
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAutomation accelerated screening of H-bond-rich iridium photosensitisers for hydrogen generation†
T. Harri
Jones
a,
Christopher F.
Bradshaw
b,
John W.
Ward
 *b and
Barry A.
Blight
*a
*b and
Barry A.
Blight
*a
aDepartment of Chemistry, University of New Brunswick, Fredericton, NB E3B 5A3, Canada. E-mail: B.Blight@unb.ca
bDepartment of Chemistry and Materials Innovation Factory, University of Liverpool, Liverpool, L69 7ZD, UK. E-mail: John.Ward@liverpool.ac.uk
First published on 28th March 2025
Abstract
In the pursuit of a stable hydrogen evolution photosensitiser, we demonstrate the incorporation of a series of our H-bond rich guanidine-styled iridium(III) complexes into a catalytic system. Using automation accelerated catalysis screening techniques we optimised our system quickly and effectively to observe how these strongly H-bonding complexes may perform. Proven to be photo-electronically suitable, and with effective electron transfer abilities evidenced by Stern–Volmer mechanistic studies, the complexes showed modest levels of H2 evolution in comparison to previously investigated photosensitisers with general formula [Ir(C^N)2(N^N)].
1. Introduction
Cyclometalated iridium complexes with the general formula [Ir(C^N)2(N^N)]+ have been shown to have high molar extinction coefficients.1–3 This property makes them desirable candidates as photosensitizers (PS) for the photocatalytic hydrogen generation.4–9 While these catalysts can show turnover numbers (TONs) as high as 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, they are typically short-lived,10 as the catalysts tend to degrade over prolonged periods.11,12 This is due to metal–ligand bonds rupturing in the presence of unstable radicals and anions, causing the cationic ligand moiety of the complex to dissociate.12–14
000, they are typically short-lived,10 as the catalysts tend to degrade over prolonged periods.11,12 This is due to metal–ligand bonds rupturing in the presence of unstable radicals and anions, causing the cationic ligand moiety of the complex to dissociate.12–14
Previous solutions to this shortcoming came from the replacement of the N^N+ ligand with a C^N ligand,15,16 but these complexes show far lower TONs (as high as 3704) than their N^N+ counterparts.16 In recent years this issue has been addressed through the development of N^N chelating ligands that can coordinate to the Ir(III) centre through both π–donor and π*–acceptor bonding interactions,17,18 maintaining the general formula [Ir(C^N)2(N^N)] around the central metal while removing the positive charge.17 This type of ligand system increases the long-term cycle stability of the catalysts by strengthening the binding of the ancillary N^N ligand to the iridium metal.17,19 Increased stability implies more longevity in the catalytic cycle, though they are yet to meet the high turnover numbers and hydrogen evolution activity of the previous generation of cationic Ir(III) catalysts.17
In recent years, we have synthesized various cyclometalated complexes with a unique N^N chelating ligand (1-(1H-benzo[d]imidazole-2-yl)-3-butylguanidine).20–24 This ligand contains a ‘guanine nucleobase mimicking’ highly donating hydrogen-bonding (H-bonding) array.22,25 These complexes contain a guanidine chelating ligand and tend to display high quantum efficiency, while also inviting the coordination of various H-bonding guests which in some cases enhance the optoelectronic properties.23 Based upon this enhancement, we believed the H-bonding array of the complex is capable of efficient energy transfer in the triplet excited state,23 and therefore can be appropriately introduced as an efficient photosensitiser into existing hydrogen evolution (HER) systems.26–28
In order to accelerate the evaluation of these complexes as PS candidates, an automation accelerated screening approach was introduced, through use of the Chemspeed automated synthesis platform (see ESI†).29,30 This allowed for high throughput screening of conditions for these catalytic systems to determine optimum parameters for the HER reaction of our complexes.30 Complexes 1–3 were previously investigated by our group for potential OLED applications and showed promising E(PS*/PS−) and E(PS+/PS*) values inspiring us to investigate them further as PS′. Complexes 4 and 5 were included in this study to gauge the effectiveness of the benzoxazole based C^N ligand's effectiveness when included in the iridium PS, as we hope to further functionalise complexes of this nature into larger supramolecular architectures.
