
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of hafnium-silica solid Lewis acids for γ-valerolactone production from biobased levulinic acid†
Margarida M. Antunes *a,
Kámilah Khana,
Patrícia Nevesa,
Auguste Fernandesb,
Mindaugas Andrulevičiusc,
Filipa Ribeirob and
Anabela A. Valente*a
*a,
Kámilah Khana,
Patrícia Nevesa,
Auguste Fernandesb,
Mindaugas Andrulevičiusc,
Filipa Ribeirob and
Anabela A. Valente*a
aDepartment of Chemistry, CICECO-Aveiro Institute of Materials, University of Aveiro, Campus Universitário de Santiago, 3810-193 Aveiro, Portugal. E-mail: margarida.antunes@ua.pt; atav@ua.pt
bCentro de Química Estrutural, Instituto Superior Técnico, Av. Rovisco Pais, 1049-001 Lisboa, Portugal
cInstitute of Materials Sciences of Kaunas University of Technology, A218, Barsusko Str. 59, Kaunas, LT-51423, Lithuania
First published on 27th May 2025
Abstract
There is growing awareness of the need to expand the γ-valerolactone (GVL) market within the biobased economy, although challenges remain in developing sustainable, green processes. In this work, the biobased top platform chemical, levulinic acid (LA), was converted to GVL under relatively mild conditions, via catalytic transfer hydrogenation (CTH) routes, using solid Lewis acid catalysts consisting of morphologically distinct EPDM and LFS type silicas and hafnium. The catalyst development was based on important requirements for process feasibility and involved several post-synthesis strategies and introduction of Hf-sites via different methods to meet superior performances. Complementary structural and molecular-level characterisation techniques were employed together with catalytic and kinetic modelling/computational studies for studying the influence of the material properties on the catalytic performances for targeting GVL. The developed heterogeneous catalysts contributed to enhanced productivity (up to 87% GVL yield, at 180 °C), were stable in consecutive runs, and also seemed promising in terms of the E factor. EPDM and LFSs were never investigated before for biomass conversion processes or any other catalytic system, to the best of our knowledge.
Introduction
Non-renewable sources of organic carbon, i.e., fossil fuels, represent more than 80% of the world's energy mix, and have largely contributed to global climate change, health and equity concerns, greatly pressing the chemical industry into the energy transition towards renewable and sustainable feedstocks.1–5 The carbon footprint of the chemical industry may be reduced via the use of renewable sources of organic carbon, obtainable from municipal/industrial residues/waste. In this sense, there is an urgent need to convert vegetable biomass derived compounds to useful biobased chemicals that may complement/substitute petrochemicals.An important biobased top platform chemical is levulinic acid (LA), which is synthesised from non-edible lignocellulosic biomass.6–8 A reference player of the LA market is GF Biochemicals Ltd.9,10 The synthesis of LA involves acid hydrolysis of hemicelluloses (C5 route) or cellulose (C6 route), under relatively mild conditions, giving predominantly pentoses such as D-xylose (C5) and the hexose D-glucose (C6).11–13 The C5 route proceeds via dehydration of D-xylose to furfural, followed by hydrogenation to furfuryl alcohol and furan ring opening to LA (Scheme 1).12,13 On the other hand, the C6 route proceeds via the isomerisation of D-glucose, dehydration to 5-(hydroxymethyl)furfural and furan ring opening to LA plus formic acid.14–16
 | ||
| Scheme 1 Vegetable biomass valorisation routes to levulinic acid and subsequent conversion to levulinic acid (LA), levulinate esters (LEs) and γ-valerolactone (GVL). | ||
The carbonyl and carboxylic functional groups of LA make it a reactive/versatile building block for several applications.11,13,17 LA can be converted to useful levulinate esters (LEs) and γ-valerolactone (GVL).13 LEs are promising for food formulations, cosmetics, fragrances, resins, polymers, plasticisers, coatings and agrochemicals.13,17,18 On the other hand, the application profile of GVL includes green solvents, liquid fuel bioadditives and green chemical intermediates.13 The conversion of LA to GVL involves hydrogenation, thus requiring a hydrogen source and adequate catalysts.19 The use of molecular hydrogen poses several concerns related to storage, transportation and equipment safety/costs.20–22 Envisaging sustainable production processes, strategies to convert LA to GVL have included the use of safe hydrogen donors (H-donors), non-noble metal catalysts to avoid expensive/scarce noble metals, and moderate operating conditions.19,20,23 Safe H-donors include secondary alcohols which, in combination with non-noble metal heterogeneous catalysts, may promote the conversion of LA to GVL via catalytic transfer hydrogenation (CTH).
Different families of heterogeneous catalysts were reported for the conversion of LA to GVL via CTH routes: mixed metal oxides, organic–inorganic hybrids, silicates and zeolites.22 Nevertheless, important requirements to be put on the CTH catalysts include: their commercial availability, cost, reduced toxicity, thermal resistance (e.g., >450 °C to remove coke – in this sense, catalysts possessing organic components, such as organic–inorganic hybrids may present drawbacks), versatility to tune the catalyst surface properties and accessibility of the active sites to enhance productivity. A study of the CTH of ethyl levulinate to GVL, reported by Luo et al.,24 compared different types of transition metals (Ti, Sn, Hf, Zr) in zeolites (20–40 days hydrothermal syntheses, using hydrofluoric acid), which showed the superiority of the Hf-catalyst, using 2-butanol as a H-donor. The superior performances of Hf-catalysts were also reported for other systems related to biomass conversion, e.g., furfural to GVL.25 In the development of CTH catalysts, scaling-up is another important factor which may be facilitated in the case of catalyst preparation via post-synthesis treatments (e.g., in relation to hydrothermal synthesis under autogenous pressure).26,27
In this work, recognising the importance of efficient processes using catalysts which fulfil the requirements for the conversion of LA to GVL via CTH routes, new hafnium/silica catalysts were developed via top-down strategies, from morphologically distinct EPDM, LFS-150 and LFS-50 silicas. EPDM consists of micron-size spheres (ca. 1.5–2.0 μm) composed of small interwoven primary entities and has macro/mesoporosity. On the other hand, the LFSs consist of aggregates of overlapping fine (20–500 nm thick) scaly-like particles with different widths and mesoporosity. The material properties were modified via post-synthesis treatments to meet superior performances for GVL production, at 180 °C. The influence of the type of silica support, modification treatments and respective conditions, hafnium loading and the type of impregnation method on the material properties and catalytic performances was studied by combining molecular level solid-state characterisation techniques of the materials (vibration spectroscopy with a base probe, NMR, XPS, etc.), catalytic/kinetic modelling and catalyst stability studies. EPDM and LFSs were never investigated for biomass conversion processes or any other catalytic system, to the best of our knowledge. These Hf-silicas are, to the best of our knowledge (based on reported characterisation studies), the first purely Lewis acidic (without intrinsic Brønsted acidity) transition metal silicates developed for the target reaction. The modified Hf-silicas developed are among the top reported fully inorganic transition metal/silicas, in terms of GVL yields for the target reaction, and seem promising in terms of the E factor.
Experimental
The suppliers and purities of the chemicals and materials used are indicated in the ESI.†Preparation and characterisation of the catalysts
The silicas EPDM-10-1000-AW (EPDM, 99.99%, AGC SI-Tech Co., Ltd.; ca. 1.5–2.0 μm spherical particles), LFS-HN-050 (LFS50, >98%, AGC; mean particle width ≅ 0.5 μm) and LFS-HN-150 (LFS150, >98%, AGC; mean particle width ≅ 1.5 μm) were subjected to different post-synthesis treatments (described below) to furnish them with adequate activity for GVL production. The materials were characterised by complementary techniques (please see details in the ESI†), namely powder X-ray diffraction (PXRD), scanning electron microscopy (SEM), energy dispersive X-ray spectroscopy (EDS), N2 sorption isotherms, diffuse reflectance (DR) UV-vis spectroscopy, 29Si (CP) MAS NMR, X-ray photoelectron spectroscopy (XPS), and attenuated total reflectance Fourier-transform infrared (ATR FT-IR) spectroscopy.The acid properties were measured by FT-IR spectroscopy of adsorbed pyridine (details in the ESI†). According to the literature, pyridine is an adequate base probe to quantify the Lewis acid sites in hafnium silicates possessing different types of Hf sites (including HfO2).28 This technique allows Lewis (L) and Brønsted acid (B) sites to be distinguished and each type to be quantified (based on the areas of the bands at ca. 1540 and 1430–1460 cm−1 which are associated with pyridine adsorbed on B and L acid sites, respectively, and using molar extinction coefficients determined in the home-made IR installation used). This technique also gives insight into the acid strength, based on the molar ratio of acid sites measured at 350 °C and 150 °C (L350/L150 and B350/B150) and considering that the acid sites of moderate strength remain adsorbed at 150 °C, and relatively strong acid sites remain adsorbed at 350 °C.
The base properties were measured by temperature programmed desorption (TPD) of CO2 as a probe molecule (CO2-TPD), using a Micromeritics Autochem II chemisorption analyser equipped with a fixed-bed U-shaped quartz flow reactor. The sample (100 mg) was pre-treated in situ at 400 °C (10 °C min−1) for 1 h, under a He flow (50 mL min−1). The reactor was cooled down to 100 °C and then fed with a gas mixture composed of 10% CO2 and 90% He (50 mL min−1), for 1 h. Afterwards, the reactor was purged with He for 1 h to remove excess/physisorbed CO2, and then the sample was heated until 800 °C (10 °C min−1) and the CO2 desorbed was analysed using a thermal conductivity detector (TCD). The amount of desorbed CO2 was calculated via deconvolution of the CO2-TPD curves, and peak integration, using calibration curves. The TPD profiles were bimodal (a peak up to 400 °C, assigned to weak (Wb) base sites plus medium strength (Mb) base sites, and another peak in the range of ca. 400–800 °C, assigned to strong base sites (Sb)). The total amount of CO2 desorbed equals Wb + Mb + Sb. The discussed base strength is based on the molar ratio Sb/(Wb + Mb).
Commercial EPDM (1.5 g) was mixed with 45 mL of an alkaline solution (base concentration, z = 0.04–1.2 M aq. TPAOH or 0.2 M aq. NaOH), in a Teflon® Erlenmeyer flask, and stirred (500 rpm) for a specified time (x = 0.5–3 h), at 65 °C. The solid was separated by filtration, washed with hot deionised water at 60 °C until neutral pH, and dried at 100 °C for 2 h, giving EPDM-zTPAx and EPDM-zNax. The commercial LFSs were pre-dried at 85 °C overnight, ground using an agate mortar/pestle, and then modified in a similar fashion to EPDM, but using a 0.2 M alkaline solution, for 3 h, at 65 °C, leading to LFS50-Na, LFS50-TPA, LFS150-Na and LFS150-TPA. Since desilication produces inorganic debris (which may cause pore blockage and affect mass transfer during the catalytic processes),47–50 the treated silicas were subjected to acid washing, as generally described in the literature (e.g., for silicate-1,45 zeolites30,37,51,52); the acid washing also eliminates alkaline cations. Specifically, 1 g of treated silica was mixed with 100 mL of 0.1 M aq. HCl in a Teflon® Erlenmeyer flask, for 5 h, at 65 °C (with stirring, 500 rpm). The solid was filtered, washed with hot deionised water (60 °C) until neutral pH, and dried at 100 °C for 2 h; the removal of Cl and Na was checked by EDS. This led to the materials (sample names starting with H-): H-EPDM-zNax, H-EPDM-zTPAx, H-LFS50-Na, H-LFS150-Na, H-LFS50-TPA, and H-LFS150-TPA. The synthesis yields were generally in the range of 55–88% and 79–99% for the Na- and TPA-treated materials, respectively.
For the WI method, 1 g of treated/untreated silica support (EPDM, H-EPDM-zNax, H-EPDM-zTPAx, EPDM-HT1.5, EPDM-HT4.5, LFS50, LFS150, H-LFS50-Na, H-LFS150-Na, H-LFS50-TPA, H-LFS150-TPA, LFS50-HT, LFS150-HT) was mixed with 0.112 mmol Hf(acac)4 in 6 mL toluene (w = 2 wt% Hf), then heated at 60 °C (with stirring) until complete evaporation of the solvent, and finally calcined as described above. Selected supports (EPDM, LFS50, LFS50-HT, LFS50-TPA, LFS50-Na, LFS150, LFS150-HT, LFS150-TPA, LFS150-HT) were impregnated in a similar fashion, but with w = 8 wt% Hf (0.448 mmolHf g−1).
Catalytic and kinetic studies
The catalytic reactions were carried out using borosilicate batch milli-reactors with a pear-shaped bottom, equipped with a Teflon® valve (for loading/emptying/purging the reactor) and a Teflon® coated magnetic stirring bar. For all catalysts, the reactors were loaded with a solution of 0.45 M LA in 2BuOH (0.75 mL) plus a catalyst (25.5 gcatalyst L−1), and then immersed in a pre-heated oil bath at 180 °C, with stirring (ca. 800 rpm, checked to avoid mass transfer limitations); this moment was taken as the initial instant of the catalytic reaction.Individual reactors were prepared for each experimental point (reaction time) and the presented results are the mean values of at least two replicates (<6% error). Freshly prepared samples (from the cooled reactors) were analysed by gas chromatography (GC) using a Thermo Scientific Trace 1300 Series GC equipped with an Agilent Technologies, Inc. capillary column (DB-5, 30 m × 0.32 mm × 0.25 μm; He as carrier gas) and a flame ionisation detector (FID). The quantification of LA and reaction products was based on response factors determined by calibration curves using internal standards (the response factor for 2-butyl levulinate (2BL) was considered as that of commercially available 1-butyl levulinate 1BL). The reaction products were identified using a Shimadzu QP 2010 ultra-GC-MS, equipped with a HT-5 GC column (25 m × 0.32 mm × 0.10 μm; He as carrier gas).
