
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of Co–Ga2O3 interfaces in methane dry reforming†
Thomas F.
Winterstein
 a,
Christoph
Malleier
a,
Asghar
Mohammadi
a,
Roham
Talei
b,
Guido
Schmitz
b,
Nicolas
Bonmassar
b,
Jesus
Andrade
c,
Marc
Armbrüster
cd and
Simon
Penner
*a
a,
Christoph
Malleier
a,
Asghar
Mohammadi
a,
Roham
Talei
b,
Guido
Schmitz
b,
Nicolas
Bonmassar
b,
Jesus
Andrade
c,
Marc
Armbrüster
cd and
Simon
Penner
*a
aInstitute of Physical Chemistry, University of Innsbruck, Innrain 52c, A-6020 Innsbruck, Austria. E-mail: simon.penner@uibk.ac.at; Tel: +4351250758003
bInstitute for Materials Science, University of Stuttgart, Heisenbergstr. 3, 70569 Stuttgart, Germany
cMaterials for Innovative Energy Concepts, Chemnitz University of Technology, Straße der Nationen 62, 09111 Chemnitz, Germany
dSolid State and Material Chemistry, Technical University Darmstadt, Peter-Grünberg Str. 12, 64287 Darmstadt, Germany
First published on 7th May 2025
Abstract
As the combination of Co with other non-noble metals is a viable way to improve the catalytic properties of Co in methane dry reforming (DRM), we studied an impregnated Co3O4/β-Ga2O3 powder catalyst to understand the influence of Ga and the catalytic role of the Co–Ga2O3 interface and the intermetallic compound CoGa in DRM. Co3O4/β-Ga2O3 undergoes a series of structural transformations during activation by reduction in hydrogen and under DRM conditions. Contact to the CO2/CH4 mixture without hydrogen pre-reduction yields CoGa2O4 spinel particles encrusting β-Ga2O3 without significant DRM activity. Hydrogen reduction transforms Co3O4/β-Ga2O3 initially to α-Co/β-Ga2O3, before it induces reactive metal–support interaction leading to the formation of bimetallic CoGa particles on β-Ga2O3. Subsequent improved DRM activity can be correlated to the decomposition of the intermetallic compound CoGa: according to operando X-ray diffraction CoGa re-transforms into α-Co/β-Ga2O3 during DRM. Hydrogen pre-reduction is a prerequisite for high DRM activity on Co3O4/β-Ga2O3, where intermediarily formed CoGa is decomposed under reaction conditions yielding a pronounced increase in the activity rivalling established noble metal and non-noble metal catalysts. A particular advantage of β-Ga2O3 is the suppression of coking and Co deactivation, as observed on a Ga-free Co/SiO2 catalyst.
1. Introduction
Dry reforming of methane (DRM) is a viable way to transform the two harmful greenhouse gases carbon dioxide (CO2) and methane (CH4) into useful syngas (i.e., a hydrogen (H2) – carbon monoxide (CO) mixture). DRM is a strongly endothermic reaction and, thus, requires rather high reaction temperatures of 600–1000 °C.1–3 The main DRM reaction (eqn (1))| CO2 + CH4 ↔ 2H2 + 2CO | (1) |
Material-wise, non-noble metal catalysts based on Ni or Co have evolved as alternatives to excellently performing, yet expensive noble metal-based catalysts.1,3–6 Whereas Ni is favored due to its outstanding catalytic DRM performance, large-scale application is hindered by sintering and coking, which diminishes the activity quickly. While slightly less active than Ni, Co offers better anti-coking properties and higher sintering stability.7–9 Thus, Co-based materials are considered as one of the most promising future DRM materials.4 Several approaches were followed in the past decade to improve the catalytic properties of Co-based catalysts, especially with respect to enhancing their oxidation resistance.4 The addition of a second metal (e.g., Ni, Ce, Fe or noble metals) can significantly improve the binding strength of adsorbed oxygen species.10–15 Specifically, co-alloying with Ni or Fe was shown to improve the dispersion of metal species and to accordingly enhance the sintering resistance, increase the amount of active sites for carbon dioxide or methane activation and beneficially influence the redox properties.16–20 As such, Co-based catalysts have been developed rapidly to improve their catalytic and physico-chemical characteristics.
As a viable strategy to improve the catalytic performance of supported Co-based catalysts in DRM, exploitation of an enhanced metal–support interaction strength has been pointed out. Derived from general principles of metal–support interaction processes,21 upon reduction such an interaction may manifest itself in three possible ways: (i) strong metal–support interaction, i.e., coating of active Co metal by a partially reduced support, (ii) electronic metal–support interaction, i.e., electron transfer between Co metal and support and (iii) reactive metal–support interaction leading to the formation of Co-based alloys or intermetallic compounds by reaction of the supported Co nanoparticles with the fully reduced support.21 Regarding the latter, research has been particularly devoted to bimetallic Co-based materials.4 Generally, alloys and intermetallic compounds are one of the most rapidly evolving materials classes with prime importance in a variety of heterogeneously catalyzed reactions.22–25
A topic that is connected to reactive metal–support interaction is the reversibility of alloy and intermetallic compound formation and their potential structural stability.26 Those compounds, also if formed by reduction of a supported metal catalyst (i.e., oxide-supported alloy or intermetallic compound particles), can be structurally unstable depending on the experimental conditions. Preferential leaching of one component may lead to decomposition and formation of an intermetallic compound – oxide (partial decomposition) or a metal–oxide interface (full decomposition).26 In either case, the alloy or intermetallic compound particles act as a catalyst precursor, which is activated upon decomposition. Although at first glance detrimental, this process can be useful, if carried out in a controlled way. Closely connected to the process of exsolution of metal particles from a host lattice,27 steered decomposition might lead to extended metal–oxide interfaces with controlled particle sizes, better-distributed or better-anchored particles with improved sinter stability or coking properties.26 This decomposition of intermetallic compounds and particles has been frequently observed in a variety of reactions,26 including methanol steam reforming (e.g., Cu51Zr14,28–30 ZnPd,31–35 GaPd2,36–40 InPd,40–42 or Cu2In (ref. 43)) or methane dry reforming (e.g., Ni5Zr,44–46 or Pd2Zr (ref. 46–48)). The common denominator is the delicate balance between high (no or only partial decomposition) and low stability (full decomposition). This was scrutinized, e.g., for Cu/In2O3,43 Pd/In2O3 (ref. 49) and Pt/In2O3 (ref. 50) previously. Catalytic-wise, such a decomposition often invokes a so-called bifunctional synergism with shared catalytic duties of reactant activation. With respect to DRM, methane activation usually proceeds on the metal surface, carbon dioxide is accordingly adsorbed and activated on acidic or basic oxide support sites.1–3 A delicate balance of the latter is important, as too strong acidic sites facilitate carbon formation through easier methane decomposition and too strong basic sites promote the Boudouard reaction and the oxidation of active metal.4
We have, thus, selected Co as the lead-element, as its DRM activity rivals that of Ni and the deactivation of Co-based materials in DRM is essentially connected to the strongly size-dependent balance between coking and cobalt oxidation.4,51 Selection of the second component is driven by the eventual co-operative interaction between the two metals and by the possibility to enter a state of reactive metal–support interaction. For Ga-based compounds, this has been already shown for Pd/Ga2O3.38,39 In due course, as Ga has a significantly higher oxidation tendency compared to Co,52 we expect an eventually formed Co–Ga intermetallic compound53,54 to at least partially decompose under DRM conditions into Co and Ga2O3. To assess the complex structural dynamics of eventual reactive metal–support interaction and to study the reversibility of Co–Ga intermetallic compound formation, we use a conventionally wet-impregnated Co3O4/β-Ga2O3 powder material. β-Ga2O3 as an amphoteric oxide with neither too strong acidic nor basic sites55 is expected to yield an optimum surface-chemical compromise to activate carbon dioxide and to facilitate the dehydrogenation of methane, thus providing high DRM activity.56 In addition, recent experiments on a Rh/Ga2O3 photothermal catalyst for DRM applications pointed out the important role of Ga2O3 defects for CO2 activation.57 The results are expected to eventually pave the way to accordingly assess the redox activation and DRM properties of single-phase Co–Ga intermetallic compounds, as pointed out for, e.g., the Cu–In system already.43 Co3O4/β-Ga2O3 is subjected to operando and in situ structural characterization by powder X-ray diffraction (PXRD), atomically resolved (scanning) transmission electron microscopy (STEM) – EDX (energy-dispersive X-ray spectroscopy) measurements and X-ray photoelectron spectroscopy, and accordingly correlated to DRM activity for establishing reliable structure–activity correlations.
