Single electron transfer (SET) and iodine-atom transfer radical addition (I-ATRA) induced cyclopropanation reaction: elucidating the role of iodine†
Received
4th March 2025
, Accepted 25th June 2025
First published on 14th July 2025
Abstract
Mechanistic studies were conducted for the visible-light-mediated cyclopropanation reaction of alkenes with diazoacetate, catalyzed by a phenalenyl-based organic hydrocarbon (PLY) and co-catalysed by iodine, using DFT. In the absence of iodine, the system proceeds towards the hydroalkylation reaction, with the aid of aryl thiol. Both reactions were studied to understand the role of iodine in promoting the formation of cyclopropanes, the key structural units in pharmacophores. Our findings suggest that the single electron transfer (SET) will generate the transient iodine radical species, whereas the iodine atom transfer radical addition (I-ATRA) will construct the iodinated acetate radical moiety for the cyclopropanation reaction. On the other hand, the proton-coupled electron transfer (PCET) mechanism induces the formation of a protonated acetate radical for the hydroalkylation reaction. The negligible back donation from the lone pair orbital of iodine to the antibonding orbital of the newly generated C–C bond makes the iodine a better leaving group, thereby making the cyclisation feasible, validating the role of iodine in the cyclopropanation reaction. Various analysis techniques such as atoms in molecule, activation strain analysis, and energy decomposition analysis reveal that the favorable non-covalent interactions and steric effects promote the regioselective anti-Markovnikov radical addition over the Markovnikov radical addition.
Introduction
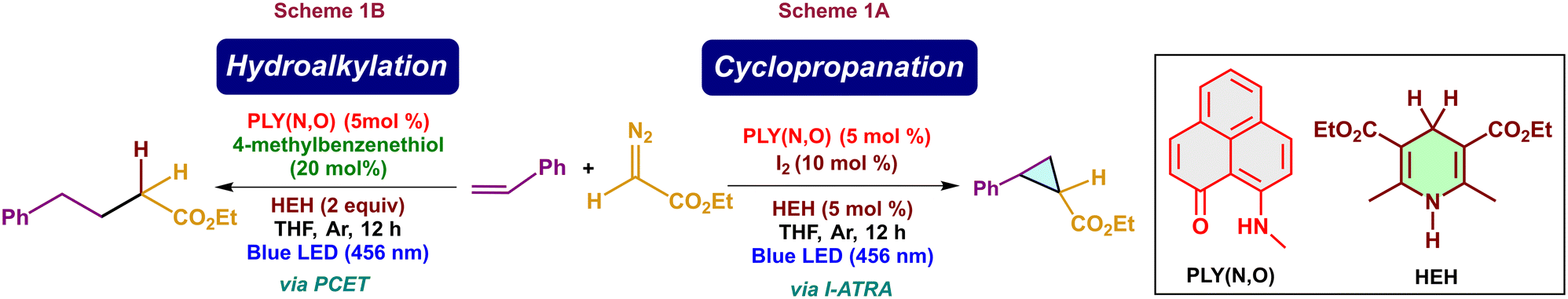
Phenalenyl (PLY), an odd alternant hydrocarbon-based photocatalyst, is a well-known organic catalyst for various reactions.1–3 This moiety has a high potential in its excited state, making it suitable for activating various inactive organic moieties without the use of any additives or exogenous oxidants.1,4 A photoexcited phenalenyl open-shell system can act as a super-reductant and possibly revolutionize the reactions which were relatively challenging with traditional photocatalysts.5 Recently, the photochemical properties of PLY were reported in certain studies.1,5 Mechanistically, one electron from the bonding molecular orbital of PLY will be excited to its non-bonding molecular orbital upon its photo-excitation. The excited PLY will then act as either an oxidant or a reductant depending upon the nature of the substrate and the reaction conditions.6 Hantzsch esters (HEH) are commonly used as terminal proton sources and electron donors in transfer hydrogenation reactions and photo-redox catalysis due to their bioinspired hydride donor properties.7 In 2024, S. K. Mandal's group showcased the iodine atom transfer radical addition (I-ATRA) method to generate cyclopropanated products using a phenalenyl-based catalyst under metal-free conditions in the presence of HEH and diazoacetate (Scheme 1A).8 Interestingly, in the absence of iodine, hydroalkylation of alkenes occurs with the aid of a thiol moiety (Scheme 1B).8 Atom transfer radical addition (ATRA) of alkenes is an excellent strategy for enabling efficient alkene functionalization.9 Reports for the cyclopropanation reactions involving the ATRA with iodine were seldom explored. Even though several metal-catalysed hydroalkylation reactions have been reported,10,11 there are only a few reports on the metal-free hydroalkylation reactions,12 which makes this reaction scheme significant.