After the reaction, the solid was separated from the reaction mixture by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 min, thoroughly washed with 2BuOH, dried overnight at 80 °C and finally calcined at 550 °C for 5 h (20 mL min−1 air flow). The contact tests (CTs) consisted of bringing into contact the solid catalyst with 2BuOH at 180 °C, under identical conditions to those for a normal catalytic test, but without LA; after 8 h, the solid was separated by centrifugation (10000 rpm, 5 min). The liquid phase was passed through a 220 nm pore size PTFE membrane, and the filtrate (LP) was transferred to a separate clean reactor. LA was added to the LP solution to give an initial LA concentration of 0.45 M, which was left to react at 180 °C (monitored by GC). Commercial HfO2 was tested as a catalyst in a hafnium molar amount equivalent to that present in the catalytic mixtures with 16Hf-silicas.
000 rpm for 5 min, thoroughly washed with 2BuOH, dried overnight at 80 °C and finally calcined at 550 °C for 5 h (20 mL min−1 air flow). The contact tests (CTs) consisted of bringing into contact the solid catalyst with 2BuOH at 180 °C, under identical conditions to those for a normal catalytic test, but without LA; after 8 h, the solid was separated by centrifugation (10000 rpm, 5 min). The liquid phase was passed through a 220 nm pore size PTFE membrane, and the filtrate (LP) was transferred to a separate clean reactor. LA was added to the LP solution to give an initial LA concentration of 0.45 M, which was left to react at 180 °C (monitored by GC). Commercial HfO2 was tested as a catalyst in a hafnium molar amount equivalent to that present in the catalytic mixtures with 16Hf-silicas.
A pseudo-homogeneous kinetic model was developed, considering a perfectly stirred, isothermal batch reactor and irreversible and first order reactions (please see details in the ESI†), which supported the discussed mechanistic proposal, and produced the kinetic constants of each step (ki).
Results and discussion
Characterisation of the materials
The SEM images of the parent EPDM show micron-size spheres (ca. 1.5–2.0 μm) composed of small interwoven primary entities (ca. 100–200 nm size) (Fig. 1a and S5†). The treatments (HT, TPAOH (Fig. S5 and S6†), NaOH (Fig. S7†)) and the WI method (Fig. 1b) did not seem to affect the morphology. However, the SS method caused some fracture of spherical particles (Fig. 1c and d), indicating the limited mechanical resistance of EPDM towards grinding processes. The elemental mappings suggested uniform Si and Hf distributions for the wHf-EPDMs prepared via the SS and WI methods (Fig. S8†).
 | ||
| Fig. 1 SEM images of EPDM (a), 2Hf-EPDM-WI (b), 2Hf-EPDM-SS (c) and 8Hf-EPDM-SS (d). | ||
Commercial EPDM exhibited a type III N2 sorption isotherm (Fig. S9a;† characteristic of materials which have weak adsorbate–adsorbent interactions) and a specific surface area (SBET) of 57 m2 g−1 (Table S1†). The pore size distributions measured by N2 sorption and Hg intrusion indicated 2–10 nm size mesopores (Fig. S10a and Table S1†) and 70–100 nm size macropores. Hence, EPDM presented bimodal mesopore/macropore distributions. Possibly, the mesopores may be intrinsically associated with the primary entities, and the macropores may be void spaces between primary entities.
The treated EPDMs exhibited similar N2 sorption isotherms to the parent EPDM (Fig. S9a†), with comparable or slightly broader mesopore size distributions (2–18 nm; Table S1†). The HT treatment (1.5 or 4.5 h duration) did not enhance the SBET (31–35 m2 g−1 for EPDM-HT1.5 and EPDM-HT4.5) (Table S1†). On the other hand, the TPAOH treatment enhanced the SBET of the EPDM-zTPAx silicas, which increased with increasing base concentration (z) in the order (keeping constant x = 3 h): 57 m2 g−1 (EPDM) < 71 m2 g−1 (EPDM-0.08TPA3 and EPDM-0.2TPA3) < 80 m2 g−1 (EPDM-0.4TPA3) < 86 m2 g−1 (EPDM-0.6TPA3) < 149 m2 g−1 (EPDM-1.2TPA3) (Table S1†). For the 0.2 M NaOH treatment, the SBET was not significantly influenced by the treatment time (x) until 1.5 h (52–53 m2 g−1 for EPDM-0.2Nax with x = 0.5 or 1.5 h), but a longer treatment (x = 3 h) enhanced the SBET (86 m2 g−1 for EPDM-0.2Na3). The acid washing of the alkaline treated EPDMs enhanced the SBET, which may be attributed to the removal of inorganic debris, e.g., 71/96 m2 g−1 for EPDM-0.2TPA3/H-EPDM-0.2TPA3; 86/167 m2 g−1 for EPDM-0.2Na3/H-EPDM-0.2Na3 (Table S1†).
The types of Si sites of the supports may change during the HT/TPA/Na treatment and/or Hf impregnation process, which was studied by 29Si MAS NMR and 29Si{1H} CP MAS NMR spectroscopy. The types of Si sites may be Qn = Si(OSi)n(OH)4−n, n = 1, 2, 3 or 4. The 29Si MAS NMR technique allows the amount of silanol groups to be calculated. On the other hand, 29Si{1H} CP MAS NMR spectroscopy is a qualitative technique that enhances the signal of the Si sites with proximal protons (e.g., Q2 and Q3 sites), and thus it is relatively sensitive to changes in the local environments of the Si sites, complementing 29Si MAS NMR spectroscopy. The 29Si MAS NMR spectrum of the parent EPDM showed a main peak at −110 ppm due to Q4 (fully condensed siloxane groups), a small shoulder centred at ca. −102 ppm due to Q3 (Si sites with one hydroxyl group) and a very small peak at −95 ppm due to Q2 (Si sites with geminal hydroxyl groups) (Fig. 2a).59–63 Consistently, 29Si{1H} CP MAS NMR spectroscopy indicated increased relative intensity of the Q3 and Q2 peaks in relation to Q4, due to the proximal hydrogen atoms (Fig. 2b).60,62 The untreated and treated EPDM supports exhibited comparable peaks, with slight enhancement in the concentration of silanols after treatment ([SiOH], which includes Q2 and Q3 sites, Table S2†): 0.4 mmol g−1 for untreated EPDM versus 0.9–1.1 mmol g−1 for the treated EPDM supports.
 | ||
| Fig. 2 29Si MAS NMR (a, c and e) and 29Si{1H} CP MAS NMR spectra (b, d and f) of selected EPDMs (a and b), LFS50s (c and d) and LFS150s (e and f). | ||
After Hf impregnation, the relative intensity of the Q3 peak decreased considerably (29Si{1H} CP MAS NMR spectra, Fig. S11†), which may be due to the interactions between Q3 sites and the hafnium precursor, possibly forming Si–O–Hf bonds (supported by XPS, discussed below). Somewhat comparable results were reported by Zhang et al.61 for Zr impregnated on SBA-15, in that Q3 sites were converted to pseudo Q4 sites of the type Si(OSi)3(OZr), although the latter were hardly distinguishable spectroscopically from the (abundant) Q4 groups (Si(OSi)4).
As opposed to 29Si (CP) MAS NMR spectroscopy, ATR FT-IR spectroscopy (Fig. S12a and b†) was not sufficiently sensitive for distinguishing silanol groups,64–66 which may be partly due to low concentration limitations and/or superimposable broad bands; e.g., the broad band centred at ca. 1065 cm−1 (assignable to Si–O–Si vibrations; Table S3†).
The DR UV-vis spectra of the Hf-EPDMs were different from that of HfO2 (Fig. S13a and b†), supporting their distinct chemical features. Specifically, monoclinic HfO2 possesses seven coordinated Hf-sites67 and exhibited an intense, single band at ca. 208 nm. On the other hand, the wHf-EPDMs exhibited a band at ca. 220 nm (like the parent support). For w ≤ 4 wt%, the wHf-EPDMs exhibited a band at ca. 200 nm (like the parent support), which decreased in relative intensity or disappeared with increasing w. These results correlated with the decreased relative intensity of the Q3 peak (mentioned above) after Hf impregnation. Somewhat consistently, according to the literature, the ca. 200 nm band may be due to defect sites of the silica framework,68–70 which may be relatively reactive with the Hf-precursor to form Si–O–Hf bonds. For w ≥ 8 wt%, a new small band appeared at ca. 260 nm (not observed for the respective support), which may be due to Hf-sites possessing distorted coordination environments and/or relatively high coordination numbers,71 or defect Hf-sites in small clusters.70 For w ≥ 8 wt% Hf, the materials additionally exhibited steep absorption as the wavelength tended to 190 nm, as well as a broad band at ca. 380 nm (Fig. S13a and b†), assignable to HfOx species72 (which may be amorphous for 8Hf-EPDMs, since these materials did not exhibit PXRD peaks characteristic of crystalline HfO2). Based on the DR UV-vis spectral features of HfO2 (intense band at relatively low wavelengths) and the PXRD studies (formation of HfO2 for higher w values), one cannot exclude the possibility of the steep absorption for w ≥ 8 wt% Hf, being associated with polynuclear hafnium oxide species. Nevertheless, based on the literature, the spectral region below 210 nm may have contributions from tetrahedral/isolated Hf-sites.25,70,71
Fig. 3a shows the XPS spectra (Hf 4f region) of selected EPDMs, namely, untreated wHf-EPDMs (8Hf-EPDM-SS, 8Hf-EPDM-WI, 16Hf-EPDM-SS) and TPA-treated 8Hf-EPDM-0.2TPA3-SS, and for comparison HfO2. Pure HfO2 exhibited two peaks at 17.3 eV and 18.9 eV due to the spin–orbit split doublets Hf 4f7/2 and Hf 4f5/2, respectively. The binding energies associated with these peaks are in good agreement with literature data for HfO2.73,74 On the other hand, the xHf-EPDMs exhibited two peaks which are slightly shifted to higher binding energies in relation to HfO2 (e.g., for 8Hf-EPDM-0.2TPA3-SS, the binding energies associated with Hf 4f7/2 and Hf 4f5/2 are 17.4 and 19.0 eV, respectively). Similar peak shifting was reported in the literature for Hf-silicates, and was attributed to the presence of framework Hf-sites in tetrahedral coordination.72,73,75,76 Accordingly, the wHf-EPDM silicates may possess tetrahedral Hf-sites. Johnson et al.28 reported different types of tetrahedral Hf-sites playing important roles in the CTH reaction of cyclohexanone with butanol, namely closed sites (fully condensed) and open sites (possessing an OH group), which may have different intrinsic activities.
 | ||
| Fig. 3 XPS spectra of (a) 8Hf-EPDM-W, 8Hf-EPDM-SS, 16Hf-EPDM-SS and 8Hf-EPDM-0.2TPA3-SS; (b) 8Hf-LFS0-SS, 8Hf-LFS50-WI, 8Hf-LFS50-HT-SS, 8Hf-LFS50-HT-WI, 8Hf-LFS50-TPA-SS, and 8Hf-LFS50-TPA-WI; (c) 8Hf-LFS150-TPA-SS and 8Hf-LFS150-TPA-WI (please see legends in a–c for sample names). The spectrum for HfO2 is included for comparison (black line in a–c). | ||
For more detailed XPS analyses, fittings were performed in the Hf 4f region for the materials 16Hf-EPDM-SS, 8Hf-EPDM-SS and bulk HfO2 (Fig. S14†). The fitted spectrum of 16Hf-EPDM-SS evidenced a shift of the Hf 4f7/2 main peak at 17.3 eV for HfO2 to 17.5 eV for 16Hf-EPDM-SS, which was accompanied by peak broadening. Hence, the spectrum of 16Hf-EPDM-SS included a new relatively low intensity Hf 4f7/2 peak at a higher binding energy of 18.2 eV (not appearing for bulk HfO2) assignable to Hf–O–Si groups (in agreement with the above discussion). The shift to higher binding energy may be explained by the higher electronegativity of silicon in relation to hafnium. Specifically, SiO– groups surrounding Hf centers (e.g., Hf(–OSi)x) may withdraw more electron density from the Hf centers than HfO– groups surrounding Hf centers (as in Hf(–OHf)x, for example). Hence, Hf–O–Si groups may lead to more electron deficient Hf centers, conferring acidity to the materials (ascertained by acid property measurements, discussed below). For 16Hf-EPDM-SS, the small shift of the main peak (ca. 0.2 eV in relation to bulk HfO2) and the weak intensity of the Hf 4f7/2 peak at 18.2 eV (Hf–O–Si) suggested that this material possessed a significant portion of HfO2 type species, which is consistent with the PXRD and DR UV-vis spectroscopy studies. On the other hand, the fitted spectra of 16Hf-EPDM-SS and 8Hf-EPDM-SS were somewhat comparable (Fig. S14†), which somewhat parallels the roughly comparable acid site concentrations for these two materials, discussed below (Table 1).