2. Methods and materials
2.1. Synthesis of materials
The Co3O4/β-Ga2O3 sample was prepared by wet impregnation. 1.8 g β-Ga2O3 were added to a Co(NO3)2·6H2O solution (referenced to 0.003394 mol Co dissolved in 150 mL water, yielding a Co concentration of 2.3 mol L−1) under constant vigorous stirring to obtain a nominal content of 10 wt% Co3O4. Subsequently, the water was slowly evaporated by heating to 60 °C at a rate of 2 K min−1. The remaining powder was collected, ground and calcined at 600 °C for 2 h in air (heating rate: 10 K min−1). PXRD patterns (cf.Fig. 2) indicate the sole presence of the Co3O4/β-Ga2O3 starting material. Co/SiO2 (nominal content 10 wt% Co3O4) was prepared the same way using Aerosil 380 powder (Degussa) and the same nominal Co3O4 loading.Bulk intermetallic CoGa was used as reference for the XPS measurements and 1 g was obtained by high-temperature synthesis. Stoichiometric amounts of Co (pieces, ChemPur 99.9%) and Ga (pellets, ChemPur, 99.999%) were mixed in a glassy carbon crucible and subjected to high-frequency heating (TruHeat HF 5005, TRUMPF Hüttinger) in inert argon atmosphere to 1250 °C. After 3 minutes, the sample was allowed to cool to ambient temperature. Characterisation by XRD resulted in the aimed-for single-phase CoGa sample with the CsCl-type of crystal structure.58
2.2. Structural characterization
Ex situ structural analysis of benchmark materials after selected treatments was carried out using a Rigaku SmartLab-SE instrument in Bragg–Brentano geometry (Co-Kα, λ = 1.7890 Å) using a D/teX Ultra 250 compound silicon strip 1D-detector (Rigaku, Tokyo, Japan). The ground sample was placed on a glass-sample holder and the patterns were recorded in a range of 2θ 5° to 90° with a step width of 0.01°. The program TOPAS 5.0 by Bruker was utilized to analyze the X-ray diffraction patterns using Rietveld refinement with a full axial model. A double-Voigt approach was applied to calculate the crystallite sizes.
In situ and operando X-ray diffraction experiments on Co3O4/β-Ga2O3 were performed using a Rigaku ReactorX on a Rigaku SmartLab XE 3 kV in Bragg–Brentano geometry (Co Kα radiation, λ = 1.17890 Å, D/teX Ultra 250 1D-detector). The reactor chamber was flushed with He with a flow rate of 40 mL min−1 for one hour before starting the experiments. These consisted of two consecutive steps. The first step was a reduction with hydrogen at 800 °C, using a gas mixture of H2/He (10% vol. H2) with a flow rate of 40 mL min−1. Before starting the reduction, an XRD scan from 2θ 5° to 100.5° was conducted. The mixture of H2/He was subsequently admitted to the system 15 min before starting to heat the sample with a temperature ramp of 25 K min−1 to 800 °C. Every minute, continuous XRD scans in the range of 2θ 48° to 52° were performed. At 800 °C, a full XRD scan was performed from 2θ 5° to 100.5°. The system was then cooled down to 50 °C with a temperature ramp of 25 K min−1, flushed with He with a flow rate of 40 mL min−1 before another full XRD scan was recorded. Subsequently, the DRM step was conducted, starting from 50 °C to at 800 °C (heating rate 5 K min−1) using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 gas mixture of CO2, CH4 and He with a flow rate of 20 mL min−1 each. During heating, every minute continuous XRD scans were performed from 2θ 48° to 52° and a full scan was performed at 800 °C. A final full XRD scan was taken after the system was cooled down to ambient temperature with 25 K min−1 and flushing with He. A mass spectrometer (GSD 350 Omnistar, Pfeiffer Vacuum) was used to follow the changes in the gas mixtures, as well to track products during the reduction and DRM steps. CO2, CO, H2O, CH4, He and H2 were assigned to m/z 44, 28, 18/17, 16, 4 and 2, respectively. The data were normalized using He as internal reference and the signals were smoothed with the Savitzky–Golay filter (20 point window and second polynomial order).
:1:1 gas mixture of CO2, CH4 and He with a flow rate of 20 mL min−1 each. During heating, every minute continuous XRD scans were performed from 2θ 48° to 52° and a full scan was performed at 800 °C. A final full XRD scan was taken after the system was cooled down to ambient temperature with 25 K min−1 and flushing with He. A mass spectrometer (GSD 350 Omnistar, Pfeiffer Vacuum) was used to follow the changes in the gas mixtures, as well to track products during the reduction and DRM steps. CO2, CO, H2O, CH4, He and H2 were assigned to m/z 44, 28, 18/17, 16, 4 and 2, respectively. The data were normalized using He as internal reference and the signals were smoothed with the Savitzky–Golay filter (20 point window and second polynomial order).
2.3. Catalytic testing
200 mg of sample powder was placed in a bed of silica wool inside a 7 mm (inner diameter) silica tube flow reactor for both H2 pre-treatment and catalytic measurements. A Linn HighTerm FRH 25/150/1100 furnace was used for all heating cycles. H2 treatments consisted of a continuous isothermal flow of 20 mL min−1 H2 at 800 °C for 10 min after heating the sample in the H2 stream from 25 °C to 800 °C with a rate of 20 K min−1. After the hydrogen pre-treatments, the samples were re-cooled in hydrogen to 25 °C before switching to the DRM mixture. For catalytic measurements CH4, CO2 and He were mixed 1:1:1 with a flow rate of 20 mL min−1 each and a heating rate of 10 K min−1 from 25 °C to 800 °C, followed by an isothermal period at 800 °C for 30 min. An external S-type thermocouple was placed in close contact to the reactor tube to ensure the correct temperature reading. The output gas was directly detected by an on-line quadrupole mass spectrometer (Balzers QME 125). CO2, CH4, CO, H2 and He were measured at their respective m/z ratios of m/z 44, 28, 16, 4 and 2. The He signal was used to normalize the gas mixture. Relevant fragmentation patterns have been considered in the analysis. To minimize condensation from potential H2O formation, silica gel was placed at the exhaust of the furnace. For the display of the catalytic data, we show the conversion of the relevant signals as a qualitative measure of the catalytic activity, as due to the ongoing structural transformations during the DRM reaction, active-site-normalized reaction rates and consequently, TOF values, cannot be reliably calculated. The conversion for each gas phase is calculated as a fraction of the mass spectrometer (MS) ion current [E−10 A] divided by the MS signal at room temperature after calibrating the flow rate of each gas phase. The measured He signal was used to correct fluctuations in the MS signals. For CO2 the calculation is as follows:Since the CO2 conversion is indicative of CO, and CH4 conversion of H2 production, the H2/CO ratio was calculated from these conversions, respectively.

2.4. (Scanning) transmission electron microscopy
We performed STEM measurements on a probe-corrected FEI Spectra300. All specimens were prepared by the drop-casting method, where a suspension consisting of isopropanol and to-be-investigated particles is dropped on a lacey carbon grid. We used high-angle annular dark field (HAADF) imaging in combination with energy-dispersive X-ray (EDX) spectroscopy (specimen-tilt independent Super-X EDS-system) for the characterization of the morphology and the chemical composition of the catalysts. For the quantitative EDXS results we used the Schreiber–Wims ionization cross-section and a parabolic background model, which are implemented into the Velox software.Scanning electron microscopy (SEM) and corresponding energy-dispersive X-ray spectroscopy measurements were performed on a field-free analytical TESCAN Clara ultra-high-resolution scanning electron microscope operated at 10 kV. EDX maps were collected using an Oxford Ultim Max 65 mm2 detector.
