
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic decarboxylative deuteration of lauric acid with heavy water for sustainable synthesis of deuterated alkanes†
Haifan Huang a,
Zihan Lina,
Akira Yamamotoa,
Yagna Bhoi Prakasha,
Kexin Zoua,
Shohichi Furukawaa,
Ken-ichi Fujitaa,
Gunik Leeb,
Jun Kumagaic and
Hisao Yoshida*a
a,
Zihan Lina,
Akira Yamamotoa,
Yagna Bhoi Prakasha,
Kexin Zoua,
Shohichi Furukawaa,
Ken-ichi Fujitaa,
Gunik Leeb,
Jun Kumagaic and
Hisao Yoshida*a
aGraduate School of Human and Environmental Studies, Kyoto University, Yoshida-nihonmatsu-cho, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: yoshida.hisao.2a@kyoto-u.ac.jp
bDepartment of Material Chemistry, Graduate School of Engineering, Nagoya University, Chikusa, Nagoya 464-8603, Japan
cInstitute of Materials and Systems for Sustainability Division of Materials Research, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8601, Japan
First published on 26th May 2025
Abstract
As a simple model system for sustainable synthesis of deuterated alkanes, photocatalytic decarboxylative deuteration of lauric acid (dodecanoic acid) was explored employing heavy water (2H2O, D2O) as the deuterium source and titanium dioxide (TiO2) photocatalysts loaded with metal cocatalysts (Au, Pt or Pd), without requiring any other reagents. The photocatalysts effectively facilitated the production of monodeuterated undecane ([2H1]undecane, C11H23D). The alkyl radical degenerated from lauric acid through decarboxylation couples with the deuterium radical generated from heavy water, resulting in the formation of the deuterated undecane. Among the tested photocatalysts, a gold-loaded TiO2 (Au/TiO2) photocatalyst pre-dried before use achieved a 15.3% yield of deuterated undecane after 3 hours of photocatalytic reaction, with the deuteration ratio (Rd) in the obtained undecane reaching 85.1%. While extending the reaction time increased the overall yield, it led to a lower Rd. The Rd did not reach 100% since the alkyl radical intermediate also reacts with lauric acid or another alkyl radical to form non-deuterated undecane (C11H24) or docosane (C22H46), respectively as byproducts. The reaction mechanism of this photocatalytic system was elucidated using ESR measurement with radical trapping. This study offers a model methodology for the efficient synthesis of deuterated compounds, with potential applications in various fields such as pharmaceuticals.
Introduction
Since the discovery of deuterium (2H, or D), an isotope of hydrogen, in 1931,1 deuterated compounds have played a crucial role in elucidating chemical reaction processes and mechanisms.2,3 Deuterium is a cost-effective, safer, and more accessible labeling alternative compared to other isotopes4 such as 13C, 14C, and 3H. Additionally, the slower dissociation rate of the bond between deuterium and carbon provides distinct properties when compared to the typical hydrogen isotope (1H). Recently, deuterium-labeled drugs have shown significant potential in drug development, as they are expected to extend the half-life of drug metabolism due to their slow reaction rate. For example, the first deuterated drug, Austedo (deuterated tetrabenazine), was approved by the US FDA in 2017 for the treatment of movement disorders.5,6 Austedo is a modified form of tetrabenazine incorporating two deuterated methyl groups (including six deuterium atoms), which results in a drug with a prolonged life-time. Given these advancements, developing effective deuteration methods is of great significance in both pharmaceutical research and production.In the past few years, considerable attention has been devoted to the development of new methods for the deuteration of organic molecules. Emerging techniques include reductive and deuterodehalogenation, photoredox-catalyzed labeling, and the development of transition metal catalytic systems.7 Recently, some outstanding deuteration reactions have also been summarized in some review articles.8–12
Several primary methods for incorporating deuterium into an organic molecule include conventional multistep synthesis and direct hydrogen isotope exchange (HIE).4,13,14 For instance, Kerr, et al.,15 used D2 as a deuterium source with homogeneous iridium catalysts to selectively deuterate indole, azaindole, and pyrrole N-heterocycles. Traditional HIE processes often require harsh conditions such as strong acids/bases,16–20 flammable gases,21–23 or multiple exchange processes24–27 to ensure efficient deuterium incorporation. In addition, some cases involve costly deuterium sources.28,29 Meanwhile, the precise and selective introduction of deuterium typically relies on functional groups like halogens, which can offer better position selectivity and efficiency than C–H/C–D exchange reactions. However, this approach still requires complex ligands and special deuterium donors like Et3SiD30 or Ph2CDOH.31 Furthermore, the large-scale applications of halogen-based methods raise environmental concerns. Therefore, there is a growing need for new, cost-effective, and environmentally friendly deuteration methodologies.
Photocatalytic synthesis has emerged as a promising approach for selective introduction of deuterium on organic molecules under mild conditions. Recent studies have reported photocatalytic dehalogenative deuteration of aryl halides using D2O or CD3CN as deuterium sources with photocatalysts such as Au/Cds,13 CdSe,32 and MOF.33 However, these methods require the use of reducing reagents. Titanium oxide (TiO2) photocatalyst, as a well-known semiconductor photocatalyst, can play a crucial role in the decomposition of organic pollutants,34–38 water splitting,39–44 and carbon dioxide reduction.45,46 TiO2 has also demonstrated promising photocatalytic performance in organic synthesis.47–51 Kraeutler and Bard reported in 1978 that TiO2 and platinated TiO2 photocatalysts can produce methane (CH4) and carbon dioxide (CO2) primarily from acetic acid (CH3COOH) aqueous solutions,52,53 which is known as photo-Kolbe reaction (eqn (1)).
| CH3COOH → CH4 + CO2 | (1) |
In this reaction, the photocatalyst facilitates both the oxidation of acetic acid by holes, generating methyl radicals (˙CH3), CO2, and protons (eqn (2)), and the reduction of protons by electrons to form hydrogen radicals (H˙, eqn (3)). These radicals then couple to produce CH4 (eqn (4)). Potential byproducts of this reaction include hydrogen and ethane, which are formed via radical homocoupling (eqn (5) and (6)).
| CH3COOH + h+ → ˙CH3 + CO2 + H+ | (2) |
| H+ + e− → H˙ | (3) |
| ˙CH3 + H˙ → CH4 | (4) |
| 2H˙ → H2 | (5) |
| 2˙CH3 → C2H6 | (6) |
Building on this reaction, our laboratory demonstrated a photocatalytic direct methylation of benzene under light irradiation using a platinum-loaded TiO2 photocatalyst (Pt/TiO2) with acetic acid as a methylation reagent.54 This study suggests that the carboxyl group in organic acid (R–COOH) can serve as a possible leaving group, enabling the generation of corresponding alkyl radicals for the chemical reaction during photocatalytic decarboxylation. Organic acids, which commonly contain carboxyl groups, thus provide additional opportunities for deuterated substrates. In recent years, several studies have explored photocatalytic decarboxylative deuteration.55–57 For example, Li et al.55 achieved precise deuteration of aliphatic carboxylic acids by synergistic photoredox and HAT catalysis, obtaining good deuterium incorporation at predicted sites.