| Sample | La (μmol g−1) | L350/L150b |
|---|---|---|
| a Acid properties (L = Lewis acid sites) measured by FT-IR of adsorbed pyridine. For 8Hf-LFS50-Na-WI, 8Hf-LFS150-TPA-WI, and 8Hf-LFS150-Na-WI, several attempts failed to form a wafer, and thus the FT-IR spectra were not recorded.b Lewis (L) acid strength based on the molar ratio L350/L150.c For this material, the amount of Brønsted (B) acid sites = 9 μmol g−1 (the remaining materials did not possess measurable B acidity). | ||
| 8Hf-EPDM-SS | 10 | — |
| 8Hf-EPDM-WI | 4 | — |
| 12Hf-EPDM-SS | 10 | — |
| 16Hf-EPDM-SS | 12 | — |
| 8Hf-EPDM-HT4.5-SS | 8 | — |
| 8Hf-EPDM-0.2TPA3-SS | 7 | — |
| 8Hf-EPDM-1.2TPA3-SS | 9 | — |
| 8Hf-EPDM-0.2Na3-SS | 12 | |
| 2Hf-LFS50-SS | 9 | — |
| 4Hf-LFS50-SS | 29 | — |
| 8Hf-LFS50-SS | 30 | 0.07 |
| 8Hf-LFS50-WI | 30 | — |
| 12Hf-LFS50-SS | 56 | 0.09 |
| 16Hf-LFS50-SS | 49 | 0.10 |
| 8Hf-LFS50-HT-SS | 37 | — |
| 8Hf-LFS50-HT-WI | 56 | — |
| 8Hf-LFS50-TPA-SSc | 48 | 0.08 |
| 8Hf-LFS50-TPA-WI | 54 | 0.02 |
| 8Hf-LFS50-Na-SS | 29 | — |
| 2Hf-LFS150-SS | 11 | — |
| 4Hf-LFS150-SS | 24 | 0.06 |
| 8Hf-LFS150-SS | 28 | — |
| 8Hf-LFS150-WI | 27 | 0.07 |
| 12Hf-LFS150-SS | 31 | 0.10 |
| 16Hf-LFS150-SS | 37 | 0.05 |
| 8Hf-LFS150-HT-SS | 45 | — |
| 8Hf-LFS150-HT-WI | 42 | — |
| 8Hf-LFS150-TPA-SS | 41 | 0.22 |
| 8Hf-LFS150-Na-SS | 24 | — |
FT-IR spectroscopy of adsorbed pyridine (as a base probe for measuring acidity) indicated that the Hf-EPDMs were solid Lewis (L) acids of moderate strength (L350/L150 = 0) and did not possess measurable Brønsted acidity (Table 1). The concentration of L acid sites did not vary considerably for w in the range of 8 to 16 wt% Hf (L = 4–12 μmol g−1) and for the different types of treatments of the 8Hf-EPDMs (L = 7–12 μmol g−1).
 | ||
| Fig. 4 PXRD patterns of wHf-LFS50s (A) and wHf-LFS150s (B). (A) 8Hf-LFS50-Na (WI (a); SS (b)); 8Hf-LFS50-TPA (WI (c); SS (d)); 8Hf-LFS50-HT (WI (e); SS (f)); wHf-LFS50-SS (w = 16 (g), 12 (h), 8 (i), 4 (j), 2 (k)); wHf-LFS50-WI (w = 8 (l), 2 (m)); HfO2 (n). (B) 8Hf-LFS150-Na (WI (a); SS (b)); 8Hf-LFS150-TPA (WI (c); SS (d)); 8Hf-LFS150-HT (WI (e); SS (f)); wHf-LFS150-SS (w = 16 (g), 12 (h), 8 (i), 4 (j), 2 (k)); wHf-LFS150-WI (w = 8 (l), 2 (m)); HfO2 (n). | ||
The SEM and STEM images of the commercial LFSs showed aggregates of fine scaly-like particles (Fig. 5 and S16†), which were somewhat larger for LFS150 than LFS50 (mean particle sizes of ca. 1.5 and 0.5 μm, respectively (technical specifications)). For each LFS, the respective untreated materials possessed somewhat comparable morphologies (Fig. 5).
 | ||
| Fig. 5 SEM images of LFS50 (a), LFS150 (b), LFS50-HT (c), LFS150-HT (d), H-LFS50-TPA (e), H-LFS150-TPA (f), H-LFS50-Na (g), and H-LFS150-Na (h). | ||
After Hf impregnation, no considerable morphological changes were observed (exemplified for the LFS50 family with the WI method, in Fig. S17†). The elemental mappings suggested uniform metal distributions for w up to at least 8 wt% Hf (Fig. S18 and S19†), but increasing further the w to 16 wt% Hf led to the appearance of intense colour spots in the hafnium mappings, assignable to HfO2 (Fig. S18j†).
The N2 sorption isotherms of the commercial LFSs showed a small uptake at low relative pressures (micropore volume was less than 0.04 cm3 g−1) and a hysteresis loop attributable to mesoporosity (Fig. S9b and c†).77 The pore size distributions of the parent LFSs were in the range of 2–10 nm (Fig. S10b and c†), and the SBET was 218 and 210 m2 g−1 for LFS50 and LFS150, respectively (Tables S4 and S5†). The treated LFSs possessed similar isotherms and slightly broader mesopore size distributions (2–20 nm) compared to the untreated LFSs (Tables S4 and S5†). The HT treatment led to enhanced SBET and Smeso; e.g., the SBET was 218/286 m2 g−1 and the Smeso was 149/284 m2 g−1 for LFS50/LFS50-HT (Table S4†). The alkaline treatments led to decreased SBET (especially using NaOH as a desilication agent), which may be due to the formation of inorganic debris (Tables S4 and S5†). However, after HCl washing of the alkaline treated silicas, the SBET and Smeso increased: e.g., the SBET was 218/269/235 m2 g−1 and the Smeso was 149/234/229 m2 g−1 for LFS50/H-LFS50-TPA/H-LFS50-Na (Table S4†), and the SBET was 210/215/249 m2 g−1 and the Smeso was 144/164/200 m2 g−1 for LFS150/H-LFS150-TPA/H-LFS150-Na (Table S5†). The impregnation of Hf led to similar or lower SBET (which seemed more pronounced for the WI method and higher Hf loadings; Tables S4 and S5†). For example, comparison of the type of impregnation method indicated that the SBET decreased from 215 m2 g−1 for H-LFS150-TPA to 193/184 m2 g−1 for 2Hf-LFS150-TPA-SS/2Hf-LFS150-TPA-WI, from 249 m2 g−1 for H-LFS150-Na to 213/174 m2 g−1 for 2Hf-LFS150-Na-SS/2Hf-LFS150-Na-WI, and from 251 m2 g−1 for LFS150-HT to 225/182 m2 g−1 for 2Hf-LFS150-HT-SS/2Hf-LFS150-HT-WI.
The 29Si MAS NMR spectra profiles of the untreated LFS supports were different from those of untreated EPDM, suggesting some differences in distributions of Si-sites with different chemical environments for the LFSs versus EPDM (Fig. 2). The spectra of the untreated LFSs showed relatively defined peaks, specifically, predominant Q4 (ca. −110 ppm), weaker Q3 (ca. −100 ppm) and very weak Q2 (ca. −90 ppm) resonances (Fig. 2c and e).62,63,78,79 The alkaline treatment led to changes in the Q4 peak profile, i.e., the less intense shoulder at ca. −110 ppm close to 120 ppm (Fig. 2c and e) suggested that a fraction of Q4 sites were reactive under alkaline conditions. Consistent with the 29Si MAS NMR data, the 29Si{1H} CP MAS NMR spectra showed enhanced relative intensity of the Q3 and Q2 peaks due to proximal hydrogen atoms (Fig. 2d and f).62 The concentration of silanol groups ([SiOH], Table S2†) was not considerably influenced by the HT treatment, but increased with the alkaline treatments (Table S2†). For example, for the NaOH treatment, [SiOH] was 1.6/3.1 mmol g−1 for LFS50/H-LFS50-Na and 1.5/2.8 mmol g−1 for LFS150/H-LFS150-Na, and for the TPAOH treatment, [SiOH] was 1.6/2.0 mmol g−1 for LFS50/H-LFS50-TPA and 1.5/2.5 mmol g−1 for LFS150/H-LFS150-TPA.
After Hf impregnation, the 29Si MAS NMR spectra showed decreased relative intensity of the Q3 peak (exemplified for untreated/treated LFS50s in Fig. S20a–d†), and [SiOH] decreased after Hf impregnation (Table S2†). Additionally, the 29Si{1H} CP MAS NMR spectra (Fig. 6 and S21†) showed decreasing relative intensity of the Q3 peak with increasing w up to 8 wt% (e.g., for wHf-LFS50-SS in Fig. S21e†). These results support that, possibly, the reaction between silanol groups and the Hf precursor occurred, forming Si–O–Hf bonds (supported by XPS, discussed below).61 Increasing further the Hf loading to 12–16 wt% did not lead to significant spectral differences (Fig. S21e†), possibly due to the formation of polynuclear HfO2.
 | ||
| Fig. 6 29Si CP {1H} MAS NMR spectra of selected untreated (a) and treated (b–d) LFS50s, before and after Hf impregnation. | ||
Based on the above studies, in general, the LFSs seemed to be more affected by the treatments than the EPDMs. A possible explanation is that the larger particles and generally lower specific surface areas of the EPDMs in relation to the LFSs may contribute to the higher robustness of the former towards chemical treatments. Nevertheless, the distinct morphological and textural features of the LFSs may lead to more favourable surface properties (e.g., acid–base properties, discussed below).
The ATR FT-IR spectra of the LFSs showed typical bands associated with external/internal, asymmetric/symmetric stretching vibrations of Si–O–Si groups (ca. 1220 cm−1,80,81 1040–1050 cm−1,65,66,69,80–82 780 cm−165,81,82) and bending mode vibrations involving Si–O bonds at ca. 420 cm−1.65,66,69,81 A somewhat broad band centred at ca. 3650 cm−1 is assignable to hydrogen-bridging hydroxyl groups (–Si–OH⋯O–Si or vicinal hydrogen bonded silanol groups),64,80 and at ca. 980 cm−1 to Si–O stretching of silanol groups (Fig. S12c–f; Table S3†).64–66 For the alkaline treated LFSs, the ca. 980 cm−1 band was generally more pronounced (Fig. S12c and e†), which correlated with the enhanced [SiOH] (Table S2†).
Somewhat in parallel with that verified for EPDM, the DR UV-vis spectra of the parent LFSs exhibited two small bands at ca. 215 and 240 nm, and their spectra were very different from that of pure HfO2 (Fig. S13c–f). For the wHf-LFSs, the relative intensity of the ca. 215 nm band (defect sites of the silica framework)68–70 decreased with increasing w until 8 wt% Hf, attributable to the reaction between defect sites of the silica framework and hafnium (possibly forming Si–O–Hf bonds, which is supported by the XPS studies discussed below). For w > 8 wt%, the ca. 215 nm band was not distinguishable, and instead a steep increasing absorption band below 220 nm, appeared, which, according to the literature, is assignable to tetrahedral/isolated Hf-sites.25,70,71 A broad band at ca. 380 nm appeared principally for the alkaline treated 8Hf-LFSs (especially using NaOH), which may be associated with hafnium oxide species.72
Fig. 3b and c show the XPS spectra (Hf 4f region) for selected wHf-LFSs. In parallel with that verified for the EPDMs, the binding energies associated with Hf 4f7/2 and Hf 4f5/2 of the untreated/treated 8Hf-LFS50 and the TPA-treated 8HF-LFS150 samples were shifted to higher binding energies in comparison with those for HfO2 (e.g., the binding energies associated with Hf 4f7/2 and Hf 4f5/2 were 17.4 and 19.1 eV, respectively, for 8Hf-LFS50-TPA-WI, and 17.5 and 19.1 eV for 8Hf-LFS150-TPA-WI). These differences may be partly due to the presence of tetrahedral Hf-sites.72,73,75,76
For more detailed XPS analyses, fittings were performed in the Hf 4f region for selected samples, namely 8Hf-LFS50-SS and 8Hf-LFS50-TPA-SS, in a similar fashion to those performed for the wHf-EPDMs discussed above (Fig. S14†). In parallel with that verified for the EPDMs, the spectra of the LFS50s showed peak broadening in comparison with the Hf 4f spectrum of pure HfO2, which may be at least partly attributed to the formation of Hf–O–Si bonds in the LFS50s. Accordingly, the LFS50s exhibited a new Hf 4f7/2 peak at ca. 18.2 eV associated with Hf–O–Si groups (besides the Hf 4f7/2 peak at ca. 17.5 eV due to the HfO2 type species). The relative intensity of the peak at ca. 18.2 eV in relation to the peak at ca. 17.5 eV was higher for 8Hf-LFS50-SS than the 8Hf-EPDM-SS counterpart, which is consistent with the higher amount of acid sites of the former (discussed below).
The fitted spectra of 8Hf-LFS50-SS and 8Hf-LFS50-TPA-SS presented similar features, in that the relative intensity of the Hf 4f7/2 peak at ca. 18.2 eV (Hf–O–Si) was slightly higher than that of the Hf 4f7/2 peak at ca. 17.5 eV (HfO2) (Fig. S14†); semiquantitative comparisons for these two materials may be erroneous due to signal/noise spectral limitations, which somewhat affects the fittings.
The acid properties measurements of selected wHf-LFSs indicated that the materials were essentially solid Lewis acids of moderate strength and, in some cases, with few relatively strong acid sites (L350/L150 < 0.22); Brønsted acidity was generally not measurable (Table 1). The amount of acid sites was generally higher for the LFSs than for the EPDM family (e.g., for w = 8, 4–12 μmol g−1 for 8Hf-EPDMs versus 24–56 μmol g−1 for 8Hf-LFSs). The smaller particle sizes and generally higher SBET of the LFSs than the EPDM counterparts may lead to favourable distributions of acid sites. For the untreated wHf-LFS50s, increasing w until 12 wt% Hf led to an increasing amount of L acid sites (9/56 μmol g−1 for 2Hf-LFS50-SS/12Hf-LFS50-SS), but increasing further w to 16 wt% did not lead to further improvement (49 μmol g−1 for 16Hf-LFS50-SS). For the untreated wHf-LF150s, the concentration of acid sites increased with w, from 11 μmol g−1 for 2Hf-LFS150-SS to 37 μmol g−1 for 16Hf-LFS150-SS.
