
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-aluminosilicate catalysts for ethylene oligomerization: recent scientific progress
Vasile Hulea

Charles Gerhardt Institute of Montpellier, University of Montpellier, CNRS, ENSCM, 1919 Rte de Mende, 34293 Montpellier Cedex 5, France. E-mail: vasile.hulea@enscm.fr
First published on 3rd July 2025
Abstract
Significant scientific effort has been made during the last decades to develop heterogeneous catalysts and processes for ethylene oligomerization. Among the reported catalysts, Ni-aluminosilicates are regarded as the most promising candidates. This paper reviews the recent advancements in ethylene conversion catalyzed by Ni-aluminosilicates. The main fundamental and practical aspects on this topic, including types of catalysts depending on the support, active nickel sites, oligomerization mechanism and kinetics and catalyst deactivation, are examined. The multi-reaction catalytic processes in which oligomerization is the key step have been also discussed.
Vasile Hulea has been full professor of heterogeneous catalysis at the National School of Chemistry of Montpellier (France) until 2023 and currently holds the position of Professor Emeritus at the “Charles Gerhardt” Institute of Montpellier. He has worked tirelessly in catalysis research for more than 40 years. His research activities focused on synthesis and characterization of microporous and mesoporous catalysts, they reactivity in acid–base and redox catalysis, and kinetic and mechanistic aspects. He developed original catalysts and friendly processes for valorisation of renewable/fossil sources and wastes (light olefin oligomerization & metathesis, methanol conversion to diesel and mild distillate, aromatics alkylation, mercaptan conversion, and mild oxidation of S-containing organic compounds). Prof. Hulea has (co)-authored more than 200 research articles, patents and book chapters in the fields of materials science and catalysis. |
1. Introduction
With an annual worldwide production of about 150 million tonnes, ethylene is a key molecule for producing other major platform molecules, end-use chemicals and polymers. Additionally, ethylene is one of the most promising bio-based chemicals, mainly because its high-volume production by bioethanol dehydration became economically feasible. The dimerization/oligomerization of ethylene is of considerable interest for the synthesis of butenes and higher olefins. This reaction can be efficiently catalysed by both homogeneous and heterogeneous catalysts containing metal species as active sites, as shown in recent reviews.1,2 Note that the first review article describes for the most part homogeneous oligomerization catalysts and only a little bit of heterogeneous catalysts,1 while the second one focused on oligomerization on metal-containing zeolites.2In line with sustainable chemistry principles, during the last few decades, research attention has been focused on the development of oligomerization processes based on heterogeneous catalysts. By far, Ni-based materials have been developed and used as heterogeneous catalysts in these research studies.
Ten years ago, I co-authored an extended review, providing knowledge on this topic at the time.3 It has been shown that Ni-exchanged microporous and mesoporous aluminosilicates are very efficient catalysts for this important application. It was also specified that, despite the scientific progress achieved at that time, some key aspects, including the nature of the Ni species, the active sites involved in the catalytic act, the oligomerization mechanism, the effect of the catalyst texture or the effect of the reaction parameters on the oligomerization, were not yet clearly elucidated. To answer the questions that remained open, a notable research effort has been conducted during the last decade.
Based on the recent literature, I try here to identify the progress attained in this scientific area. The first section discusses the main families of Ni-aluminosilicate catalysts used in ethylene oligomerization, paying principal attention to their composition and texture. In the next two major sections, I examine the nature of the active sites and identify them, as well as the elementary reactions involved in the oligomerization mechanism. Then, the effect of the contaminants and the parameters on the oligomerization process are evaluated. Finally, the multi-reaction processes, such as fuel or propylene production, where ethylene oligomerization is a crucial step, are discussed. The major contributions of my group in the field are underscored in this review.