2. Results and discussion
2.1. Synthesis and characterisation
The synthesis of complexes 1–5 and supporting ligands can be found in the supporting information (see section SIII†). All compounds were characterised through a combination of 1H and 13C NMR, ESI+-MS and absorption/emission spectroscopy.2.2. Photophysical properties
The UV-vis spectra of complexes 1–5 in CH3CN are shown in Fig. 2.The absorption spectra share similar bands to those observed in other charge neutral Ir(C^N)2(N^N) complexes.17,22,23 Absorptions from 350–450 nm are caused by spin-allowed metal to ligand charge transfer and ligand to ligand charge transfer 1MLCT/1LLCT. The weak bands extending beyond 450 nm can be attributed to the spin forbidden 3MLCT/3LLCT transitions.31 The sharper absorbances above 350 nm are assigned to ligand centred singlet state (1LC) π–π* transitions.
Complex 3 showed the largest absorption above 350 nm, due to the cyclometalating ligands extended π-system producing more efficient (1LC) π–π* transitions. Complex 1 and 2, based upon ppy and F2ppy ligands show very similar absorptions from above 260 nm, though complex 1 has a sharper absorbance at wavelengths lower than this due to the electron withdrawing fluorine atoms present on the phenyl moiety of the ligand. Complexes 4 and 5 have the larger absorbances at wavelengths above 350 nm showing stronger 1MLCT/1LLCT bands and forbidden 3MLCT/3LLCT transitions. Furthermore, UV-vis spectra were taken in a variety of solvents before and after prolonged excitation by blue (465 nm) and UV light (365 nm), the results of these experiments can be found in ESI† SVI (Table 1).
| Complex | λ exc (nm) | λ em (nm) | Φ PL (%) | τ (μs) | k r (× 105 s−1) | k nr (× 105 s−1) |
|---|---|---|---|---|---|---|
| a All complexes were prepared and measured in argon saturated CH3CN at 298 K. b PLQYs for Ir(III) complexes, determined using a FS5 SC-30 integrating sphere module. c Decay lifetimes measured using TCSPC excited at 365 nm. d The radiative (kr) rate constant was calculated as kr = ΦPL/τPL. e The nonradiative (knr) rate constant was calculated as knr = (1 − kr = ΦPL/τPL). | ||||||
| 1 | 266, 320, 368 | 473, 498 | 89 | 1.84 | 4.8 | 0.6 |
| 2 | 264, 335, 391 | 500, 522 | 67 | 0.68 | 9.9 | 4.9 |
| 3 | 294, 382, 450 | 547, 583 | 12 | 0.02 | 60.0 | 440.0 |
| 4 | 316, 363, 401 | 534, 576 | 30 | 1.75 | 1.7 | 4.0 |
| 5 | 316, 365, 399 | 530, 568 | 34 | 1.92 | 1.8 | 3.4 |
The emission and tuneable properties of complexes 1–3 have been previously characterised in CHCl3 and within PMMA films.22,23 In this study we wished to investigate the quantum efficiencies and energetics of the complexes in a polar solvent system. This was to observe any possible effects upon the guanidine ligand's hydrogen-bonding array when saturated with an H-bond acceptor species like CH3CN.32–34 Fortunately, we saw quantum efficiencies and lifetimes comparable with our previous studies, suggesting minimal quenching by saturation of the H-bond donor ligands.
Complexes 4 and 5 are reported for the first time, and they both show dual emission peaks in the yellow region. Complex 5 exhibits a slightly higher quantum efficiency at 34% compared to complex 4's 30%, while the lifetimes of the complexes 4 and 5 were also very similar (1.92 and 1.75 μs respectively). Most of the complexes we present in this paper have low non-radiative decay showing efficient emissive properties and a high probability of inter-system crossing events, thus high contributions of phosphorescent emission.35,36 Complex 3 is the only candidate we proposed herein with a high non-radiative decay (knr) which is consistent with previous studies of iridium complexes with similar ligand systems.23
