
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced electrocatalytic hydrogenation of levulinic acid to value-added chemical platforms†
Pol
Vilariño
ab,
Elvira
Gómez
 ab and
Albert
Serrà
*ab
ab and
Albert
Serrà
*ab
aGrup d'Electrodeposició de Capes Primes i Nanoestructures (GE-CPN), Departament de Ciència de Materials i Química Física, Universitat de Barcelona, Martí i Franquès, 1, E-08028, Barcelona, Catalonia, Spain. E-mail: a.serra@ub.edu
bInstitute of Nanoscience and Nanotechnology (IN2UB), Universitat de Barcelona, Barcelona, Catalonia, Spain
First published on 2nd June 2025
Abstract
The electrocatalytic hydrogenation (ECH) of levulinic acid (LA) has been identified as a sustainable and energy-efficient route for the production of high-value chemicals, including γ-valerolactone (GVL) and valeric acid (VA). This study explores the electrochemical reduction of LA using electrodeposited Cu-, Ni-, and Ru-based catalysts, including their binary (CuNi, CuRu, NiRu) and ternary (CuNiRu) systems, under both acidic and alkaline conditions. Catalysts were prepared by electrodeposition from new developed formulations. Among the electrocatalysts studied, Ni-rich deposits exhibited superior performance, with CuNi and CuNiRu catalysts achieving faradaic efficiencies above 80%, LA conversion rates exceeding 85%, and GVL selectivity as high as 94% in acidic media. Electrochemical analyses revealed that the reaction pathway and product distribution were strongly influenced by catalyst composition and solution pH, with acidic conditions favouring higher conversion efficiencies and selectivity toward GVL. Conversely, alkaline media gave rise to diminished reaction rates and a shift toward VA production. In acidic medium, reusability tests assessed the long-term stability of CuNi-based catalysts, with moderate performance degradation over multiple cycles and negligible catalyst leaching. A comparative analysis with state-of-the-art electrocatalysts highlights the competitive advantages of the developed materials, particularly in terms of efficiency and selectivity. The findings emphasise the potential of electrodeposited Ni-rich deposits for scalable, cost-effective, and environmentally friendly biomass conversion, advancing the prospects of electrochemical LA valorisation as a viable alternative to conventional hydrogenation methods.
Introduction
The global challenge of climate change has led to a heightened focus on identifying sustainable and renewable sources of energy and chemicals.1,2 The substantial reliance on fossil fuels not only amplifies greenhouse gas emissions but also underscores the vulnerability of supply chains that are heavily reliant on finite resources.3 This critical situation underscores the urgent need to explore alternative feedstocks and innovative processes capable of meeting industrial demands while minimizing environmental impact. Among these alternatives, biomass—a renewable and widely available resource derived from agricultural and forestry residues—has emerged as a transformative solution.4,5 The utilization of biomass holds the potential to substitute for petroleum-derived feedstocks and to serve as the foundation for bio-based chemicals and fuels, thereby paving the way for a circular and sustainable bioeconomy.4,5Among the various chemicals that can be derived from biomass, levulinic acid (LA) is of particular significance as a pivotal platform compound due to its structural versatility and potential to be upgraded into a wide range of valuable products.6–8 LA is produced via the acid-catalysed hydrolysis of lignocellulosic biomass, which is primarily composed of cellulose and hemicellulose, LA can be obtained in high yields from a variety of carbohydrate sources.9 LA's molecular structure, which features both keto and carboxyl functional groups, renders it a suitable precursor for the production of biofuels, solvents, pharmaceuticals, plastics, herbicides, and flavour agents. As a renewable and bio-based precursor, LA has become a cornerstone of research aimed at reducing reliance on traditional petroleum-based compounds.10,11
The transformation of levulinic acid into gamma-valerolactone (GVL) is particularly promising due to GVL's low toxicity, biodegradability, and potential as a bio-based solvent and renewable fuel additive.12–14 The compound has applications spanning industrial, pharmaceutical, and energy sectors, making it a focal point for sustainable innovation.15–17 However, conventional methodologies for the conversion of LA to GVL, which rely on homogeneous and heterogeneous catalytic hydrogenation, present significant challenges. These conventional methods frequently require elevated temperatures and high pressures, in addition to the use of expensive and unsustainable precious-metal-based catalysts, resulting in energy-intensive and costly processes that hinder scalability.18–20
In contrast, electrochemical methods could provide a more sustainable and efficient alternative for the conversion of LA.21–23 These approaches enable transformations to occur under mild conditions, obviating the need for external reducing agents, and could permit precise control over product selectivity.21–23
In light to these limitations, electrocatalytic hydrogenation (ECH) of LA has emerged as a promising alternative for the sustainable conversion of LA into GVL,24,25 by enabling conversion under mild conditions and obviating the necessity for external reducing agents. Electrocatalytic approaches could confer considerable advantages in terms of energy efficiency, environmental impact, and process control. Furthermore, the capacity to precisely modulate reaction conditions allows for enhanced selectivity and efficiency in product formation.26 Notably, the ECH of LA has shown promise in producing not only GVL but also other value-added derivatives, such as valeric acid (VA).24,26 However, the efficiency and scalability of ECH are constrained by the availability of suitable electrocatalysts.9,25,26
Research has emphasized the critical role of both the electrode material and the reaction medium in enhancing the efficiency and selectivity of the electrochemical conversion process. Various electrocatalysts, including lead, copper, or nickel, have been studied, exhibiting disparate levels of efficiency and selectivity. While lead electrodes have exhibited high conversion rates and selectivity, the environmental concerns associated with lead, coupled with the lower efficiency of other electrodes, underscore the necessity for new and optimized electrocatalytic materials.23,27 The optimisation of electrode material is a pivotal factor in determining the efficiency, selectivity, and sustainability of electrocatalytic processes. This highlights the pressing need for innovative electrocatalysts that seamlessly integrate high catalytic activity with environmental safety and economic viability.23,27,28
In order to address the aforementioned challenges, the present study investigates the electrochemical reduction of LA using advanced electrode materials, including Cu, Ni, and Ru, as well as their binary and ternary systems. A particular emphasis is placed on nickel-rich deposits, given their demonstrated capacity to attain high catalytic performance.29,30 The study objective is the development of efficient and selective processes for the sustainable transformation of biomass-derived LA into high-value chemicals like GVL. The research begins with the electrosynthesis of electrocatalysts via electrodeposition, a versatile method that allows for precise control over the composition, structure, and morphology of the prepared deposits. Subsequently, the catalytic performance of these materials was systematically evaluated in the electrochemical reduction of LA. The study explores key performance metrics, including conversion, efficiency, product selectivity, and catalyst stability, under varying reaction conditions using aqueous acidic and alkaline media. In contrast to previous studies focusing primarily on Pb-based systems or mid-pH conditions, this work offers a comparative analysis of electrochemical LA conversion under strongly acidic and alkaline environments using industrially relevant catalysts.
Experimental section
Electrosynthesis and characterization of electrocatalysts
A total of seven aqueous solutions were freshly prepared as described in Table 1, using NiCl2·6H2O (Merck, 99.9%, CAS: 7791-20-0), CuCl2·2H2O (Sigma-Aldrich, >99.0%, CAS: 10125-13-0), RuCl3·3H2O (Sigma-Aldrich, >99.0%, CAS: 13815-94-6), and HOC(COONa)(CH2COONa)2·2H2O (Sigma-Aldrich, >99.0%, CAS: 6132-04-3). Furthermore, NaCl (Sigma-Aldrich, >99.0%, CAS: 7647-14-5) was employed to regulate the ionic strength. The ionic strength of each solution was adjusted to 2.30 M, thereby ensuring consistent conditions for comparative studies across samples. Prior to each experiment, the pH of the solution was adjusted to 3.0 with hydrochloric acid. All solutions were prepared using deionized Milli-Q water. The temperature was maintained at 25 °C during the experiments.| Ni | Ru | Cu | NiRu | NiCu | RuCu | NiRuCu | |
|---|---|---|---|---|---|---|---|
| NiCl2/M | 0.30 | 0.00 | 0.00 | 0.30 | 0.30 | 0.00 | 0.30 |
| RuCl3/M | 0.00 | 0.02 | 0.00 | 0.02 | 0.00 | 0.02 | 0.02 |
| CuCl2/M | 0.00 | 0.00 | 0.05 | 0.00 | 0.05 | 0.05 | 0.05 |
| C6H5Na3O7/M | 0.20 | 0.20 | 0.20 | 0.20 | 0.20 | 0.20 | 0.20 |
| NaCl/M | 0.20 | 0.98 | 0.95 | 0.08 | 0.05 | 0.83 | 0.00 |
The solutions under consideration were formulated with the aim of attaining Ni-rich electrocatalysts. To this end, the nickel(II) concentration was elevated to 15 times the level of ruthenium and 6 times the level of copper. This high nickel concentration was used due to the electrochemical inert behaviour of nickel. Additionally, due to the electrocatalytic character of nickel towards hydrogen reaction, the reduction of nickel overlaps with the onset of hydrogen evolution, rendering this process competitive. Ruthenium(III) concentration, an expensive and high-value material, was proposed attending, also, to economic considerations.