On the other hand, a comparative study of the type of impregnation method for the 8Hf-LFSs indicated a roughly comparable or higher amount of acid sites for the WI method. While the WI method involves liquid–solid mixtures, the SS method involves solid–solid mixtures, which may impact the acidity. Specifically, factors such as differences in mass transfer phenomena during the impregnation process may influence the distribution of acid sites.
Regarding the types of treatments, the HT- and TPA-treated 8Hf-LFSs possessed enhanced acidity in relation to the untreated 8Hf-LFSs (e.g., 30/57 μmol g−1 for 8Hf-LFS50-SS/8Hf-LFS50-TPA-SS and 30/54 μmol g−1 for 8Hf-LFS50-WI/8Hf-LFS50-TPA-WI, Table 1). On the other hand, a comparative study of the TPA- versus Na-treated 8Hf-LFSs indicated that the former possessed an enhanced amount of acid sites (29/57 μmol g−1 for 8Hf-LFS50-Na-SS/8Hf-LFS50-TPA-SS versus 24/41 μmol g−1 for 8Hf-LFS150-Na-SS/8Hf-LFS150-TPA-SS). At least for the LFS50 family, the higher SBET of the HT- and TPA-treated supports than that of the untreated LFS50 and Na-treated supports possibly led to favourable distribution of acid sites (Tables 1 and S4†). Nevertheless, additional factors such as surface chemistry of the supports may impact acid properties. For example, although no direct correlation could be established with the molar ratio (SiOH groups of the support)/Hf, one cannot exclude the possibility of this influencing the distributions of acid sites.
Based on the above complementary characterisation studies, at least some acid sites seem to be related to Hf–O–Si bonds. The acid strength may depend on the structure of the Hf sites. It is very challenging to unambiguously assign a specific structure of Hf sites to a specific acid strength. Literature studies for fully inorganic hafnium silicates reported Hf sites with different chemical environments (coordination spheres) and in different locations of the catalyst's structure.28,83 Otomo et al.83 reported an extensive study entirely dedicated to the characterisation (combining techniques) of the structure and acidity of Hf sites in Hf-BEA type materials, which suggested the presence of Hf sites such as: tetrahedral open sites (Hf(OH)(SiO)3), tetrahedral closed sites (Hf(SiO)4), sites associated with bridging Si–(OH)–Hf groups, Hf sites in extra-framework HfOx species, and Hf–OH groups. It was hypothesised that the acid strength may follow the order: open and closed Hf sites > extra-framework HfOx species > Hf–OH.83 Nevertheless, it is possible that the populations of Hf sites and their strengths differ for different families of materials.
The base properties were measured for selected Hf-LFS50s (Table 2). Besides acidity, the materials possessed base properties, i.e., they are amphoteric and may act as bifunctional acid–base catalysts. The higher amount of base sites than of acid sites (Tables 1 and 2) may be at least partly related to the fact that the acid and base probe molecules possess very different features (e.g., reactivity and molecular dimensions, which may impact factors such as steric hindrance and extension of the titration reactions). In general, the materials possessed more stronger (Sb) base sites than weak plus moderate base sites (Wb + Mb) (i.e., Sb/(Wb + Mb) > 1). Increasing the Hf loading from 2 to 12 wt% led to increasing amounts of base sites, without affecting considerably the base strength (Sb/(Wb + Mb) in the range of 2.0–2.8). A further increase of w from 12 to 16 wt% Hf impacted negatively the amount of base sites (which decreased from 326 to 237 μmol g−1, Table 2). This trend paralleled the trend of acid sites concentration as a function of w (Table 1). The HT- and (especially) TPA-treated materials (namely 8Hf-LFS50-HT-SS and 8Hf-LFS50-TPA-SS) possessed more base sites than the Na-treated 8Hf-LFS50-Na-SS (Table 2); also, this trend paralleled the trend of acid sites concentration (Table 1). The TPA treatment seemed to favour the formation of some stronger base sites (Sb/(Wb + Mb) = 5.9 versus 2.5–2.7 for the Na and HT treatments), which paralleled the fact that stronger L acid sites were verified for 8Hf-LFS50-TPA-SS and not for the Na- and HT-treated materials (Table 1). Hence, 8Hf-LFS50-TPA-SS seemed to possess some strong acid–base sites.
| Sample | Total Base sites (μmol g−1) | Sb/(Wb + Mb)a |
|---|---|---|
| a Base strength is based on the molar ratio Sb/(Wb + Mb), where Wb = amount of weak base sites, Mb = amount of medium strength base sites, and Sb = amount of strong base sites. | ||
| 8Hf-EPDM-SS | 51 | 0.6 |
| 2Hf-LFS50-SS | 49 | 2.0 |
| 4Hf-LFS50-SS | 201 | 2.7 |
| 8Hf-LFS50-SS | 220 | 2.8 |
| 12Hf-LFS50-SS | 326 | 2.5 |
| 16Hf-LFS50-SS | 237 | 2.3 |
| 8Hf-LFS50-HT-SS | 242 | 2.7 |
| 8Hf-LFS50-TPA-SS | 476 | 5.9 |
| 8Hf-LFS50-Na-SS | 215 | 2.5 |
For comparison with the LFS type materials, the base properties of 8Hf-EPDM-SS were measured, which indicated that the latter possessed a lower amount and strength of base sites (Table 2); this matched the acid properties in that 8Hf-EPDM-SS possessed lower acid sites concentration than 8Hf-LFS50-SS (Table 1).
Catalytic studies
According to the literature, the secondary alcohols 2-propanol and 2-butanol are among the most frequently employed H-donors with considerable potential for the target CTH process.22,84 Some literature studies reported somewhat higher selectivity to biobased products using 2BuOH: e.g., furfural to GVL73 and other biobased products,85,86 and xylose to LEs.87 This partly motivated the choice of 2BuOH in the present study. Additionally, 2BuOH may be produced from LA88 and other components derived from vegetable biomass, e.g., lignocelluloses,89,90 or in the wine industry,91 which is attractive for integrated biorefinery processes and a circular bioeconomy.
The first stage of the catalyst development involved studying the influence of the types of treatments of the silica supports at a relatively low Hf loading of 2 wt%; subsequently, the influence of the Hf loading (w) was studied for selected supports.
In the catalytic studies of the 2Hf-silicas the mass of the catalyst and Hf loading was kept approximately constant (w = 2 wt% Hf). Hence, the discussed trends of LA conversion parallel those of catalytic activity (based on the mass of the catalyst) or turnover frequencies (based on the Hf loading). The same applies for the discussed trends of GVL yields in that they parallel those of GVL formation rates (based on the mass of the catalyst). Hence, for the sake of simplicity, LA conversions and GVL yields are presented in the figures for discussion.
The LA reaction without a catalyst gave 2-butyl levulinate (2BL) as a sole product in 37% yield at 39% conversion, at 24 h and 180 °C. The silica supports without Hf were ineffective in converting LA to GVL, leading to 2BL in 37–48% yield at 39–49% conversion, at 24 h (Fig. S22†); the carbon balances closed in >97% (Fig. S23†). Pure HfO2 was also sluggish, leading to a 46% 2BL yield and less than 2% GVL yield at 51% conversion, at 24 h. The GVL production was considerably augmented using the Hf-silicas (up to 87% yield). Hence, the developed catalysts possessed adequate Hf-sites that promoted the target reaction (which, as mentioned above, may be tetrahedral Hf sites72,92,93).
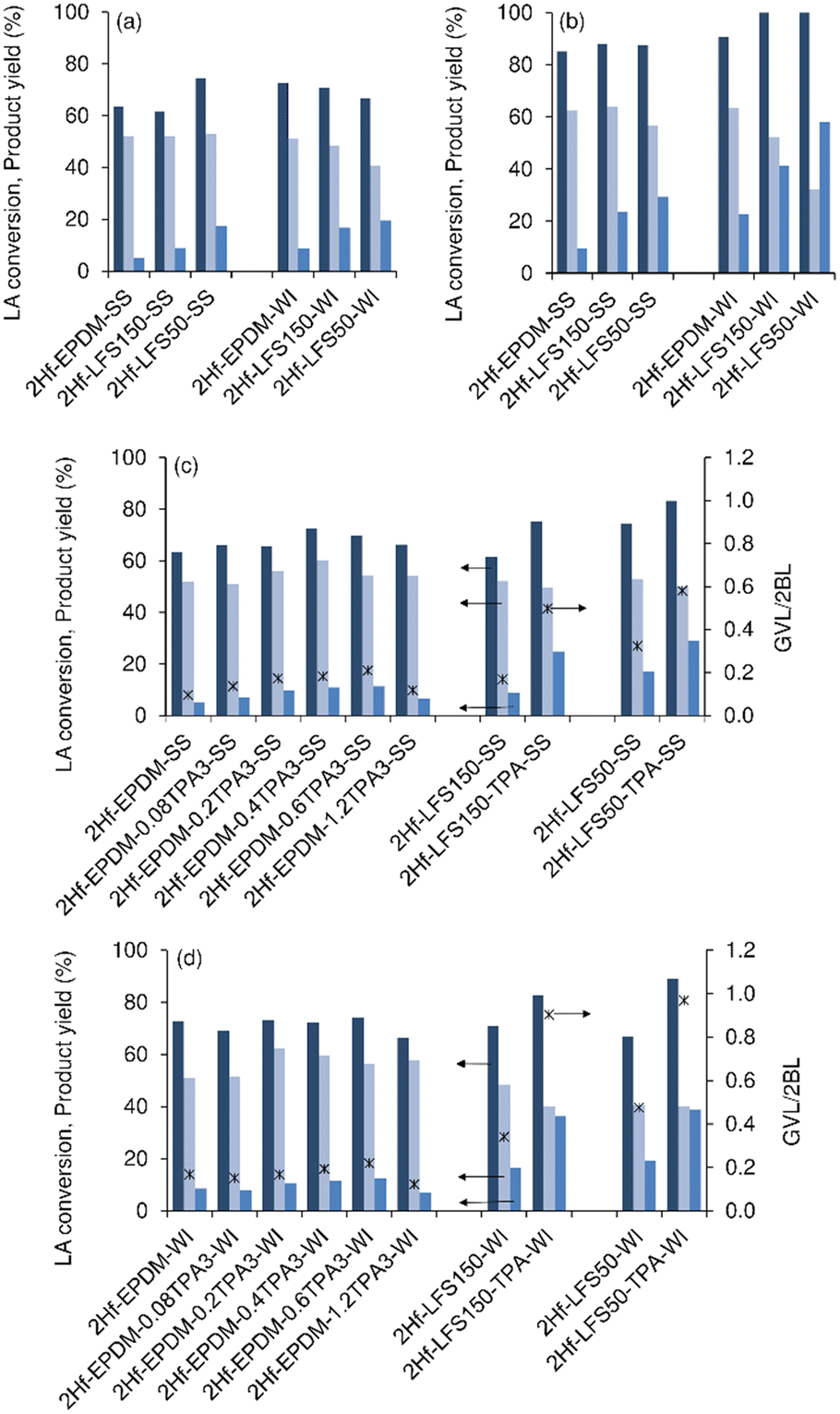 | ||
| Fig. 7 Influence of the TPAOH treatment and Hf impregnation method (SS or WI) on the catalytic performances of the 2Hf-silicas for LA conversion (first dark blue bars) to 2BL (second light blue bars) and GVL (third blue bars), and the GVL/2BL molar ratio (*). Catalytic results for the untreated 2Hf-silicas at 24 h (a) and 48 h (b), and comparisons with the TPA-treated 2Hf-silicas at 24 h (c and d). Reaction conditions: 0.45 M LA in 2BuOH, 25.5 gcat L−1, 180 °C. | ||
For each support, the WI impregnation method generally led to higher LA conversions and GVL yields at 48 h than the SS method (Fig. 7b): e.g., 58%/29% for 2Hf-LFS50-WI/2Hf-LFS50-SS (at 100%/87% conversion). These results suggested that the WI method may favour the formation of Hf-sites with relatively high intrinsic activity.
 | ||
| Fig. 8 Influence of the Na-treatment and type of impregnation method (SS (a and c) or WI (b and d) method) on the catalytic performances of the 2Hf-silicates for LA conversion (first dark blue bars) to 2BL (second light blue bars) and GVL (third blue bars), and GVL/2BL molar ratio (*), at 24 h (a and b) and 48 h (c and d). Reaction conditions: 0.45 M LA in 2BuOH, 25.5 gcat L−1, 180 °C. | ||
In general, the alkaline treated materials led to higher LA conversions and GVL yields than the untreated counterparts, especially for 2Hf-LFSs (Fig. 7c and d, S24† and 8). Fig. 7c and d and S24† show the influence of the TPAOH concentration on the performance of the treated 2Hf-EPDMs. Until 0.6 M TPAOH (0.6TPA), the GVL yield at 48 h was slightly enhanced in relation to the untreated counterparts, e.g., 9%/31% yield (at 85%/100% conversion) for 2Hf-EPDM-SS/2Hf-EPDM-0.6TPA3-SS, and 23%/30% yield (at 91%/97% conversion) for 2Hf-EPDM-WI/2Hf-EPDM-0.6TPA-WI. However, a further increase in TPAOH concentration to 1.2 M (1.2TPA) did not significantly enhance the GVL yields (21%/28% for 2Hf-EPDM-1.2TPA-WI/2Hf-EPDM-1.2TPA-SS).
On the other hand, the alkaline treatment time of 1.5 or 3 h did not impact significantly the catalytic performance of the 2Hf-EPDMs (Fig. S26†). Hence, in subsequent studies, the LFSs were treated with 0.2 M base for 3 h, for further catalytic studies.