2.3. Electrochemical properties
Complexes 1–5 were studied by cyclic voltammetry experiments (see ESI† section SV) and the electrochemical data is reported in Table 2. Each complex exhibits irreversible reduction peaks between −1.45 and −1.31 V. There is little variation in these reduction peaks suggesting that these irreversible reduction peaks should be ascribed to the cyclometalating C^N/C^N− one electron reduction process.37 The Eox is generally ascribed to the Ir(IV)/Ir(III) one electron oxidation process. Eox values vary significantly from 0.93 V to 0.63 V; the values tend to decrease as either the π-system of ligands extends or electron withdrawing groups are removed.38 These changes show that the nature of the cyclometalating C^N ligands have significant implications for the ligand field strength of the metal centre.39–41| Complex | E red (V) | E ox (V) | E 0–0 (eV) | E (PS*/PS−) (V) | E (PS+/PS*) (V) |
|---|---|---|---|---|---|
| a E ox and Ered values were measured by CV in argon saturated CH3CN at 298 K, E0–0 values were estimated from the intersection of the emission-excitation spectra as shown in Fig. 1. b Rehm–Weller calculated E(PS+/PS*) = Eox − E0–0; E(PS*/PS−) = Ered + E0–0. | |||||
| 1 | −1.45 | 0.93 | 2.74 | 1.29 | −1.81 |
| 2 | −1.40 | 0.75 | 2.64 | 1.24 | −1.89 |
| 3 | −1.31 | 0.63 | 2.45 | 1.14 | −1.82 |
| 4 | −1.35 | 0.77 | 2.47 | 1.12 | −1.70 |
| 5 | −1.35 | 0.75 | 2.50 | 1.15 | −1.75 |
We also calculated the photo-redox excited state oxidation E(PS+/PS*) and reduction E(PS*/PS−) potentials through the Rehm–Weller equations, estimating the feasibility of electron transfer during an HER cycle.42 The E(PS*/PS−) values range from 1.29–1.12 eV as (see Table 2) all of these are more positive than the triethylamine (TEA) redox couple E(TEA+/TEA) (+0.93 V).3,8 This difference indicated that all our complexes can accept electrons from the chosen ideal electron donor (TEA). Additionally, the E(red) values for all our complexes are lower than the reduction redox couple of our chosen co-catalyst [Co(bpy)3(PF6)2] (−0.95 V vs. NHE).2,3,17 The data from these redox couples informed us that our complexes should effectively evolve hydrogen when paired with donor TEA and co-catalyst [Co(bpy)3(PF6)2] through the reductive quenching process. This pathway is indicated as the relationship between the PS, ED and catalyst follows the trend; E(PS*/PS−) > E(ED+/ED) and E(PS−/PS) < E(cat/cat−) < E(H+/H2).3,8,17
 | ||
| Fig. 1 H-bond rich cyclometalated iridium complexes 1–5 studied herein as photosensitisers for hydrogen generation. | ||
 | ||
| Fig. 2 a) UV-vis spectra of complexes 1–5 in CH3CN (1.0 × 10−5 M, 298 K). b) Normalised emission-excitation spectra of complexes 1–5 in CH3CN (1.0 × 10−5 M, 298 K), the solid lines represent the emission while the dashed lines represent the absorption. All emission spectra were excited at 300 nm. | ||
2.4. Computational investigation
For further insight into the photophysical and electronic properties of the complexes, time dependent density functional theory (DFT) calculations were undertaken at the B3LYP level of theory with the (6-31G**/LANL2DZ) basis sets.43 Butyl chains on the guanidine ligands were truncated to methyl groups to limit computational cost, likewise the decene chains of complex 5 were shortened to hexyl chains.Frontier energies and the HOMO–LUMO distributions are shown in Fig. 3. In all complexes the HOMO resides on the N^N benzimidazole guanidine ligand's π-orbitals with residuals on the iridium's dπ orbitals. This is due to the high concentration of nitrogen within the guanidine moiety of the