The electrochemical study was conducted using a potentiostat–galvanostat (Metrohm Autolab PGSTAT204), with data acquisition and control facilitated by NOVA 2.1 software. A three-electrode configuration was utilized, comprising a platinum electrode as the counter electrode, an Ag|AgCl|Cl− electrode as the reference electrode, and a glassy carbon electrode (GC) as the working electrode. Prior to each experiment, the GC electrode was polished to a mirror finish using alumina powders of decreasing particle size (3.75 μm and 1.87 μm) on polishing pads. Following each polishing stage, the electrode was rinsed with deionized water to eliminate any residual alumina particles. Subsequently, the electrode was subjected to sonication in Milli-Q water for a period of two minutes, with the objective of dislodging any residual particles or impurities that may have remained on the surface. All electrochemical experiments were performed under an argon atmosphere. Cyclic voltammetry (CV) was employed to analyse the electrochemical behaviour in each bath, while potentiostatic chronoamperometry (CA) was used for electrodeposit preparation.
The morphology and elemental composition of the electrodeposited films were characterized using field-emission scanning electron microscopy (FE-SEM, JEOL-7100), equipped with energy-dispersive X-ray spectroscopy (EDS). Furthermore, the elemental composition and total amount of deposits were quantified using inductively coupled plasma optical emission spectrometry (ICP-OES, Optima 8300, PerkinElmer) for ppm-level detection and inductively coupled plasma mass spectrometry (ICP-MS, NexIon 2000, PerkinElmer) for ppb-level analysis, in order to calculate the faradaic efficiency.
Electrocatalytic reduction of levulinic acid
The electrochemical reduction of LA was conducted using the same potentiostat–galvanostat system and three-electrode configuration. The bare GC or the electrodeposited metals on GC were used as the working electrode, an Ag/AgCl|Cl− as a reference electrode, and a platinum spiral as a counter electrode. The electrochemical media were prepared according to two different conditions, as illustrated in Table 2, which included acidic and alkaline aqueous solutions. In all conditions, the LA concentration was fixed at 0.5 M. The solutions were maintained at 25 °C throughout the experiments, with a total reaction volume of 15 mL. Acidic conditions were achieved by the addition of sulfuric acid, while alkaline conditions were achieved using sodium hydroxide.| Conditions | Acidic conditions | Alkaline conditions |
|---|---|---|
| Solvent | H2O | H2O |
| LA/M | 0.50 | 0.50 |
| H2SO4/M | 0.50 | 0.00 |
| NaOH/M | 0.00 | 1.00 |
| KCl/M | 0.50 | 0.50 |
| pH | 0.00 | 14.00 |
| Applied potential/V | −1.40 and −1.80 | −1.80 and −2.00 |
| Volume/mL | 15 | 15 |
| Charge/C | 1500 | 1500 |
In order to evaluate the ECH performance of LA on GC and electrodeposited materials, linear sweep voltammograms (LSV) were recorded within a potential range of 0.00 V to −2.00 V at 50 mV s−1, both in presence and absence of LA. The potentials applied in subsequent chronoamperometric experiments of the ECH process were selected based on the LSV results.
The electrochemical media after the ECH process were analysed using high-performance liquid chromatography (HPLC) with UV-vis detection. Two distinct HPLC setups were utilized depending on the analyte of interest: for γ-valerolactone (GVL), a XBridge C18 column (3.5 μm, 4.5 × 50 mm) was used with a 10 μL injection volume, isocratic elution with a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v mixture of water and acetonitrile as the mobile phase, and detection at 210 nm using an ACQ-PSA Instrument Setup; for LA and VA, an Aminex HPX-87H column (300 × 7.8 mm) was employed with a column temperature of 60 °C, an internal column temperature of 145 °C, a 50 μL injection volume, and an isocratic mobile phase consisting of a 9:1 v/v mixture of water and acetonitrile with 10 mM sulfuric acid, with UV-vis detection at 210 nm. These setups enabled precise characterisation and quantification of the species present in the reaction mixture, ensuring a comprehensive evaluation of the electrochemical reduction process.
:1 v/v mixture of water and acetonitrile as the mobile phase, and detection at 210 nm using an ACQ-PSA Instrument Setup; for LA and VA, an Aminex HPX-87H column (300 × 7.8 mm) was employed with a column temperature of 60 °C, an internal column temperature of 145 °C, a 50 μL injection volume, and an isocratic mobile phase consisting of a 9:1 v/v mixture of water and acetonitrile with 10 mM sulfuric acid, with UV-vis detection at 210 nm. These setups enabled precise characterisation and quantification of the species present in the reaction mixture, ensuring a comprehensive evaluation of the electrochemical reduction process.
For the most effective materials and conditions, five consecutive reusability cycles were conducted without any intermediate treatment of the electrocatalyst to assess its recyclability and stability. Following each cycle, the concentration of dissolved catalyst in the electrochemical medium was quantitatively analysed using ICP-OES (Optima 8300, PerkinElmer) for ppm-level and ICP-MS (NexIon 2000, PerkinElmer) for ppb-level analysis.
Results and discussion
Role of citrate and chloride in the proposed formulation for Cu, Ni, and Ru on glassy carbon
The selection of chloride- and citrate-based electrolytes was strategically made to approach the reduction potential of the selected metal ions, which is a key factor under the codeposition of metals.Citrate (Cit) and chloride complexation play a crucial role in governing metal ion reduction kinetics and deposit composition. The stability constants (logβ) of metal–citrate complexes, which quantify the interaction strength between metal ions and citrate ligands, are tabulated between 5.2 and 6.0 for the [NiCit]− complex and 6.2–6.4 for the [CuCit]− complex.31–33 While specific values for Ru(III)–citrate complexes are not available in the literature, Ru(III) is expected to exhibit a higher affinity for multidentate ligands such as citrate compared to Cu(II) and Ni(II). This increased affinity influence its deposition potential, requiring more energy to be deposited, resulting in its electrodeposition at more negative potentials.31,33–35
The stability constants (logβ) for the chloride complexes [NiCl4]2− and [CuCl4]2− are 3.0–4.1 and 7.5–8.5, respectively. However, data concerning Ru(III)–chloride complexation is scarce, despite Ru(III)'s propensity to form stable chloride complexes, which significantly impact its chemical stability and reactivity during deposition.36–38
From the data and literature, the interplay between citrate and chloride is proposed in order to facilitate the codeposition. Moreover, the known modulator effect of chloride presence due to its complexation of the three metals, as well as the benefit of hydroxylated species from citrate, can contribute to obtaining homogeneous deposits and reducing the significant electrocatalytic towards hydrogen reaction of Ni and Ru. Through the careful counterbalance of bath composition and complexation equilibria, we propose the bath compositions included in Table 1. As outlined previously in the experimental section, the electrochemical baths were formulated with a view to producing Ni-rich electrocatalysts when binary or ternary systems were prepared.
 | ||
| Fig. 1 Cyclic voltammograms (CV) recorded on GC electrode at various potential limits in a (a) copper (II) solution, (c) nickel (II) solution, and (e) ruthenium (III) solution, at 50 mV s−1 at 25 °C. FE-SEM micrographs of (b) copper deposits obtained at −0.39 V vs. Ag|AgCl|Cl−, (d) nickel deposits obtained at −0.90 V vs. Ag|AgCl|Cl−, and (f) ruthenium deposits obtained at −0.59 V vs. Ag|AgCl|Cl−. Q = 36 mC. Scale bar: 200 nm. | ||
Fig. 1b, shows the FE-SEM images of copper deposits prepared at −0.39 V demonstrated that the deposit consists of well-distributed large grains. The formation of these grains is likely facilitated by the superficial mobility of the copper grains on glassy carbon. In comparison, at −0.30 V, smaller grain sizes were observed, reflecting differences in nucleation and growth dynamics under less negative deposition conditions (Fig. S1†).39,40 Furthermore, faradaic efficiencies (Fig. S1a†) exceeded 85% in all cases, with values above 90% at lower overpotentials.