2.5. X-ray photoelectron spectroscopy
Chemical analysis of the near-surface region was carried out using a Thermo Fisher X-ray spectrometer with Al-Kα radiation. For all measurements, samples were fixed on stainless steel holders using conductive carbon tape. Qualitative analysis was based on the Ga 2p, Co 2p, O 1s and C 1s high-resolution spectra. Chemical shifts were internally calibrated to the O 1s component at 531.8 eV. Fitting of the spectra by different Co 2p components and oxidation states was performed using literature-reported constraints for the full-width-at-half maximum and the binding energies, where applicable.59 Background correction has been done using Tougaard-type functions.3. Results and discussion
3.1. Dry reforming performance of Co3O4/β-Ga2O3
We have performed methane dry reforming experiments on two different samples: Co3O4/β-Ga2O3 with and without hydrogen pre-reduction and referenced the catalytic activity to a corresponding Ga-free Co3O4/SiO2 catalyst with the same nominal Co3O4 loading (Fig. 1). Without hydrogen pre-reduction (Fig. S1†), the sample is hardly active: limited catalytic activity is observed at around 700 °C, and conversions of CO2 and CH4 of only 10% and 5% are reached after the 30 min isothermal section at 800 °C. The deviation of the CO2 and CH4 profiles already indicate a competing reverse water–gas shift activity, most likely mediated by the β-Ga2O3 support. Reference DRM profiles of β-Ga2O3, SiO2, the blank reactor filled with quartz wool and Co3O4 are provided in Fig. S2.† No activity is observed for the first three. Panel d in Fig. S2† (pure Co3O4) reveals that the situation is more complex: as anticipated, Co3O4 itself is DRM active starting at around 740 °C. CO2 and CH4 conversions of ca. 50% and 40% are observed. Beyond 760 °C, the catalyst strongly deactivates by coking. If supported on β-Ga2O3, the activity of Co3O4 is also affected by the ongoing transformation into the DRM-inactive CoGa2O4 spinel phase (see Fig. S2e†). Synthesis temperatures for this spinel have been reported as low as 700 °C,60 so the active DRM window directly coincides with the formation temperature of the spinel phase. Previous work has shown, that β-Ga2O3 is an efficient water–gas shift catalyst above 600 °C, capable of catalyzing this reaction via two separate reaction mechanisms: a formate-bound one, and a more vacancy-dominated pathway.61 Hydrogen reduction prior to DRM operation (Fig. 1a and b) changes the scenario drastically. We have performed hydrogen reduction at two different maximum temperatures: 1 h at 550 °C and 10 min at 800 °C, respectively. As will be discussed in the context of Fig. 2, the difference between the two pre-treatments is that reduction at 550 °C causes only reduction of Co3O4 to Co metal, whereas the latter induces the formation of a CoGa intermetallic compound before DRM operation. Starting with the DRM activity in the first DRM cycle after hydrogen pre-reduction at 800 °C (Fig. 1b), after a period of slow activation between 500 °C and 650 °C, the material becomes very active. CO2 and CH4 conversion levels of 80% and 70% are reached in the isothermal period at 800 °C, respectively. The considerably higher CO2 conversion is once again notable, getting increasingly more pronounced as the reaction temperature increases. This again points to the selectivity-spoiling reverse water–gas shift reactivity of β-Ga2O3.61 Put into literature perspective, within the framework of slightly different experimental conditions, the Co3O4/β-Ga2O3 catalyst rivals similar Co-based catalysts modified with either noble- or non-noble metals. Specifically, Co3O4/β-Ga2O3 after hydrogen reduction consistently outperforms Ru-Co catalysts supported on TiO2 or α-Al2O3 (for the former, CO2 and CH4 conversions of 75% and 55% were reported at 850 °C respectively; for the latter CO2 and CH4 conversions of 35% and 29% were reported at 900 °C (ref. 4)) and equals those supported on mesoporous SiO2 (CH4 conversion 74% (ref. 4)). In due course, the performance of Co3O4/β-Ga2O3 matches or exceeds Fe-doped Co alloy particles supported on activated carbon materials or those exsolved from mixed or layered perovskite materials like GdCo0.5Fe0.5O3 (CH4 and CO2 conversions both below 30% at 800 °C (ref. 62)) or PrBaFeCoO5+δ (CH4 and CO2 conversion of <20% and 36% at 900 °C, respectively4). | ||
| Fig. 1 Methane dry reforming profiles of Co3O4/β-Ga2O3 during DRM operation after hydrogen reduction at 550 °C for 1 h (panel a) and after prior hydrogen reduction at 800 °C for 10 min (panel b). Panel c shows the DRM performance of Co/SiO2 after initial hydrogen reduction at 450 °C for 10 min. Sample mass: 200 mg each, GHSV: 18 L g−1 h−1, heating rate: 10 K min−1. The respective H2/CO ratios are highlighted on the right axis. | ||
 | ||
| Fig. 2 XRD patterns of Co3O4/β-Ga2O3 in panel a: as-calcined state, panel b: after hydrogen reduction treatment at 550 °C, panel c: after hydrogen reduction treatment at 800 °C and panel d: after DRM operation at 800 °C without prior hydrogen, panel e: after DRM operation at 800 °C with prior hydrogen reduction at 800 °C, panel f: after DRM operation at 800 °C with prior hydrogen reduction at 550 °C. Structure ICSD-numbers and references: α-Co 136039,63 ε-Co 76633,64 CoO 9865,65 CoGa2O4 172183,66 CoGa 657494,67 β-Ga2O3 3423423,68 Co3O4 36256.69 | ||
As will be discussed in the context of the operando XRD experiments in Fig. 4, the CoGa intermetallic compound is stable up to ca. 550 °C, before it decomposes into metallic Co and additional β-Ga2O3. The strong acceleration of catalytic activity at around 600 °C in the DRM profiles of Fig. 1b (first cycle) is, therefore, associated with the transition from CoGa/β-Ga2O3 to Co/β-Ga2O3.
After pre-reduction at 550 °C, the respective DRM profile (Fig. 1a) exhibits a much lower onset temperature of 520 °C and conversion levels of both CO2 and CH4 are much higher at comparable temperatures. According to the structural characterization of the benchmark states (Fig. 2) and the TEM analysis and the in situ and operando stability tests (cf.Fig. 3, 4, 6 and 7), DRM after pre-reduction at 550 °C probes metallic Co on β-Ga2O3, whereas DRM after pre-reduction at 800 °C reveals the activity of the CoGa compound during DRM operation up to ca. 550 °C. At higher reaction temperatures – as after pre-reduction at 550 °C – Co/β-Ga2O3 is probed (we have accordingly assessed the Co crystallite sizes after the respective hydrogen treatments at 550 °C/800 °C and subsequent DRM runs to be 28 nm and 35 nm, respectively. The respective SEM data indicate comparable Co particle sizes of around 50–100 nm after both treatments, excluding substantial Co crystallite and particle size effects). To test this hypothesis, we have subjected the 800 °C pre-reduced sample to a second consecutive DRM cycle (i.e., in the Co/β-Ga2O3 state after decomposition of CoGa in the first DRM run). As anticipated, the respective DRM profile (dashed lines in Fig. 1b) matches not only that after a pre-reduction at 550 °C, but also that of a Ga-free Co3O4/SiO2 reference catalyst after pre-reduction (Fig. 1c, hydrogen pre-reduction at 450 °C for 10 min). The latter reaches final conversion levels of 84% (CO2) and 76% (CH4) at 800 °C, respectively. A closer look at the DRM profiles indicates a two-fold beneficial role of β-Ga2O3: it lowers the initial onset temperature by about 50 °C (400 °C vs. 450 °C on Co3O4/SiO2) and it suppresses catalyst deactivation in the isothermal section at 800 °C at least in the studied time-on-stream regime. While Co3O4/SiO2 shows clear signs of deactivation after the pre-reduction-DRM cycle, the latter is virtually absent for Co3O4/β-Ga2O3, irrespective of the nature of the hydrogen pre-treatment. As will be discussed in the context of Fig. S3 and S4,† this is essentially due to the different coking behavior. On the basis of several SEM images after a hydrogen treatment at 800 °C and a subsequent DRM run, Co particles sizes between 50 nm and 150 nm have been determined on Co3O4/SiO2 and Co3O4/β-Ga2O3, ruling out substantial particle size or Co dispersion-related effects on DRM activity or coking resilience.
 | ||
| Fig. 3 In situ XRD patterns collected on Co3O4/β-Ga2O3 during reduction in hydrogen: panel a: patterns collected during the reduction step with associated mass spectra of m/z 18, 17 and 2; panel b: XRD patterns at 800 °C during reduction and after the reduction step on the sample cooled to 50 °C. Gas mixture: H2/He (10% vol. H2) with a flow rate of 40 mL min−1. Heating rate: 25 K min−1. The dotted lines in the inset are the calculated XRD patterns of the modified lattice parameters due to thermal expansion. Structure references: Co (fcc)70 β-Ga2O3,71 CoGa,72 Co80Ga20.73 | ||
 | ||
| Fig. 4
Operando XRD patterns collected on Co3O4/β-Ga2O3 during DRM after the hydrogen reduction step shown in Fig. 5: panel a: patterns collected during DRM with the associated mass spectra of m/z 28, 18 and 2; panel b: XRD patterns at 800 °C during DRM and after DRM operation and cooling the sample to room temperature. Gas mixture: 1:1:1 ratio of CO2, CH4 and He with a flow rate of 20 mL min−1 each. Heating rate: 5 K min−1. The dotted lines in the inset are the calculated XRD patterns of the modified lattice parameters due to thermal expansion. Structure references: Co (fcc),70 Co (hcp),74 β-Ga2O3,71 CoGa,72 Co80Ga20.73 | ||
3.2. Structural characterization of selected benchmark states after reduction and DRM operation