ligand.23 HOMO energies range from −4.75 eV in complex 1 to −4.45 eV in complexes 4 and 5. Complex 1's significantly lower HOMO energy can be ascribed to the electron withdrawing effects of the fluorides on the cyclometalating ligands leading to a destabilised LUMO and an increased HOMO–LUMO gap causing a blue shift.23 Our studies also show the LUMOs of the complexes reside on the cyclometalating ligands. This tends to align with our previous studies on guanidine type iridium complexes22,23 along with previous studies on the electronic distribution of the HOMO-LUMO in nitrogen rich Ir(C^N)2(N^N) complexes.17,18
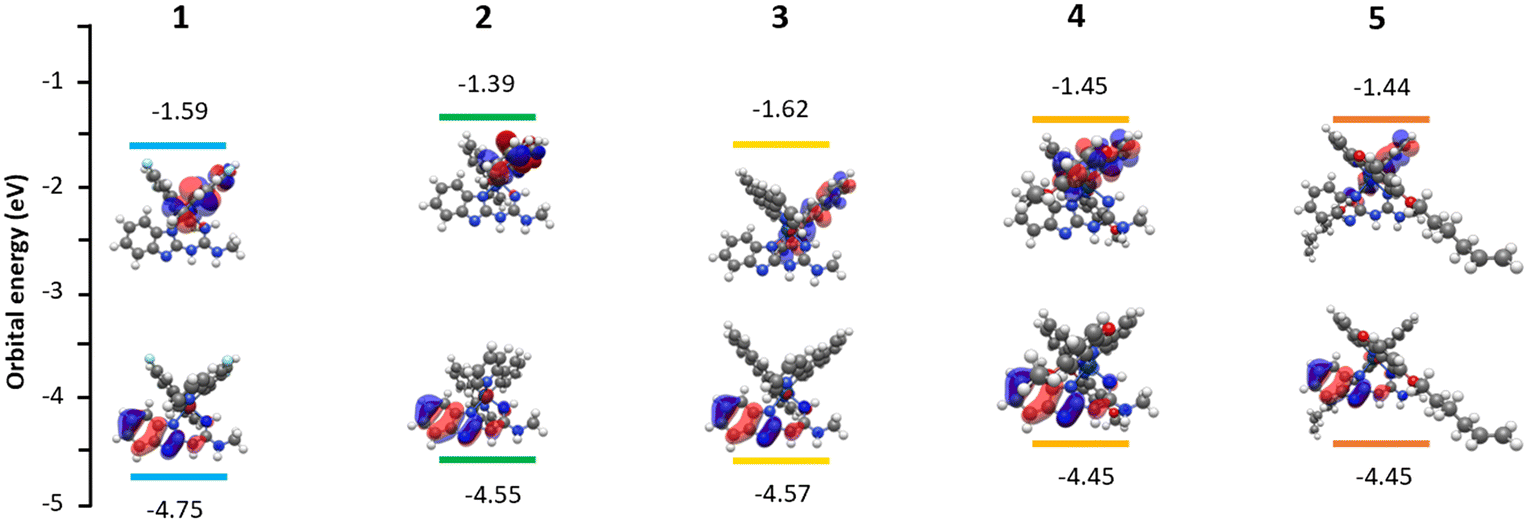 | ||
| Fig. 3 Molecular orbital diagrams, with TD-DFT calculated HOMO–LUMO gap energies for complexes 1–5. | ||
To further investigate the nature of the excited states of the complexes a TD-DFT approach was applied. The first five triplet states were calculated for each complex, estimating major orbital transitions and the electronic nature and energies of the T1–T5 states. The first 25 excited singlet states were also simulated to predict absorption properties and plotted into a UV-vis prediction using Gaussum. A full detailed breakdown of major orbital contributions for computationally predicted absorptions and emissions is available in the ESI† (see section SX).
The variation in the cyclometalating ligands and their electronic structure leads to significant differences in the predicted emission profiles. This agrees with our photophysical measurements, larger ligands with extended π-systems tend to lead to a smaller bandgap and a red shift in emission. Despite significant variations there are still common trends, the T1 and T2 excited states tend to involve the HOMO−4, HOMO−3, HOMO−2 and HOMO all contributing to the LUMO and LUMO+1. The aforementioned HOMOs reside on the iridium dπ orbitals and the π-orbitals of the benzimidazole guanidine ligand.
2.5. Accelerated hydrogen screening
After probing the photo-redox properties of the complexes, we began investigating the hydrogen generation potential. To accelerate the optimisation of the catalytic systems, we decided to incorporate automation accelerated screening techniques inspired by previous studies in the discovery of iridium catalysts.2,7,29 Using our automated approach, we performed all 43 screening experiments in 36 hours, including preparation of stock solutions, photolysis, and GC analysis. Each experiment was performed twice, totalling 86 experiments in all, to ensure consistency in the results (Table S3†). Carefully selected condition variations were informed by previous literature and comparable studies into metallic photo-redox systems,2–5,8,10,17 while the robotic platform was used for the preparation of all samples under an inert atmosphere. A full breakdown of all variations in the condition screening can be found in the ESI† (see section SIX).During the screen, complex 2 was chosen as the photosensitiser to develop the optimum conditions to test all our catalysts, additionally there would be no variation in co-catalyst. We chose to focus on complex 2 and co-catalyst [Co(bpy)3(PF6)2] due to the abundance of literature on similar complexes as photoredox catalysts for hydrogen evolution.2,3,17 Additionally, we kept our electrolyte of LiCl constant for the same reason, though we did experiment with the concentration of the LiCl electrolyte. Control experiments were also performed during the screening, such as removal of photosensitiser and co-catalyst. All controls gave expected results of negligible, or no hydrogen gas generated when catalysts or electron donors were removed from the system.
