medvedev, M. M. Abu-Omar, J. M. Caruthers and W. N. Delgass, ACS Catal., 2018, 8(11), 10407–10418 Search PubMed.
- W. Gruszka, L. C. Walker, M. P. Shaver and J. A. Garden, Macromolecules, 2020, 53(11), 4294–4302 CrossRef CAS.
- G. W. Coates, P. D. Hustad and S. Reinartz, Angew. Chem., Int. Ed., 2002, 41(13), 2236–2257 CrossRef CAS.
- P. Wang, Y. Wang, H. Neumann and M. Beller, Chem. – Eur. J., 2023, 29(8), e202203342 CrossRef CAS PubMed.
- D. Chakraborty, A. Rodriguez and E. Y. X. Chen, Macromolecules, 2003, 36(15), 5470–5481 CrossRef CAS.
- K. Udomsasporn, S. Haesuwannakij, P. Piromjitpong, P. Chumsaeng and K. Phomphrai, Dalton Trans., 2020, 49(41), 14378–14382 RSC.
- J. E. Báez, A. Ramírez-Hernández and Á. Marcos-Fernández, Polym. Adv. Technol., 2010, 21(1), 55–64 CrossRef.
- S. Penczek and J. Pretula, ACS Macro Lett., 2021, 10(11), 1377–1397 CrossRef CAS PubMed.
- E. Farrow, Y. Sarazin, D. L. Hughes and M. Bochmann, J. Organomet. Chem., 2004, 689(24), 4624–4629 CrossRef CAS.
- W. Meelua, M. Linnolahti and J. Jitonnom, RSC Adv., 2024, 14(17), 11715–11727 RSC.
- W. Meelua, T. Wanjai and J. Jitonnom, Sci. Rep., 2024, 14(1), 3952 CrossRef CAS PubMed.
- E. Kraka, W. Zou and Y. Tao, WIREs Comput. Mol. Sci., 2020, 10(5), e1480 CrossRef CAS.
- L. S. Boffa and B. M. Novak, Chem. Rev., 2000, 100(4), 1479–1494 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00170f |
| This journal is © The Royal Society of Chemistry 2025 |