 |
| | Scheme 1 Cyclopropanation (Scheme 1A) and hydroalkylation (Scheme 1B) reactions of alkenes with diazoacetate in the presence of PLY(N,O) catalyst. | |
Herein, we intend to explore the mechanism of the cyclopropanation reaction of alkenes with diazoacetate catalysed by a phenalenyl-based (PLY) reduced organic hydrocarbon via a photo-redox process. In particular, we became interested in exploring the mode of operation of co-catalyst iodine, the mechanism of the generation of the transient iodine radical, iodine atom transfer radical addition, and the origin of selectivity in the cyclopropanation reaction. In addition, we will be analysing the mechanism, the role of additives, and factors promoting the hydroalkylation reaction as well.
Computational methodology
All density functional theory (DFT) calculations were conducted using Gaussian 16 (Revision C.01) quantum chemical programs.13 Geometries were optimised using the hybrid functional B3LYP along with Grimme's D3 dispersion correction and the Becke–Johnson damping scheme using the 6-31+G(d,p) basis set for all atoms, except for Br and iodine, for which the Stuttgart–Dresden double-ζ (SDD) and LANL2DZ basis sets with an effective core potential (ECP) were employed, respectively. Frequency calculations were also performed with optimisation to ensure that the stationary points corresponded to either minima or a transition state. Intrinsic reaction coordinate calculations were conducted on the transition states to confirm their connections to the expected minima. The effect of the solvent was evaluated using the SMD continuum solvation model in tetrahydrofuran (ε = 7.42) solvent using the same functional but a different basis set, def2-TZVPP for all the atoms except for Br and iodine.13 For those, the SDD and LANL2DZ basis sets with an effective core potential (ECP) were employed, respectively. Excited-state calculations were performed with the time-dependent DFT (TD-DFT) method. To elucidate the fine electronic features of significant transition states, we have carried out topological analysis of electron density using the atoms in molecule (AIM) formalism. Activation strain analysis was conducted to understand the contribution of distortion and interaction energies by the participating fragments in the transition states.13 The activation strain energy can be written as the sum of interaction (ΔEint) and distortion (ΔEdis) energies.| | |
ΔE‡act = ΔEint + ΔEdis
| (1) |
The component-wise distribution to the interaction energy (ΔEint) was further analysed using the second-generation energy decomposition analysis (EDA), which is based on absolutely localised molecular orbitals (ALMO-EDA) using Q-Chem 6.2.13 The interaction energy between two fragments is dissected according to the following equation.| | |
ΔEint = ΔEpauli + ΔEelstat + ΔEpol + ΔEct + ΔEdisp
| (2) |
According to eqn (2), the interaction energy (ΔEint) is composed of Pauli repulsion (ΔEpauli), electrostatic interaction (ΔEelstat), polarisation (ΔEpol), charge transfer (ΔEct) and dispersion (ΔEdisp). Therefore, the activation strain energy can be written as:| |
 | (3) |
The sum of distortion energy (ΔEdis) and Pauli repulsion (ΔEpauli) can be considered as the contribution to steric effects (ΔEsteric), while the polarization (ΔEpol) and charge transfer (ΔEct) contributes to the orbital interactions (ΔEorbital). The ALMO-EDA calculations were performed at the B3LYP-GD3BJ/def2-TZVPP-SDD(Br),SMD(THF) level of theory. NBO (natural bonding orbital) analysis was carried out using NBO 7.0 software.13 Spin density plots and NBO plots are generated using Chemcraft software.13 The non-covalent interactions present in the selective transition states were plotted using interaction region indicator analysis by Multiwfn version 3.8(dev).13 IRI isosurfaces were rendered by visual molecular dynamics (VMD) software.13 The discussion in the manuscript is presented on the basis of the Gibbs free energies obtained at 298.15 K.