In this study, we explored a simple and sustainable reaction model system for photocatalytic decarboxylative deuteration, using lauric acid (LA) as the substrate and D2O as the deuterium source to produce deuterated alkanes (eqn (7)). The reaction was conducted with a metal-loaded TiO2 photocatalyst under light irradiation, without requiring any other reagents.
| C11H23COOH + D2O → C11H23D + HDO + CO2 | (7) |
The successful production of the deuterated product was confirmed. In this reaction, alkyl radicals such as undecyl radical (˙C11H23) and deuterium radical (D˙) are formed from LA and heavy water prior to the formation of the deuterated undecane (C11H23D), denoted as C11-d in the present study (eqn (8)–(10)) although non-deuterated undecane (C11H24, denoted as C11-h) and docosane (C22H46, denoted as C22) were also formed as dominant byproducts (eqn (11) and (12)).
| C11H23COOH + h+ → ˙C11H23 + CO2 + H+ | (8) |
| D+ + e− → D˙ | (9) |
| ˙C11H23 + D˙ → C11H23D (C11-d) | (10) |
| ˙C11H23 + H˙ → C11H24 (C11-h) | (11) |
| 2˙C11H23 → C22H46 (C22) | (12) |
ESR spectroscopy provided insights into the possible reaction mechanism of this photocatalytic system. These findings demonstrate a straightforward, green, and cost-effective strategy for synthesizing deuterated compounds using organic acid and heavy water, including potential applications in the production of deuterated pharmaceuticals.
Results
Photocatalytic reaction tests with M/TiO2 photocatalysts
The results of the photocatalytic reaction tests are summarized in Fig. 1A and Table S1.† After photoirradiation (wavelength > 360 nm) for 30 min, the target product, deuterated undecane (C11-d), was obtained when TiO2 or metal-loaded TiO2 photocatalysts were used. The quantity of C11-d contains one-deuterium undecane (C11-d1) and little doubly deuterated undecane (C11-d2, see Fig. S3†). In addition, non-deuterated undecane (C11-h) and docosane (C22) were detected as the dominant byproducts. Isomers of C22 were observed, though the amounts were very tiny. Further details can be found in the ESI.† Under certain conditions, oily products, likely resulting from successive polymerization, were observed. Regarding the gas-phase products, CO2 and H2 were detected; however, accurate quantitative analysis remained technically challenging. | ||
| Fig. 1 Results of the reaction tests for photocatalytic decarboxylation of LA in the presence of heavy water: (A) results with the TiO2 and M(0.1)/TiO2 samples (M = Au, Pt, and Pd). The green, light blue, and orange bars represent. YC11-d, YC11-h, and YC22, respectively. Dark red triangles indicate Sd (selectivity to C11-d), and light blue squares show Rd (selectivity to C11-d in the obtained undecanes). The reaction time was 0.5 h. Data are taken from Table S1;† see the caption of Table S1† for other reaction conditions. (B) Time course of Ytotal, Sd, and Rd with the Au(0.1)/TiO2 sample. (C) Time courses of YC11-d, YC11-h, and YC22 with the Au(0.1)/TiO2 catalysts. Detailed conditions for panels B and C are shown in Table S3.† (D) Results of the photocatalytic reaction tests with Au(x)/TiO2 samples with different Au loading amounts (x wt%). The reaction time was 3 h. See also Table S4.† | ||
Compared with the bare TiO2 photocatalyst, the Au, Pt, and Pd metal-loaded TiO2 samples exhibited higher photocatalytic activity for decarboxylation of LA as expected (Fig. 1A). Among the M(0.1)/TiO2 photocatalysts, the Au(0.1)/TiO2 sample gave lower total yield (Yx) and C11-d yield (YC11-d) but higher selectivity toward C11-d (Sd) compared to the Pt(0.1)/TiO2 and Pd(0.1)/TiO2 samples (Table S1†). The Pt(0.1)/TiO2 sample showed the highest overall photocatalytic activity (Ytotal) but the lowest selectivity to C11-d, suggesting that the higher photocatalytic activity may lead to an increased concentration of alkyl radicals, promoting the formation of C22, thus decreasing Sd value. This sample was employed for the mechanistic study as mentioned later. The Pd(0.1)/TiO2 sample achieved the highest YC11-d (6.68%), though it exhibited a close value of Sd compared to Au(0.1)/TiO2 sample due to the formation of a significant amount of by-products, C11-h and C22.
The selectivity of deuterated n-undecane (Rd), defined as the ratio of C11-d to the total n-undecane (C11-d and C11-h) obtained, is a key parameter, as the separation of C11-d from the mixture of undecane is very challenging. As shown in Fig. 1A, the Au(0.1)/TiO2 sample exhibited the highest Rd. In contrast, the Pt(0.1)/TiO2 and Pd(0.1)/TiO2 samples showed comparatively lower Rd values. As discussed later, since Rd is determined by the reaction selectivity of the generated alkyl radical toward deuterium radical (D˙) or other organic species, the highest Rd value is attributed to the properties of the Au cocatalyst. The reasons why the highest Rd did not reach 100% with any of the photocatalysts will be discussed later.