To identify benchmark phases during hydrogen reduction and DRM operation, Fig. 2 shows that after initial calcination (panel a), Co3O4 (space group: Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m)69 on β-Ga2O3 (space group: C2/m68) is present. The diffractograms after reduction and DRM already indicate the high propensity of Co3O4/β-Ga2O3 to undergo structural transformations. According to XRD (panel d), during DRM operation without prereduction Co3O4/β-Ga2O3 transforms into CoGa2O4/Ga2O3. CoGa2O4 is a spinel-type bimetallic oxide (space group Fdm)75 with reported dominant oxygen vacancy chemistry, enabling rich electrochemical applications.60,66,76 As shown in Fig. S1,† this structural transformation is not associated with significant DRM activity. After hydrogen reduction at 550 °C, Co3O4 is reduced to metallic cubic Co (panel b). Hydrogen reduction at 800 °C, however, allows to enter a state of reactive metal–support interaction (panel c, RMSI)21via formation of the intermetallic compound CoGa (space group: Pmm67) on β-Ga2O3 (an XRD pattern of the synthesized, untreated CoGa intermetallic compound is provided in Fig. S5†). The latter term is used to classify reactions between supported elemental nanoparticles and the support, usually leading to irreversible oxide reduction and formation of intermetallic compound particles on the supporting oxide surface. This type of reactive metal–support interaction is a frequently observed phenomenon in catalysis and concerns both noble- and non-noble metal containing catalysts. Recently, the term has also been introduced to account for according changes under non-reducing conditions.77 Although initially restricted to easy reducible oxides, the term is also used to describe the associated changes on hard-to-reduce oxides like β-Ga2O3. Specifically, Pt, Pd, Rh and Ni particles have been all found to enter such a state if supported on β-Ga2O3, resulting in GaPt2, GaPd2, GaPd, Ga3Rh5 and GaNi3. For many of those systems, distinct structure–activity correlations have been established.21
m)69 on β-Ga2O3 (space group: C2/m68) is present. The diffractograms after reduction and DRM already indicate the high propensity of Co3O4/β-Ga2O3 to undergo structural transformations. According to XRD (panel d), during DRM operation without prereduction Co3O4/β-Ga2O3 transforms into CoGa2O4/Ga2O3. CoGa2O4 is a spinel-type bimetallic oxide (space group Fdm)75 with reported dominant oxygen vacancy chemistry, enabling rich electrochemical applications.60,66,76 As shown in Fig. S1,† this structural transformation is not associated with significant DRM activity. After hydrogen reduction at 550 °C, Co3O4 is reduced to metallic cubic Co (panel b). Hydrogen reduction at 800 °C, however, allows to enter a state of reactive metal–support interaction (panel c, RMSI)21via formation of the intermetallic compound CoGa (space group: Pmm67) on β-Ga2O3 (an XRD pattern of the synthesized, untreated CoGa intermetallic compound is provided in Fig. S5†). The latter term is used to classify reactions between supported elemental nanoparticles and the support, usually leading to irreversible oxide reduction and formation of intermetallic compound particles on the supporting oxide surface. This type of reactive metal–support interaction is a frequently observed phenomenon in catalysis and concerns both noble- and non-noble metal containing catalysts. Recently, the term has also been introduced to account for according changes under non-reducing conditions.77 Although initially restricted to easy reducible oxides, the term is also used to describe the associated changes on hard-to-reduce oxides like β-Ga2O3. Specifically, Pt, Pd, Rh and Ni particles have been all found to enter such a state if supported on β-Ga2O3, resulting in GaPt2, GaPd2, GaPd, Ga3Rh5 and GaNi3. For many of those systems, distinct structure–activity correlations have been established.21
Panels e and f reveal that upon subsequent DRM operation metallic Co is stable (referring to hydrogen pre-reduction at 550 °C, panel e) and that CoGa is unstable and decomposes into cubic α-Co/β-Ga2O3 with trace amounts of hexagonal ε-Co present after cooling (panel f). The details of the Rietveld refinement are summarized in Table S1.†
The respective PXRD patterns for the Co/SiO2 catalyst (Fig. S3†) – apart from the consistent presence of metallic Co and SiO2 in the as-calcined and hydrogen pre-reduced/DRM state – indicate the formation of graphitic carbon species after DRM operation. Connecting to the catalytic results in Fig. 1, this explains the deactivation feature in the isothermal section for Co/SiO2 – in striking contrast to Co3O4/β-Ga2O3.
As the PXRD patterns after DRM operation already indicate a different coking behavior and revealed (partially) crystalline graphite-like carbon species on Co3O4/SiO2 after DRM operation in contrast to Co3O4/β-Ga2O3, SEM analysis was carried out to pinpoint the exact location of these carbon species (Fig. S4†). After hydrogen pre-reduction at 550 °C (Fig. S4b,† in comparison to the as-calcined state shown in Fig. S4a†), the EDX spectra of Co-L, Ga-K and C-K clearly reveal that no coking of the Co particles during DRM operation took place. In contrast, the same treatment of Co3O4/SiO2 induces significant coking (Fig. 4Sc–e†). In Fig. S4c,† three areas with Co particles are marked. A clearly visible halo-like feature surrounding the Co particles is visible, which by analysis of the EDX spectra is identified as carbon encapsulating the Co particles. Additionally, extended flake-like carbon features are often observed decorating areas with Co particles (Fig. S4d and e†). Correlating the PXRD and SEM results, we infer that the Co particles on SiO2 give rise to deactivation by formation of graphite species arising from methane decomposition. Beneficial DRM operation using β-Ga2O3 as a support for Co particles might be traced back to the presence of CO2 during the dehydrogenation of light alkanes (e.g., methane, ethane, propane or ethylbenzene).57,78–80 CO2 has been reported to act in a dual role: favor the dehydrogenation process, but in terms of decoking, at the same time, it facilitates the reaction by accelerating the Boudouard reaction. Specifically on β-Ga2O3, depending on the degree of reduction, a more vacancy-dominated pathway involving unsaturated Ga sites, or surface-bound heterolytic dissociation has been put forward.56 In the latter pathway, the alkane (e.g., propane) is activated by heterolytic dissociation at Ga2O3 sites to form Ga-H and Ga-OR species, whereby the latter are decomposed to the products.56
3.3. In situ and operando powder X-ray diffraction studies of Co3O4/β-Ga2O3 during hydrogen reduction and DRM operation
Fig. 1 already revealed, that the reduction pre-treatment leads to a strong increase in DRM activity, especially in the temperature window 650 °C to 800 °C. We, thus, performed additional in situ and operando PXRD measurements to follow the structural evolution and to correlate the activity increase with potential structural transformations.The initial state of the sample is a mixture of β-Ga2O3 and Co3O4 (Fig. 3). During the reduction step, Co3O4 transforms into α-Co with the Cu-type of crystal structure (Fig. 3a). The reduction of Co3O4 starts around 450 °C, indicated by water formation (m/z 18 and 17) and decreasing hydrogen (m/z 2) concentration, as well as by the diffuse intensity in the XRD pattern around the reflection position for Co, reaching the highest rate at around 600 °C. This effect is coupled to a broadening and intensity increase of the β-Ga2O3 reflections, which are likely due to defect formation and further crystallisation, respectively. Over time and temperature the slope of the signal for m/z 17/18 and 2 changes. This is accompanied by a reduction of the full-width-at-half-maximum of the Co reflection at 51.5°, which is shifted to lower 2θ due to thermal expansion. Subsequently, the water and hydrogen levels decrease further as the temperature stabilized at 800 °C (Fig. S6†). However, during further heating, the Co reflection is further shifting to lower 2θ. While this could be assigned to thermal expansion, this would not fit the observation under DRM conditions (see below), where the reflection is shifted to higher 2θ values with temperature. More likely, the shift is a combination of thermal expansion and the beginning of RMSI, leading to the incorporation of Ga into the Co lattice and, thus, the formation of a Co(Ga) alloy. Indeed, the reflection matches the one of the substitutional alloy Co80Ga20 (Fig. 3b).73 The appearance of the substitutional alloy at the beginning of the RMSI stage is in accordance with the Co–Ga phase diagram.81 With time, the composition gets more Ga-rich, as more elemental Ga is formed by reduction, until the intermetallic compound CoGa appears (Fig. 3b). A trace of Co80Ga20 is still present (at 51.1°), most likely due to diffusion limitation in larger particles.
During DRM, both Co80Ga20 and CoGa are, therefore, present at the beginning of heating (Fig. 4a). Up to 550 °C the reflections shift to lower 2θ values due to thermal expansion and the intensity of Co80Ga20 decreases due to the further ongoing RMSI process Following the m/z 2 and 28 profiles, indicating catalytic activity in DRM, the conversion of the CH4/CO2 mixture starts at around 450 °C, thus at a temperature when the material is mostly in the state of CoGa/β-Ga2O3. With increasing temperature, the activity for H2 and CO formation is increasing until the decomposition of the intermetallic compound starts (550 °C) allowing to assign the catalytic activity to CoGa/β-Ga2O3.
Above 550 °C, the CoGa reflections decrease in intensity and shift to higher 2θ values, while the reflections of Co80Ga20 gain intensity. This indicates a loss of Ga from CoGa by oxidation to Ga2O3 resulting in the decomposition of the intermetallic compound. Also Co80Ga20 is further oxidized until full loss of Ga, shown by the shift of the Co80Ga20 reflections to higher 2θ with temperature due to loss of Ga.81 At 800 °C, a mixture of cubic and hexagonal Co coexists on β-Ga2O3 (Fig. 4b), which is maintained upon cooling to ambient temperature. However, at room temperature hexagonal Co is the most stable allotrope, whereas cubic Co is at temperatures higher than 425 °C. While this could be explained by slow transition kinetics, stabilization by the support (e.g., by epitaxy) or by stabilization of Ga in the lattice, the latter can be ruled out by Co–Ga phase diagram as the transition temperature is independent of the Ga-content of the alloy.81 Moreover, the loss of CoGa leads to a complete change of the selectivity pattern. The formation of CO continues, while the hydrogen formation rate decreases, which can be explained by the reverse water–gas shift activity of β-Ga2O3.61 Upon reaching 800 °C and during the isothermal section, the catalytic activity decreases – likely by slight sintering (Fig. S6†).
The XRD patterns during cooling down after reduction and DRM are highlighted in Fig. S7 and S8,† respectively, and revealed no structural changes.