Several screened conditions identified clear trends; one notable example is pH. Variation of pH revealed an obvious optimum pH of 10. We hypothesised that this was due to the basic nature of the guanidine moiety of complex 2 being proton saturated at low pH and unable to transfer electrons. However, a UV-vis titrations with the catalyst in both organic solvent and water/DMSO mixtures showed little impact on absorption capacity. Further review of the literature revealed that NEt3 is not an appropriate electron donor under acidic conditions as shown by Pellegrin et al.44 Solvent variation showed that organic solvents miscible with water are necessary for hydrogen generation. We observed DMF/H2O to be the best preforming solvent mixture, yet we chose DMSO/H2O for optimum conditions to reduce the excessive use of hazardous DMF.45 Select results of screening experiments can be seen in Fig. 4.
 | ||
| Fig. 4 a) Rate of hydrogen evolution determined from volume of hydrogen generated over a 5-hour period, all conditions were kept consistent with initial screen parameters (see ESI,† S7) while pH was varied as specified. b) Rate of hydrogen evolution determined from volume of hydrogen generated over a 5-hour period, solvent mixture varied while all other conditions were consistent with initial screen. | ||
After the screening optimum conditions were established, and all 5 complexes were tested under these conditions, the results of which (and the nature of the conditions) are shown in Fig. 5. The hydrogen generated in our system was less than anticipated based upon the energetics of our complexes and previously established literature on charge neutral Ir(C^N)2(N^N) photosensitisers. Although we did not expect turnover numbers comparable to cationic iridium complexes with N^N type ligands, based on our electrochemical and photo-physical studies we anticipated a more efficient photocatalytic performance than we observed.
 | ||
| Fig. 5 Hydrogen generation rate of complexes 1–5 in the optimum conditions informed the screening of complex 2. Reaction conditions: 20 μM iridium complex, [Co(bpy)3(PF6)2] 1 mM, DMSO:H2O (1:1, 5 mL), 0.35 M NEt3, LiCl 0.3 M, and pH 10, samples were irradiated for 5 h under an oriel solar simulator 1.0 sun. | ||
Complexes 1 and 2 performed best under the optimum conditions showing the highest rate of hydrogen evolution, due to their E(PS*/PS−) values being significantly more positive than the TEA redox couple E(TEA+/TEA) (+0.93 V).3,8,10 Complexes 3–5 all show similar rates of hydrogen generation, consistent with their E(PS*/PS−) values being similar ranging from 1.12–1.15 V. These values are closer to the TEA redox couple making the oxidation of TEA less energetically favourable with these photosensitisers.
2.6. Quenching kinetics and photo-stability
To understand why our hydrogen generation numbers were lower than expected, we performed quenching studies with both our co-catalyst [Co(bpy)3(PF6)2] and sacrificial electron donor NEt3. Stern-Volmer constants (ksv) were determined from the fit of PL0/PL = 1 + ksv[NEt3]; kq's were determined from the resolved Stern–Volmer equation kq = ksv/τ0. Table 3 shows the summary of these results.| Complex | k sv[NEt3]a M−1 | k q[NEt3]b M−1 s−1 | k sv[Co(bpy)3(PF6)2]a M−1 | k q[Co(bpy)3(PF6)2]b M−1 s−1 |
|---|---|---|---|---|
| a k sv calculated from the fit of the steady-state Stern–Volmer equation (PL0/PL = 1 + ksv[NEt3]). b k q Was calculated from kq = ksv/τ0. | ||||
| 1 | 4 × 103 | 2 × 109 | 9 × 104 | 5 × 1010 |
| 2 | 4 × 103 | 5 × 109 | 5 × 104 | 8 × 1010 |
| 3 | 3 × 103 | 2 × 1011 | 6 × 105 | 3 × 1013 |
| 4 | 5 × 103 | 3 × 109 | 1 × 104 | 6 × 109 |
| 5 | 5 × 103 | 3 × 109 | 1 × 104 | 8 × 109 |
k q values tend to stay consistent in the presence of molar additions of electron donor, NEt3, with little variation and kq's remaining around 109 except for complex 3. Complex 3's high quenching constants for both NEt3 and [Co(bpy)3(PF6)2 may be due to the high non-radiative decay (knr) of the complex. This higher non-radiative decay may allow for faster electron transfer and a larger quenching constant. It also explains the underperformance of complex 3 as a PS, as appropriate kq values for PS compounds with good Stern–Volmer behaviour tend to lie between 108 and 1010.46 The kq's for complexes 1 and 2 being above 1010 also may explain the lower activity, as kq's above this value show potential for static quenching as well as dynamic. This evidences that multiple mechanisms of electron transfer may be involved leading to a lower efficiency of the proposed catalytic cycle.46 The kq's also observe a general trend of shorter lifetime complexes having higher quenching constants, though there is a notable exception to this trend when complex 1 is in the presence of NEt3. Complexes 4 and 5 have the lowest kq in the presence of the cobalt co-catalyst and second and third lowest in the presence of NEt3, where their kq's were determined to be near identical. These lower kq's explain the rather modest catalytic activity from these complexes.