Fig. 1c displays the CV profiles for nickel deposition on the GC. During the cathodic sweep, a reduction current emerged at approximately −0.80 V, which is consistent with the electrochemical reduction of Ni(II) ions to metallic Ni(0). In the reverse scan, a characteristic nucleation loop was observed, which is indicative of the nucleation and growth process of metal electrodeposition. The formation of bubbles occurred during the scan at high negative potentials, related to hydrogen codeposition, a reaction favoured by the freshly deposited nickel.41–43 In the positive sweep, an oxidation peak emerged at approximately −0.10 V, associated with the partial reoxidation of metallic nickel to Ni(II). As previously reported, this asymmetric signal actually comprised two overlapping oxidation peaks, corresponding to hydrogen-poor α-Ni and hydrogen-rich β-Ni.44–46 Moreover, the charge involved in the oxidation peak was dependent to the extent of nickel deposition, as reflected in the curves recorded increasing the negative potential limits.
As shown in Fig. 1d and S2b–d,† FE-SEM micrographs of nickel electrodeposits revealed distinct morphological differences. Deposits prepared at −0.90 V exhibited larger grain sizes and apparent surface roughness, suggesting a slow nucleation rate at this potential. Conversely, at more negative potentials (−1.00 V and −1.20 V), deposits became fine and more homogeneous. The finer grain structure suggested enhanced uniformity and compactness of the deposit. Importantly, at −0.90 V, the faradaic efficiency exceeded 90%, indicating that nearly all the applied charge was invested in nickel reduction (Fig. S2a†). However, at more negative potentials, efficiency declined significantly, attributed to the hydrogen evolution, which became more favoured under more negative potentials. This phenomenon was promoted by intrinsic nickel electrocatalytic activity for hydrogen evolution, rendering the process less efficient at negative potentials.
Fig. 1e shows the cyclic voltammograms from the ruthenium solution. In the cathodic scan, two distinct reduction peaks were observed at approximately 0.65 V, corresponding to the reduction of Ru(III) to Ru(II). At more negative potentials, approximately −0.50 V, a second reduction current, corresponding to the reduction of Ru(II) to Ru(0). Hydrogen evolution became significant at more negative potentials, competing with Ru deposition.47 In the positive scan, a broad oxidation potential range was observed, corresponding to the reoxidation of Ru(0). Notably, at the more positive potentials a sharp oxidation peak is recorded, close to the onset of oxygen evolution.
FE-SEM micrographs of Ru deposits (Fig. 1f, S3b and c†) revealed a rounded grains leading homogeneous coverage, but with a significant number of grain boundaries, which is consistent with previous reports on Ru electrodeposition.48–50 Elemental mapping confirmed a uniform distribution of Ru throughout the deposit. Furthermore, Fig. S3a† demonstrates that the faradaic efficiencies of all deposits prepared remained above 70%. However, at preparation conditions of high negative potentials, a substantial drop in efficiency was observed.
The electrochemical conversion of LA to value-added chemicals, such as GVL and VA, was systematically studied to assess catalysts performance under various pH conditions. A series of single-metal deposits were prepared at −0.33 V, −0.90 V, and −0.59 V using Cu, Ni, and Ru solutions, respectively, to investigate their electrocatalytic potential for the ECH of LA.
LSV measurements (Fig. 2a and b) were conducted to investigate the electrochemical behaviour of each catalyst in the presence and absence of LA.
 | ||
| Fig. 2 Linear sweep voltammograms (LSV) recorded within a potential range of 0.0 V to −2.0 V vs. Ag|AgCl|Cl− at 50 mV s−1, in the absence (dashed line) and presence (solid line) of 0.5 M of LA, using Cu, Ni, Ru, and bare GC electrodes in (a) acidic and (b) alkaline media. Conversion percentages and product selectivity toward GVL and VA in (c) acidic and (d) basic media. All experiments were performed in triplicate, with values reported as the mean ± standard deviation. | ||
Across both acidic and alkaline media, Ru-based electrodes demonstrated the most pronounced current response, followed by Ni-based electrodes. Notably, acidic conditions facilitated higher current densities. In acidic medium (Fig. 2a), the addition of 0.5 M LA resulted in a positive shift in the onset potential for the reduction current: from −0.34 V to −0.30 V for the Ru-electrode, from −0.61 V to −0.44 V for the Ni-based electrode, from −0.95 V to −0.56 V for the Cu-based electrode, and from −1.09 V to −0.84 V for the GC electrode. These shifts, accompanied by a significant increase in current density, suggested that ECH of LA was more favourable than the simultaneous hydrogen evolution reaction on all electrode materials.
In contrast, in alkaline media (Fig. 2b) the processes exhibited low current densities and a different electrochemical response, involving slower reaction rates. The onset potential shifts were consistent with those observed in acidic media, but the lower current densities suggest that reaction kinetics were hindered, possibly due to differences in intermediate adsorption processes and/or stabilization of key reaction intermediates.
According to the results from the LSV evaluation, electrolysis experiments were conducted. In acidic media, the ECH of LA was investigated at −1.4 V and −1.8 V for all electrode materials, whereas in alkaline conditions, the selected potentials were −1.8 V and −2.0 V. Electrolysis were carried out for a total charge of 1500 C, with significant variation in reaction duration across different catalysts and reaction environments. The results show the impact of both catalyst composition and reaction medium on the efficiency and stability of LA electrochemical hydrogenation.
In acidic media, the electrolysis time ranged from 50 min to 210 min, depending on the electrode material. In contrast, the electrolysis process in alkaline media required a significant longer duration for all the catalysts. Furthermore, in alkaline media, evidence of catalyst degradation was observed, particularly for Cu-based electrodes, likely due to the highly aggressive nature of the medium. The combination of hydroxide ion activity and prolonged exposure to high potentials accelerated surface corrosion and structural instability, thereby reducing their catalytic durability.
Regarding product distribution, in acidic media, the conversion of LA primarily resulted in the selective formation of GVL as the main product, regardless of the applied potential (Fig. 2c). At −1.4 V, the LA conversion was observed for Cu (90.7 ± 2.0%), followed closely by Ni (89.2 ± 1.8%) and Ru (88.1 ± 1.6%), while GC exhibited a slightly lower conversion (83.3 ± 1.2%). Despite the relatively small differences in conversion rates, product selectivity varied significantly among the catalysts. Notably, GC and Cu did not produce any detectable amounts of GVL or VA, indicating that while these catalysts effectively convert LA, they do not promote its further transformation into the desired products. In contrast, Ni produced GVL and VA, achieving a yield for GVL of 51.2 ± 1.5% and for VA of 12.9 ± 1.4%, while Ru exhibited even higher selectivity, producing 60.0 ± 1.2% GVL and 18.1 ± 1.3% VA. The findings indicate that Ru demonstrates superior selectivity under the tested electrochemical conditions, likely attributable to its enhanced hydrogenation and ring-closure kinetics.51,52 However, as demonstrated in numerous reports in the literature, Ni-based catalysts – particularly when supported on acidic materials such as SiO2, zeolites as H-ZSM-5, or mesoporous silica – can also effectively catalyse both hydrogenation and lactonization steps in the thermal hydrocyclization of LA to GVL. Consequently, the ring-closure activity is not exclusive to Ru, and Ni plays a well-established role in GVL formation via both acid-mediated and metal-catalysed mechanisms.53–55
When the applied potential was established at −1.8 V, LA conversion efficiency declined slightly for all catalysts. However, Ru still exhibited the highest conversion (85.1 ± 1.8%). Despite this reduction in conversion, GVL formation improved significantly for Ru, reaching 72.0 ± 1.2%, the highest GVL yield observed under the tested conditions. Ni also maintained a high GVL yield (55.1 ± 1.2%). The VA yield for Ru remained stable at 18.1 ± 1.2%, whereas Ni experienced a slight decline to 11.0 ± 1.2%. Interestingly, GC and Cu, on which at −1.4 V did not produce any GVL or VA, exhibited discrete VA formation at −1.8 V, with yields of 1.2 ± 0.6% and 0.8 ± 0.5%, respectively. This suggests a subtle shift in reaction selectivity at more negative potentials, though their overall selectivity toward VA remained very low.