Results from blank reaction tests with the Au(0.5)/TiO2 sample (Table S2†) indicated that both photoirradiation and the presence of a photocatalyst were essential for the reactions to occur, confirming that the deuteration proceeds photocatalytically. The time course study showed that the total yield of the products monotonously increased with time while the Sd and Rd decreased with time (Fig. 1B, Table S3†). The product distribution slightly varied with time (Fig. 1C, Table S3†). The YC11-h and YC22 continuously increased, whereas the increase in the YC11-d slowed down with time. A possible explanation for this variation with time could be the change in the hydrophilic property of the photocatalyst surface due to the adsorption of hydrophobic products. Notably, the amount of main products decreased after 12 h of reaction, indicating that some successive reactions may happen to convert C11-d into other products. Based on these results, the reaction time was set to 3 hours for all subsequent reaction tests. A recyclability test was also carried out, which led to a slight decrease in the C11-d yield and Rd value. Details are described in the ESI,† Fig. S4. To improve the durability of the catalysts, improvements are required in the process of the catalyst recycling, catalyst surface modification, and improving the property of the loaded co-catalyst metals. By using a flow reactor, it might be possible to avoid these problems.58
Since the Au(0.1)/TiO2 sample showed the best Rd value among the three M(0.1)/TiO2 samples (Fig. 1A), additional Au(x)/TiO2 samples were tested with varying Au loading, and the results were presented in Fig. 1D and Table S4.† The Au(0.5)/TiO2 sample demonstrated the best performance, achieving a high YC11-d (15.7%) and a high Rd (80%) under these conditions. It is observed that the Au(1.0)/TiO2 sample exhibited lower activity compared to the Au(0.5)/TiO2 sample, as shown in Fig. 1D and Table S4.† This decline in performance at higher loadings may be attributed to the aggregation of Au nanoparticles (NPs), which is known to occur when the metal content exceeds a certain threshold. At higher metal loadings, the particle size of metal NPs tends to increase, leading to a reduced surface area. This decrease in surface area may, in turn, lower the efficiency of electron transfer required for the reduction, thus decreasing the overall photocatalytic activity. This phenomenon has been reported in similar systems and highlights the importance of optimizing metal dispersion for maintaining catalytic efficiency.59–61
Mechanistic studies
As mentioned above, the Rd could not reach 100% with any of the photocatalysts under these conditions. This suggests that hydrogen (1H) from other compounds or materials reacted with the photocatalytically generated undecyl radical (eqn (11)). To increase the Rd and to elucidate the reaction mechanism, additional experiments were conducted as described below.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH3CN = 2:3, 1:1, 3:2. v/v) were examined (Fig. 2A). To maintain a constant total reactant volume of 5 mL, 2 mL, 2.5 mL, and 3 mL of D2O were used, corresponding to 40, 50, 60% of total. Higher D2O proportions resulted in better YC11-d and higher Rd values. However, since LA does not fully dissolve in solutions with higher concentrations of D2O, experiments using more than 3 mL of D2O were not conducted in the present study. From these results, a high concentration of D2O is beneficial for producing deuterated compounds.
:CH3CN = 2:3, 1:1, 3:2. v/v) were examined (Fig. 2A). To maintain a constant total reactant volume of 5 mL, 2 mL, 2.5 mL, and 3 mL of D2O were used, corresponding to 40, 50, 60% of total. Higher D2O proportions resulted in better YC11-d and higher Rd values. However, since LA does not fully dissolve in solutions with higher concentrations of D2O, experiments using more than 3 mL of D2O were not conducted in the present study. From these results, a high concentration of D2O is beneficial for producing deuterated compounds.
 | ||
| Fig. 2 Results of photocatalytic reaction tests with the Au(0.5)/TiO2 sample. (A) Different ratios of D2O and CH3CN. (B) Dependence on light intensity. (C) Various pre-treatments, (a) without any pre-treatment, (b) with pre-irradiation in the presence of D2O, (c) with pre-heating and pre-irradiation in the presence of D2O, and (d) with pre-heating and pre-irradiation in the absence of D2O as a standard condition. (D) Various liquid phases: (a) D2O/CH3CN, (b) D2O/CD3CN, (c) H2O/CD3CN, and (d) H2O/CH3CN. Other reaction conditions for panels A, B, C and D are provided in the captions of Tables S5–S8,† respectively. | ||
Subsequently, the photocatalytic reaction was carried out at varying light intensities (Fig. 2B). The YC11-d increased with light intensity, while YC11-h initially increased until the intensity reached 21 mW cm−2, after which it decreased at a higher light intensity. In contrast, YC22 remained relatively stable across different light intensities. These results suggest that the production of C11-d is enhanced by light intensity, while the radical coupling process that produces C22 is not significantly influenced by light intensity. At higher light intensities, more alkyl radicals are utilized to form C11-d rather than C11-h. In addition, Rd increased as the light intensity was increased, and further discussion of this trend can be found in the succeeding section.
The results are shown in Fig. 2C and Table S7.† Although pre-irradiation without pre-heating enhanced the photocatalytic activity and selectivity for deuterated undecane (Sd), it did not improve Rd (Fig. 2Cb). In contrast, pre-heating led to higher YC11-d and an improved Rd, with values of 15.3% and 85.1%, respectively (Fig. 2Cb and Cc). Heating the sample in an argon flow at 450 K effectively should remove adsorbed organic matter and water, reducing the formation of C11-h and enhancing Rd. Furthermore, when pre-heating was performed, pre-irradiation (with or without D2O) did not significantly impact Rd (Fig. 2Cc and Cd). The best result with the highest Rd (85.1%) among them was achieved under the conditions shown in Fig. 2Cc. These findings highlight that removing initially adsorbed impurities on the photocatalyst surface improves Rd, although complete Rd remains still unachieved.