Results and discussion
The reaction begins with the excitation of the phenalenyl (PLY (N,O)) (1) catalyst to its singlet excited state S1, upon light irradiation. It subsequently relaxes to the long-lived triplet state (2) with an energy of 43.5 kcal mol−1 (Scheme 2 and Fig. 1). The triplet PLY (N,O) species (2) will accept an electron from the Hantzsch ester (HEH) (3) via a single electron transfer (SET) pathway. The energy barrier for the SET was found to be 18.1 kcal mo1−1, calculated using the Marcus–Hush theory.13 This results in the generation of the Hantzsch ester radical cation (4) along with the phenalenyl radical anion (5). The reverse SET, i.e., the formation of the PLY radical cation14 and the Hantzsch ester radical anion, was found to be energetically unfavorable (Fig. S2†), which is supported by the experimental findings.8
 |
| | Scheme 2 Proposed reaction scheme for the phenalenyl (PLY) catalysed cyclopropanation reaction. | |
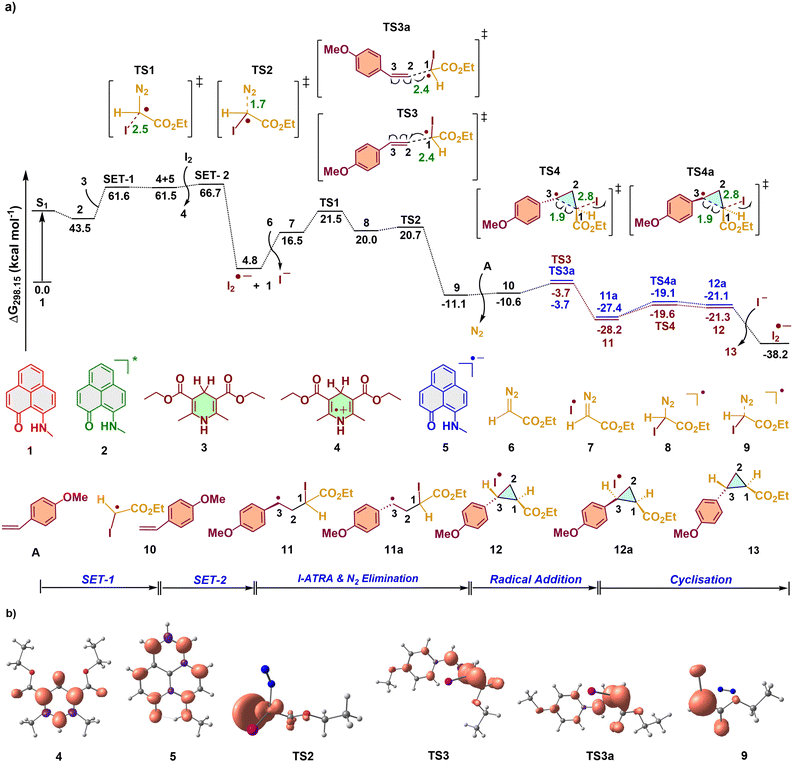 |
| | Fig. 1 a) Computed Gibbs free energy profile for the phenalenyl (PLY) catalyzed cyclopropanation reaction of alkenes with diazoacetate. b) Spin density plot of various key intermediates and transition states involved in the reaction mechanism (isosurface value = 0.005 au). | |
A possible energy transfer process from the species 2 was also examined (Fig. 2a).15 The TD-DFT calculations followed by the visualization of Kohn–Sham molecular orbitals show that the electronic excitations from the HOMO−1 to the LUMO of the diazoacetate present in the T3 triplet excited state populate the π* orbital of nitrogen, which corresponds to its release, to generate the carbene species (further details are provided in Table S2, ESI†). The energy required for this transition is 111.3 kcal mol−1. However, the energy gap between the triplet state and the S0 state of the PLY catalyst was calculated to be 43.5 kcal mol−1, which is insufficient for the energy transfer process to occur (Fig. 2a).13,16 These findings strongly support that the reaction doesn't involve the generation of carbene intermediates through energy transfer, reinforcing the experimental observations.8 The reaction will then bifurcate into two mechanistic pathways for the cyclopropanation and hydroalkylation reactions depending upon the experimental conditions. The presence of iodine prefers the cyclopropanation reaction, whereas in its absence, a hydroalkylation product will be generated with an aryl thiol moiety and an excess amount of the Hantzsch ester.