As shown in Fig. 2d, in alkaline conditions at −1.8 V, the highest conversion recorded was for Ru (97.1 ± 1.2%), followed closely by Ni (96.4 ± 1.3%), Cu (92.0 ± 1.8%), and GC (90.2 ± 1.4%). Despite these high conversion rates, GVL formation was observed only on Ni (49.4 ± 1.1%) and Ru (51.3 ± 1.4%), suggesting that these catalysts effectively facilitate the hydrogenation and lactonization steps required for GVL production. Cu and GC, in contrast, did not produce any detectable amounts of GVL, further confirming their lack of catalytic character for this transformation. Regarding VA yield, Ru exhibited moderate VA formation (8.8 ± 1.5%), while GC produced a slightly higher VA yield (13.7 ± 0.9%). Cu and Ni did not generate any VA.
At −2.0 V, LA conversion declined for all catalysts showing GC the highest conversion (88.5 ± 1.5%). This diminution in conversion suggests that excessive hydrogenation or competing side reactions became more pronounced as more negative was the applied potential. It can thus be concluded that at negative potentials and strong alkaline conditions, catalyst deactivation and degradation is a probable risk. Similar to the results obtained at −1.8 V, GVL production was also observed only using Ni (42.5 ± 1.3%) and Ru (41.1 ± 1.9%), although both yields were lower than those recorded at −1.8 V. Interestingly, VA formation was observed exclusively with Ru (13.8 ± 1.8%). This suggests that Ru remained uniquely active in VA formation, possibly due to its superior ability to facilitate the sequential hydrogenation of GVL into VA.51,52
Overall, these results highlight Ru as the most effective catalyst for promoting both GVL and VA formation in acidic and alkaline media, with Ni also showing strong selectivity for GVL production. However, at more negative potentials, the increasing competition from HER reduces overall catalytic efficiency, particularly affecting GVL yield. These findings underscore the importance of optimizing both catalyst selection and applied potential to enhance LA conversion and product selectivity.
The observed differences in product distribution can be explained by considering the electrochemical mechanisms operating under acidic and alkaline conditions (Fig. 3 and S5†). In acidic aqueous media (pH < 4), LA undergoes proton-coupled electron transfer, which facilitates the selective reduction of its carbonyl group to form 4-HPA. This intermediate then spontaneously cyclizes to GVL, a transformation promoted by the high proton concentration, favourable leaving group properties of water, and the inherent thermodynamic stability of the five-membered lactone ring. Additionally, the protonated state of the carboxylic acid group at low pH supports efficient intramolecular esterification. This mechanism is especially effective at temperatures above 20–25 °C, where both the hydrogenation and lactonization kinetics are enhanced, leading to high selectivity and yield of GVL.22,24,26
 | ||
| Fig. 3 Proposed electrochemical reaction pathway for the hydrogenation of levulinic acid (LA) to γ-valerolactone (GVL) and valeric acid (VA) in acidic media. LA is first reduced via proton-coupled electron transfer to 4-hydroxypentanoic acid (4-HPA). Under acidic conditions, 4-HPA undergoes intramolecular esterification to form GVL, driven by favourable thermodynamic and kinetic factors. Alternatively, further hydrogenation of 4-HPA leads to the formation of VA. Reaction conditions, pH, and catalyst choice influence the selectivity between GVL and VA. | ||
In contrast, under strongly alkaline conditions (pH > 9), the reaction mechanism shifts markedly. LA exists as the levulinate anion, which affects its adsorption characteristics and limits proton availability. The electrochemical reduction of the carbonyl group proceeds more slowly due to decreased proton-coupled electron transfer efficiency and potential electrostatic repulsion at the cathode. Additionally, lactonization is suppressed because the carboxylate group is a poor electrophile and hydroxide is a poor leaving group. While deeper hydrogenation to VA might be expected under such conditions, our experiments at pH 14 did not detect VA formation on either Cu or Ni electrodes, even under high reducing potentials.22,24,26 This lack of VA formation in alkaline media can be attributed to several factors: (i) the suppression of proton availability at high pH hinders proton-coupled electron transfer steps essential for the hydrogenation of GVL or intermediates into VA; (ii) the hydrogen evolution reaction, which is strongly favoured on Cu and Ni surfaces at the required negative potentials, competes with LA reduction. This further suppresses the selective reduction of carbonyl groups to form VA; (iii) surface corrosion or oxide formation under alkaline and high-potential conditions may passivate active sites; and lastly, (iv) the LA reduction pathway may shift under alkaline conditions, with intermediates being less stable or diverted toward side products rather than VA. Despite these constraints, moderate GVL formation was still observed for Ni and Ru catalysts, indicating that some degree of ring closure may proceed via surface-mediated or residual proton pathways.
 | ||
| Fig. 4 Cyclic voltammograms (CV) recorded at various potential limits in a (a) CuNi solution, (b) CuRu solution, (c) NiRu solution, and (d) CuNiRu solution on a GC electrode, with a scan rate of 50 mV s−1 at 25 °C. FE-SEM micrographs of (e) CuNi deposits obtained at −0.90 V vs. Ag|AgCl|Cl−, (f) CuRu deposits obtained at −0.70 V vs. Ag|AgCl|Cl−, (g) NiRu deposits obtained at −0.90 V vs. Ag|AgCl|Cl−, and (h) CuNiRu deposits obtained at −1.00 V vs. Ag|AgCl|Cl−. Q = 36 mC. Scale bar: 200 nm. | ||
During the anodic sweep, several oxidation peaks were identified. The first peak corresponded to the oxidation of Cu(0) to Cu(I), followed by a second oxidation current associated with the oxidation of α-Ni, β-Ni, and the CuNi system. The dominant contribution depended on the limit potential—at more negative potentials, it primarily originated from CuNi deposits. Finally, at more positive potentials, Cu(I) underwent oxidation to Cu(II). These oxidation features confirmed the codeposition of Cu(0), Ni(0), and the CuNi system, offering valuable insights into the electrochemical stability and composition of the electrodeposited CuNi.
Chronoamperometric experiments were performed at varying applied potentials to analyse the deposit morphology and elemental composition. FE-SEM micrographs of the CuNi deposits (Fig. 4e and S6†) revealed a homogeneous and smooth morphology. Furthermore, as shown in Table 3, the application of more negative potentials resulted in the formation of Ni-rich deposits. The faradaic efficiency decreased from 82% at −0.80 V to 67% at −1.00 V, suggesting a trade-off between deposition rate and hydrogen evolution.
| CuNi | |||
|---|---|---|---|
| E/V | Ni/at% | Cu/at% | FE/% |
| −0.80 | 20.5 ± 0.4 | 79.5 ± 0.4 | 82 |
| −0.90 | 65.0 ± 0.8 | 35.0 ± 0.8 | 79 |
| −1.00 | 85.0 ± 0.6 | 15.0 ± 0.6 | 67 |
| CuRu | |||
|---|---|---|---|
| E/V | Cu/at% | Ru/at% | FE/% |
| −0.60 | 71.4 ± 0.6 | 28.6 ± 0.6 | 55 |
| −0.70 | 55.0 ± 0.7 | 45.0 ± 0.7 | 47 |
| NiRu | |||
|---|---|---|---|
| E/V | Ru/at% | Ni/at% | FE/% |
| −0.90 | 15.5 ± 0.3 | 84.5 ± 0.3 | 62 |
| −1.00 | 5.2 ± 0.6 | 94.8 ± 0.6 | 54 |
| CuNiRu | ||||
|---|---|---|---|---|
| E/V | Cu/at% | Ni/at% | Ru/at% | FE/% |
| −0.80 | 33.3 ± 0.8 | 26.4 ± 0.8 | 40.3 ± 0.8 | 66 |
| −1.00 | 16.3 ± 0.7 | 56.5 ± 0.7 | 27.2 ± 0.7 | 59 |
| −1.50 | 9.8 ± 1.1 | 73.7 ± 1.1 | 16.4 ± 1.1 | 42 |
Fig. 4b displays the CVs for a CuRu solution. During the cathodic scan, an initial small reduction signal at 0.50 V indicated the reduction of Ru(III) to Ru(II). This was followed by the reduction of Cu(II) to metallic copper near −0.30 V. A subsequent reduction signal around −0.60 V corresponded to the start of codeposition of Cu(0) and Ru(0). During the anodic scan, distinct oxidation peaks were observed. The first peak corresponded to the oxidation of Cu(0) to Cu(I), followed by the oxidation of CuRu, and finally, a third peak associated with the transition of Cu(I) to Cu(II).