3.4. Nanoscale structure of Co3O4/β-Ga2O3 after hydrogen reduction and DRM operation
Electron microscopy analysis of the as-calcined Co3O4/β-Ga2O3 sample (Fig. 5) in essence confirms the XRD patterns (cf.Fig. 2). The individual β-Ga2O3 grains are covered by irregular ∼50 nm-sized oxidized Co3O4 grains (panel a). The majority of Co, therefore, is oxidized (Fig. 5, panel b–g). Interestingly, all observed areas with oxidized Co are located in direct proximity to Ga-oxide with a lower oxygen content than Ga2O3 indicating the presence of a second Ga-phase with less oxygen Ga2O3−x, as shown on the left side in Fig. 5, panel f, where the Ga content is ∼60 at% and O ∼ 40 at%. In line with the electron microscopy results, partial formation of reduced Ga2O3−x species (e.g., Ga2O) upon thermal annealing and/or evaporation is a well-documented phenomenon.82 | ||
| Fig. 5 (Scanning) transmission electron microscopy analysis of as-calcined Co3O4/β-Ga2O3. Panel a: HRTEM of a β-Ga2O3 grain covered by Co3O4 particles. Panel b: Overlay of Co-K (green) and Ga-K (red) EDX mappings. Panels c–e: elemental EDX mappings of O-K (blue), Co-K (red), and Ga-K (green), respectively. Panels f and g: elemental line profiles along the orange and green-framed arrows shown in panel e, respectively. Standard deviations in panel f of O, Co, and Ga are ±2 at%, ±2 at%, and ±1 at%, respectively. Standard deviations in panel g of O, Co, and Ga are ±3 at%, ±2 at%, and ±3 at%, respectively. | ||
After hydrogen reduction at 800 °C (Fig. 6) – corroborating the XRD results – the Co3O4 particles are reduced and bimetallic Co–Ga particles are formed, which are identified by XRD as the intermetallic compound CoGa. At the nanoscale, all individual CoGa particles are covered by an oxygen-deficient Ga2O3−x layer (cf. line profile in Fig. 6j). Given the propensity of Ga2O3 to readily lose oxygen upon thermal annealing, we might tentatively explain the formation of this layer by formation of mobile Ga2O3−x species upon hydrogen reduction and the according encapsulation of the CoGa particles by Ga2O3−x. Electron microscopy analysis directly confirms the reactive metal–support interaction state of Co on β-Ga2O3 inferred from the ex situ and in situ XRD experiments. After a subsequent DRM treatment up to 800 °C, the structural situation gets more complex (Fig. 7): corroborating the XRD patterns, also EDX measurements (Fig. 7, panel a, d and e) confirm that the CoGa intermetallic particles are decomposed into mostly metallic Co particles embedded in the Ga2O3 or Ga2O3−x matrix and no signs of coking are detectable. Most interestingly, Fig. 7 panel f reveals on many metallic Co particles a Ga2O3 layer in close proximity. This specific Ga2O3 species is a direct result of the decomposition of the CoGa particles and creates an additional Co–Ga2O3 interface apart from the interface created by Co socketed into the main supporting Ga2O3 matrix. Note that this state is entirely different from the one that is entered by a direct DRM treatment without hydrogen pre-reduction (Fig. S9 and S10†). In that case, XRD indicates the presence of a CoGa2O4 spinel phase and EDX measurements reveal, that the Ga2O3 surface is encrusted with CoGa2O4 particles a few tens of nanometers thick. The stoichiometric evolution of Ga2O3 to CoGa2O4 is visualized by the line profile in Fig. S10,† panel f.
 | ||
| Fig. 6 (Scanning) transmission electron microscopy analysis of the hydrogen pre-reduced Co3O4/β-Ga2O3. Panel a: mesoscale HAADF overview image. Panels b–d: elemental Co-K (red), O-K (blue) and Ga-K (green) EDX mappings. The white frame in panels b and d indicate Co–Ga intermetallic formation. Panel e: nanoscale HAADF image highlighting a single Co–Ga nanoparticle. Panels f–i: elemental Ga-K (green), O-K (blue), Co + Ga (red + green) and Co-K (red) EDX mappings, respectively. Panel j: line profiles along the arrow shown in panel i. Standard deviations in panel j of O, Co, and Ga are ±2.22 at%, ±2.15 at%, and ±3.42 at%, respectively. | ||
 | ||
| Fig. 7 (Scanning) transmission electron microscopy analysis of the DRM-treated Co3O4/β-Ga2O3 with prior hydrogen reduction at 800 °C. Panel a: HAADF overview image highlighting metallic nanoparticles supported on Ga2O3. Panel b: Ga–Co overlay of individual elemental mappings. Panels c–e: elemental O-K (blue), Ga-K (green), and Co-K (red) EDX mappings, respectively. Panel f: zoomed-in overlay of Co and Ga signals from the yellow-framed area in panel e, showcasing a Co nanoparticle with Ga2O3 particles positioned on its surface. | ||
3.5. Surface chemical characterization by X-ray photoelectron spectroscopy
In light of the substantial bulk structural and chemical changes, we monitored the according changes in the near-surface region by ex situ X-ray photoelectron spectroscopy (Fig. 8). We show the changes of the Ga 2p and Co 2p region as a function of treatment, referenced to the spectra of unsupported single-phase CoGa (panel a). The latter shows a single narrow Ga 2p3/2 signal at a binding energy of 1116.5 eV, typical of metallic Ga.83–85 A strong metallic component is also seen in the respective Co 2p spectrum (light green fit, main Co 2p3/2 at 778.4 eV).58 In addition, an oxidized Co 2p3/2 component with its satellite features at 780.4 eV is also seen, matching that of CoO.58 With respect to the overall shape and binding energy, all other Ga 2p spectra (panels b–e) are similar. They are dominated by a strongly oxidized Ga3+ component at ca. 1117.7 eV,83–85 irrespective of the treatment, in clear agreement with the electron microscopy analysis. Even after hydrogen reduction (panel c), only oxidized Ga is seen, which matches the ca. 5 nm thick oxygen-deficient GaxOy layer covering the intermetallic CoGa particles (cf.Fig. 6). The corresponding Co 2p spectrum (right side of panel c) reveals that Co is almost entirely shielded also due to the CoGa-covering GaxOy layer. The according peak data are detailed in Table S2.† In accordance with STEM-EDX (Fig. 7) and operando XRD (Fig. 4), the Co 2p spectrum in panel e features both a metallic and an oxidic Co component.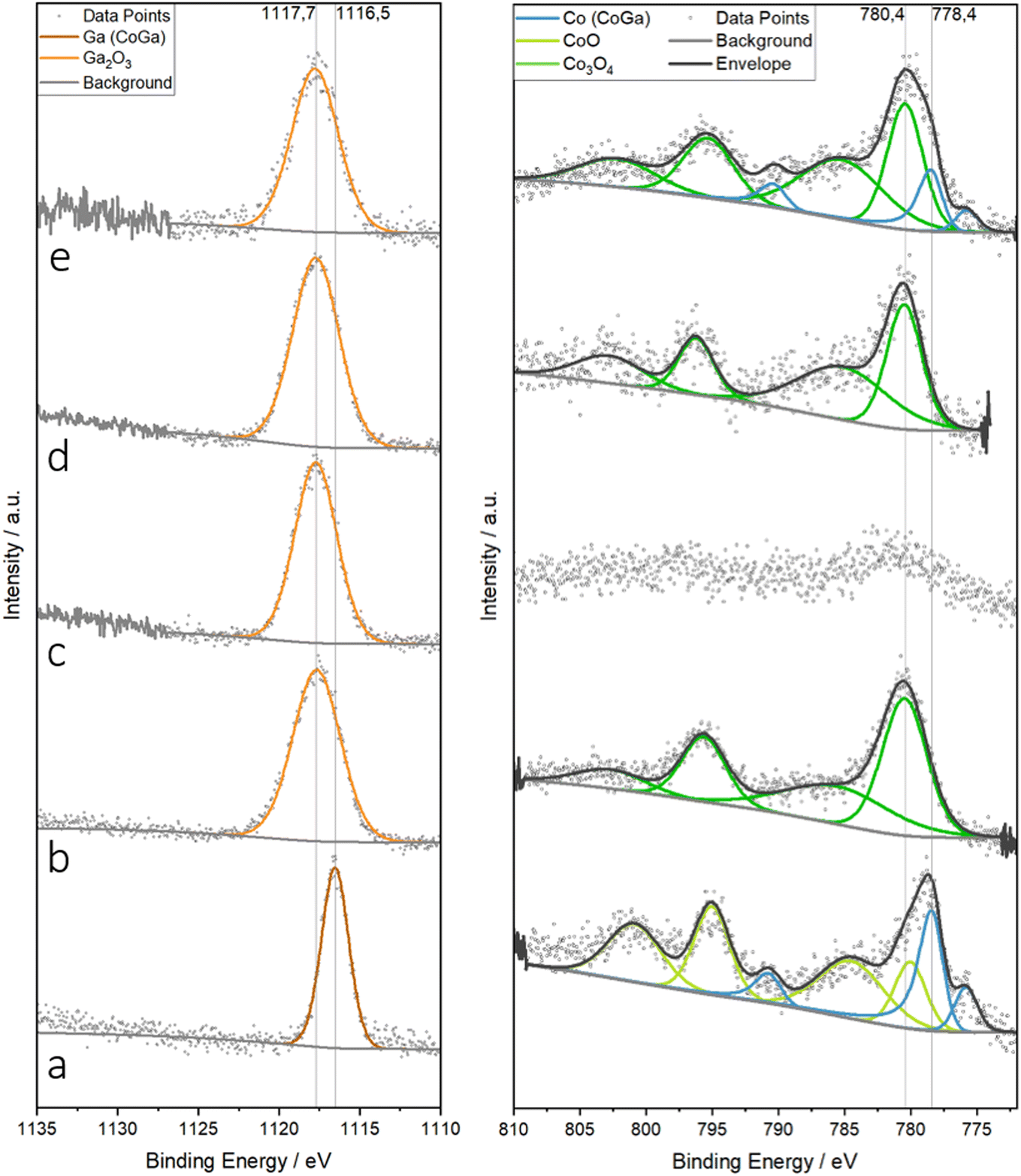 | ||
| Fig. 8 High-resolution X-ray photoelectron spectra of the Ga 2p (left) and Co 2p (right) region of Co3O4/β-Ga2O3 after selected treatments in comparison (panel b–e) in comparison with a CoGa intermetallic compound (panel a). Panel a: CoGa intermetallic compound in the as-prepared state. Panel b: Co3O4/β-Ga2O3 as calcined. Panel c: Co3O4/β-Ga2O3 after hydrogen reduction at 800 °C. Panel d: Co3O4/β-Ga2O3 after DRM. Panel e: Co3O4/β-Ga2O3 after DRM with prior hydrogen reduction at 800 °C. Binding energies of the metallic and oxidized surface states are marked by dashed lines. | ||
4. Conclusions