 |
| | Fig. 2 a) The Kohn–Sham molecular orbitals of diazoacetate. The energies of the molecular orbitals are provided in electron volts (eV). b) Calculated Gibbs free energies for the competing SET-2, PCET-A, and PCET-B processes. c) NCI plot for the cyclopropanation radical recombination transition state for (i) trans (TS4) and (ii) cis (TS4a) isomers. d) Natural bond orbital (NBO) analysis (isosurface value = 0.03 au) of transition states TS4b and TS4c. The donor–acceptor interaction energies (kcal mol−1) calculated via second order perturbation theory (SOPT) and orbitals involved are also provided. Gibbs free energy barriers are calculated with respect to the intermediate before the corresponding transition states. LP stands for the lone pair orbitals while BD* denotes the antibonding orbitals. | |
Cyclopropanation reaction
In the presence of I2, the reaction can proceed via three competitive pathways (Fig. 2b): a) PLY (N,O) catalyst regeneration from the phenalenyl radical anion (5) by transferring an electron to I2 through another SET process (SET-2). This leads to the generation of the key I2 radical anion in the reaction medium; b) the proton-coupled electron transfer pathway involving species 4, 5, and diazoacetate (6), which can generate Hantzsch ester radical intermediate 14, PLY catalyst 1 and protonated diazoacetate radical 15 (PCET-A); c) the PCET pathway involving 4, 5 and I2 (PCET-B), which leads to the formation of Hantzsch ester radical intermediate 14, PLY catalyst 1, hydrogen iodide (HI), and an iodine radical. Through PCET-A, the iodine radical cannot be generated, due to which the cyclopropanation will not be possible (details are provided below). Even though the iodine radical is formed via PCET-B, due to the difficulty in the dissociation of the proton from HI, the regeneration of cocatalyst I2 won't take place, which will affect the catalytic system of the cyclopropanation reaction. So, mechanistically, the reaction will only proceed through SET-2. From Fig. 2b, it is also evident that the energy of the intermediates formed after SET-2 is more stabilized than the intermediates formed after PCET-A and PCET-B. Moreover, the barrier for the SET-2 pathway is also very small (5.2 kcal mol−1) which makes it more feasible. The I2 radical anion, which is generated via SET-2, will be the source of the iodine atom radical, the key species involved in the iodine atom transfer radical addition (I-ATRA) mechanism with the diazoacetate (6). This occurs with an energy barrier of 21.5 kcal mol−1 (TS1, Fig. 1a). Subsequently, an expulsion of N2 occurs from the iodine-incorporated diazoacetate radical species (8) via TS2 with an energy barrier of 20.7 kcal mol−1. The iodinated acetate radical can then undergo a radical addition reaction with the olefinic moiety (A) in an anti-Markovnikov fashion to generate the intermediates 11 and 11a via two iso-energetic transition states, TS3 and TS3a (7.4 kcal mol−1 with respect to intermediate 9). The high yielding p-methoxystyrene (A) was used as the olefinic substrate for this study.8 The spin density plots for these transition states suggest that the radical is mainly located on the C1 carbon of the iodinated acetate radical (Fig. 1b). Another possible Markovnikov radical addition (MRA) transition state was found to exhibit a high energy barrier (TS3m).13 The cyclisation of the radical intermediate 11 can lead to the construction of cyclo-propane product 12 with a trans configuration via TS4. The other isomer 11a, undergoes cyclization, generating the cis-configured cyclopropane 12a via TS4a. The difference in the Gibbs free energy between the transition states for the formation of these cis and trans isomers is 0.5 kcal mol−1, which corresponds to the experimentally observed diastereomeric ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.8
:1.8
To gain additional insights into the origin of the energy difference between the diastereomeric transition states, non-covalent interactions (NCIs) were plotted. We found that TS4 is well decorated with a set of strong attractive non-covalent interactions, which includes two CH---O, two CH---π, and I---O interactions, whereas the stabilizing interactions found in TS4a are CH---O and two CH---π interactions. Hence the larger number of interactions present in TS4 can be accounted for as the reason for the extra stability. Upon the cyclisation, the eliminated iodine radical can further combine with the iodine anion which is already present in the system, to form the I2 radical anion. This species will then transfer an electron back to the triplet PLY (N,O) species (2) (via SET-1a with an energy barrier of 15.4 kcal mol−1), thereby generating phenalenyl radical anion 5 for the next catalytic cycle. That is, the Hantzsch ester is only required to initiate the first catalytic cycle, after which the generated iodine radical anion will initiate subsequent cycles by providing electrons to the triplet PLY (N,O) species (2). This justifies the use of the Hantzsch ester in a reduced amount for the cyclopropanation reaction. The Hantzsch ester radical cation 4 will be converted to Hantzsch pyridine B with the aid of the I2 radical anion at the end of the reaction.13
For the cyclopropanation reaction, after the SET process, the highest barrier in the mechanistic pathway corresponds to the iodine atom transfer radical addition (I-ATRA) transition state, TS1. In an attempt to describe the reason behind the ease of formation of cyclopropane in the presence of I2, we modelled transition states TS4b and TS4c with styrene as the olefinic substrate (Fig. 2d). The barrier for the transition state TS4b was determined to be 11.3 kcal mol−1, while TS4c was found to have a quite high barrier of 61.1 kcal mol−1 (with respect to the intermediate before the corresponding transition state), indicating that TS4b is more favorable. NBO analysis revealed that the donation from the lone pair orbital (LP) of hydrogen to the antibonding (BD*) orbital of the C1–C3 bond is stronger in magnitude in TS4c, which is quantified using second-order perturbation theory.17 This interaction suggests that the back donation will abate the formation of the C1–C3 bond due to the difficulty in removing the hydrogen radical, making the cyclisation less favorable. Meanwhile in the case of TS4b, the donation from the lone pair orbital (LP) of the iodine radical to the antibonding (BD*) orbital of the C1–C3 bond is not much pronounced, suggesting the stability of the iodine radical for its easier removal. The ease of the construction of the iodine radical can also be explained by the large size of the atom, which allows the electron-deficient orbital to spread out over a larger volume. This distribution can reduce the electron–electron repulsion and stabilize the radical, where the case is reversed for the hydrogen radical.