When H2O was used, no deuterated alkane was detected regardless of the isotopic property of acetonitrile (Fig. 2Dc and Dd), confirming that D2O is the sole deuteration source. In addition, it was observed that the YC11-h was higher than the combined yields of YC11-d and YC11-h obtained with D2O (Fig. 2Da and Db). This result indicates an isotopic effect. Considering that the C11-h obtained in the presence of D2O (Fig. 2Da) is produced via the reaction of an alkyl radical with an organic compound, the YC11-h in the absence of D2O (Fig. 2Dd) should include C11-h formed in the same way. Thus, the kH/kD value for C11 production with H2O or D2O can be estimated to be 1.58. Similarly, the amount of C22 obtained was higher, further supporting an isotopic effect with a kH/kD of 1.74. These findings suggest that the reaction rate is slower when breaking the D–OD bond in D2O (eqn (13) and (14)) compared to the H–OH bond in H2O, indicating that the activation of D2O to form either radical species (eqn (13) or (14)) is the rate-determining steps (RDS) in this reaction system. However, the separate study revealed that the formation of ˙OD radical (eqn (14)) would be minor due to the presence of LA.62 Thus, the observed RDS would be the water reduction (eqn (13)). The plausible pathways for LA oxidation by the ˙OD radical are likely minor (eqn (15) and (16)).
| D2O + e− → D˙ + OD− | (13) |
| D2O + h+ → ˙OD + D+ (minor) | (14) |
| C11H23COOH + ˙OD → ˙C11H23 + CO2 + HDO (minor) | (15) |
| C11H23COOH + ˙OD → ˙C11H22COOH + HDO (minor) | (16) |
As mentioned, the Rd did not reach 100% even under the optimized conditions. This suggests that another hydrogen (1H) source contributing to the formation of C11-h should still exist within the reaction system. Remaining possible hydrogen sources are the organic molecules, such as LA and the produced undecane. It is proposed that the undecyl radical, photocatalytically generated from LA, could react with another LA (eqn (17)) or the produced alkanes (eqn (18)). The former will give deuterated or non-deuterated LA and the latter can give doubly deuterated undecane (C11H22D2).
| ˙C11H23 + C11H23COOH → C11H24 (C11-h) + ˙C11H22COOH | (17) |
| ˙C11H23 + C11H23D → C11H24 (C11-h) + ˙C11H22D | (18) |
In addition, one may consider that protons (H+) formed from LA (eqn (8)) would be reduced by photoexcited electrons, potentially contributing to the generation of C11-h and thus decreasing the Rd. However, it cannot be ignored that the amount of LA (50 μmol) is limited, whereas D2O (56 mmol) is in excess. The dissociated protons from LA react with the excess D2O to form deuterated hydronium ions (D2HO+), which can subsequently release D+ instead of H+, as shown in eqn (19). This suggests that protons (H+) dissociated from LA are unlikely to be reduced by photoexcited electrons to generate H· radicals, and therefore contribute negligibly to the formation of C11-h. Therefore, the pathway shown in eqn (20) highly a minor contributor to the overall product formation.
| H+ + D2O → HDO + D+ | (19) |
| ˙C11H23 + H˙ → C11H24 (C11-h) (minor route) | (20) |
Therefore, achieving complete selectivity to deuterated alkane would be difficult when using these kinds of organic acids like LA. To further increase the Rd value, the reaction rate of radical coupling between the alkyl radical and deuterium radical (eqn (10)) should be improved by further optimization of reaction conditions and improvement in photocatalyst properties.
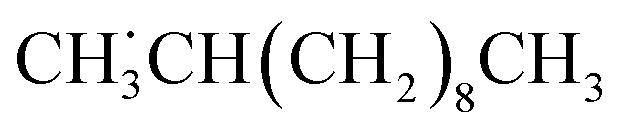 , is the most stable among the alkyl radicals.
, is the most stable among the alkyl radicals.Radical intermediate species photocatalytically generated from LA were detected by using ESR measurement with a radical trapping method. A spin trapping reagent, DBNBS, trapped the radical species generated from LA with the Pt(0.1)/TiO2 photocatalyst and formed some kinds of radical adducts. The spectrum is shown together with the calculated spectra in Fig. 3. Simulation of the obtained ESR spectra revealed that the spectrum consisted of two components: the major one (93.8%) was an adduct trapping secondary radical, while the minor one (6.2%) was the adduct trapping primary radical. In our previous work,62 ESI-MS analysis clarified the presence of two major secondary radicals formed from LA: one of these radicals was a secondary alkyl radical, represented as  (x = 0–4), and another contained a carboxy group, represented as
(x = 0–4), and another contained a carboxy group, represented as 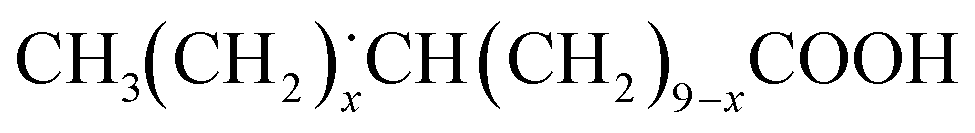 (0 ≤ x ≤ 9). These facts evidenced that the primary and the secondary alkyl radicals generated from LA were present under the presence conditions. When the radical species containing a carboxy group reacts with a deuterium radical (D˙), it forms deuterated LA, meaning that it does not directly contribute to the formation of deuterated undecane in the initial stage. On the other hand, the primary and the secondary alkyl radicals can serve as the reaction intermediates, possibly forming the corresponding deuterated undecane.
(0 ≤ x ≤ 9). These facts evidenced that the primary and the secondary alkyl radicals generated from LA were present under the presence conditions. When the radical species containing a carboxy group reacts with a deuterium radical (D˙), it forms deuterated LA, meaning that it does not directly contribute to the formation of deuterated undecane in the initial stage. On the other hand, the primary and the secondary alkyl radicals can serve as the reaction intermediates, possibly forming the corresponding deuterated undecane.
 | ||
| Fig. 3 Observed ESR spectrum of DBNBS-adduct-trapping radical species in supernatants of the resulting mixture after the photocatalytic reaction (a, black), and calculated spectra of primary radicals (b, red), secondary radicals (c, blue) and the sum of simulated spectra (d, overlapped to the observed spectrum), along with the representative spin adduct structures of carboxy secondary radical (e), alky secondary radical (f), and alkyl primary radical (g). The photocatalytic reaction was carried out in a mixed solvent of water (0.6 mL) and acetonitrile (0.4 mL), containing 10 mM of lauric acid (C11H23COOH), 10 mM of DBNBS, and 10 mg of the Pt(0.1)/TiO2 photocatalyst. The simulation parameters for hyperfine coupling constants are ANO = 1.37 mT, AH = 1.13 mT for the primary radical; ANO = 1.37 mT, AH = 0.71 mT for the secondary radicals, respectively. | ||
Among the possible secondary alkyl radicals, the one with the radical center at the second carbon from the terminal 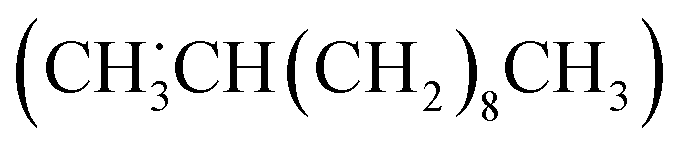 would be the most stable radical as mentioned above (Table S9†). Since the primary radical initially generated from LA is thermodynamically unfavorable, radical rearrangement reaction likely occurs. This rearrangement involves an intramolecular hydrogen atom transfer, which converts the primary radical into a more stable secondary radical with the radical center at the second carbon from the terminal (eqn (21)).