2. Ni-aluminosilicate catalysts for ethylene oligomerization
Metal-containing microporous and mesoporous aluminosilicates are efficient catalysts for many industrial and academic chemical applications. Among them, Ni-containing aluminosilicates have proven their high catalytic potential in reactions such as selective hydrogenation and methane reforming or low olefin oligomerization. While the hydrogenation and reforming reactions require metallic nickel as catalytic centers,4,5 the oligomerization of olefins is activated by the isolated ionic Ni sites.1,3 Ni-aluminosilicates are considered the most promising heterogeneous catalysts for ethene oligomerization carried out at moderate temperatures, without any co-catalysts and activators. The catalysts are typically obtained by ion exchange or impregnation of different supports. Table 1 lists the main families of Ni-containing catalysts prepared, characterized and used during the last decade in ethylene oligomerization.| Catalyst | Support topology | Reaction conditions | Main aim of the study | Ref. |
|---|---|---|---|---|
| Ni-microporous materials | ||||
| Ni-Beta | Beta zeolite | 180 °C, 0.1 MPa | Active sites, mechanism | 6 |
| Ni-Beta | Beta zeolite | 120 °C, 0.1 MPa | Active sites, mechanism | 7 |
| Ni-Beta | Beta zeolite | 180 °C, 0.1 MPa | Catalyst deactivation | 8 |
| Ni-Beta | Beta and ZSM-5 zeolites | 200 °C, 3.5 MPa | Beta vs. ZSM-5: crystal morphology effect | 9 |
| Ni-ZSM-5 | ||||
| Ni-Beta | Beta zeolite | 30–300 °C, 0.1 MPa | Subcritical and supercritical conditions, coke formation | 10 |
| Ni-Beta | Beta zeolite | 200 °C, 3.5 MPa | Active sites | 11 |
| Ni-Beta | Beta zeolite | 120 °C, 3.5 MPa | Active sites, Beta vs. MCM-41 and SIRAL: effect of the support | 12 |
| Ni-Beta | Beta zeolite | 120 °C, 3.5 MPa | Active sites, Beta vs. ASA and Al2O3 effect of the support | 13 |
| Ni-Beta | Beta zeolite | 225 °C, 1.1 MPa | Active sites | 14 |
| Ni-Beta | Beta zeolite | 100 °C, 2.8 MPa | Active sites, mechanism | 15 |
| Ni-Beta | Beta and ZSM-5 zeolites | 300 °C, 0.1 MPa | Beta vs. ZSM-5: effect of pore topology, Ni sites, and acid sites | 16 |
| Ni-ZSM-5 | ||||
| Ni-Beta | Beta and FAU zeolites | −30, −15 °C, 0.1–2.4 MPa | Beta and FAU vs. MCM-41: Ni site deactivation, working at low T | 17 |
| Ni-FAU | ||||
| Ni-Beta | Beta zeolite | 250 °C, 2.5–3.5 MPa | Ni–SiO2–Al2O3 vs. Ni-Beta, kinetic study | 18 |
| Ni-Beta | Beta zeolite | 30–190 °C, 3.5–6.5 MPa | Fuel production | 19 |
| Ni-Beta | Beta zeolite | 50–190 °C, 0.85–2.56 MPa | Effect of parameters | 20 |
| Ni-Beta | Beta zeolite | 120, 250 °C, 3.0–3.5 MPa | Kinetic study | 21 |
| Ni-Beta | Beta zeolite | 50–100 °C, 0.5–2.8 MPa | Kinetic study | 22 |