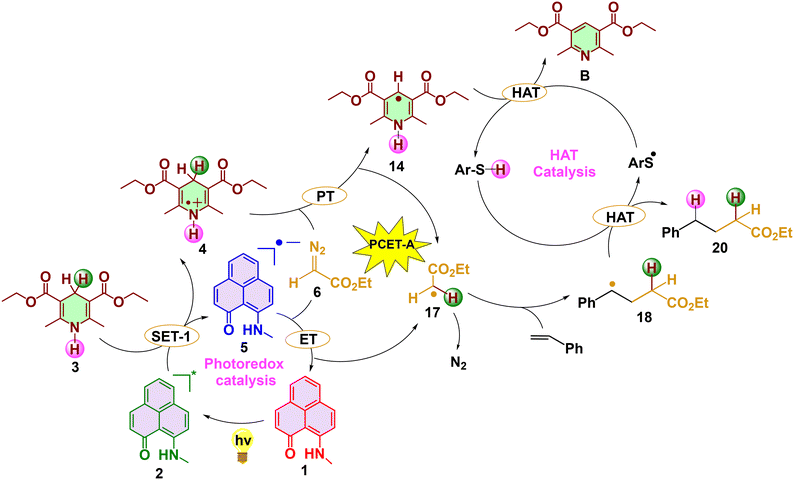
Hydroalkylation reaction
Experimentally, in the absence of I2, the reaction proceeds to form a hydroalkylated product with a higher concentration of the Hantzsch ester. We were intrigued to study the mechanism of this reaction, especially to find the role of the additional Hantzsch ester. The energetics of the hydroalkylation reaction is as follows (Scheme 3 and Fig. 3a). After the formation of the PLY(N,O) radical anion 5 and Hantzsch ester radical cation 4, since I2 is absent, the reaction will follow a proton-coupled electron transfer (PCET-A) mechanism to regenerate the catalyst. In this process, a proton from the C4 position of the Hantzsch ester radical cation 4 and an electron from the PLY radical anion 5 will be transferred to the diazoacetate 6 in a concerted manner. This favorably regenerates the photocatalyst along with the protonated diazoacetate radical 15. Instead of the PCET mechanism, we have looked into the energetics of the stepwise pathways that include either the proton transfer (PT) followed by the electron transfer (ET) or the ET followed by the PT. From the square scheme (Fig. S3†), it is clearly evident that the PCET mechanism is more favorable compared to the stepwise mechanism.18,19 This PCET-A will drive the expulsion of N2 to generate the protonated acetate radical 17 via the transition state TS5 with a barrier of 18.6 kcal mol−1. This also proves that it is the PCET process, instead of hydrogen atom transfer radical addition (H-ATRA), that paves the way for the generation of the protonated acetate radical in the hydroalkylation reaction. Afterwards, the protonated acetate radical undergoes radical addition with the styrene in an anti-Markovnikov fashion (via TS6 with an energy barrier of 10.4 kcal mol−1, with respect to the stable intermediate 17), generating a stable intermediate with a new C–C bond involving secondary radical 18. Here, styrene (A1) was used as the olefinic substrate for the mechanistic study. The stability of the species 18, despite being a radical species, can be attributed to the delocalization of its radical within the phenyl ring of the styrene, as seen in the spin density plot (Fig. 3b). From the mechanistic study of the hydroalkylation reaction, it is evident that the radical addition transition state TS6 is regioselective (Fig. 3a). With this optimized transition state in hand, we evaluated the other possible Markovnikov radical addition (MRA) transition state between styrene and the diazoacetate radical, leading to the formation of a primary radical intermediate. We found that the corresponding transition state TS6a traverses an energy barrier of 16.3 kcal mol−1, which is higher than TS6 by 5.9 kcal mol−1 (Fig. 3a) (with respect to the stable intermediate 17). In order to analyze the influence of the alkene substituent in the regioselective transition state, we also considered propene as the reacting partner with the acetate moiety. The trend is the same as that with the styrene, where the anti-Markovnikov radical addition (AMRA) (TS6b) is favored over the Markovnikov radical addition (MRA) (TS6c) (Fig. 4a), which is in agreement with the experimental findings.8
 |
| | Scheme 3 Proposed reaction scheme for the phenalenyl (PLY) catalysed hydroalkylation reaction. | |
 |
| | Fig. 3 a) Computed Gibbs free energy profile diagram for the phenalenyl (PLY) catalyzed hydroalkylation reaction of alkenes with diazoacetate. b) Spin density plots of various key intermediates and transition states involved in the reaction mechanism (isosurface value = 0.005 au). | |