We have highlighted the potential of a Co3O4/β-Ga2O3 material as a promising DRM catalyst, rivalling known noble and/or non-noble metals-modified Co-based catalysts in terms of activity and coking resilience. In due course, we showed that while activation of Co3O4/β-Ga2O3 directly in the DRM mixture is only possible to a limited extent due to the formation of a surface-bound CoGa2O4 spinel phase, hydrogen pre-reduction yields reduction of the initial Co3O4 particles on β-Ga2O3 to α-Co metal and further to CoGa on β-Ga2O3. Hence, Co is able to enter a reactive metal–support interaction stage (RMSI) on β-Ga2O3. This also leads to coverage of the individual CoGa particles by a strongly oxygen-deficient GaxOy layer, arising from the transport of mobile Ga–O species onto the intermetallic particles. Oxygen-deficient Ga sites are already present in the as-calcined state, but are more abound after hydrogen reduction, as evidenced by STEM/EDX. Intermetallic compound formation is reversed by subsequent treatment in a DRM mixture, causing decomposition of CoGa into cubic α-Co particles on β-Ga2O3, i.e., after CoGa decomposition, the formed Co–Ga2O3 interface remains exposed and accessible to reactants, based on electron microscopy data. These two phases do not form the slightly active CoGa2O4 spinel, thus leading to excellent activity of the material. The decomposition temperature of CoGa can be directly related to the strong increase in DRM activity by operando XRD measurements. In comparison to SiO2, β-Ga2O3 shows a better overall DRM activity with respect to coking resilience within the limitation of comparing activity without actual TOF values, which are inaccessible due to the in situ formation of the active sites.These studies in turn pave the way to the potential redox activation of isolated Co–Ga intermetallic compounds. Similar to the Cu–In system, due the selective oxidation of Ga, we expect the oxidative decomposition of Co–Ga intermetallic compounds with varying stoichiometry, e.g., CoGa or CoGa3, and a potentially improved DRM performance due to beneficial extents of Co–Ga2O3 phase boundary dimensions and the accordingly higher activity, stability and anti-coking behavior.
Data availability
Data for this article are available at Researchdata at https://doi.org/10.48323/fv702-2t003.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was conducted within the framework of the Research Focus “Functional Materials” at the University of Innsbruck. This research was funded by the Austrian Science Fund (FWF) https://doi.org/10.55776/P35770. R. T., N. B. and G. S. acknowledge support by the Deutsche Forschungsgemeinschaft (DFG, 41/1096-1 FKGG and CRC1333/C09, grant 358283783) in the acquisition of the aberration-corrected microscope, R. T. and G. S. are also grateful for project funding by DFG through CRC1333/C09, grant 358283783. M. A. and J. A. thank the Deutsche Forschungsgemeinschaft for financial support (grants 517014885 and INST 270/316-1 FUGG). N. B. thanks the research network Advanced Materials Innovation and Characterization (AMiCA) for technical support of the Spectra300. For open access purposes, the authors have applied a CC BY public copyright license to any author-accepted manuscript version arising from this submission.References
- N. Aramouni, J. Touma, B. Tarboush, J. Zeaiter and M. Ahmad, Catalyst design for dry reforming of methane: Analysis review, Renewable Sustainable Energy Rev., 2018, 82, 2570–2585 CrossRef CAS.
- A. Hussein and K. Polychronopoulou, A Review on the Different Aspects and Challenges of the Dry Reforming of Methane (DRM) Reaction, Nanomaterials, 2022, 12, 3400 CrossRef.
- L. Sandoval-Diaz, R. Schlögl and T. Lunkenbein, Quo Vadis Dry Reforming of Methane?—A Review on Its Chemical, Environmental, and Industrial Prospects, Catalysts, 2022, 12, 465 Search PubMed.
- Y. Sun, Y. Zhang, X. Yin, C. Zhang, Y. Li and J. Bai, Recent Advances in the design of high-performance cobalt-based catalysts for dry reforming of methane, Green Chem., 2024, 26, 5103–5126 RSC.
- L. Baharudin, N. Rahmat, N. Othman, N. Shah and S. Syed-Hassan, Formation, control, and elimination of carbon on Ni-based catalyst during CO2 and CH4 conversion via dry reforming process: A review, J. CO2 Util., 2022, 61, 102050 CrossRef CAS.
- Z. Alipour, V. Borugadda, H. Wang and A. Dalai, Syngas production through dry reforming: A review on catalysts and their materials, preparation methods and reactor type, Chem. Eng. J., 2023, 452, 139416 CrossRef CAS.
- J. Li, J. Li, Q. Zhu and H. Li, Magnetic field acceleration of CO2 reforming of methane over novel hierarchical Co/MgO catalyst in fluidized bed reactor, Chem. Eng. J., 2018, 350, 496–506 CrossRef CAS.
- K. Takanabe, K. Nagaoka, K. Nariai and K. Aika, Influence of reduction temperature on the catalytic behavior of Co/TiO2 catalysts for CH4/CO2 reforming and its relation with titania bulk crystal structure, J. Catal., 2005, 230, 75–85 CrossRef CAS.
- J. Park, S. Yeo, T. Kang, H. Shin, I. Heo and T. Chang, Effect of Zn promoter on catalytic activity and stability of Co/ZrO2 catalyst for dry reforming of CH4, J. CO2 Util., 2018, 23, 10–19 CrossRef CAS.
- J. Liu, Y. Zhang, Z. Liang, G. Zhang, Y. Wang, Y. Zhao, G. Li and Y. Lv, Enhancing the dry reforming of methane over Ni-Co-Y/WC-AC catalyst: Influence of the different Ni/Co ratio on the catalytic performance, Fuel, 2023, 335, 1277082 Search PubMed.
- Y. Sun, G. Zhang, Y. Xu, Y. Zhang, Y. Lv and R. Zhang, Comparative study on dry reforming of methane over Co-M (M = Ce, Fe, Zr) catalysts supported on N-doped activated carbon, Fuel Process. Technol., 2019, 192, 1–12 CrossRef CAS.
- A. Paksoy, C. Akdag, B. Caglayan and A. Aksoylu, Kinetic and mechanistic features of carbon dioxide reforming of methane over Co–Ce/ZrO2 catalysts, Int. J. Chem. Kinet., 2019, 51, 138–145 CrossRef CAS.
- M. Cheng, J. Zhang, C. Zhang, J. Wang, Y. Jiao, Y. Chen and X. Li, Synergistic effect of bimetal for steam reforming of n-decane over M-M/CaO–Al2O3 (M = Fe, Co, Ni) catalysts under low S/C condition, Int. J. Hydrogen Energy, 2022, 47, 11799–11812 CrossRef CAS.
- L. Chen, Q. Huang, Y. Wang, H. Xiao, W. Liu, D. Zhang and T. Yang, Tailoring performance of Co–Pt/MgO–Al2O3 bimetallic aerogel catalyst for methane oxidative carbon dioxide reforming: Effect of Pt/Co ratio, Int. J. Hydrogen Energy, 2019, 44, 19878–19889 CrossRef CAS.
- S. Shah, M. Xu and K. Gilliard-Abdulaziz, Exsolution of Embedded Ni–Fe–Co Nanoparticles: Implications for Dry Reforming of Methane, ACS Appl. Nano Mater., 2021, 4, 8628–8636 Search PubMed.
- Z. Wu, B. Yang, S. Miao, W. Liu, J. Xie, S. Lee, M. Pellin, D. Xiao, D. Su and D. Ma, Lattice Strained Ni-Co alloy as a High-Performance Catalyst for Catalytic Dry Reforming of Methane, ACS Catal., 2019, 9, 2693–2700 CrossRef CAS.
- L. Lyu, M. Shengene, Q. Ma, J. Sun, X. Gao, H. Fan, J. Zhang and T. Zhao, Synergy of macro-meso bimodal pore and Ni-Co alloy for enhanced stability in dry reforming of methane, Fuel, 2022, 310, 122375 CrossRef CAS.