The alkaline treated 2Hf-LFSs led to higher LA conversions and GVL yields than the alkaline treated 2Hf-EPDM counterparts (0.2 M base), in parallel with that verified for the untreated 2Hf-LFSs versus the untreated 2Hf-EPDMs. These results correlated, for example, with the generally higher SBET for the alkaline treated 2Hf-LFSs (up to 263 m2 g−1; Tables S4 and S5†) than the 2Hf-EPDM counterparts (up to 192 m2 g−1, Table S1†). A comparative study of the type of base (0.2 M NaOH or TPAOH) indicated that, in general, NaOH led to lower GVL yields and GVL/2BL molar ratios than TPAOH; the differences were less pronounced for the 2Hf-EPDMs (Fig. 7c and d and 8 and S24†).
A comparative study of the type of impregnation method for the alkaline treated (0.2 M base) 2Hf-silicas indicated that, in general, the WI method impacted more positively the catalytic performance than the SS method (in parallel with that verified for the untreated silicates), especially for the treated 2Hf-LFSs (Fig. 7c and d and S24† and 8). For example, the GVL yield at 48 h was 60–68% (at 100% conversion) for 2Hf-LFS150-TPA-WI/2Hf-LFS50-TPA-WI versus 39–43% (at 95–97% conversion) for 2Hf-LFS150-TPA-SS/2Hf-LFS50-TPA-SS (Fig. S24 and S25†).
Regarding the treatments, the HT treatment of the 2Hf-LFSs (using the SS or WI method) led to higher GVL yields than the untreated counterparts, albeit to a smaller extent than that observed for the alkaline treated versus untreated counterparts (Fig. S25 and S27†). For example (comparing HT-treated versus untreated LFS50s), 2Hf-LFS50-HT-SS/2Hf-LFS50-HT led to 35%/29% GVL yields (87–93% conversion), whereas 2Hf-LFS50-HT-WI/2Hf-LFS50-WI led to 67%/58% GVL yields (100% conversion), at 48 h (Fig. S25 and S27†). The catalytic performances may be due to the complex interplay of several factors such as acidity, texture/morphology and surface chemistry.
 | ||
| Fig. 9 Catalytic activity and GVL formation rate for the 8Hf-LFS50s (a) and 8Hf-LFS150s (b) prepared via the SS or WI impregnation method. Reaction conditions: 0.45 M LA in 2BuOH, 25.5 gcat L−1, 180 °C. | ||
For the untreated silicas, increasing the w from 2 to 12 wt% enhanced the LA conversion to GVL (Fig. 9); e.g., the GVL yield at 48 h was 44%/67%/78% (at > 99% conversion) for (w = 12 wt%) 12Hf-EPDM-S/12Hf-LFS150-SS/12Hf-LFS50-SS, compared to the 9%/24%/29% yields (at ca. 85% conversion) for (w = 2 wt%) 2Hf-EPDM-SS/2Hf-LFS50-S/2Hf-LFS150-SS. These results correlated with the higher amount of acid sites of the catalysts with higher w (in the range of 2–12 wt%, Table 1). However, for w > 12 wt%, the GVL yield decreased, likely due to the formation of HfO2 species (based on PXRD (Fig. S3† and 4B and D) and elemental mappings (Fig. S18†)). The catalytic results also correlated with the base sites concentration (exemplified for wHf-LFS50-SS in Table 2), which increased with w in the range of 2–12 wt% Hf and then decreased. Huang et al.92 reported for fully inorganic hafnium silicates (natural clay halloysite nanotubes), the possible involvement of Hf–O Lewis acid–base pairs in the CTH of LA to GVL. Hence, one cannot rule out the possibility of the LA reaction being promoted by acid–base sites of the prepared materials.
A comparative study of the 8Hf-silicas versus the 2Hf-silica counterparts indicated higher GVL yields for the former (Fig. S28†). On the other hand, in parallel with that observed for the untreated 2Hf-silicas, for the untreated 8Hf-silicas the LA conversion and GVL yields increased in the order 8Hf-EPDMs < 8Hf-LFS150s < 8Hf-LFS50s (Fig. S28†). A similar trend was observed for the GVL formation rates and catalytic activities expressed per unit mass of the catalyst (Fig. 10 and S29†), as well as for the turnover frequency (TOF) based on the Hf loading (since, as mentioned above, the mass of the catalyst was kept constant). These results correlated with the higher SBET of the 8Hf-LFSs (161–202 m2 g−1) versus 8Hf-EPDMs (109–117 m2 g−1; Tables S1, S4 and S5†) and the higher concentration of acid sites of the untreated 8Hf-LFSs (28–30 μmol g−1, Table 1) versus 8Hf-EPDM counterparts (4–10 μmol g−1, Table 1). Moreover, the results also correlated with the higher amount of base sites of 8Hf-LFS50-SS in relation to the EPDM counterpart (Table 2).
 | ||
| Fig. 10 Impact of the Hf loading (SS method) on the performances of the wHf-silicates for LA conversion (dark blue bars) to 2BL (light blue bars) and GVL (blue bars), and GVL/2BL molar ratio (*), at 24 h (a) and 48 h (b). Reaction conditions: 0.45 M LA in 2BuOH, 25.5 gcat L−1, 180 °C. | ||
For the treated 8Hf-silicas, superior performances of the LFSs to the EPDMs were also verified (Fig. S28†). For the 8Hf-EPDMs, the type of treatment did not considerably influence the GVL yields (Fig. S28a and b†). In contrast, for the 8Hf-LFSs, the TPA- and HT-treatments seemed generally more favourable than the Na-treatment; e.g., 69%/74% GVL yields at 24 h (100% LA conversion) for 8Hf-LFS50-TPA-SS/8Hf-LFS50-HT-SS versus 33% yield (79% conversion) for 8Hf-LFS50-Na-SS, and 62/68% yields at 24 h (100% conversion) for 8Hf-LFS150-TPA-SS/8Hf-LFS150-HT-SS versus 48% yield (82% conversion) for 8Hf-LFS150-Na-SS (Fig. S28c–f†). A similar trend was observed in comparing the GVL formation rates and catalytic activities (and TOF) of catalysts (Fig. 10 and S29†). The higher GVL yields for the HT- and TPA-treated 8Hf-LFSs in relation to the Na-treated counterparts correlated with the higher concentration of acid sites of the former: 37/57 μmol g−1 for 8Hf-LFS50-HT-SS/8Hf-LFS50-TPA-SS versus 30/29 μmol g−1 for 8Hf-LFS50-SS/8Hf-LFS50-Na-SS, and for the LFS150s, 41/45 μmol g−1 for 8Hf-LFS150-TPA-SS/8Hf-LFS150-HT-SS versus 24 μmol g−1 for 8Hf-LFS150-Na-SS (Table 1). An overall comparison of the 8Hf-silicas indicated that the GVL yields tended to increase with increasing concentration of Lewis (L) acid sites (Fig. 11a).
 | ||
| Fig. 11 For all 8Hf-silicates: relationship between the GVL yields at 24 h and the (a) Lewis acidity or (b) kinetic constants (+) of GVL formation (k2 + k3), and relationship between the Lewis acidity and the (c) kinetic constants (−) of LA conversion (k1 + k3) or (d) kinetic constants (*) of GVL formation (k2 + k3). Experimental reaction conditions: 0.45 M LA in 2BuOH, 25.5 gcat L−1, 180 °C. | ||
The superior catalytic results (higher GVL yields) also seemed to correlate with the higher base sites concentration of the catalysts (exemplified for the 8Hf-LFS50s in Table 2). No clear correlation could be established between the catalytic results and the base strengths, e.g., the molar ratio Sb/(Wb + Mb) was similar for 8Hf-LFS50-HT-SS and 8Hf-LFS50-Na-SS (Table 2), whereas the latter catalyst performed inferiorly.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of α-angelica lactone, giving GVL.96 In alcohol medium, the esterification of LA gives levulinate esters (e.g., 2BL), which, in turn, may be hydrogenated to 4-hydroxypentanoate intermediates (e.g., 2-butyl 4-hydroxypentanoate (BHP)), followed by cyclisation to GVL.96 In this work, α-angelica lactone was not formed, suggesting that route (i) prevails. The intermediates BHP and HPA were not detected, possibly due to their high reactivity.95,97 Tan et al.97 did not experimentally detect the 4-hydroxypentanoate ester, but its formation was supported by theoretical calculations.97 To gain further insights into the reaction mechanism, the kinetic curves were analysed. Fig. 12 shows the kinetic profiles for the untreated 8Hf-silicas, at 180 °C. The 2BL yields reached a maximum and then decreased, whereas the GVL yield continued increasing reaching 83–87% for 8Hf-LFS50s, 65–70% for 8Hf-LFS150s and 44–48% for 8Hf-EPDMs, at 72 h. For example, for 8Hf-LFS150-SS and 8Hf-LFS150-WI, the 2BL yield reached 45–46% at 24 h and 32 h, respectively, and then decreased to 23–25% at 72 h, and for the 8Hf-EPDMs, the 2BL yield reached 52–60% at 32 h and then decreased to 44–47% at 72 h. The kinetic profiles for the treated 8Hf-LFSs were, in general, similar, i.e., the 2BL yield reached a maximum (Fig. S30b and e and S31b and e†) and then decreased with the concomitant increase of GVL yields (Fig. S30c and f and S31c and f).
C bond of α-angelica lactone, giving GVL.96 In alcohol medium, the esterification of LA gives levulinate esters (e.g., 2BL), which, in turn, may be hydrogenated to 4-hydroxypentanoate intermediates (e.g., 2-butyl 4-hydroxypentanoate (BHP)), followed by cyclisation to GVL.96 In this work, α-angelica lactone was not formed, suggesting that route (i) prevails. The intermediates BHP and HPA were not detected, possibly due to their high reactivity.95,97 Tan et al.97 did not experimentally detect the 4-hydroxypentanoate ester, but its formation was supported by theoretical calculations.97 To gain further insights into the reaction mechanism, the kinetic curves were analysed. Fig. 12 shows the kinetic profiles for the untreated 8Hf-silicas, at 180 °C. The 2BL yields reached a maximum and then decreased, whereas the GVL yield continued increasing reaching 83–87% for 8Hf-LFS50s, 65–70% for 8Hf-LFS150s and 44–48% for 8Hf-EPDMs, at 72 h. For example, for 8Hf-LFS150-SS and 8Hf-LFS150-WI, the 2BL yield reached 45–46% at 24 h and 32 h, respectively, and then decreased to 23–25% at 72 h, and for the 8Hf-EPDMs, the 2BL yield reached 52–60% at 32 h and then decreased to 44–47% at 72 h. The kinetic profiles for the treated 8Hf-LFSs were, in general, similar, i.e., the 2BL yield reached a maximum (Fig. S30b and e and S31b and e†) and then decreased with the concomitant increase of GVL yields (Fig. S30c and f and S31c and f).
 | ||
| Fig. 12 Kinetic model fitting (dashed lines) to the experimental results (points) for the LA conversion (a and d), 2BL yield (b and e) and GVL yield (c and f) versus time, for the untreated 8Hf-silicates, prepared via the SS (a–c) or WI (e–g) impregnation method. Specifically, 8Hf-LFS50s – blue, 8Hf-LFS150s – orange and 8Hf-EPDMs – grey. Reaction conditions: 0.45 M LA in 2BuOH, 25.5 gcat L−1, 180 °C. | ||
Based on these mechanistic considerations and literature data, the reaction mechanism for the LFSs and EPDMs is proposed in Scheme 2. The overall mechanism involves the acid-catalysed esterification of LA to 2BL (k1) and the subsequent catalytic transfer hydrogenation (CTH) of 2BL to GVL (k2). In parallel with the esterification route, LA may be converted to GVL (k3) without the intermediate formation of levulinate esters (LEs).94,95,97–99 The possible formation of by-products (DLA; k4) is contemplated because the material balances did not close in 100% (Fig. S23†). Huang et al.100 proposed a simpler one-step mechanism for a Hf-polyacrylonitrile nanofiber (Hf@PAN-TP) catalyst; in that study, the LE yields were less than 10%, at 170 °C, and LA esterification was not considered.
 | ||
| Scheme 2 Mechanistic proposal for LA conversion to GVL production, in 2BuOH at 180 °C. | ||
A kinetic model was developed, which fitted reasonably well the experimental data for the 8Hf-silicas, indicating that the proposed mechanism is plausible (Fobj = 1.10 × 10−3–2.93 × 10−2; Fig. 12 and S30 and S31†). The calculated kinetic constants ki are given in Table S6.† In general, the lowest kinetic constant was that of the decomposition of LA (k4) (<5.89 × 10−4 L gcat−1 h−1, Table S6†). Hence, the developed catalysts were effective for selectively targeting GVL. The kinetic constants for LA conversion (k1 + k3), as well as for GVL formation (k2 + k3), tended to somewhat increase with the concentration of L acid sites (Fig. 11c and d).
Although the catalysts possessed Lewis acid sites and negligible/no Brønsted acid sites, one cannot fully exclude the possible involvement of Brønsted acidity in steps that may benefit from Brønsted acid sites, such as the esterification of LA to 2BL (e.g., reported in ref. 101–103). Esterification leads to the formation of water as a co-product, which may interact with Lewis acid sites. The Lewis acid sites may polarize water molecules, inducing the in situ formation of Brønsted acidity. For example, Omata and Nambu104 reported the catalytic role of the water molecules polarized at Lewis acid sites of transition metal oxides (studies involving isotopes), and the polarized water molecules acted as Brønsted acid sites in cumene cracking which is a reaction that has been generally understood to proceed on Brønsted acid sites.
As mentioned above, besides the acid sites, the base sites may be involved in the CTH reaction of LA to GVL. According to literature studies (e.g., for Zr-containing heterogeneous catalysts), the oxygen-containing base sites (e.g., oxygen bridges) may facilitate the deprotonation of the alcohol H-donor (via dissociation of the O–H bond). On the other hand, the carbonyl group (possessing an electron-rich O-atom) of LA may be activated by (electron-deficient) Lewis acid Hf-sites (please see the discussion of the XPS studies). Then, a hydrogen transfer step may occur between the dissociated O–H (of the alcohol H-donor) and the activated LA, leading to 4-hydroxypentanoate intermediates, which finally convert to GVL.94,103,105–107 Accordingly, one cannot rule out the possibility of the catalytic results correlating with both acid and base site concentrations of the prepared materials (as discussed above).