would be the most stable radical as mentioned above (Table S9†). Since the primary radical initially generated from LA is thermodynamically unfavorable, radical rearrangement reaction likely occurs. This rearrangement involves an intramolecular hydrogen atom transfer, which converts the primary radical into a more stable secondary radical with the radical center at the second carbon from the terminal (eqn (21)).
| ˙CH2(CH2)9CH3 (primary alkyl radical) → CH3˙CH(CH2)8CH3 (secondary alkyl radical) | (21) |
Therefore, the major deuterated undecane (C11-d) obtained in the present system would be 2-deuterated n-undecane with deuterium incorporated at the second carbon from the terminal, CH3CHD(CH2)8CH3 (eqn (22)). This selectivity is determined thermodynamically. The 1-deuterated n-undecane would be the minor C11-d product (eqn (23)).
| CH3˙CH(CH2)8CH3 + D˙ → CH3CHD(CH2)8CH3 | (22) |
| ˙CH2(CH2)9CH3 + D˙ → CH3(CH2)9CH2D | (23) |
On the other hand, the main isomer of docosane (C22) obtained was n-docosane, which should be formed via the radical–radical coupling of the primary undecyl radical (Scheme S2a†). This could be explained by the difference in the reaction rate, including the migration rate. Two primary undecyl radicals initially produced via decarboxylation at the oxidation sites on the TiO2 photocatalyst can immediately combine prior to the intramolecular hydrogen atom transfer (eqn (21)). On the other hand, the rate of crosscoupling between the undecyl radical and deuterium radical (D˙) including the mass transfer rate would be relatively slow so that the intramolecular hydrogen atom transfer (eqn (21)) would occur before the crosscoupling of the radicals, producing CH3CHD(CH2)8CH3 (eqn (22)).
Proposed reaction mechanisms
 | ||
| Scheme 1 Proposed mechanism for the production of deuterated undecane in the photocatalytically decarboxylation of LA in the presence of D2O; formation of LA radical cation intermediates through (a) the direct hole oxidation or (b) indirect oxidation facilitated by ˙OD radical, and successive alternative reactions of the LA radical cation for (c) the production of deuterated undecane and (d) the production of deuterated lauric acid, the latter of which does not directly produce deuterated undecane, and (e–g) three reaction pathways of alkyl radical. | ||
Upon photoirradiation, the TiO2 photocatalyst generates electron–hole pairs. In the presence of a metal cocatalyst, the generated charge carries are spatially separated to some extent, with the electrons migrating to the cocatalyst and the holes reaching the TiO2 surface. A molecule of LA (C11H23COOH) adsorbed on the TiO2 surface is oxidized by a photogenerated hole, resulting in the formation of an unstable LA cation radical, [C11H23COOH]+˙ (Scheme 1a). Alternative minor oxidation process is mediated by OD radical (˙OD) to form the LA cation radical (Scheme 1b). On the other hand, D2O is reduced to deuterium radicals (D˙) and OD− by photoexcited electrons on the metal cocatalyst (eqn (15)), which is suggested by the observation of isotope effect. This generation of hydroxy anions through reduction is balanced by the formation of protons via oxidation mentioned below.
The cation radical subsequently releases a proton (H+), leading to the formation of either a carboxyl radical, [C11H23COO]˙ (Scheme 1c) or an LA radical, ˙C11H22COOH (Scheme 1d). Among the two routes, the former directly generates a molecular CO2 and an undecyl radical, ˙C11H23, where the initially produced primary undecyl radical, ˙CH2(CH2)9CH3, changes to the secondary radical, 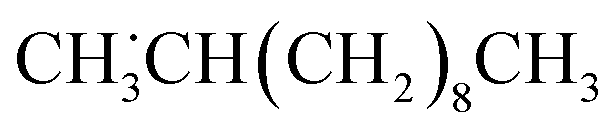 as a result of intramolecular hydrogen transfer prior to the final radical coupling step due to its higher thermodynamic stability as discussed above. Combining with deuterium radical, the target deuterated n-undecane featuring deuterium substitution at the second carbon from the terminal carbon, CH3CHD(CH2)8CH3 is formed (Scheme 1c). This is the major product C11-d in this system. The latter, the LA radical, does not directly contribute to the undecane formation (Scheme 1d).
as a result of intramolecular hydrogen transfer prior to the final radical coupling step due to its higher thermodynamic stability as discussed above. Combining with deuterium radical, the target deuterated n-undecane featuring deuterium substitution at the second carbon from the terminal carbon, CH3CHD(CH2)8CH3 is formed (Scheme 1c). This is the major product C11-d in this system. The latter, the LA radical, does not directly contribute to the undecane formation (Scheme 1d).
Three reaction pathways of alkyl radicals (˙C11H23) are summarized in Scheme 1e–g. Only when the formed alkyl radical reacts with ˙D (or D2O), the deuterated alkane is formed (Scheme 1e). On the other hand, when it reacts with LA or alkyl radical, byproducts (C11-h or C22, respectively) are formed. To improve the Rd value, suppressing pathway f and increasing the pathway e are options worth considering. Furthermore, increasing the proportion of D2O in the system or improving the hydrophilicity of the catalyst surface are both possible effective methods to enhance the Rd. Additionally, reducing the occurrence of pathway g is also beneficial for improving Sd.