- J. Sasson Bitters, T. He, E. Nestler, S. Senanayke, J. Chen and C. Zhang, Utilizing bimetallic catalysts to mitigate coke formation in dry reforming of methane, J. Energy Chem., 2022, 68, 124–142 CrossRef CAS.
- B. Safaviniaa, Y. Wang, C. Jiang, C. Roman, P. Darapanemi, J. Larraviere, D. A. Cullen, K. Dooley and J. Dorman, Enhancing CexZr1–xO2 Activity for Methane Dry Reforming Using Subsurface Ni Dopants, ACS Catal., 2020, 10, 4070–4079 CrossRef.
- Y. Zhu, N. Jin, R. Liu, X. Sun, L. Bai, H. Tian, X. Ma and X. Wang, Bimetallic BaFe2MAl9O19 (M = Mn, Ni, and Co) hexaaluminates as oxygen carriers for chemical looping dry reforming of methane, Appl. Energy, 2020, 258, 114070 CrossRef CAS.
- S. Penner and M. Armbrüster, Formation of Intermetallic Compounds by Reactive Metal–Support Interaction: A Frequently Encountered Phenomenon in Catalysis, ChemCatChem, 2015, 7, 374–392 CrossRef CAS.
- Y. Nakaya and S. Furukawa, Catalysis of Alloys: Classification, Principles, and Design for a Variety of Materials and Reactions, Chem. Rev., 2022, 123, 5859–5947 CrossRef PubMed.
- M. Armbrüster, Intermetallic compounds in catalysis – a versatile class of materials meets interesting challenges, Sci. Technol. Adv. Mater., 2020, 21, 303–322 CrossRef.
- P. Adamski, H. Zhang, S. Kaur, X. Chen, C. Liang and M. Armbrüster, Selective Hydrogenation of α,β-Unsaturated Aldehydes Over Intermetallic Compounds – A Critical Review, Chem. Mater., 2024, 36, 10383–10407 CrossRef CAS.
- Y. Yang and M. Wei, Intermetallic compound catalysts: synthetic scheme, structure characterization and catalytic application, J. Mater. Chem. A, 2020, 8, 2207–2221 RSC.
- S. Penner and P. D. K. Nezhad, Steering the Catalytic Properties of Intermetallic Compounds and Alloys in Reforming Reactions by Controlled in Situ Decomposition and Self-Activation, ACS Catal., 2021, 11, 5271–5286 CrossRef CAS.
- Z. Sun, C. Hao, S. Toan, R. Zhang, H. Li, Y. Wu, H. Liu and Z. Sun, Recent advances in exsolved perovskite oxide construction: exsolution theory, modulation, challenges, and prospects, J. Mater. Chem. A, 2023, 11, 17961–17976 RSC.
- L. Mayr, B. Klötzer, D. Schmidmair, N. Köpfle, J. Bernardi, S. Schwarz, M. Armbrüster and S. Penner, Boosting Hydrogen Production from Methanol and Water by in situ Activation of Bimetallic Cu−Zr Species, ChemCatChem, 2016, 8, 1778–1781 CrossRef CAS.
- S. Penner, T. Götsch and B. Klötzer, Increasing Complexity Approach to the Fundamental Surface and Interface Chemistry on SOFC Anode Materials, Acc. Chem. Res., 2020, 53, 1811–1821 CrossRef CAS PubMed.
- L. Mayr, N. Köpfle, B. Klötzer, T. Götsch, J. Bernardi, S. Schwarz, T. Keilhauer, M. Armbrüster and S. Penner, Microstructural and Chemical Evolution and Analysis of a Self-Activating CO2-Selective Cu–Zr Bimetallic Methanol Steam Reforming Catalyst, J. Phys. Chem. C, 2016, 120, 25395–25404 CrossRef CAS.
- M. Friedrich, S. Penner, M. Heggen and M. Armbrüster, High CO2 selectivity in methanol steam reforming through ZnPd/ZnO teamwork, Angew. Chem., Int. Ed., 2013, 52, 4389–4392 CrossRef CAS PubMed.
- M. Heggen, S. Penner, M. Friedrich, R. Dunin-Borkowski and M. Armbrüster, Formation of ZnO Patches on ZnPd/ZnO during Methanol Steam Reforming: A Strong Metal–Support Interaction Effect?, J. Phys. Chem. C, 2016, 120, 10460–10465 CrossRef CAS.
- C. Rameshan, et al., Hydrogen Production by Methanol Steam Reforming on Copper Boosted by Zinc-Assisted Water Activation, Angew. Chem., Int. Ed., 2012, 51, 3002–3006 CrossRef CAS PubMed.
- M. Armbrüster, M. Behrens, K. Föttinger, M. Friedrich, E. Gaudry, S. K. Matam and H. Sharma, The Intermetallic Compound ZnPd and Its Role in Methanol Steam Reforming, Catal. Rev.: Sci. Eng., 2013, 55, 289–367 CrossRef.
- C. Rameshan, Steam reforming of methanol on PdZn near-surface alloys on Pd(1 1 1) and Pd foil studied by in-situ XPS, LEIS and PM-IRAS, J. Catal., 2010, 276, 101–113 CrossRef CAS.
- L. Mayr, et al., The catalytic properties of thin film Pd-rich GaPd2 in methanol steam reforming, J. Catal., 2014, 309, 231–240 CrossRef CAS.
- A. Haghofer, K. Föttinger, F. Girgsdies, D. Teschner, A. Knop-Gericke, R. Schlögl and G. Rupprechter, In situ study of the formation and stability of supported Pd2Ga methanol steam reforming catalysts, J. Catal., 2012, 286, 13–21 CrossRef CAS.
- S. Penner, H. Lorenz, W. Jochum, M. Stöger-Pollach, D. Wang, C. Rameshan and B. Klötzer, Pd/Ga2O3 methanol steam reforming catalysts: Part I. Morphology, composition and structural aspects, Appl. Catal., A, 2009, 358, 193–202 CrossRef CAS.
- S. Penner, H. Lorenz, W. Jochum, M. Stöger-Pollach, D. Wang, C. Rameshan and B. Klötzer, Pd/Ga2O3 methanol steam reforming catalysts: Part II Catalytic selectivity, Appl. Catal., A, 2009, 358, 203–210 CrossRef.
- C. Rameshan, H. Lorenz, M. Armbrüster, I. Kasatkin, B. Klötzer, T. Götsch, K. Ploner and S. Penner, Impregnated and Co-precipitated Pd–Ga2O3, Pd–In2O3 and Pd–Ga2O3–In2O3 Catalysts: Influence of the Microstructure on the CO2 Selectivity in Methanol Steam Reforming, Catal. Lett., 2018, 148, 3062–30771 CrossRef CAS PubMed.
- H. Lorenz, S. Turner, O. I. Lebedev, G. van Tendeloo, B. Klötzer, C. Rameshan, K. Pfaller and S. Penner, Pd–In2O3 interaction due to reduction in hydrogen: Consequences for methanol steam reforming, Appl. Catal., A, 2010, 374, 180–188 CrossRef CAS.
- N. Köwitsch, et al., Proving a Paradigm in Methanol Steam Reforming: Catalytically Highly Selective InxPdy/In2O3 Interfaces, ACS Catal., 2020, 11, 304–312 CrossRef.
- K. Ploner, et al., Reactive metal-support interaction in the Cu-In2O3 system: intermetallic compound formation and its consequences for CO2-selective methanol steam reforming, Sci. Technol. Adv. Mater., 2019, 25, 356–366 CrossRef PubMed.
- L. Haug, et al., Zirconium Carbide Mediates Coke-Resistant Methane Dry Reforming on Nickel-Zirconium Catalysts, Angew. Chem., Int. Ed., 2022, 61, e202213249 CrossRef CAS PubMed.
- L. Haug, et al., Pivotal Role of Ni/ZrO2 Phase Boundaries for Coke-Resistant Methane Dry Reforming Catalysts, Catalysts, 2023, 13, 804 CrossRef CAS.
- N. Köpfle, L. Mayr, D. Schmidmair, J. Bernardi, A. Knop-Gericke, M. Hävecker, B. Klötzer and S. Penner, A Comparative Discussion of the Catalytic Activity and CO2-Selectivity of Cu-Zr and Pd-Zr (Intermetallic) Compounds in Methanol Steam Reforming, Catalysts, 2017, 7, 53 CrossRef.
- N. Köpfle, et al., Zirconium-Assisted Activation of Palladium To Boost Syngas Production by Methane Dry Reforming, Angew. Chem., Int. Ed., 2018, 57, 14613–14618 CrossRef PubMed.
- N. Köpfle, et al., Zirconium-Assisted Activation of Palladium To Boost Syngas Production by Methane Dry Reforming, Catalysts, 2020, 10, 1000 CrossRef.
- N. Köwitsch, L. Thoni, B. Klemmed, A. Benad, P. Paciok, M. Heggen, I. Köwitsch, M. Mehring, A. Eychmüller and M. Armbrüster, Proving a Paradigm in Methanol Steam Reforming: Catalytically Highly Selective InxPdy/In2O3 Interfaces, ACS Catal., 2021, 11, 304–312 CrossRef.