 |
| | Fig. 4 a) Regioselective radical addition transition states for the propene and styrene substrates and the topological analysis of the electron density using the atoms in molecules (AIM) formalism. Bond critical points are marked in green color and their estimated strength using the Espinosa formulation is provided in parentheses. The relative Gibbs free energies in kcal mol−1 for the Markovnikov radical addition (MRA) transition states TS6a and TS6c are calculated with respect to the anti-Markovnikov radical addition (AMRA) transition states TS6 and TS6b, respectively; b) activation strain analysis for the transition states TS6, TS6a, TS6b, and TS6c. The relative energies in kcal mol−1 for the MRA transition states TS6a and TS6c are calculated with respect to the AMRA transition states TS6 and TS6b respectively. c) Contribution of different types of interactions obtained from the EDA for various para-substituted styrene and propene. The contributions of various components to the interaction energy, such as orbital interaction (ΔΔEorbital), dispersion interaction (ΔΔEdisp), electrostatic interaction (ΔΔEelstat), and steric effects (ΔΔEsteric), are calculated from the energy difference between anti-Markovnikov and Markovnikov radical addition transition states. Positive values indicate that the effect is promoting Markovnikov addition, while negative values indicate that the effect is promoting the anti-Markovnikov radical addition transition state. | |
These regioselective transition states were further analyzed using the atoms in molecule (AIM) formalism (Fig. 4a). The total number of bond critical points was considered to evaluate the total stabilization due to the non-covalent interactions (NCIs).13 From Fig. 4a, it is evident that the bond critical points are only present in TS6 (two π(phenyl)---HC interactions and one π(phenyl)---O interaction) and TS6b (CH---O interactions), while the non-covalent interactions are totally absent in the Markovnikov radical addition transition states TS6a and TS6c. Therefore, the NCIs present in TS6 and TS6b can be stated as one of the major reasons behind the stability of these transition states.13 In order to gain additional insights into the regioselective radical addition transition states, activation-strain analysis was carried out (Fig. 4b). Using this, the distortion and interaction energies between the two fragments present in the transition state were calculated. It was observed that the total distortion energy, which has a destabilizing effect, is smaller for both TS6 and TS6b compared to their respective Markovnikov radical addition transition states TS6a and TS6c. The interaction energy was also found to be stabilizing for both TS6 and TS6b.
To illustrate the component-wise contribution of interaction energy of the alkene substrate with the acetate radical to the overall regioselectivity, energy decomposition analysis (EDA) was conducted (Fig. 4c). For all the substitutions, the dominant effect that promotes the AMRA is the steric effect (ΔΔEsteric). Along with the steric effects, in the substitutions where R = H, F, Br, and OMe, dispersion interactions (ΔΔEdisp) were also found to be promoting the AMRA. However, for R = Me only the steric effects (ΔΔEsteric) promoted the anti-Markovnikov one. Since the sum of promoting interactions is greater than others, the AMRA is preferred for all substitutions. In the case where the propene is the reacting substrate, steric effects (ΔΔEsteric) along with the dispersion interactions (ΔΔEdisp) play the role of promoting the AMRA transition state.