The FE-SEM micrographs in Fig. 4f and S7† provide insights into the morphological evolution of CuRu deposits as a function of the applied potential. The deposits prepared at −0.60 V exhibited a rough and non-homogeneous surface structure with visible cracks (Fig. S7c†). This suggests that deposition involves both simultaneous and sequential nucleation, leading to a patchy, interconnected morphology. At more negative potentials, such as −0.70 V, the deposits showed increased density and roughness. As the potential became more negative, the formation of cracks became more pronounced. The increased driving force at negative potentials may have facilitated faster nucleation and growth, yielding a rougher, more compact film with a homogeneous CuRu distribution.
As shown in Table 3, an increase in the applied potential resulted in deposits with a higher ruthenium content, emphasizing the impact of potential on elemental composition. The efficiency gradually declined with more negative potentials, reaching 47% at −0.70 V, likely due to increased hydrogen coevolution.
Fig. 4c presents the CVs for a NiRu solution. During the cathodic scan, an initial peak at 0.65 V corresponded to the reduction of Ru(III) to Ru(II), followed by the reduction of Ru(II) to metallic ruthenium at −0.50 V. As the potential became more negative, the current changed its slope, evidencing the concurrent codeposition of Ni and Ru.
At potentials more negative than −1.00 V, hydrogen evolution was evident, competing with metal deposition and affecting the morphology of the NiRu film while reducing the overall process efficiency. During the anodic scan, partial re-oxidation of ruthenium back into solution was identified by oxidation current at approximately 1.0 V. However, no distinct signal corresponding to NiRu oxidation was detected before oxygen evolution became evident.
Fig. 4g presents FE-SEM micrographs (see also Fig. S8†), illustrating the morphological evolution of NiRu deposits. At −0.90 V, the deposits exhibited a granular structure. Further lowering the potential to −1.0 V resulted in more compact and uniform deposits, suggesting an increased nucleation rate and improved growth.
As shown in Table 3, decreasing the applied potential led to a higher nickel content in the deposits, emphasizing the potential-dependent composition of the NiRu alloy. However, deposition efficiency declined to 54% at −1.00 V due to the increasing significance of hydrogen evolution.
Fig. 4d displays the CVs for the ternary CuNiRu system. During the negative scan, the signals corresponding to the sequential appearance of the currents related to the reduction of Ru(III) and Cu(II) species, followed by the Ni(II), were observed. As in the previously described systems, hydrogen evolution became evident near −1.00 V.
During the positive scan, several overlapping oxidation signals were observed. As the scan was extended, these signals became more complex.
The FE-SEM micrographs (Fig. 4h and S9†) show the morphology of the ternary CuNiRu deposits obtained at varying deposition potentials. At −0.80 V, a granular coating with a uniform distribution of grains across the surface was observed. However, at −1.0 V, a semi-spongy morphology appeared, characterized by grains that did not fully coalesce. At −1.20 V, a more compact morphology V was observed, with small separation between grains compared to −1.0 V.
As shown in Table 3, the atomic composition varied with the applied potential, exhibiting a general trend of increasing Ni content at more negative potentials. At −0.80 V, the deposit consisted of 33.3% Cu, 26.4% Ni, and 40.3% Ru, with a faradaic efficiency of 66%. As the potential became more negative, Ni incorporation increased, lowering Cu and Ru content. At −1.50 V, a substantial shift occurred, with Ni becoming the predominant element (73.7% Ni), while the Ru content decreased to 16.4% and Cu reached its lowest value at 9.8%. The faradaic efficiency significantly decreased to 42% at −1.50 V.
 | ||
| Fig. 5 Linear sweep voltammograms (LSV) recorded within a potential range of 0.0 V to −2.0 V vs. Ag|AgCl|Cl− at a scan rate of 50 mV s−1, in the absence (dashed line) and presence (solid line) of 0.5 M of LA, using CuRu, CuNi, NiRu, and CuNiRu electrodes in (a) acidic and (b) basic media. Conversion percentages and product selectivity toward GVL and VA in (c) acidic and (d) basic media. All experiments were performed in triplicate, with values reported as the mean ± standard deviation. | ||
LSV measurements (Fig. 5a and b) were used as an initial assessment of the electrochemical behaviour of the analysed catalytic systems. The study revealed significant performance variations depending on the catalyst composition and reaction medium.
In acidic media, the NiRu electrode exhibited the high current density, although the differences compared to CuRu, CuNiRu, and CuNi were not significant. As shown in Fig. 5a, the addition of 0.5 M LA caused a positive shift in the onset potential for the reduction current: from −0.32 V to −0.28 V for the CuRu electrode, from −0.56 V to −0.36 V for the CuNi electrode, from −0.35 V to −0.31 V for the NiRu electrode, and from −0.34 V to −0.27 V for the CuNiRu electrode. Under alkaline conditions, all systems exhibited a minor current density compared to their performance in acidic media. This diminution was attributed to the diminished availability of protons in the alkaline environment, which may slow the electrochemical hydrogenation process and potentially changes the reaction mechanism toward alternative intermediates or pathways. As shown in Fig. 5b, the onset potential of reduction also shifted in the presence of LA: from −0.95 V to −0.83 V for the CuRu electrode, from −1.21 V to −1.14 V for the CuNi electrode, from −0.98 V to −0.86 V for the NiRu electrode, and from −1.16 V to −1.02 V for the CuNiRu electrode.
Notably, in alkaline media, the CuRu electrode maintained the highest current density. Overall, the LSV data indicated that higher current densities in acidic media could correlate with faster reaction kinetics during electrolysis, whereas the lower current densities in alkaline media reflected slower reaction rates and the consequent extended electrolysis durations.
Following the LSV analysis, electrolysis experiments were conducted under both acidic and alkaline conditions at −1.4 and −1.8 V in acidic media, and at −1.8 and −2.0 V in alkaline conditions.
In acidic media, GVL was the dominant product across all catalytic systems (Fig. 5c). At −1.4 V, all catalyst achieved conversion rates above 85%. Although CuRu showed the lowest conversion among the tested catalysts, the overall variation in conversion rates was relatively small. However, significant differences in product selectivity were observed. CuNiRu produced the highest GVL yield (85.0 ± 1.2%), followed closely by CuNi (79.3 ± 2.1%), whereas NiRu (55.1 ± 1.8%) and CuRu (42.5 ± 1.5%) exhibited lower GVL yield. Regarding VA formation, the relatively low VA yields across all catalysts indicate that the reaction pathway primarily favours GVL formation under these conditions, with only limited further hydrogenation to VA.
When the applied potential was −1.8 V, LA conversion followed mixed trends. CuNi achieved the highest conversion (94.1 ± 1.9%), and its GVL yield increased markedly to 90.9 ± 0.8%, surpassing all other catalysts. CuNiRu also retained a high GVL yield (85.2 ± 1.2%), while NiRu (59.0 ± 1.2%) and CuRu (58.0 ± 1.2%) showed modest improvements compared to −1.4 V. This enhanced GVL yield at a more negative potential suggests that increased hydrogen availability facilitates the hydrogenation of intermediates, thereby promoting GVL formation. The VA yield continues being moderate, although NiRu exhibited a notable increase to (7.8 ± 0.9%) from 3.2 ± 1.5% attained at −1.4 V. CuNiRu (1.8 ± 0.8%) and CuNi (1.9 ± 0.7%) produced only minimal VA.