The undecyl radicals possibly react with some other chemical species, such as lauric acid, undecane, H˙ radical, to form the undecane-h (eqn (21), (22) and (25)) and with undecyl radical to give docosane (Scheme S2†), which reduced the Rd value down to 85% even in the best conditions in the present study. Like we discussed in the previous section, because of the large amount of D2O and small amount of LA, the H+ or H radicals formed from LA are exposed to a large amount of heavy water and they are unlikely to be involved in the reaction. Therefore, the main reason for the incomplete selectivity, Sd and Rd are the reaction of the undecyl radical with other organic matters in the reaction system, such as LA, undecane, and undecyl radical.
Additionally, when the alkyl carboxylic radical (˙C11H22COOH) combines with a deuterium radical (D˙) in route B, deuterated LA is formed. This may give doubly deuterated undecane (C11H22D2), the presence of which was suggested by the GC-MS analysis (see Fig. S3†). Although C11-d2 compounds were usually formed only in trace amounts, under the conditions where their formation was particularly pronounced, they accounted for up to 30% of the total C11-d compounds. This result confirms that the formation of deuterated LA molecule (C11H22DCOOH), which provides further support for the proposed Scheme 1(d). The formation of C11-h is attributed to the reaction between alkyl radicals and LA molecule.
Experimental
Catalyst preparation
All metal-loaded titanium oxide (M/TiO2) photocatalysts were prepared by a photodeposition method with a commercial TiO2 sample (Ishihara Sangyo Kaisha, ST-01, anatase, 300 m2 g−1). Aqueous solutions of metal precursors, HAuCl4 (Nacalai Tesque, 99%), H2PtCl6 (Nacalai Tesque, 99%), and PdCl2 (Kishida Chemical, 99%), were used at concentrations of 9.89, 7.53, and 6.37 g L−1 as Au, Pt, and Pd, respectively.The preparation of the Au(0.5)/TiO2 sample is described as a representative example as follows. First, 2.0 g of TiO2 powder was dispersed in 300 mL of ion-exchanged water and irradiated with a xenon lamp (PE300BUV, 300 W) for 30 min under magnetic stirring. Subsequently, 100 mL of methanol and 1 mL of the HAuCl4 solution (9.894 g L−1 as Au) were added to the suspension. The mixture was stirred for 15 min without irradiation, followed by stirring for 1 h under light irradiation. After irradiation, the suspension was filtered and washed with over 500 mL of deionized water. The wet residue was dried in an electric oven at 353 K for 12 h, yielding a purple-colored powder of the Au(0.5)/TiO2 sample. Here, x in M(x)/TiO2 represents the metal loading amount as weight percent (wt%).
Other M(x)/TiO2 photocatalysts were synthesized using similar procedures.
Synthesis of monodeuterated n-undecane with Grignard reagent
The n-[1-2H1]undecane (CH3(CH2)9CH2D) as a reference was synthesized by the Grignard reagent (see ESI,† Scheme S1).Photocatalytic reaction tests
The photocatalytic reaction test was conducted in a closed system using a capped 20 mL quartz tube. Typically, 50 mg of the photocatalyst was introduced and subjected to pre-heating at 450 K using a metal bath for 30 min in an argon gas flow (15 ml min−1) to remove adsorbed water. Then the catalyst underwent pre-irradiation with a xenon lamp (PE300BUV, 300 W) for 15 min to clean the catalyst surface. Following the pre-heating and pre-irradiation, 5 mL of reactant solution containing LA (50 μmol), D2O (3 mL), and acetonitrile (CH3CN, 2 mL) was added to the quartz tube. The tube was sealed with a white silicone septum stopper and Parafilm, the air was purged from the tube using an argon flow (15 ml min−1) for 30 min, and the contents were stirred under light irradiation for the desired duration (typically, 180 min) using an optical filter that permitted light with wavelengths >360 nm to promote the photocatalytic reaction. The reaction is carried out at an ambient temperature of 25 °C.Optimization experiments were conducted under various conditions, including reaction time (Fig. 1), D2O concentration, light intensity, pretreatment conditions, and deuterium source (Fig. 2).
After the reaction, the liquid phase was filtered through a syringe equipped with a PTFE membrane filter and analyzed qualitatively and quantitatively using gas chromatography with GC-MS (Shimadzu QP-2020) and GC-FID (Shimadzu GC-2014), with n-decane (C10H22) as an internal standard. Details of the analysis for deuterated products and byproducts are described in the ESI.† The gas-phase products were detected by GC-TCD prior to the liquid-phase analysis.
The yield of product, Yx (%) (x = C11-d, C11-h), YC22 (%), the selectivity to deuterated undecane, Sd (%), and the ratio of deuterated undecane in the obtained undecane, Rd, were defined as follows (eqn (24)–(27)):
| Yx (%) = 100 × Ax/Ainitial LA | (24) |
| YC22 (%) = 100 × 2 × AC22/Ainitial LA | (25) |
| Sd (%) = 100 × AC11-d/(AC11-d + AC11-h + 2 × AC22) | (26) |
| Rd (%) = 100 × AC11-d/(AC11-d + AC11-h) | (27) |
| Ytotal (%) = 100 × (AC11-d + AC11-h + 2 × AC22)/Ainitial LA | (28) |
Analyses of radical intermediates
Electron spin resonance (ESR) spectra of spin adducts were recorded at room temperature by a JES-RE1X spectrometer (JEOL Co. Ltd, Japan) equipped with a TE102 cavity using a quartz flat cell as described.62 The Pt(0.1)/TiO2 photocatalyst powder (10 mg) was put into a disposable test tube (D-10S, Nichiden-Rika Glass Co., Ltd., 75 mm height, o.d. = 10 nm, i.d = 8 mm) and pre-irradiated with a high-pressure Hg lamp (Ushio UI-501C) for 30 min to purify the catalyst surface. After the pre-irradiation, a reaction mixture of ultrapure water (H2O, 0.6 mL), acetonitrile (CH3CN, 0.4 mL), and LA (10 mM) were added to the test tube. Subsequently, 0.01 mM of DBNBS (3,5-dibromo-4-nitrosobenzenesulfonate sodium salt (C6H2NO4SBr2Na), Kyoto Spin Lab, ≥95%) was introduced as a spin-trapping reagent. The sample was then irradiated through an optical filter (wavelength ≥350 nm) for 10 min. Following the photoirradiation, the mixture was centrifuged at 1.0 × 104 rpm for 5 min, and the supernatant was used for ESR measurement. Manganese (Mn2+) dispersed in magnesium oxide (JEOL, Co. Ltd., Japan) served as the reference for sensitivity and g-value calibration. The ESR spectra of DBNBS spin adducts were simulated using EasySpin 6.0.0 software on MATLAB®, employing the “garlic” core function without additional perturbations.Conclusions
In this study, we successfully demonstrated a simple and sustainable method for the synthesis of deuterated alkanes using lauric acid (C11H23COOH) as an example. Metal-loaded TiO2 photocatalysts facilitate photocatalytic decarboxylation of carboxylic acid, followed by successive deuteration using heavy water (D2O), yielding deuterated alkane (deuterated undecane, C11H23D, C11-d). Compared with the limited C11-d yield and deuteration selectivity achieved with the bare TiO2 sample, the Au(0.5)/TiO2 photocatalyst sample showed a significantly improved performance, achieving a high yield (YC11-d = 15.3%) and a deuterated undecane ratio in the obtained undecane (Rd = 85.1%) in the present best conditions (Fig. 2Cc).C11H23COOH molecule adsorbed on the TiO2 surface is oxidized by a photogenerated hole, resulting in a LA cation radical, which subsequently generates an undecyl radical, ˙C11H23. Combining with a deuterium radical, the target deuterated n-undecane is formed.