It is also important to evaluate the catalytic performances in terms of green chemistry metrics. For example, the non-productive decomposition (NPdec) of 2BuOH (i.e., not used for GVL formation), which reflects the hydrogen atom transfer efficiency, may enhance the E factor. For the 8Hf-LFSs, the NPdec of 2BuOH increased with time, giving sec-(butoxy)butane (BBu) in up to 0.11% yield at 72 h (Fig. S32†). Theoretically, one may consider the molar ratio BBu/GVL = 0 as indicative of 100%hydrogen atom transfer efficiency (higher BBu/GVL, indicating lower efficiency). Since the highest BBu yield was reached at 81% GVL yield, corresponding to a relatively low ratio BBu/GVL of 0.06, it seemed that the hydrogen atom transfer was rather efficient.
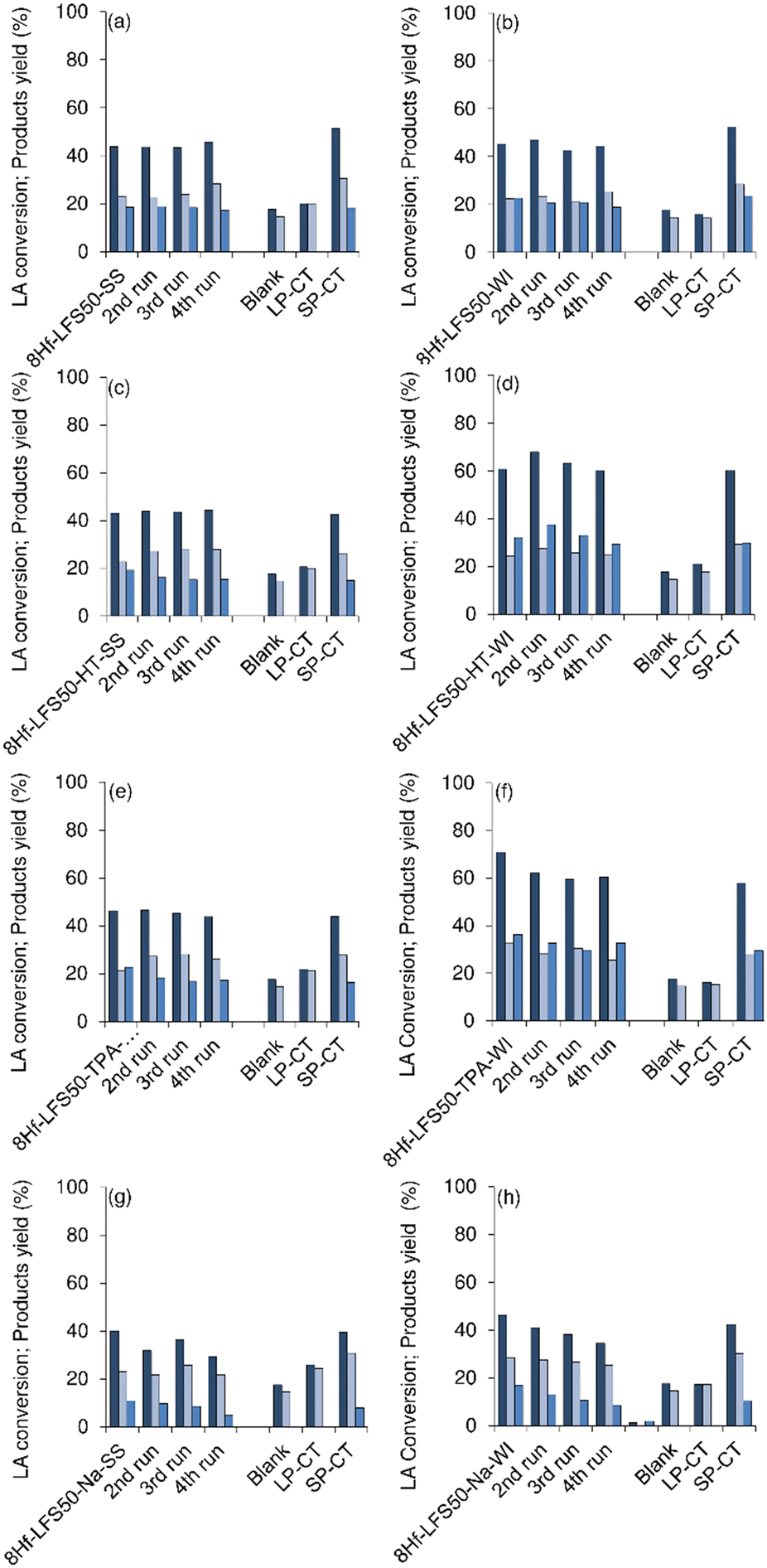 | ||
| Fig. 13 Catalytic stability of 8Hf-LFS50-SS (a), 8Hf-LFS50-WI (b), 8Hf-LFS50-HT-SS (c), 8Hf-LFS50-HT-WI (d), 8Hf-LFS50-TPA-SS (e), 8Hf-LFS50-TPA-WI (f), 8Hf-LFS50-Na-SS (g) and 8Hf-LFS50-Na-WI (h) in the conversion of LA (first dark blue bars) to 2BL (second light blue bar) and GVL (third blue bar) at 180 °C at 8 h. The tests without a catalyst and the liquid and solid phases of the contact tests are included. Reaction conditions: 0.45 M LA in 2BuOH and 25.5 gcat L−1. | ||
The liquid phases of the CTs (please see details in the Experimental section) were sluggish and gave roughly comparable results to the blank test without a catalyst (no GVL was formed; Fig. 13 and S33†), suggesting that the materials performed as heterogeneous catalysts. Moreover, the solid phases of the CTs (SP-CT) led to comparable results to the respective original solids. Consistently, the Si/Hf molar ratios were similar for the original and respective used solids (Table S7†), suggesting that no metal leaching occurred.
In general, the catalytic performances remained similar in consecutive runs, excluding the Na-treated samples which seemed to suffer from partial loss of activity (Fig. 13 and S33†). The morphology of the used catalysts was essentially preserved (Fig. S35 and S36†), and no crystalline structural modifications were observed, with the exception of 8Hf-LFS50-Na-SS and 8Hf-LFS50-Na-WI which seemed to exhibit somewhat pronounced reflections at ca. 28.5 and 31.6 2° 2θ, characteristic of HfO2 (Fig. S34Be and g†). These results, together with the fact that HfO2 was sluggish, suggested that the partial drop of performance of the Na-treated materials may be due to HfO2 species.
Only one hafnium/silica was previously reported. Specifically, some of us reported hydrothermally synthesised mesoporous Hf-TUD-1 (Si/Hf = 50; ca. 0.31 mmolHf gsupport−1), which led to a 29% GVL yield, at 200 °C/24 h (2BuOH as a H-donor; entry 7).110 Using the same LA initial concentration, LA/catalyst ratio, and H-donor, but at a lower temperature of 180 °C, higher GVL yields were reached for 8Hf-LFS50-SS, 8Hf-LFS50-HT-SS and 8Hf-LFS50-TPA-SS (62–74%; entries 1–6) versus 29% for Hf-TUD-1(50) at 200 °C (entry 7). Even for a lower Hf loading than that of Hf-TUD-1, e.g., 4Hf-LFS50-S (ca. 0.22 mmolHf g−1), the GVL yield was 47% at 24 h/180 °C (Fig. 10a) which is still greater than the 29% yield for Hf-TUD-1(50) at 200 °C. Moreover, as discussed below, Hf-TUD-1 is outperformed by the Hf-LFS50s in terms of the E factor, which is important for green processes.
There is very limited literature of fully inorganic transition metal/silicas for the target reaction via CTH routes. The reported studies are limited to ZrO2 impregnated on mesoporous silica SBA-15 (functionalised or not),99,108,111–114,116 SnO2 on SBA-15,101,115 and Zr on mesoporous silica KIT.109 Fair comparisons are difficult to establish due to the great differences in reaction conditions used between the different studies. Hence, the following discussion is not intended to rank catalysts, but instead, to highlight relevant differences from catalytic and green chemistry perspectives.
A chlorinated tin precursor (dimethyldichlorostannane) in p-xylene was used to prepare SnO2/SBA-15 (0.31 mmolSn g−1; entry 15), which led to an 81% GVL yield at 8 h/110 °C.101 However, tin may present some concerns. Based on LD50 (rat, oral; higher values indicate lower toxicity), zirconium and hafnium metal-based powders have similar LD50 (e.g., toxicological information for CAS. No. 7440-67-7 and 7440-58-6, Sigma-Aldrich, pointing out 5000 mg kg−1 for both), whereas tin-based powders present greater toxicity (e.g., CAS. No. 7440-31-5, ThermoFisher, LD50 of 2000 mg kg−1). Hence, from a toxicological perspective, the choice of Hf- or Zr-catalysts may be preferable to Sn-catalysts.
A zirconium n-butoxide precursor was used to prepare ZrO2-SBA-15 with 10 wt% Zr, which led to a very high GVL yield of 91% at 3 h/150 °C (entry 8).111 However, 10 wt% Zr loading corresponds to ca. 1.1 mmolZr g−1, which is ca. 2.5 times greater than the Hf loading of the 8Hf-LFS50s (0.448 mmolHf g−1). Moreover, less than half the initial concentration of LA was used, which is less demanding than the conditions used for the 8Hf-LFS50s (0.2 M versus 0.45 M LA for the 8Hf-LFS50s). Zirconium acetylacetonate was used to prepare ZrO2-SBA-15 with an even higher metal loading of ca. 23 wt% Zr, for LA conversion at higher temperatures of 250–310 °C, which initially led to a 96% GVL yield that decreased to ca. 80% after 20 h on-stream, at 250 °C (entry 9).112
Fig. 14 shows the reported GVL yields for the reaction parameters temperature and initial LA/catalyst mass ratio (LA/Cat), as well as the E factor, for previously reported fully inorganic transition metal/silicas (data in Table S8†). The GVL yields for the 8Hf-LFS50s were among the highest values (Fig. 14a and b). Regarding the E factor, it is important to include the contribution of the NPdec of the H-donor to waste generation. However, the previous studies (Fig. 14c) did not mention or report data of the NPdec of the H-donor. Thus, the E factor was calculated as follows from the data of the literature studies: E factor = [(initial mass of LA) − (mass of bioproducts)]/(mass of bioproducts), in which bioproducts = GVL + levulinate esters (whenever quantitative data were reported). This formula for the E factor considers that all LA that was not converted to bioproducts is possible waste and neglects the NPdec of the H donor. Nevertheless, for the 8Hf-LFSs, the E factor was calculated taking into account the NPdec of the H donor (putting more demanding requirements on the 8Hf-LFSs), i.e., E factor = [(initial mass of LA) − (mass of bioproducts) + (mass of BBu)]/(mass of bioproducts). Fig. 14c compares the E factor for the different catalysts, indicating that the modification treatments led to Hf-silicas (e.g., 8Hf-LFS50-HT-SS, 8Hf-LFS50-TPA-WI) which seemed promising in terms of the E factor.