After the regioselective radical addition step, a hydrogen atom transfer (HAT) process occurs from the aryl thiol species ArSH to the intermediate 18 via TS7, forming the final product, ethyl 4-phenylbutanoate 20 (with an energy barrier of 11.9 kcal mol−1 with respect to the stable intermediate 18). In the final step, the thiol (ArS) radical will abstract a hydrogen radical from 14 to regenerate aryl thiol species ArSH via TS8 with an energy barrier of 0.8 kcal mol−1 (with respect to the stable intermediate 21). This results in the generation of Hantzsch pyridine B. The Hantzsch ester was used in excess in this reaction since it is recovered as Hantzsch pyridine at the end of every catalytic cycle.
Conclusions
A comprehensive mechanistic study was conducted for the cyclopropanation and hydroalkylation reactions between olefins and diazoacetate with a phenalenyl (PLY) catalyst. The key intermediates and transition states involved in the catalytic cycle of both reactions were found by using density functional theory. The TD-DFT calculations followed by the visualization of Kohn–Sham molecular orbitals discarded the possibility of energy transfer between PLY and diazoacetate for the generation of transient carbene. A single electron transfer process from the PLY(N,O) radical anion to the iodine generates the transient iodine radical, which is involved in the subsequent iodine atom transfer radical addition step to generate the key iodinated acetate radical in the cyclopropanation reaction. However, the proton-coupled electron transfer process generates the protonated acetate radical during the hydroalkylation reaction. The experimentally observed diastereomeric ratio for the trans and cis cyclopropanes was validated through the modelling of respective transition states. NBO analysis revealed that the back donation from the lone pair orbital (LP) of hydrogen to the antibonding (BD*) orbital of the generated C–C bond is stronger in magnitude, whereas this similar interaction was less pronounced in iodine. This makes the cyclisation difficult with the removal of the hydrogen radical compared to the iodine radical, which in turn proves the role of iodine in the cyclopropanation reaction. The Hantzsch ester was used in excess in the hydroalkylation reaction since it is recovered as Hantzsch pyridine at the end of each catalytic cycle, unlike cyclopropanation, where the Hantzsch ester is only required for the initiation of the first catalytic cycle. The reason behind the stability of the anti-Markovnikov radical addition was decoded by various computational tools like AIM analysis, EDA, the activation-strain model, etc. It was observed that the non-covalent interactions and steric effects contribute to the stability of the anti-Markovnikov radical addition transition state over Markovnikov radical addition. Overall, the co-catalyst, additives, and mode of electron transfer play a prominent role in the generation of cyclopropanes, which are the key structural units in pharmacophores.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†
Author contributions
R. K. conceived and directed the projects. K. A. U., A. S. B. and S. K. did the computational calculations. R. K. and K. A. U. wrote the manuscript. All the authors discussed the results and commented on the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
R. K. gratefully acknowledges the Science and Engineering Research Board (SERB), Government of India (File Number: SRG/2022/000307) and the Scheme for Transformational and Advanced Research in Sciences (STARS) MoE project (File Number: STARS-2/2023-0829) for the financial support. K. A. U. and A. S. B. thank IIT Palakkad for the financial support. S. K. is grateful to the Department of Science and Technology (DST), Government of India, for the DST Women in Science and Engineering Post-Doctoral Fellowship (WISE-PDF) (Project Reference No. DST/WISE-PDF/CS-73/2023). The authors are also grateful to IIT Palakkad for the use of the Chandra high performance computing cluster and other computational facilities.
Notes and references
- P. P. Sen, N. Saha and S. Raha Roy, Investigating the Potency of a Phenalenyl-Based Photocatalyst under the Photoelectrochemical Condition for Intramolecular C-S Bond Formation, ACS Catal., 2024, 14, 907–920 CrossRef CAS.
- V. Pathania, V. Jyoti Roy and S. Raha Roy, Transforming Non-Innocent Phenalenyl to a Potent Photoreductant: Captivating Reductive Functionalization of Aryl Halides through Visible-Light-Induced Electron Transfer Processes, J. Org. Chem., 2022, 87, 16550–16566 CrossRef CAS.
- J. Ahmed, P. Sreejyothi, G. Vijaykumar, A. Jose, M. Raj and S. K. Mandal, A New Face of Phenalenyl-Based Radicals in the Transition Metal-Free C–H Arylation of Heteroarenes at Room Temperature: Trapping the Radical Initiator via C–C σ-Bond Formation, Chem. Sci., 2017, 8, 7798–7806 RSC.