- N. Köwitsch, L. Thoni, B. Klemmed, A. Benad, P. Paciok, M. Heggen, A. Eychmüller and M. Armbrüster, Unprecedented Catalytic Activity and Selectivity in Methanol Steam Reforming by Reactive Transformation of Intermetallic In-Pt Compounds, J. Phys. Chem. C, 2021, 125, 9809–9817 CrossRef.
- S. Chen, J. Zaffran and B. Yang, Dry reforming of methane over the cobalt catalyst: Theoretical insights into the reaction kinetics and mechanism for catalyst deactivation, Appl. Catal., B, 2020, 270, 118859 CrossRef CAS.
- Y. Wong, H. Halim, N. Khairudin, T. Pham, S. Putra, Y. Hamamoto, K. Inagaki, I. Hamada, A. Mohamed and Y. Morikawa, Dry Reforming of Methane on Cobalt Catalysts: DFT-Based Insights into Carbon Deposition Versus Removal, J. Phys. Chem. C, 2021, 125, 21902–21913 CrossRef CAS.
- P. Eckerlin and H. Kandler, Crystal Symmetry Tables, in Structure Data of Elements and In-termetallic Phases, ed. K. H. Hellwege and A. M. Hellwege, Springer, Berlin, 1971, p. 24 Search PubMed.
- P. Feschotte and P. Eggimann, Les systemes binaires cobalt-gallium et nickel-gallium-étude compare, J. Less-Common Met., 1979, 63, 15–30 CrossRef CAS.
- W. Jochum, S. Penner, R. Kramer, K. Föttinger, G. Rupprechter and B. Klötzer, Defect formation and the water–gas shift reaction on β-Ga2O3, J. Catal., 2008, 256, 278–286 CrossRef CAS.
- X. Han, Y. Yang, R. Chen, J. Zhou, X. Yang, X. Wang and H. Ji, One-dimensional Ga2O3-Al2O3 nanofibers with unsaturated coordination Ga: Catalytic dehydrogenation of propane under CO2 atmosphere with excellent stability, J. Colloid Interface Sci., 2024, 666, 76–87 CrossRef CAS PubMed.
- Y. Li, D. Li, H. Liu, Y. Lei, R. Zhao, D. He, Z. Zheng, H. Luo and A. Liu, In situ fabricating a Rh/Ga2O3 photothermal catalyst for dry reforming of methane, Catal. Sci. Technol., 2024, 14, 2722 RSC.
- H. Ipser, A. Mikula and W. Schuster, Lattice Parameter and Melting Behavior of the Ternary B2-Phase in the Co-Ga-Ni System, Monatsh. Chem., 1989, 120, 283–289 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson, R. St and C. Smart, Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- D. Liu, X. Mo, K. Li, Y. Liu, J. Wang and T. Yang, The performance of spinel bulk-like oxygen-deficient CoGa2O4 as an air-cathode catalyst in microbial fuel cell, J. Power Sources, 2017, 359, 355–362 CrossRef CAS.
- W. Jochum, S. Penner, R. Kramer, K. Föttinger, G. Rupprechter and B. Klötzer, Hydrogen on polycrystalline β-Ga2O3: Surface chemisorption, defect formation, and reactivity, J. Catal., 2008, 256, 268–277 CrossRef CAS.
- T. F. Shesko, T. A. Kryuchkova, L. V. Yafarova, E. M. Borodina, Y. M. Serov, T. A. Zvereva and A. G. Cherednichenko, Gd-Co-Fe perovskite mixed oxides as catalysts for dry reforming of methane, Sustainable Chem. Pharm., 2022, 30, 100897 CrossRef.
- A. Khort, S. Roslyakov and P. Loginov, Solution combustion synthesis of single-phase bimetallic nanomaterials, Chem. Phys. Lett., 2021, 26, 100727 CAS.
- A. Taylor and R. W. Floyd, Precision measurements of lattice parameters of non-cubic crystals, Acta Crystallogr., 1950, 3, 285–289 CrossRef CAS.
- S. Sasaki, K. Fujino and Y. Takeuchi, X-ray determination of electron-density distributions in oxides, MgO, MnO, CoO, and NiO, and atomic scattering factors of their constituent atoms, Proc. Jpn. Acad., 1979, 55, 43–48 CrossRef CAS.
- Z. Xu, S. Yan, Z. Shi, Y. Yao, P. Zhou, H. Wang and Z. Zou, Adjusting the Crystallinity of Mesoporous Spinel CoGa2O4 for Efficient Water Oxidation, ACS Appl. Mater. Interfaces, 2016, 8, 12887–12893 CrossRef CAS PubMed.
- H. Ipser and A. Mikula, On the ternary B2 -phase in the Al - Co - Ga system, Monatsh. Chem., 1992, 123, 509–513 CrossRef CAS.
- S. Geller, Crystal Structure of β-Ga2O3, J. Chem. Phys., 1960, 33, 676–684 CrossRef CAS.
- J. P. Picard, G. Baud, J. P. Besse and R. Chevalier, Croissance cristalline et etude structurale de Co3O4, J. Less-Common Met., 1980, 75, 99–104 CrossRef CAS.
- B. S. S. Sudhakar Panday and P. Jeevanandam Daniel, Synthesis of nanocrystalline Co–Ni alloys by precursor approach and studies on their magnetic properties, J. Magn. Magn. Mater., 2011, 323, 2271–2280 CrossRef.
- A. A. Reinert, C. Payne, L. Wang, J. Ciston, Y. Zhu and P. G. Khalifah, Synthesis and Characterization of Visible Light Absorbing (GaN)1–x(ZnO)x Semiconductor Nanorods, Inorg. Chem., 2013, 52, 8389–8398 CrossRef CAS.
- D. M. R. L. Cascio and H. Bakker, Change in Magnetisation of Co50Ga50 Due to Atomic Disorder Induced by Quenching from High Temperatures, Phys. Status Solidi A, 1993, 135, 611–619 CrossRef.
- K. Schubert, H. L. Lukas, H. G. Meissner and S. Bhan, Zum Aufbau der Systeme Kobalt-Gallium, Palladium-Gallium, Palladium-Zinn und verwandter Legierungen, Z. Metallkd., 1959, 50, 534–540 CAS.
- A. O. Pan, G. W. D. Spratt, L. Tang and D. E. Laughlin, Magnetic properties and microstructure of evaporated Co oxide tape media, J. Magn. Magn. Mater., 1996, 155, 309–311 CrossRef.
- A. Nakatsuka, Y. Ikeda, N. Nakayama and T. Mizota, Inversion parameter of the CoGa2O4 spinel determined from single-crystal x-ray data, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, i109–i111 CrossRef CAS.
- M. T. Greiner, L. Chai, M. g. Helander, W. Tang and Z. Lu, Transition Metal Oxide Work Functions: The Influence of Cation Oxidation State and Oxygen Vacancies, Adv. Funct. Mater., 2012, 22, 455–4568 CrossRef.
- S. Li, G. Wang, H. Lv, Z. Lin, J. Liang, X. Liu, Y. Wang, Y. Huang, G. Wang and Q. Li, Constructing Gradient Orbital Coupling to Induce Reactive Metal-Support Interaction in Pt-Carbide Electrocatalysts for Efficient Methanol Oxidation, J. Am. Chem. Soc., 2024, 146, 17659–17668 CrossRef CAS.
- Z. Shen, J. Liu, H. Xu, Y. Yue, W. Hua and W. Shen, Dehydrogenation of ethane to ethylene over a highly efficient Ga2O3/HZSM-5 catalyst in the presence of CO2, Appl. Catal., A, 2009, 356, 148–153 CrossRef CAS.
- L. Liang, S. Xiong and Y. Xu, Low Content Ga2O3 Enables the Direct Methane Conversion, ACS Omega, 2024, 9, 25027–25033 CrossRef CAS PubMed.
- H. Li, Y. Yue, C. Miao, Z. Xie, W. Hua and Z. Gao, Dehydrogenation of ethylbenzene and propane over Ga2O3–ZrO2 catalysts in the presence of CO2, Catal. Commun., 2007, 8, 1317–1322 CrossRef CAS.
- K. M. Wunsch and E. Wachtel, Defektstruktur und Gitterparameter von Co-Ga-Legierungen mit B2-Struktur, Int. J. Mater. Res., 1982, 73, 311–315 CrossRef CAS.
- S. Penner, B. Klötzer, B. Jenewein, F. Klauser, X. Liu and E. Bertel, Growth and stability of Ga2O3 nanospheres, Thin Solid Films, 2008, 516, 4742–4749 CrossRef CAS.
- S. C. Ghosh, M. C. Biesinger, R. R. LaPierre and P. Kruse, X-ray photoelectron spectroscopic study of the formation of catalytic gold nanoparticles on ultraviolet-ozone oxidized GaAs(110) Substrates, J. Appl. Phys., 2007, 101, 114322 CrossRef.
- H. A. Budz, M. C. Biesinger and R. R. LaPierre, Passivation of GaAs by octadecanethiol self-assembled monolayers deposited from liquid and vapor phases, J. Vac. Sci. Technol., B, 2009, 27, 637 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Perkin-Elmer Corp, Eden Prairie, MN, 1992 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00179j |
| This journal is © The Royal Society of Chemistry 2025 |