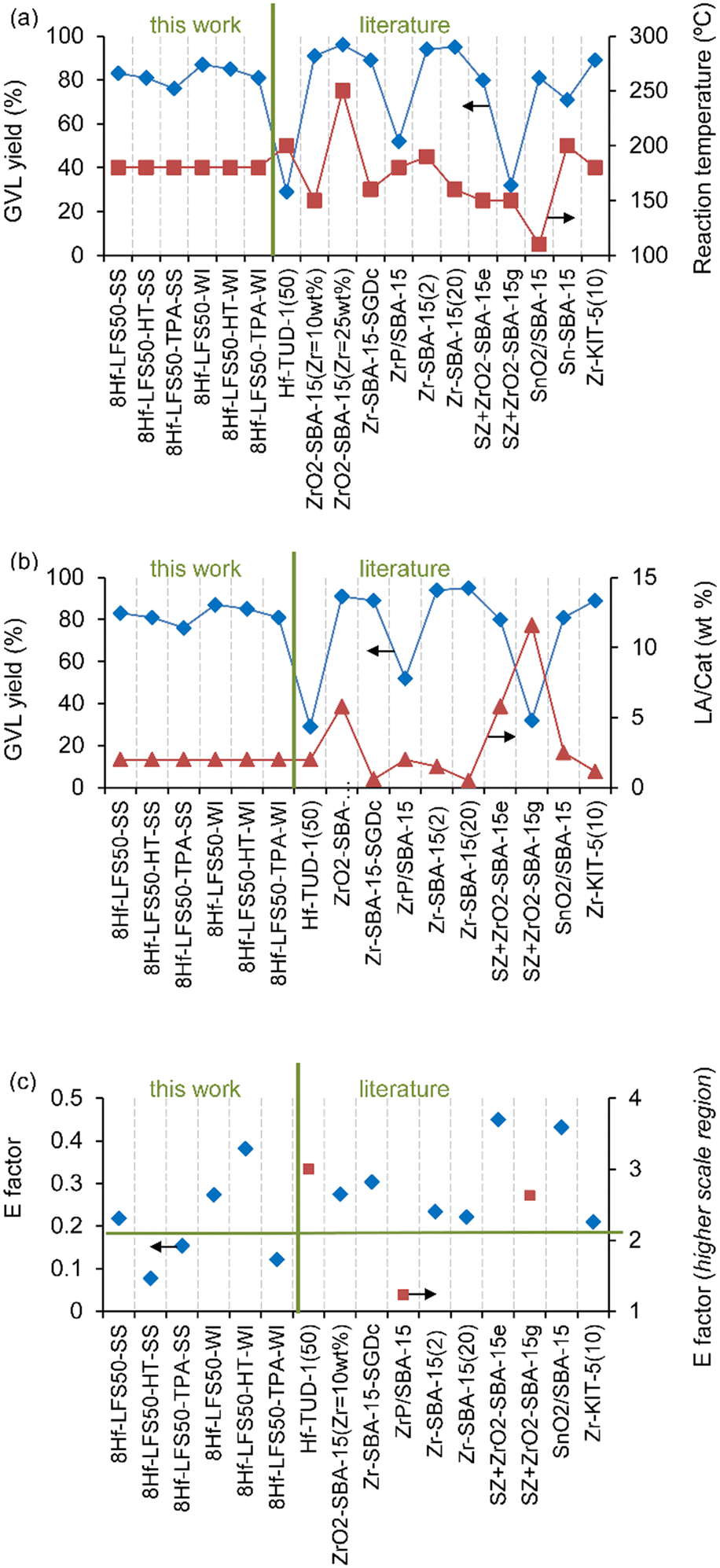 | ||
| Fig. 14 Comparisons to literature data: GVL yields at different LA reaction temperatures (a) and LA/catalyst mass ratios (LA/Cat) (b), and E factor (c), for previously reported fully inorganic transition-metal containing silicas (data in Table S8†). The thick black horizontal line in (c) marks the lowest E factor (0.17) of the literature survey. | ||
Conclusions
The catalytic transfer hydrogenation (CTH) of the biobased platform chemical levulinic acid (LA) to γ-valerolactone (GVL) was promoted by Lewis acid heterogeneous Hf-silica catalysts (2-butanol as a H-donor, at 180 °C). The catalysts were prepared from hafnium acetylacetonate and morphologically distinct EPDM and LFS silicas. These Hf-silicas are the first purely Lewis acidic (without measurable Brønsted acidity) transition metal/silicas developed for the target reaction (to the best of our knowledge, based on reported characterisation studies). The materials were modified in a versatile fashion to meet superior performances for green reactions/processes. The modifications involved alkaline or surface hydroxylation (HT) treatments of the supports (and optimisation of the conditions), and solid state (SS) or wet (WI) impregnation methods of different Hf loadings. In general, superior performances were observed for the LFSs (up to 87% GVL yield for Hf-LFSs, and up to 48% for Hf-EPDMs), subjected to alkaline or HT treatment, and ca. 8 wt% Hf loading seemed to be a good compromise. Kinetic modelling supported an overall mechanism in which GVL was produced from LA, with or without the intermediate formation of 2-butyl levulinate.Modified Hf-silicas were developed and are among the top previously reported fully inorganic transition metal/silicas, in terms of GVL yields for the target reaction, and seemed promising in terms of the E factor. It is, nevertheless, worth mentioning that, considering the catalyst requirements pointed out in the Introduction and for the sake of conciseness, the literature survey did not contemplate other families of materials, such as materials containing carbonaceous matter (organocatalysts, MOFs, carbons, etc.).
Given the complex interplay of several factors of the material properties influencing the catalytic performance, challenges remain in establishing more in-depth structure–activity relationships, where it would be desirable to better distinguish (qualitatively and quantitatively) the Hf-sites with different electronic/chemical environments along the catalyst's surface, and then use, for example, computational chemistry to compare their different intrinsic activities in the CTH of LA. In these studies, it would also be interesting to relate the structures of the Hf-sites to the acid strength by combining more characterisation techniques.
Data availability
Most data supporting this article have been included as part of the ESI† and other data are available from the corresponding authors upon reasonable request.Author contributions
MMA (investigation, conceptualisation, formal analysis, methodology, supervision, validation, visualisation, writing – original draft, writing – review and editing); KK (formal analysis, investigation); PN (formal analysis, investigation, validation); AF (formal analysis, investigation, validation); MA (formal analysis, investigation, validation); FR (resources); AAV (conceptualisation, resources, supervision, validation, visualisation, project administration, writing – review and editing).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was developed within the scope of the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020 (DOI 10.54499/UIDB/50011/2020), UIDP/50011/2020 (DOI 10.54499/UIDP/50011/2020) & LA/P/0006/2020 (DOI 10.54499/LA/P/0006/2020), financed by national funds through FCT/MCTES (PIDDAC), UIDB/00100/2020 (DOI 10.54499/UIDB/00100/2020), UIDP/00100/2020 (DOI 10.54499/UIDP/00100/2020) and LA/P/0056/2020 (DOI 10.54499/LA/P/0056/2020) in the scope of CQE-IMS (Centro de Química Estrutural, Institute of Molecular Sciences). The positions held by M. M. A. and A. F. were funded by national funds (OE), through FCT, I. P., in the scope of the framework contract foreseen in the numbers 4, 5 and 6 of article 23 of the Decree-Law 57/2016 of 29 August, changed by Law 57/2017 of 19 July (for M. M. A.: https://doi.org/10.54499/DL57/2016/CP1482/CT0062). The NMR spectrometers are part of the National NMR Network (PTNMR) and are partially supported by Infrastructure Project No. 022161 (co-financed by FEDER through COMPETE 2020, POCI and PORL and FCT through PIDDAC). The authors greatly acknowledge the AGC Si-Tech Co., Ltd. company for kindly supplying the three commercial samples, EPDM-10, LFS-50 and LFS-150.References
- R. Perkins and H. Edwardes-Evans, Fossil fuels ‘stubbornly’ dominating global energy despite surge in renewables: Energy Institute, https://www.spglobal.com/commodityinsights/en/market-insights/latest-news/oil/062623-fossil-fuels-stubbornly-dominating-global-energy-despite-surge-in-renewables-energy-institute, (accessed 21 February 2025).
- A. Hassan, S. Z. Ilyas, A. Jalil and Z. Ullah, Environ. Sci. Pollut. Res., 2021, 28, 21204–21211 CrossRef PubMed.
- R. Lei, S. Feng and T. Lauvaux, Environ. Res. Lett., 2020, 16, 014006 CrossRef.
- E. Maibach, J. Kotcher and L. Patel, J. Commun. Healthcare, 2024, 17, 194–196 CrossRef PubMed.
- R. V. Asase, Q. N. Okechukwu and M. N. Ivantsova, Environ. Dev. Sustain., 2024 DOI:10.1007/s10668-024-04992-w.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass, Volume I, US Department of Energy, 2004, p. 76, DOI:10.2172/15008859.
- V. Ghorpade and M. Hanna, in Cereals, ed. G. M. Campbell, C. Webb and S. L. McKee, Springer Nature Link, Boston, MA, 1997, pp. 49–55 Search PubMed.
- D. J. Hayes, S. Fitzpatrick, M. H. B. Hayes and J. R. H. Ross, in Biorefineries-Industrial Processes and Products: Status Quo and Future Directions, ed. B. Kamm, P. R. Gruber and M. Kamm, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, pp. 139–164 Search PubMed.
- A. Kumar, D. Z. Shende and K. L. Wasewar, Mater. Today: Proc., 2019, 29, 790–793 Search PubMed.
- C. Jaiswal, Levulinic Acid Market Research Report Information By Technology (Hydrolysis Production Process, Biofine Production Process, and Others), By Application (Food Additives, Pharmaceuticals, Cosmetic & Personal Care, Agriculture, Plasticizer, and Others), https://www.marketresearchfuture.com/reports/levulinic-acid-market-1639, (accessed 21 February 2025).
- S. Kang, J. Fu and G. Zhang, Renewable Sustainable Energy Rev., 2018, 94, 340–362 CrossRef CAS.
- L. D. Mthembu, R. Gupta and N. Deenadayalu, Waste Biomass Valorization, 2023, 14, 1–22 CrossRef CAS.
- S. F. Bazoti, C. Bonatto, T. Scapini, A. F. Camargo, H. Treichel and D. de Oliveira, Biofuels, Bioprod. Biorefin., 2023, 17, 1068–1084 CrossRef CAS.
- M. Sajid, U. Farooq, G. Bary, M. M. Azim and X. Zhao, Green Chem., 2021, 23, 9198–9238 RSC.
- C. Antonetti, D. Licursi, S. Fulignati, G. Valentini and A. M. R. Galletti, Catalysts, 2016, 6, 1–29 CrossRef.
- D. D. M. Di Bucchianico, Y. Wang, J. C. Buvat, Y. Pan, V. C. Moreno and S. Leveneur, Green Chem., 2022, 24, 614–646 RSC.
- P. Gautam, S. Barman and A. Ali, Biofuels, Bioprod. Biorefin., 2022, 16, 1095–1115 CrossRef CAS.
- J. Nelson Appaturi, J. Andas, Y. K. Ma, B. Lee Phoon, S. Muazu Batagarawa, F. Khoerunnisa, M. Hazwan Hussin and E. P. Ng, Fuel, 2022, 323, 124362 CrossRef CAS.
- M. Khalid, M. Granollers Mesa, D. Scapens and A. Osatiashtiani, ACS Sustainable Chem. Eng., 2024, 16494–16517 CrossRef CAS PubMed.
- A. Shivhare, A. Kumar and R. Srivastava, ChemCatChem, 2021, 13, 59–80 CrossRef CAS.
- L. Ye, Y. Han, J. Feng and X. Lu, Ind. Crops Prod., 2020, 144, 112031 CrossRef CAS.
- A. Osatiashtiani, A. F. Lee and K. Wilson, J. Chem. Technol. Biotechnol., 2017, 92, 1125–1135 CrossRef CAS.
- S. Dutta, I. K. M. Yu, D. C. W. Tsang, Y. H. Ng, Y. S. Ok, J. Sherwood and J. H. Clark, Chem. Eng. J., 2019, 372, 992–1006 CrossRef CAS.
- H. Y. Luo, D. F. Consoli, W. R. Gunther and Y. Román-Leshkov, J. Catal., 2014, 320, 198–207 CrossRef CAS.
- B. Tang, S. Li, W. C. Song, Y. Li and E. C. Yang, Sustainable Energy Fuels, 2021, 5, 4724–4735 RSC.
- H. Sun, P. Peng, Y. Wang, C. Li, F. Subhan, P. Bai, W. Xing, Z. Zhang, Z. Liu and Z. Yan, J. Porous Mater., 2017, 24, 1513–1525 CrossRef CAS.
- P. C. Lai, C. Y. Hsieh, C. H. Chen and Y. C. Lin, Catalysts, 2017, 7, 259 CrossRef.
- B. A. Johnson, J. R. Di Iorio and Y. Román-Leshkov, J. Catal., 2021, 404, 607–619 CrossRef CAS.
- J. Zhao, H. Li, Y. Wang, R. Zhu, S. Sun, J. Zhang, C. Li and Mei Hong, Catal. Commun., 2023, 175, 106619 CrossRef CAS.
- D. Verboekend, G. Vilé and J. Pérez-Ramírez, Adv. Funct. Mater., 2012, 22, 916–928 CrossRef CAS.
- Y. Han, K. Larmier, M. Rivallan and G. D. Pirngruber, Microporous Mesoporous Mater., 2024, 365, 112906 CrossRef CAS.
- J. Fals, J. F. Garcia-Valencia, E. Puello-Polo, F. Tuler and E. Márquez, Molecules, 2024, 29, 3085 CrossRef CAS PubMed.
- G. Dashtpeyma, S. R. Shabanian, J. Ahmadpour and M. Nikzad, Chem. Eng. Res. Des., 2022, 178, 523–539 CrossRef CAS.
- V. Van-Dúnem, A. P. Carvalho, L. M. D. R. S. Martins and A. Martins, ChemCatChem, 2018, 10, 4058–4066 CrossRef.
- A. Ruggiu, P. Parpot, I. C. Neves, A. P. Carvalho, M. G. Cutrufello, A. M. Fonseca, A. Martins and E. Rombi, Microporous Mesoporous Mater., 2016, 379, 113300 CrossRef.
- S. Abelló, A. Bonilla and J. Pérez-Ramírez, Appl. Catal., A, 2009, 364, 191–198 CrossRef.
- D. Verboekend, S. Mitchell, M. Milina, J. C. Groen and J. Pérez-Ramírez, J. Phys. Chem. C, 2011, 115, 14193–14203 CrossRef CAS.
- J. Kowalska-Kuś, A. Held and K. Nowińska, ChemCatChem, 2020, 12, 510–519 CrossRef.
- A. Aloise, A. Marino, F. Dalena, G. Giorgianni, M. Migliori, L. Frusteri, C. Cannilla, G. Bonura, F. Frusteri and G. Giordano, Microporous Mesoporous Mater., 2020, 302, 110198 CrossRef CAS.
- Z. Hong, G. Zhao, C. Xiong, W. Jia, F. Huang and Z. Zhu, Appl. Catal., A, 2021, 612, 117983 CrossRef CAS.
- P. Wang, T. Chen, Z. Qiu, W. Yao, P. Liu, Y. Zhang, Y. Song, Q. Cui and X. Bao, Fuel, 2024, 368, 131593 CrossRef CAS.
- Y. Chen, X. Zhu, X. Wang and Su Yunpeng, Chem. Eng. J., 2021, 419, 129641 CrossRef CAS.
- C. Dai, A. Zhang, L. Li, K. Hou, F. Ding, J. Li, D. Mu, C. Song, M. Liu and X. Guo, Chem. Mater., 2013, 25, 4197–4205 CrossRef CAS.
- D. Verboekend and J. Pérez-Ramírez, Chem. – Eur. J., 2011, 17, 1137–1147 CrossRef CAS PubMed.
- T. Li, F. Krumeich, J. Ihli, Z. Ma, T. Ishikawa, A. B. Pinar and J. A. Van Bokhoven, Chem. Commun., 2019, 55, 482–485 RSC.
- X. Meng, M. Zhang, C. Chen, C. Li, W. Xiong and M. Li, Appl. Catal., A, 2018, 558, 122–130 CrossRef CAS.