In alkaline conditions, at −1.8 V, LA conversion remained high across all catalysts: CuRu (97.1 ± 1.9%), CuNi (96.0 ± 1.3%), NiRu (94.7 ± 1.7%), and CuNiRu (92.9 ± 1.2%). Despite these high conversion rates, GVL yield varied significantly, reflecting differences in catalytic selectivity. CuNiRu led the highest GVL yield (52.0 ± 1.2%), followed by CuRu (50.3 ± 0.8%). CuNi produced a moderate GVL yield (41.3 ± 1.2%), whereas NiRu (28.7 ± 1.8%) showed the lowest GVL selectivity. For VA yield, CuNi exhibited the highest formation (6.2 ± 0.3%), followed by CuRu (4.2 ± 0.7%), NiRu (3.5 ± 1.4%), and CuNiRu (2.8 ± 1.3%). These results indicate that while CuNiRu is highly effective for GVL production, it does not significantly promote further hydrogenation toward VA. The lower VA yields compared to GVL suggest that the reaction pathway primarily favours GVL formation in alkaline media (Fig. 5d).
At −2.0 V, LA conversion remained high, with CuNiRu exhibiting the highest value (93.6 ± 1.2%), followed closely by CuRu (93.3 ± 1.2%), NiRu (92.8 ± 1.2%), and CuNi (91.2 ± 1.2%). Although CuNi showed a slight decrease in conversion compared to −1.8 V, conversion rates for the other catalysts remained relatively stable. GVL yield increased across all catalysts at −2.0 V. The highest GVL yield was achieved by CuRu (55.8 ± 1.8%), followed by CuNi (43.3 ± 0.9%), CuNiRu (37.8 ± 2.3%), and NiRu (34.7 ± 2.1%). This increase suggests that hydrogenation is partially more efficient at −2.0 V, although HER and other side reactions may compete with LA reduction, limiting further improvements in selectivity. In relation to the VA yield at −2.0 V, the utilization of NiRu (6.2 ± 1.8%) and CuRu (5.4 ± 2.1%) resulted in enhanced VA yields compared to the yield obtained at −1.8 V. Conversely, CuNiRu (4.9 ± 1.4%) and CuNi (4.8 ± 0.9%) remained at moderate levels. Additionally, in alkaline medium, catalyst degradation was notably more pronounced during electrolysis process. The corrosive nature of the alkaline environment was a contributing factor to surface deterioration and, by extension, the deactivation of the catalyst.
These results demonstrate that an acidic medium is the more suitable environment for achieving optimal conversion rates, selectivity, and efficiency. Among the synthesised catalysts, CuNi and CuNiRu demonstrate the greatest effectiveness for GVL production. These insights can be a guidance for optimizing catalyst selection and reaction conditions in biomass-derived platform molecule transformations.
The reusability of CuNi- and CuNiRu-based electrodes in acidic medium was evaluated through multiple electrolysis cycles performed at a fixed applied potential of −1.8 V and under constant charge. The results are summarized in Fig. 6.
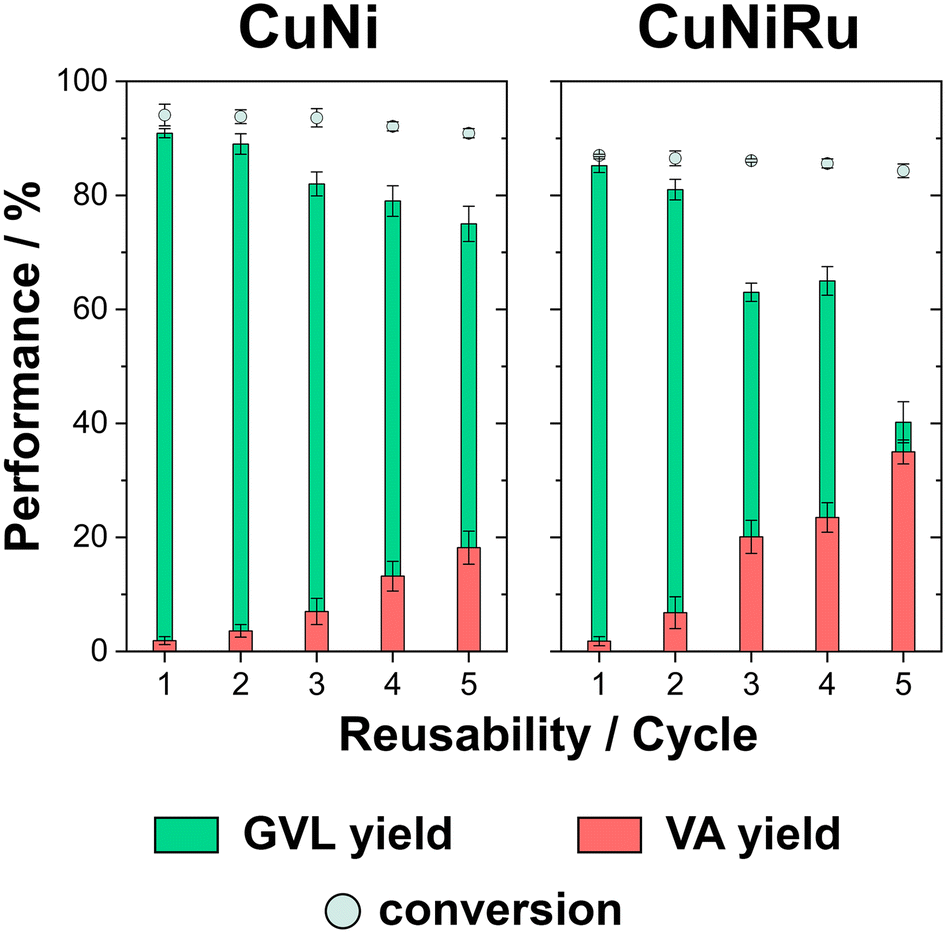 | ||
| Fig. 6 Conversion percentages and product selectivity toward GVL and VA in acidic during five reusability cycles at a fixed applied potential of −1.8 V vs. Ag|AgCl|Cl− and a constant circulated charge density for CuNi-based electrode and CuNiRu-based electrode. All experiments were performed in triplicate, with values reported as the mean ± standard deviation. | ||
The CuNi-based electrode exhibited remarkable stability during successive electrolysis cycles, initially achieving a conversion efficiency of 94.1 ± 1.9%, with a GVL yield of 90.9 ± 0.8% and negligible VA production. Notably, the conversion efficiency remained above 90% throughout the electrolysis cycles, despite the electrolysis time increase with the number of cycles. This performance was achieved without any catalyst rejuvenation between cycles. However, a gradual decline in GVL selectivity was observed, with the yield decreasing from 90.9 ± 0.8% to 75.0 ± 3.1%, while VA production increased. These results suggest that while the CuNi catalyst maintained high conversion efficiency, it underwent partial deactivation over time, likely due to surface degradation or compositional changes from extended operation.
The CuNiRu-based electrode exhibited slightly lower initial activity compared to the CuNi catalyst, with a conversion efficiency of approximately 87% and a GVL yield of 85.2 ± 1.2% (Fig. 6). Notably, the conversion efficiency of CuNiRu remained consistent and above 80% throughout the reusability cycles. Additionally, the electrolysis time required to achieve the overall charge increased as the cycle number rose. For CuNiRu catalyst, GVL selectivity decreased significantly, with the yield dropping from 85.2 ± 1.2% to 40.2 ± 3.6%, while VA production increased notably to 35.0% by the fifth cycle.
The reusability experiments also evaluated the leaching behaviour of CuNi- and CuNiRu-based electrodes after the fifth consecutive electrolysis cycle using ICP analysis. For the CuNi-based electrode, 9.8% of the catalyst mass was dissolved, with Ni being the most leached element. Greater degradation was detected for the CuNiRu-based electrode, with 27.2% of the initial catalyst mass being leached, with Ni once again being the predominant element. Nevertheless, the relatively stable conversion efficiencies observed for both materials suggest that possible re-electrodeposition processes resulting from leaching did not play a predominant role in influencing the overall catalytic performance.