3. Conclusions
Five iridium complexes have been investigated as photosensitisers for hydrogen production. The optoelectronic and photoredox properties were determined through a mixture of analytical and computational techniques. Electrochemical data showed suitable photoredox properties for use in reductive quenching mechanism for water splitting.3 Automation accelerated screening techniques were then employed to determine the optimum conditions for hydrogen generation for complexes 1–5 using complex 2 as a basis for the studies.Upon investigation into the hydrogen generation abilities of all our complexes, we found that although they had the capacity to behave as photosensitisers in the optimised cycle, they showed lower catalytic activity than their optoelectronic and electrochemical profiles would suggest. Quenching studies did confirm that the complexes can accept/donate electrons from co-catalysts and electron donor species but also revealed that some of the compounds may have multiple quenching behaviours (static and dynamic). These quenching studies did show that overall, these iridium complexes are capable of electron transfer with quencher species. This implies that they may yet have applications in other photocatalytic cycles or with different co-catalysts.
Additionally, we hypothesise that with minor modification this series of complexes may have applications in photoredox/hydrogen-bonding cooperative catalysis, due to the guanidine moiety of our complexes. This moiety may allow our complexes to act as a hybrid photosensitiser and hydrogen-bonding catalyst, imparting stereoselective control through the guanidine ligand.46–48
Data availability
The raw data that underpins this work can be accessed through the University of New Brunswick's data repository: https://doi.org/10.25545/KSKKT5.Author contributions
THJ conceptualisation and completed experimental methodology, computational methodology, investigation, validation, formal analysis, writing of manuscript. Experimental implementation, validation CFB. JWW supervised the automation and photocatalysis work, analysed the data and results, writing of the manuscript. BAB conceptualisation, supervision, analysed the data and results, funding procurement, writing of manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Andrew Cooper at the University of Liverpool for the use of the Chemspeed Technologies Sweigher platform, Oriel Solar Simulator 94123A, and Shimadzu GC-2010 Plus GC with HS-20 Headspace Sampler, and Rob Clowes and Dr Sriram Vijayakrishnan for providing training. This work made use of shared equipment located at the Materials Innovation Factory (MIF); created as part of the UK Research Partnership Innovation Fund (Research England) and co-funded by the Sir Henry Royce Institute. We would like to thank Johnson Matthey plc for the award of the platinum group metal materials used in this research. We gratefully acknowledge the University of New Brunswick, and its Department of Chemistry for financial and technical support. This work was fully supported by Natural Science and Engineering Council of Canada (NSERC; DG RGPIN-2018-04021, DG RGPIN-2024-04793) and New Brunswick Foundation for Innovation (NBIF; RAI_2022_074).Notes and references
- D. N. Tritton, F.-K. Tang, G. B. Bodedla, F.-W. Lee, C.-S. Kwan, K. C.-F. Leung, X. Zhu and W.-Y. Wong, Coord. Chem. Rev., 2022, 459, 214390 CrossRef CAS.
- J. I. Goldsmith, W. R. Hudson, M. S. Lowry, T. H. Anderson and S. Bernhard, J. Am. Chem. Soc., 2005, 127, 7502–7510 CrossRef CAS PubMed.
- S. Fan, X. Zong, P. E. Shaw, X. Wang, Y. Geng, A. R. G. Smith, P. L. Burn, L. Wang and S.-C. Lo, Phys. Chem. Chem. Phys., 2014, 16, 21577–21585 RSC.
- Y. Wang, Y. Huang, S. Chen, J. Gao, Y. Zhang, Y. Duan and P. Deng, Inorg. Chem., 2023, 62, 7212–7219 Search PubMed.
- S.-C. Yiu, P.-Y. Ho, Y.-Y. Kwok, X. He, Y. Wang, W.-H. Yu, C.-L. Ho and S. Huang, Chem. – Eur. J., 2022, 28, e202104575 Search PubMed.