The major product was identified as deuterated n-undecane with deuterium substitution predominantly occurring at the second carbon from the terminal carbon, CH3CDH(CH2)8CH3. Mechanistic investigations suggested that the limited selectivity arose from undesirable side reactions of the radical intermediate with other organic species, including the substrate, the derived radical species, and products. Further investigation to overcome these points is still desired.
These findings highlight a valuable strategy for synthesizing deuterated compounds by using heavy water (D2O) as the deuterium source, providing a pathway for simple and highly selective deuteration reactions with broad potential applications.
Data availability
The authors confirm that the data supporting the findings of this study are available within the manuscript and its ESI.† Raw data that support the findings of this study are available from the corresponding author, upon reasonable request.Author contributions
HH: investigation, methodology, and writing – original draft; ZL: investigation and methodology; AY: investigation and funding acquisition; BYP, KZ, SF, KF, GL and JK: investigation; HY: conceptualization, supervision, writing – review & editing, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Prof. Naoki Komatsu of Kyoto University for his kind discussion. This study was financially supported by the Grant-in-Aid for Scientific Research (A) (22H00274) and the Grant-in-Aid for the Promotion of Joint International Research (Fostering Joint International Research (B), 20KK0116) from the Japan Society for the Promotion of Science (JSPS).References
- H. Kragh, Stud. Hist. Philos. Sci. B: Stud. Hist. Philos. Mod. Phys., 2012, 43, 176–183 Search PubMed.
- C. S. Elmore, Annu. Rep. Med. Chem., 2009, 44, 515–534 CAS.
- Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L. Colletti, I. W. Davies and D. W. C. Macmillan, Science, 2017, 358, 1182–1187 CrossRef CAS PubMed.
- W. Ou, C. Qiu and C. Su, Chin. J. Catal., 2022, 43, 956–970 CrossRef CAS.
- S. H. DeWitt and B. E. Maryanoff, Biochemistry, 2018, 57, 472–473 CrossRef CAS PubMed.
- C. Scnmidt, Nat. Biotechnol., 2017, 35, 493–495 CrossRef PubMed.
- S. Kopf, F. Bourriquen, W. Li, H. Neumann, K. Junge and M. Beller, Chem. Rev., 2022, 122, 6634–6718 CrossRef CAS PubMed.
- H. Li, M. Shabbir, W. Li and A. Lei, Chin. J. Chem., 2024, 42, 1145–1156 CrossRef CAS.
- G. Prakash, N. Paul, G. A. Oliver, D. B. Werz and D. Maiti, Chem. Soc. Rev., 2022, 51, 3123–3163 RSC.
- N. Li, Y. Li, X. Wu, C. Zhu and J. Xie, Chem. Soc. Rev., 2022, 51, 6291–6306 RSC.
- P. L. Norcott, Chem. Commun., 2022, 58, 2944–2953 RSC.
- R. Zhou, L. Ma, X. Yang and J. Cao, Org. Chem. Front., 2021, 8, 426–444 RSC.
- Y. Dong, Y. Su, L. Du, R. Wang, L. Zhang, D. Zhao and W. Xie, ACS Nano, 2019, 13, 10754–10760 CrossRef CAS PubMed.
- A. Di Giuseppe, R. Castarlenas and L. A. Oro, C. R. Chim., 2015, 18, 713–741 CrossRef CAS.
- W. J. Kerr, D. M. Lindsay, P. K. Owens, M. Reid, T. Tuttle and S. Campos, ACS Catal., 2017, 7, 7182–7186 CrossRef CAS.
- P. Ranjan, S. Pillitteri, E. V. Van Der Eycken and U. K. Sharma, Green Chem., 2020, 22, 7725–7736 RSC.
- J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744–7765 CrossRef CAS PubMed.
- Y. Chang, A. Yesilcimen, M. Cao, Y. Zhang, B. Zhang, J. Z. Chan and M. Wasa, J. Am. Chem. Soc., 2019, 141, 14570–14575 CrossRef CAS PubMed.
- A. Martins and M. Lautens, Org. Lett., 2008, 10, 4351–4353 CrossRef CAS PubMed.
- F. Bourriquen, N. Rockstroh, S. Bartling, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2022, 61, e202202423 CrossRef CAS PubMed.
- T. Kurita, K. Hattori, S. Seki, T. Mizumoto, F. Aoki, Y. Yamada, K. Ikawa, T. Maegawa, Y. Monguchi and H. Sajiki, Chem. – Eur. J., 2008, 14, 664–673 CrossRef CAS PubMed.
- W. N. Palmer and P. J. Chirik, ACS Catal., 2017, 7, 5674–5678 CrossRef CAS PubMed.
- V. Pfeifer, T. Zeltner, C. Fackler, A. Kraemer, J. Thoma, A. Zeller and R. Kiesling, Angew. Chem., Int. Ed., 2021, 60, 26671–26676 CrossRef CAS PubMed.