In conclusion, the reusability experiments highlighted the different stability and performance characteristics of CuNi- and CuNiRu-based electrodes under acidic conditions. While the CuNi electrode exhibited stability, with consistent conversion efficiencies above 90%, it demonstrated gradual selectivity loss for GVL, likely due to partial deactivation from surface degradation and catalyst leaching. Conversely, the CuNiRu electrode exhibited slightly lower initial activity but maintained conversion efficiencies above 80% throughout the cycles, despite a more pronounced decline in GVL selectivity and a significant increase in VA production. The higher dissolution rate, particularly of Ni, observed for CuNiRu underscores its susceptibility to structural and compositional degradation during prolonged operation.56
To further investigate the role of surface composition in catalyst deactivation and selectivity loss, we performed an additional experiment in which fresh CuNi and CuNiRu layers were electrodeposited onto the spent electrodes following five catalytic cycles. The regenerated electrodes exhibited a remarkable recovery in both LA conversion and GVL selectivity, comparable to that of the freshly prepared materials. This result confirms that catalyst deactivation is largely attributable to surface depletion or restructuring – particularly the partial dissolution of Ni – as supported by ICP-OES analysis. These findings underscore the importance of maintaining the optimal metal composition at the surface to preserve activity and selectivity. Accordingly, strategies aimed at mitigating Ni leaching are essential for improving long-term catalyst stability. These may include modifying the electrodeposition conditions, incorporating corrosion-resistant supports (e.g., carbon nanotubes, graphene oxide), or introducing stabilizing dopants (e.g., Co, Cr, Mo) known to suppress Ni dissolution under acidic conditions. Such approaches could prolong catalyst life while maintaining high selectivity for GVL formation.
The findings of this study represent a significant advancement in the efficiency and selectivity of LA conversion to GVL. Previous studies have shown that LA conversion is possible in acidic media using various catalysts; however, these approaches often suffer from low selectivity and efficiency or require prohibitively expensive catalysts. Compared to previous research, our prepared catalysts achieved high selectivity to GVL versus VA, whereas previous reports indicated a maximum of 75% without achieving high efficiency.7,19,57,58
For instance, Pb/CFs electrodes exhibited a VA selectivity of up to 92%, while Cd/CFs electrodes achieved an LA conversion of 57%, ranking them among the best ECH electrodes reported. Additionally, previous research with Al-based catalysts yielded a GVL-to-VA selectivity ratio of 1:3, while GC-based methods achieved 100% selectivity to GVL but only 30% conversion.59 Pb-based catalysts produced a 1:1 GVL-to-VA selectivity ratio with a conversion rate of 55%, whereas graphite-based catalysts achieved 100% selectivity to GVL but just 25% conversion.7,19,21,28,57,59
Notably, this study, as in others, evidences that the optimal medium for this conversion was acidic medium, given the low kinetics and conversions observed under alkaline conditions. To the best of our knowledge, this is the first study to evaluate and develop a catalyst that maintained relatively high performance over multiple cycles, demonstrating superior efficiency and stability compared to state-of-the-art catalysts. Although further work is needed to optimize the preparation of the selected catalyst to achieve morphological and structural characteristics that maintain its compositional integrity and prevent the leaching of the components.
Conclusions
This study provides a comprehensive evaluation of electrodeposited Cu-, Ni-, and Ru-based catalysts, along with their binary and ternary alloys, for the electrocatalytic hydrogenation (ECH) of levulinic acid (LA) to γ-valerolactone (GVL) and valeric acid (VA). The findings of the study demonstrate that the composition of the electrode and the conditions of the electrolyte have a critical influence on the catalytic efficiency, product selectivity, and the stability of the catalyst.Among the monometallic catalysts, Ru-based electrodes exhibited the highest current densities, particularly in acidic conditions, leading to enhanced conversion efficiencies. However, the cost and potential scarcity of Ru limit its industrial scalability. Conversely, Ni-based electrodes exhibited favourable performance, exhibiting both reduced cost and enhanced stability, albeit with moderate selectivity. Conversely, Cu-based electrodes exhibited diminished catalytic efficiency and selectivity, particularly in alkaline conditions, underscoring the importance of alloying strategies to enhance their performance.
The introduction of binary and ternary alloy catalysts has been shown to significantly improve electrocatalytic efficiency and selectivity. The most notable finding was that the CuNi and CuNiRu alloys exhibited outstanding catalytic activity, achieving faradaic efficiencies in excess of 80%, LA conversion rates in excess of 85%, and GVL selectivity as high as 94% in acidic media in the first cycle. These findings underscore the pivotal role of Ni in facilitating efficient hydrogenation pathways, while leveraging the electronic modifications of Cu and the catalytic properties of Ru. The observed synergistic effect in these alloyed catalysts could be attributed to enhanced electron transfer, improved stability, and optimised adsorption properties for intermediates involved in the hydrogenation of LA.
The reaction environment was found to have a significant impact on the catalytic performance and product distribution. In the presence of acidic conditions, the selectivity towards GVL was found to be notably higher, and the reaction kinetics were observed to be enhanced. Conversely, alkaline conditions resulted in reduced reaction rates, prolonged electrolysis times, and an increased propensity for VA formation, which was attributed to variations in intermediate adsorption and reaction pathways. Additionally, alkaline media led to higher catalyst degradation in the first cycle.
Stability and reusability studies confirmed the durability of CuNi- and CuNiRu-based electrodes, which maintained high conversion rates and selectivity over multiple electrolysis cycles. The CuNi electrode, in particular, exhibited remarkable stability, retaining a conversion efficiency above 90% after five cycles, with moderate catalyst leaching. However, the selectivity for GVL gradually declined over repeated use, suggesting possible surface modifications or partial compositional changes. The CuNiRu electrode also demonstrated sustained performance, but with slightly higher catalyst dissolution, particularly for Ni, indicating that additional stabilization strategies may be required to further enhance long-term durability. A comparative assessment with previously reported electrocatalysts highlights the advantages of the developed materials. While Pb- and Cd-based catalysts have demonstrated moderate LA conversion efficiencies, their toxicity and environmental concerns limit their practical application. Conversely, the CuNi and CuNiRu alloys presented in this study offer a non-toxic, scalable, and highly efficient alternative, with superior selectivity and long-term stability.
Data availability
The data supporting this study's findings are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Ministerio de Ciencia e Innovación (MICIN) is acknowledged through Grant TED2021-129898B-C22 funded by NextGenerationEU and MICIU/AEI/10.13039/501100011033. The authors also would like to express their gratitude to the Departament de Recerca i Universitats, particularly the Departament d'Acció Climàtica, Alimentació i Agenda Rural, as well as the Fons Climàtic de la Generalitat de Catalunya (project 2023 CLIMA 00009 AGAUR) for their support. Authors thank the CCiT-UB for the use of their equipment.Notes and references
- A. Serrà, R. Artal, L. Philippe and E. Gómez, Langmuir, 2021, 37, 4666–4677, DOI:10.1021/acs.langmuir.1c00461.
- V. I. Timokhin, M. Regner, A. H. Motagamwala, C. Sener, S. D. Karlen, J. A. Dumesic and J. Ralph, ACS Sustainable Chem. Eng., 2020, 8(47), 17427–17438, DOI:10.1021/acssuschemeng.0c05651.
- D. Ryan Georgianna and S. P. Mayfield, Nature, 2012, 488, 329–335, DOI:10.1038/nature11479.
- H. Wang, Y. Wu, S. Guo, C. Dong and M. Ding, Mol. Catal., 2020, 497, 111218, DOI:10.1016/j.mcat.2020.111218.
- H. Wang, Y. Wu, T. Jin, C. Dong, J. Peng, H. Du, Y. Zeng and M. Ding, Mol. Catal., 2020, 498, 111267, DOI:10.1016/j.mcat.2020.111267.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595, 10.1039/c3gc37065h.
- S. Dutta, I. K. M. Yu, D. C. W. Tsang, Y. H. Ng, Y. S. Ok, J. Sherwood and J. H. Clark, Chem. Eng. J., 2019, 372, 992–1006, DOI:10.1016/j.cej.2019.04.199.
- K. Yan, C. Jarvis, J. Gu and Y. Yan, Renewable Sustainable Energy Rev., 2015, 51, 986–997, DOI:10.1016/j.rser.2015.07.021.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558, 10.1039/c1cs15147a.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963, 10.1039/c3gc41935e.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513, 10.1039/c004654j.
- X. Li, P. Jia and T. Wang, ACS Catal., 2016, 6, 7621–7640, DOI:10.1021/acscatal.6b01838.
- W. R. H. Wright and R. Palkovits, ChemSusChem, 2012, 5, 1657–1667, DOI:10.1002/cssc.201200111.
- R. Bujaldón, A. Fons, J. Garcia-Amorós, C. Vaca, J. Nogués, M. J. Esplandiu, E. Gómez, B. Sepúlveda and A. Serrà, Adv. Sci., 2025, 2416153, DOI:10.1002/advs.202416153.