- J.-G. Cai, Z.-T. Yu, Y.-J. Yuan, F. Li and Z.-G. Zou, ACS Catal., 2014, 4, 1953–1963 Search PubMed.
- S. DiLuzio, V. Mdluli, T. U. Connell, J. Lewis, V. VanBenschoten and S. Bernhard, J. Am. Chem. Soc., 2021, 143, 1179–1194 Search PubMed.
- G. B. Bodedla, D. N. Tritton, X. Chen, J. Zhao, Z. Guo, K. C.-F. Leung, W.-Y. Wong and X. Zhu, ACS Appl. Energy Mater., 2021, 4, 3945–3951 CrossRef CAS.
- F. Gärtner, S. Denurra, S. Losse, A. Neubauer, A. Boddien, A. Gopinathan, A. Spannenberg, H. Junge, S. Lochbrunner, M. Blug, S. Hoch, J. Busse, S. Gladiali and M. Beller, Chem. – Eur. J., 2012, 18, 3220–3225 Search PubMed.
- D. R. Whang, K. Sakai and S. Y. Park, Angew. Chem., Int. Ed., 2013, 52, 11612–11615 Search PubMed.
- L. L. Tinker, N. D. McDaniel, P. N. Curtin, C. K. Smith, M. J. Ireland and S. Bernhard, Chem. – Eur. J., 2007, 13, 8726–8732 Search PubMed.
- Y.-J. Yuan, Z.-T. Yu, H.-L. Gao, Z.-G. Zou, C. Zheng and W. Huang, Chem. – Eur. J., 2013, 19, 6340–6349 Search PubMed.
- S. H. Wadman, J. M. Kroon, K. Bakker, R. W. A. Havenith, G. P. M. van Klink and G. van Koten, Organometallics, 2010, 29, 1569–1579 CAS.
- F.-M. Hwang, H.-Y. Chen, P.-S. Chen, C.-S. Liu, Y. Chi, C.-F. Shu, F.-I. Wu, P.-T. Chou, S.-M. Peng and G.-H. Lee, Inorg. Chem., 2005, 44, 1344–1353 CrossRef CAS PubMed.
- Z.-T. Yu, Y.-J. Yuan, J.-G. Cai and Z.-G. Zou, Chem. – Eur. J., 2013, 19, 1303–1310 CAS.
- Y.-J. Yuan, Z.-T. Yu, J.-G. Cai, C. Zheng, W. Huang and Z.-G. Zou, ChemSusChem, 2013, 6, 1357–1365 Search PubMed.
- Y. Zhou, P. He, X.-F. Mo, C. Liu, Z.-L. Gan, H.-X. Tong and X.-Y. Yi, Inorg. Chem., 2021, 60, 6266–6275 Search PubMed.
- Y. Wang, X. Zhao, Y. Zhao, T. Yang, X. Liu, J. Xie, G. Li, D. Zhu, H. Tan and Z. Su, Dyes Pigm., 2019, 170, 107547 CrossRef CAS.
- J. A. Flores, J. G. Andino, N. P. Tsvetkov, M. Pink, R. J. Wolfe, A. R. Head, D. L. Lichtenberger, J. Massa and K. G. Caulton, Inorg. Chem., 2011, 50, 8121–8131 Search PubMed.
- B. Balónová and B. A. Blight, Front. Chem., 2021, 9, 712698 Search PubMed.
- S. J. Thomas, B. Balónová, J. Cinatl Jr., M. N. Wass, C. J. Serpell, B. A. Blight and M. Michaelis, ChemMedChem, 2020, 15, 349–353 Search PubMed.
- B. Balónová, D. R. Martir, E. R. Clark, H. J. Shepherd, E. Zysman-Colman and B. A. Blight, Inorg. Chem., 2018, 57, 8581–8587 CrossRef PubMed.
- B. Balónová, T. H. Jones, A. E. True, S. M. Hetherington and B. A. Blight, RSC Adv., 2024, 14, 34288–34297 RSC.
- W. Da, T. H. Jones, J.-B. Lin and B. A. Blight, Can. J. Chem., 2023, 101, 177–185 CrossRef CAS.
- M. Chahkandi, B. Madani Khoshbakht and M. Mirzaei, Comput. Theor. Chem., 2016, 1095, 36–43 CAS.
- S. Sobottka, M. Nößler, A. L. Ostericher, G. Hermann, N. Z. Subat, J. Beerhues, M. Behr-van der Meer, L. Suntrup, U. Albold, S. Hohloch, J. C. Tremblay and B. Sarkar, Chem. – Eur. J., 2020, 26, 1314–1327 CAS.