- Z. Yu, W. Wang and L. Chen, J. Labelled Compd. Radiopharm., 2011, 54, 352–356 CrossRef CAS.
- C. Zhang, Z. Shen, L. Tian and L. Chen, J. Labelled Compd. Radiopharm., 2012, 55, 401–405 CrossRef CAS.
- M. Fang and K. R. Cadwallader, J. Agric. Food Chem., 2013, 61, 3580–3588 CrossRef CAS PubMed.
- G. Srinivas, V. K. P. Unny, K. Mukkanti and B. M. Choudary, J. Labelled Compd. Radiopharm., 2013, 56, 325–329 CrossRef CAS PubMed.
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 3022–3047 CrossRef CAS PubMed.
- J. Martin, J. Eyselein, S. Grams and S. Harder, ACS Catal., 2020, 10, 7792–7799 CrossRef CAS.
- C. S. Donald, T. A. Moss, G. M. Noonan, B. Roberts and E. C. Durham, Tetrahedron Lett., 2014, 55, 3305–3307 CrossRef CAS.
- M. Kuriyama, N. Hamaguchi, G. Yano, K. Tsukuda, K. Sato and O. Onomura, J. Org. Chem., 2016, 81, 8934–8946 CrossRef CAS PubMed.
- C. Liu, Z. Chen, C. Su, X. Zhao, Q. Gao, G. H. Ning, H. Zhu, W. Tang, K. Leng, W. Fu, B. Tian, X. Peng, J. Li, Q. H. Xu, W. Zhou and K. P. Loh, Nat. Commun., 2018, 9, 80 CrossRef PubMed.
- T. Luo, Z. Wang, Y. Chen, H. Li, M. Peng, F. Tuna, E. J. L. McInnes, S. J. Day, J. An, M. Schröder and S. Yang, Angew. Chem., Int. Ed., 2023, 62, e202306267 CrossRef CAS PubMed.
- C. S. Turchi and D. F. Ollis, J. Catal., 1990, 122, 178–192 CrossRef CAS.
- T. Yang, J. Peng, Y. Zheng, X. He, Y. Hou, L. Wu and X. Fu, Appl. Catal., B, 2018, 221, 223–234 CrossRef CAS.
- L. B. Reuterglrdh and M. Langphasuk, Chemosphere, 1997, 35, 585–596 CrossRef.
- Y. Sari, P. L. Gareso, B. Armynah and D. Tahir, Int. J. Hydrogen Energy, 2024, 55, 984–996 CrossRef CAS.
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- J. C. Smart, B. L. Pinsky, M. F. Fredrich and V. W. Day, J. Am. Chem. Soc., 1979, 101, 4373–4374 CrossRef.
- K. Yamaguti and S. Sato, J. Chem. Soc., Faraday Trans. 1, 1985, 81, 1237–1246 RSC.
- T. Kawai and T. Sakata, Chem. Phys. Lett., 1980, 72, 87–89 CrossRef CAS.
- S. Sato and J. M. White, Chem. Phys. Lett., 1980, 72, 83–86 CrossRef CAS.
- H. Eidsvåg, S. Bentouba and P. Vajeeston, et al., Molecules, 2021, 26, 1687 CrossRef PubMed.
- H. Abdullah, M. M. R. Khan, H. R. Ong and Z. Yaakob, J. CO2 Util., 2017, 22, 15–32 CrossRef CAS.
- A. Hammad, A. Anzai, X. Zhu, A. Yamamoto, D. Ootsuki, T. Yoshida, A. EL-Shazly, M. Elkady and H. Yoshida, Catal. Lett., 2020, 150, 1081–1088 CrossRef CAS.
- H. Yuzawa and H. Yoshida, Chem. Lett., 2013, 42, 1336–1343 CrossRef.
- D. Friedmann, A. Hakki, H. Kim, W. Choi and D. Bahnemann, Green Chem., 2016, 18, 5391–5411 RSC.
- S. Kohtani, E. Yoshioka, K. Saito, A. Kudo and H. Miyabe, Catal. Commun., 2010, 11, 1049–1053 CrossRef CAS.
- K. Imamura, T. Yoshikawa, K. Hashimoto and H. Kominami, Appl. Catal., B, 2013, 134–135, 193–197 CrossRef CAS.
- N. Hoffmann, Aust. J. Chem., 2015, 68, 1621–1639 CrossRef CAS.
- B. Kraeutler and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 2239–2240 CrossRef CAS.
- B. Kraeutler and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 5985–5992 CrossRef CAS.
- K. Zou, A. Yamamoto and H. Yoshida, Catal. Today, 2024, 425, 114354 CrossRef CAS.
- N. Li, Y. Ning, X. Wu, J. Xie, W. Li and C. Zhu, Chem. Sci., 2021, 12, 5505–5510 RSC.
- Y. He, H. Yang, D. Gao, J. Ma, Y. Shao, G. An and G. Li, Chin. J. Org. Chem., 2021, 41, 4725–4731 CrossRef CAS.
- Z. Huang, Z. Zhao, C. Zhang, J. Lu, H. Liu, N. Luo, J. Zhang and F. Wang, Nat. Catal., 2020, 3, 170–178 CrossRef CAS.
- H. Yoshida, Adv. Energy Sustainability Res., 2025, 2400439 CrossRef.
- S. Park, J. Jeong, K. I. Fujita, A. Yamamoto and H. Yoshida, J. Am. Chem. Soc., 2020, 142, 12708–12714 CrossRef CAS PubMed.
- H. Yuzawa, M. Aoki, K. Otake, T. Hattori, H. Itoh and H. Yoshida, J. Phys. Chem. C, 2012, 116, 25376–25387 CrossRef CAS.
- H. Yuzawa, J. Kumagai and H. Yoshida, J. Phys. Chem. C, 2013, 117, 11047–11058 CrossRef CAS.
- G. Lee, R. Maruyama, K. Zou, H. Huang, K. Oyama, H. Yoshida and J. Kumagai, J. Phys. Chem. C, 2025, 129, 402–414 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. Nakatsuji, et al., Gaussian 16, Vers. Revision C.02, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- K. Zou, A. Yamamoto and H. Yoshida, Int. J. Hydrogen Energy, 2024, 91, 989–996 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00316d |
| This journal is © The Royal Society of Chemistry 2025 |