- R. Caicedo-Realpe and J. Pérez-Ramírez, Microporous Mesoporous Mater., 2010, 128, 91–100 CrossRef CAS.
- D. Verboekend, A. M. Chabaneix, K. Thomas, J. P. Gilson and J. Pérez-Ramírez, CrystEngComm, 2011, 13, 3408–3416 RSC.
- X. H. Vu, T. T. Truong, U. Armbruster and A. Martin, React. Kinet., Mech. Catal., 2018, 124, 437–452 CrossRef CAS.
- P. del Campo, P. Beato, F. Rey, M. T. Navarro, U. Olsbye, K. P. Lillerud and S. Svelle, Catal. Today, 2018, 299, 120–134 CrossRef CAS.
- H. Chen, H. Cheng, F. Zhou, K. Chen, K. Qiao, X. Lu, P. Ouyang and F. Jie, J. Anal. Appl. Pyrolysis, 2018, 131, 76–84 CrossRef CAS.
- C. Fernandez, I. Stan, J. P. Gilson, K. Thomas, A. Vicente, A. Bonilla and J. Pérez-Ramírez, Chem. – Eur. J., 2010, 16, 6224–6233 CrossRef CAS PubMed.
- D. M. Gomes, P. Neves, M. M. Antunes, A. J. S. Fernandes, M. Pillinger and A. A. Valente, Catalysts, 2022, 12, 1513 CrossRef CAS.
- T. Kawai and K. Tsutsumi, Colloid Polym. Sci., 1998, 276, 992–998 CrossRef CAS.
- R. Maddalena, C. Hall and A. Hamilton, Thermochim. Acta, 2019, 672, 142–149 CrossRef CAS.
- Y. Hui, Y. Zhang, Y. Luo, J. Li, Y. Wang, T. Gao, J. Xia, S. Wang and S. Zhang, Res. Chem. Intermed., 2021, 47, 521–532 CrossRef CAS.
- E. Ekinci, M. Oruç, N. Oktar and K. Murtezaoğlu, Catal. Lett., 2024, 154, 3776–3786 CrossRef CAS.
- A. Kumar, R. Police, S. Basavaraju and D. Valluri, Bull. Mater. Sci., 2015, 38, 227–234 CrossRef.
- L. Munguía-Cortés, I. Pérez-Hermosillo, R. Ojeda-López, J. M. Esparza-Schulz, C. Felipe-Mendoza, A. Cervantes-Uribe and A. Domínguez-Ortiz, J. Mex. Chem. Soc., 2017, 61, 273–281 Search PubMed.
- M. M. Antunes, A. F. Silva, C. D. Bernardino, A. Fernandes, F. Ribeiro and A. A. Valente, Molecules, 2021, 26, 7203 CrossRef CAS PubMed.
- J. Zhang, Z. Ma, J. Jiao, H. Yin, W. Yan, E. W. Hagaman, J. Yu and S. Dai, Microporous Mesoporous Mater., 2010, 129, 200–209 CrossRef CAS.
- S. H. Kim, O. H. Han, J. K. Kim and K. H. Lee, Bull. Korean Chem. Soc., 2011, 32, 3644–3649 CrossRef.
- M. Muroya and K. Yaguchi, Prog. Org. Coat., 1997, 31, 153–156 CrossRef.
- T. Karbowiak, M. A. Saada, S. Rigolet, A. Ballandras, G. Weber, I. Bezverkhyy, M. Soulard, J. Patarin and J. P. Bellat, Phys. Chem. Chem. Phys., 2010, 12, 11454–11466 RSC.
- D. M. Widjonarko, J. Jumina, I. Kartini and Nuryono, Indones. J. Chem., 2014, 14, 143–151 CrossRef CAS.
- S. M. Shawky, A. A. Abo-AlHassan, H. Lill, D. Bald, S. FEL-Khamisy and E.-Z. M. Ebeid, J. Nanosci. Curr. Res., 2016, 1, 103 Search PubMed.
- L. C. Gallington, Y. Ghadar, L. B. Skinner, J. K. R. Weber, S. V. Ushakov, A. Navrotsky, A. Vazquez-Mayagoitia, J. C. Neuefeind, M. Stan, J. J. Low and C. J. Benmore, Materials, 2017, 10, 1290 CrossRef PubMed.
- M. Qasim, J. Ananthaiah, S. Dhara, P. Paik and D. Das, Adv. Sci., Eng. Med., 2014, 6, 965–973 CrossRef CAS.
- I. A. Rahman, P. Vejayakumaran, C. S. Sipaut, J. Ismail and C. K. Chee, Mater. Chem. Phys., 2009, 114, 328–332 CrossRef CAS.
- Y. Zhang, L. Qi, A. Lund, P. Lu and A. T. Bell, J. Am. Chem. Soc., 2021, 143, 8352–8366 CrossRef CAS PubMed.
- Z. A. Pour, D. G. Boer, S. Fang, Z. Tang and P. P. Pescarmona, Catalysts, 2021, 11, 1346 CrossRef.
- B. Tang, S. Li, W. C. Song, E. C. Yang, X. J. Zhao, N. Guan and L. Li, ACS Sustainable Chem. Eng., 2019, 7, 16329–16343 CrossRef CAS.
- M. M. Antunes, A. F. Silva, A. Fernandes, M. Pillinger, F. Ribeiro and A. A. Valente, J. Catal., 2022, 406, 56–71 CrossRef CAS.
- A. A. Sokolov, E. O. Filatova, V. V. Afanas'Ev, E. Y. Taracheva, M. M. Brzhezinskaya and A. A. Ovchinnikov, J. Phys. D: Appl. Phys., 2009, 42, 035308 CrossRef.
- S. Lee, D. J. Yun, S. W. Rhee and K. Yong, J. Mater. Chem., 2009, 19, 6857–6864 RSC.
- Y. B. Huang, Y. J. Luo and F. Wang, Nanomaterials, 2019, 9, 1128 CrossRef PubMed.
- C. Schlumberger and M. Thommes, Adv. Mater. Interfaces, 2021, 8, 2002181 CrossRef CAS.
- L. K. H. Pham, S. Kongparakul, P. Reubroycharoen, M. Ding, G. Guan, D. V. N. Vo, N. Chanlek, C. N. Van and C. Samart, Top. Catal., 2023, 66, 22–33 CrossRef CAS.
- X. Liu, G. Qian, Z. Jiao, M. Wu and H. Zhang, Chem. – Eur. J., 2017, 23, 8066–8072 CrossRef CAS PubMed.
- E. F. Vansant, P. Van Der Voort and K. C. Vrancken, in Studies in Surface Science and Catalysis, ed. E. F. Vansant, P. Van Der Voort and K. C. Vrancken, Elsevier B. V., 1995, vol. 93, pp. 59–77 Search PubMed.
- Y. Tao, H. Kanoh and K. Kaneko, J. Am. Chem. Soc., 2003, 125, 6044–6045 CrossRef CAS PubMed.
- K. F. M. G. J. Scholle, W. S. Veeman, P. Frenken and G. P. M. van der Velden, Appl. Catal., 1985, 17, 233–259 CrossRef CAS.
- R. Otomo, T. Nakamura and Y. Kamiya, Microporous Mesoporous Mater., 2024, 378, 113246 CrossRef CAS.
- X. Li, X.-L. Shi, J. Wang, K. Shi and W. Qiang, J. Ind. Eng. Chem., 2024, 138, 17–33 CrossRef CAS.
- M. M. Antunes, S. Lima, P. Neves, A. L. Magalhães, E. Fazio, A. Fernandes, F. Neri, C. M. Silva, S. M. Rocha, M. F. Ribeiro, M. Pillinger, A. Urakawa and A. A. Valente, J. Catal., 2015, 329, 522–537 CrossRef CAS.
- M. M. Antunes, P. Neves, A. Fernandes, S. Lima, A. F. Silva, M. F. Ribeiro, C. M. Silva, M. Pillinger and A. A. Valente, Catal. Sci. Technol., 2016, 6, 7812–7829 RSC.
- L. F. Veiros, Â. Prazeres, P. J. Costa, C. C. Romão, F. E. Kühn and M. José Calhorda, Dalton Trans., 2006, 1383–1389 RSC.
- A. S. Azzahra, Rodiansono, I. F. Nata, K. C. Sembiring, I. B. Adilina and A. Afandi, Sustainable Energy Fuels, 2025, 9, 1279–1292 RSC.
- J. P. C. Pereira, L. A. M. van der Wielen and A. J. J. Straathof, Bioresour. Technol., 2018, 256, 187–194 CrossRef CAS PubMed.
- J. P. C. Pereira, W. Overbeek, N. Gudino-Reyes, E. Andrés-García, F. Kapteijn, L. A. M. Van Der Wielen and A. J. J. Straathof, Ind. Eng. Chem. Res., 2019, 58, 296–305 CrossRef CAS PubMed.
- G. Speranza, S. Corti, G. Fontana, P. Manitto, A. Galli, M. Scarpellini and F. Chialva, J. Agric. Food Chem., 1997, 45, 3476–3480 CrossRef CAS.
- R. Huang, Y. Wang, F. Chen, H. Liu, R. Zhang, W. Jia, L. Peng, Y. Sun and J. Zhang, Chem. Eng. J., 2024, 497, 154537 CrossRef CAS.
- B. Bohigues, S. Rojas-Buzo, M. Moliner and A. Corma, ACS Sustainable Chem. Eng., 2021, 9, 15793–15806 CrossRef CAS PubMed.
- R. Zhao, S. Kasipandi, C. H. Shin and J. W. Bae, ACS Catal., 2023, 13, 12711–12722 CrossRef CAS.
- M. T. Jayakumari and C. Kanakkampalayam Krishnan, Appl. Catal., A, 2023, 663, 119318 CrossRef CAS.
- R. Xu, K. Liu, H. Du, H. Liu, X. Cao, X. Zhao, G. Qu, X. Li, B. Li and C. Si, ChemSusChem, 2020, 13, 6461–6476 CrossRef CAS PubMed.
- H. Tan, S. Rong, Z. Zong, P. Zhang, R. Zhao, F. Song, H. Cui, Z. N. Chen, W. Yi and F. Zhang, Phys. Chem. Chem. Phys., 2023, 25, 18215–18223 RSC.
- V. B. Khajone, S. U. Raut, S. A. Deshmukh, K. J. Bhansali, K. R. Balinge, P. N. Muskawar and P. R. Bhagat, Biomass Convers. Biorefin., 2023, 13, 9107–9117 CrossRef CAS.
- A. Merenda, S. A. Orr, Y. Liu, B. Hernández Garcia, A. Osatiashtiani, G. Morales, M. Paniagua, J. A. Melero, A. F. Lee and K. Wilson, ChemCatChem, 2023, 15, e202201224 CrossRef CAS.
- R. Huang, H. Liu, J. Zhang, Y. Cheng, L. He and L. Peng, Renewable Energy, 2022, 200, 234–243 CrossRef CAS.
- S. Xu, D. Yu and P. Tian, RSC Adv., 2017, 7, 1026–1031 RSC.
- F. Wang, Z. Chen, H. Chen, T. A. Goetjen, P. Li, X. Wang, S. Alayoglu, K. Ma, Y. Chen, T. Wang, T. Islamoglu, Y. Fang, R. Q. Snurr and O. K. Farha, ACS Appl. Mater. Interfaces, 2019, 11, 32090–32096 CrossRef CAS PubMed.
- L. Zhang, B. Zhou, Y. Hong, Q. Wu, J. Qiu, J. Chen and X. Zeng, Renewable Energy, 2024, 236, 121453 CrossRef CAS.
- K. Omata and T. Nambu, Appl. Catal., A, 2020, 607, 117812 CrossRef CAS.
- J. Song, B. Zhou, H. Zhou, L. Wu, Q. Meng, Z. Liu and B. Han, Angew. Chem., Int. Ed., 2015, 54, 9399–9403 CrossRef CAS PubMed.
- M. Chia and J. A. Dumesic, Chem. Commun., 2011, 47, 12233–12235 RSC.
- Z. Xue, J. Jiang, G. Li, W. Zhao, J. Wang and T. Mu, Catal. Sci. Technol., 2016, 6, 5374–5379 RSC.
- R. Zhao and J. W. Bae, Catal. Today, 2024, 435, 114718 CrossRef CAS.
- J. He, H. Li, Y. Xu and S. Yang, Renewable Energy, 2020, 146, 359–370 CrossRef CAS.
- M. M. Antunes, A. F. Silva, A. Fernandes, F. Ribeiro, P. Neves, M. Pillinger and A. A. Valente, Front. Chem., 2022, 10, 1–21 Search PubMed.
- Y. Kuwahara, W. Kaburagi, Y. Osada, T. Fujitani and H. Yamashita, Catal. Today, 2017, 281, 418–428 CrossRef CAS.
- S. S. Enumula, V. R. B. Gurram, M. Kondeboina, D. R. Burri and S. R. R. Kamaraju, RSC Adv., 2016, 6, 20230–20239 RSC.
- V. V. Sychev, Y. N. Zaitseva, A. O. Eremina, O. V. Shabanova, S. D. Kirik, V. N. Panchenko and O. P. Taran, Chemistry, 2022, 15, 137–155 Search PubMed.
- Y. H. Zhou, Y. J. Luo, Y. T. Lin and Y. B. Huang, ChemistrySelect, 2018, 3, 11071–11080 CrossRef.
- S. Kumaravel, S. Thiripuranthagan, M. Durai, E. Erusappan and T. Vembuli, New J. Chem., 2020, 44, 8209–8222 RSC.
- S. Ostovar, H. Saravani and D. Rodríguez-Padrón, Renewable Energy, 2021, 178, 1070–1083 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00307e |
| This journal is © The Royal Society of Chemistry 2025 |