- S. Sil, A. Santha Bhaskaran, S. Chakraborty, B. Singh, R. Kuniyil and S. K. Mandal, Reduced-Phenalenyl-Based Molecule as a Super Electron Donor for Radical-Mediated C-N Coupling Catalysis at Room Temperature, J. Am. Chem. Soc., 2022, 144, 22611–22621 CrossRef CAS.
- P. Datta, T. Goswami, N. Kandoth, A. Banik, J. Ahmed, A. S. Bhaskaran, R. Saha, R. Kuniyil, H. N. Ghosh and S. K. Mandal, Generation of Photoinduced Phenalenyl-Based Radicals: Towards Designing Reductive C−C Coupling Catalysis, ChemPhotoChem, 2023, 7, e202300033 CrossRef CAS.
- D. H. Reid, The Chemistry of the Phenalenes, Q. Rev., Chem. Soc., 1965, 19, 274–302 RSC.
- D. L. Zhu, Q. Wu, H. Y. Li, H. X. Li and J. P. Lang, Hantzsch Ester as a Visible-Light Photoredox Catalyst for Transition-Metal-Free Coupling of Aryl halides and Aryl sulfinates, Chem. – Eur. J., 2020, 26, 3484–3488 CrossRef CAS PubMed.
- P. Datta, D. Roy, D. Jain, S. Kumar, S. Sil, A. Bhunia, J. Dasgupta and S. K. Mandal, Uncovering the On-Pathway Reaction Intermediates for Metal-Free Atom Transfer Radical Addition to Olefins through Photogenerated Phenalenyl Radical Anion, ACS Catal., 2024, 14, 3420–3433 CrossRef CAS.
- J. M. Muñoz-Molina, T. R. Belderrain and P. J. Pérez, Atom Transfer Radical Reactions as a Tool for Olefin Functionalization – On the Way to Practical Applications, Eur. J. Inorg. Chem., 2011, 3155–3164 CrossRef.
- X. X. Wang, X. Lu, Y. Li, J. W. Wang and Y. Fu, Recent Advances in Nickel-Catalyzed Reductive Hydroalkylation and Hydroarylation of Electronically Unbiased Alkenes, Sci. China:Chem., 2020, 63, 1586–1600 CrossRef CAS.
- B. Liu and Q. Liu, Cobalt-Catalyzed Hydroalkylation of Alkenes and Alkynes: Advantages and Opportunities, ChemCatChem, 2024, 16, e202301188 CrossRef CAS.
- P. Miao, R. Li, X. Lin, L. Rao and Z. Sun, Visible-Light Induced Metal-Free Cascade Wittig/Hydroalkylation Reactions, Green Chem., 2021, 23, 1638–1641 RSC.
- Kindly refer to the ESI†.
- R. Paira, B. Singh, P. K. Hota, J. Ahmed, S. C. Sau, J. P. Johnpeter and S. K. Mandal, Open-Shell Phenalenyl in Transition Metal-Free Catalytic C−H Functionalization, J. Org. Chem., 2016, 81, 2432–2441 CrossRef CAS.
- T. Langletz, C. Empel, S. Jana and R. M. Koenigs, Stereoconvergent, photocatalytic cyclopropanation reactions of β-substituted styrenes with ethyl diazoacetate, Tetrahedron Chem, 2022, 3, 100024–100030 CrossRef CAS.
- A. K. Sharma and F. Maseras, The Subtle Mechanism of Nickel-Photocatalyzed C(SP3)-H Cross-Coupling, Inorg. Chem., 2024, 63, 13801–13806 CrossRef CAS PubMed.
- F. Weinhold, C. R. Landis and E. D. Glendening, What Is NBO Analysis and How Is It Useful?, Int. Rev. Phys. Chem., 2016, 35, 399–440 Search PubMed.
- R. Tyburski, T. Liu, S. D. Glover and L. Hammarstrom, Proton-Coupled Electron Transfer Guidelines, Fair and Square, J. Am. Chem. Soc., 2021, 143, 560–576 CrossRef CAS.
- A. J. Göttle and M. T. M. Koper, Proton-Coupled Electron Transfer in the Electrocatalysis of CO2 Reduction: Prediction of Sequential vs. Concerted Pathways Using DFT, Chem. Sci., 2016, 8, 458–465 RSC.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Athul Santha Bhaskaran
,
Athul Santha Bhaskaran