- S. Masaoka, Y. Mukawa and K. Sakai, Dalton Trans., 2010, 39, 5868–5876 Search PubMed.
- D. Kim and T. S. Teets, Chem. Phys. Rev., 2022, 3, 021302 Search PubMed.
- T. F. Jaramillo, A. Ivanovskaya and E. W. McFarland, J. Comb. Chem., 2002, 4, 17–22 Search PubMed.
- W. Zhang, M. Yu, T. Liu, M. Cong, X. Liu, H. Yang, Y. Bai, Q. Zhu, S. Zhang, H. Gu, X. Wu, Z. Zhang, Y. Wu, H. Tian, X. Li, W.-H. Zhu and A. I. Cooper, Nat. Synth., 2024, 3, 595–605 Search PubMed.
- P.-N. Lai, C. H. Brysacz, M. K. Alam, N. A. Ayoub, T. G. Gray, J. Bao and T. S. Teets, J. Am. Chem. Soc., 2018, 140, 10198–10207 CAS.
- A. Ganguly, S. Jana, S. Ghosh, S. Dalapati and N. Guchhait, Spectrochim. Acta, Part A, 2013, 112, 237–244 Search PubMed.
- Y. Liu, X. Tao, F. Wang, J. Shi, J. Sun, W. Yu, Y. Ren, D. Zou and M. Jiang, J. Phys. Chem. C, 2007, 111, 6544–6549 CrossRef CAS.
- Y. Zhou, G. Baryshnikov, X. Li, M. Zhu, H. Ågren and L. Zhu, Chem. Mater., 2018, 30, 8008–8016 CrossRef CAS.
- Q. Peng, Q. Shi, Y. Niu, Y. Yi, S. Sun, W. Li and Z. Shuai, J. Mater. Chem. C, 2016, 4, 6829–6838 Search PubMed.
- R. E. Harding, S.-C. Lo, P. L. Burn and I. D. W. Samuel, Org. Electron., 2008, 9, 377–384 Search PubMed.
- H. J. Bae, J. Chung, H. Kim, J. Park, K. M. Lee, T.-W. Koh, Y. S. Lee, S. Yoo, Y. Do and M. H. Lee, Inorg. Chem., 2014, 53, 128–138 Search PubMed.
- M. Hebda, G. Stochel, K. Szaciłowski and W. Macyk, J. Phys. Chem. B, 2006, 110, 15275–15283 CrossRef CAS PubMed.
- A. J. Wilkinson, H. Puschmann, J. A. K. Howard, C. E. Foster and J. A. G. Williams, Inorg. Chem., 2006, 45, 8685–8699 CAS.
- X. Qu, Y. Liu, G. Godefroid, Y. Si, X. Shang, X. Wu and Z. Wu, Eur. J. Inorg. Chem., 2013, 2013, 3370–3383 CrossRef CAS.
- H.-R. Tsai, K.-Y. Lu, S.-H. Lai, C.-H. Fan, C.-H. Cheng and I.-C. Chen, J. Phys. Chem. C, 2011, 115, 17163–17174 Search PubMed.
- A. Rosspeintner, D. R. Kattnig, G. Angulo, S. Landgraf and G. Grampp, Chem. – Eur. J., 2008, 14, 6213–6221 Search PubMed.
- Assessment of the “6-31+G** + LANL2DZ” Mixed Basis Set Coupled with Density Functional Theory Methods and the Effective Core Potential: Prediction of Heats of Formation and Ionization Potentials for First-Row-Transition-Metal Complexes | The Journal of Physical Chemistry A, https://pubs.acs.org/doi/10.1021/jp807643p, (accessed 14 December 2024).
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CAS.
- Dimethylformamide [MAK Value Documentation, 2016] - Hartwig - Major Reference Works - Wiley Online Library, https://onlinelibrary.wiley.com/doi/full/10.1002/3527600418.mb6812e6017, (accessed 14 December 2024).
- X. L. Soto and J. R. Swierk, ACS Omega, 2022, 7, 25532–25536 Search PubMed.
- C. Jiang, W. Chen, W.-H. Zheng and H. Lu, Org. Biomol. Chem., 2019, 17, 8673–8689 CAS.
- D. Uraguchi, Y. Kimura, F. Ueoka and T. Ooi, J. Am. Chem. Soc., 2020, 142, 19462–19467 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00176e |
| This journal is © The Royal Society of Chemistry 2025 |