- K. Yan, Y. Yang, J. Chai and Y. Lu, Appl. Catal., B, 2015, 179, 292–304, DOI:10.1016/j.apcatb.2015.04.030.
- F. D. Pileidis and M. M. Titirici, ChemSusChem, 2016, 9, 562–582, DOI:10.1002/cssc.201501405.
- G. Dautzenberg, M. Gerhardt and B. Kamm, Holzforschung, 2011, 65, 439–451, DOI:10.1515/HF.2011.081.
- M. J. Gilkey and B. Xu, ACS Catal., 2016, 6, 1420–1436, DOI:10.1021/acscatal.5b02171.
- Y. Kuwahara, W. Kaburagi, Y. Osada, T. Fujitani and H. Yamashita, Catal. Today, 2017, 281, 418–428, DOI:10.1016/j.cattod.2016.05.016.
- D. Fegyverneki, L. Orha, G. Láng and I. T. Horváth, Tetrahedron, 2010, 66, 1078–1081, DOI:10.1016/j.tet.2009.11.013.
- G. V. Burmakina, D. V. Zimonin, T. A. Kenova, V. V. Verpekin, V. V. Sychev, N. A. Zos'ko and O. P. Taran, Curr. Res. Green Sustainable Chem., 2023, 7, 100379, DOI:10.1016/j.crgsc.2023.100379.
- Y. Zhang and Y. Shen, Int. J. Hydrogen Energy, 2022, 47, 22763–22774, DOI:10.1016/j.ijhydene.2022.05.080.
- F. W. S. Lucas, Y. Fishler and A. Holewinski, Green Chem., 2021, 23, 9154–9164, 10.1039/d1gc02826j.
- T. R. Dos Santos, P. Nilges, W. Sauter, F. Harnisch and U. Schröder, RSC Adv., 2015, 5, 26634–26643, 10.1039/c4ra16303f.
- J. C. Yu, M. M. Baizer and K. Nobe, J. Electrochem. Soc., 1988, 135, 83–87, DOI:10.1149/1.2095595.
- P. Nilges, T. R. Dos Santos, F. Harnisch and U. Schröder, Energy Environ. Sci., 2012, 5, 5231–5235, 10.1039/c1ee02685b.
- Y. Zhang and Y. Shen, Appl. Catal., B, 2024, 343, 123576, DOI:10.1016/j.apcatb.2023.123576.
- H. Wu, J. Song, C. Xie, Y. Hu, P. Zhang, G. Yang and B. Han, Chem. Sci., 2019, 10, 1754–1759, 10.1039/c8sc03161d.
- V. Vij, S. Sultan, A. M. Harzandi, A. Meena, J. N. Tiwari, W. G. Lee, T. Yoon and K. S. Kim, ACS Catal., 2017, 7, 7196–7225, DOI:10.1021/acscatal.7b01800.
- S. De, J. Zhang, R. Luque and N. Yan, Energy Environ. Sci., 2016, 9, 3314–3347, 10.1039/c6ee02002j.
- O. Y. Zelenin, Russ. J. Coord. Chem., 2007, 33, 346–350, DOI:10.1134/S1070328407050065.
- G. R. Hedwig, J. R. Liddle and R. D. Reeves, Aust. J. Chem., 1980, 33, 1685–1693, DOI:10.1071/CH9801685.
- T. B. Field, J. L. McCourt and W. A. E. McBryde, Can. J. Chem., 1974, 52, 3119–3124, DOI:10.1139/v74-458.
- T. B. Field, J. Coburn, J. L. McCourt and W. A. E. McBryde, Anal. Chim. Acta, 1975, 74, 101–106, DOI:10.1016/S0003-2670(01)82783-5.
- M. R. Pitluck, B. D. Pollard and D. T. Haworth, Anal. Chim. Acta, 1987, 197, 339–342, DOI:10.1016/S0003-2670(00)84747-9.
- A. N. Srivastva, N. Singh, J. Singh and A. Singh, Stability Constants of Metal Complexes in Solution, ed. A. N. Srivastva, IntechOpen, Rijeka, 2019 Search PubMed.
- M. T. Weller, Inorganic chemistry, Oxford University Press, Oxford, 6th edn, 2014 Search PubMed.
- W. Horwitz, J. AOAC Int., 2020, 48, 870, DOI:10.1093/jaoac/48.4.870a.
- M. Cortés, E. Gómez, A. Pérez-Rodríguez, C. Serre and E. Vallés, J. Electroanal. Chem., 2008, 619–620, 176–182, DOI:10.1016/j.jelechem.2008.04.011.
- A. Roldan, E. Gómez, S. Pané and E. Vallés, J. Appl. Electrochem., 2007, 37, 575–582, DOI:10.1007/s10800-006-9288-7.
- E. Gómez, R. Pollina and E. Vallés, J. Electroanal. Chem., 1995, 386, 45–56, DOI:10.1016/0022-0728(95)03817-Z.
- P. Sebastian, M. I. Giannotti, E. Gómez and J. M. Feliu, ACS Appl. Energy Mater., 2018, 1, 1016–1028, DOI:10.1021/acsaem.7b00177.
- K. Eiler, H. Krawiec, I. Kozina, J. Sort and E. Pellicer, Electrochim. Acta, 2020, 359, 136952, DOI:10.1016/j.electacta.2020.136952.
- W. G. Proud, E. Gomez, E. Sarret, E. Valles and C. Müller, J. Appl. Electrochem., 1995, 25, 770–775, DOI:10.1007/BF00648632.
- E. Gómez, C. Muller, W. G. Proud and E. Vallés, J. Appl. Electrochem., 1992, 22, 872–876, DOI:10.1007/BF01023732.
- Y. Lin and J. R. Selman, J. Electrochem. Soc., 1993, 140, 1299–1303, DOI:10.1149/1.2220974.
- D. Kutyła, K. Kołczyk, P. Żabiński, R. Kowalik, A. Kwiecińska and K. Skibinska, Russ. J. Electrochem., 2020, 56, 214–221, DOI:10.1134/S1023193520030064.
- K. M. Kost, D. E. Bartak, B. Kazee and T. Kuwana, Anal. Chem., 1990, 62, 151–157, DOI:10.1021/ac00201a013.
- D. K. Oppedisano, L. A. Jones, T. Junk and S. K. Bhargava, J. Electrochem. Soc., 2014, 161, D489–D494, DOI:10.1149/2.0441410jes.
- M. Jayakumar, K. A. Venkatesan, R. Sudha, T. G. Srinivasan and P. R. Vasudeva Rao, Mater. Chem. Phys., 2011, 128, 141–150, DOI:10.1016/j.matchemphys.2011.02.049.
- A. V. Mironenko, M. J. Gilkey, P. Panagiotopoulou, G. Facas, D. G. Vlachos and B. Xu, J. Phys. Chem. C, 2015, 119, 6075–6085, DOI:10.1021/jp512649b.
- O. A. Petcuta, N. C. Guzo, M. Bordeiasu, A. Nicolaev, V. I. Parvulescu and S. M. Coman, Catalysts, 2025, 15(1), 80, DOI:10.3390/catal15010080.
- V. Mohan, V. Venkateshwarlu, C. V. Pramod, B. D. Raju and K. S. R. Rao, Catal. Sci. Technol., 2014, 4, 1253–1259, 10.1039/c3cy01072d.
- V. Mohan, C. Raghavendra, C. V. Pramod, B. D. Raju and K. S. Rama Rao, RSC Adv., 2014, 4, 9660–9668, 10.1039/c3ra46485g.
- M. Varkolu, V. Velpula, S. Ganji, D. R. Burri and S. R. Rao Kamaraju, RSC Adv., 2015, 5, 57201–57210, 10.1039/c5ra10857h.
- F. Safizadeh, E. Ghali and G. Houlachi, Int. J. Hydrogen Energy, 2015, 40, 256–274, DOI:10.1016/j.ijhydene.2014.10.109.
- X. L. Du, L. He, S. Zhao, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, Angew. Chem., Int. Ed., 2011, 50, 7815–7819, DOI:10.1002/anie.201100102.
- S. S. R. Gupta and M. L. Kantam, Catal. Today, 2018, 309, 189–194, DOI:10.1016/j.cattod.2017.08.007.
- A. M. Hengne and C. V. Rode, Green Chem., 2012, 14, 1064–1072, 10.1039/c2gc16558a.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00319a |
| This journal is © The Royal Society of Chemistry 2025 |