| Ni-Beta | Beta and ZSM-5 zeolites | 180, 200 °C, 0.25, 3 MPa | Ni-Beta vs. Ni-ZSM-5: active sites | 23 |
| Ni-ZSM-5 | ||||
| Ni-Beta | Beta zeolite | 180 °C, 0.1 MPa | Beta heteroatom composition; X-Beta, X = Al, Ga, Fe | 24 |
| Ni-Beta | Beta zeolite | 250 °C, 3 MPa | Beta heteroatom composition; X-Beta, X = Sn, Ge, Hf, Zr, Ti | 25 |
| Ni-Beta | Beta zeolite | 30–120 °C, 1.1–5.3 MPa | Subcritical or supercritical ethylene | 26 |
| Ni-ZSM-5 | ZSM-5 zeolite | 300 °C, 3.5 MPa | Effect of the incorporation procedure on the nature of the active sites | 27 |
| Ni-ZSM-5 | ZSM-5 | 250 °C, 2 MPa | Relationship between activity and acid site distribution | 28 |
| Ni-ZSM-5 | ZSM-5 | 300–400 °C, 0.1 MPa | Deactivation mechanism | 29 |
| Ni-MgY | FAU zeolite | 25 °C | 1-Butene production, dimerization mechanism DFT calculation | 30 |
| Ni-FAU | FAU zeolite | 300–350 °C, 3.5 MPa | Fuel production, cascade reactions | 31 |
| H-ZSM-5 | ||||
| Ni-MCM-22 | MWW zeolite | 300 °C, 2 MPa | Synergetic effect of acid and nickel sites | 32 |
| 450 °C, 0.1 MPa | ||||
| Ni-ERB-1 | MWW zeolite | 300 °C, 2 MPa | Synergetic effect of acid and nickel sites | 32 |
| 450 °C, 0.1 MPa | ||||
| Ni-SSZ-24 | SSZ-24 zeolite | No reaction | Mechanism, DFT | 33 |
| 150 °C, 3 MPa | Mechanism, kinetic, DFT-MD | 34 | ||
| Ni-ETS-10 | ETS-10 zeolite | 180 °C, 0.5 MPa | New catalysts | 35 |
| Ni-CIT-6 | BEA zeolite | |||
| Ni-ordered mesoporous catalysts | ||||
|---|---|---|---|---|
| Ni-AlSBA-15 | AlSBA-15 | 80 °C, 3 MPa | ETP (ethylene to propylene), metathesis catalyst: MoO3–SiO2–Al2O3 | 36 |
| Ni-AlSBA-15 | AlSBA-15 | 210 °C, 1 MPa | Fuel production, Ni-AlSBA-15 + Amberlyst-35 | 37 |
| Ni-AlSBA-15 | AlSBA-15 | 150 °C, 3.5 MPa | 38 | |
| Ni-AlSBA-15 | AlSBA-15 | 250 °C, 1.5 MPa | Ni-AlSiO2 vs. Ni-AlSBA-15 diluted stream | 39 |
| Ni-AlSBA-15 | AlSBA-15 | 150 °C, 3.5 MPa | Mechanism | 40 |
| Ni-AlSBA-15 | AlSBA-15 | 150–350 °C, 0.1–2 MPa | Fuel production | 41 |
| Ni-AlSBA-15 | AlSBA-15 | 30–120 °C, 1.1–5.3 MPa | Subcritical or supercritical ethylene | 26 |
| Ni-AlSBA-15 | AlSBA-15 | 300 °C, 1.15 MPa | Effect of the catalyst morphology | 42 |
| Ni-AlKIT-6 | AlKIT-6 | 60 °C, 3 MPa | ETP, metathesis catalyst: ReOx/Al2O3 | 43 |
| Ni-AlKIT-6 | AlKIT-6 | 60–120 °C, 3 MPa | ETP, metathesis catalyst: WOx/KIT-6 | 44 |
| Ni-AlKIT-6 | AlKIT-6 | 60–120 °C, 0.1–3 MPa | ETP, metathesis catalyst: ReOx/Al2O3 | 45 |
| Ni-AlKIT-6 | AlKIT-6 | 40–120 °C, 0.1–2 MPa | Kinetic study | 46 |
| Ni-AlKIT-6 | AlKIT-6 | 120 °C, 4 MPa | Effect of calcination temperature | 47 |
| Ni-AlMCM-41 | AlMCM-41 | 250 °C, 2 MPa | Relationship between activity and acid site distribution | 28 |
| Ni-AlMCM-41 | AlMCM-41 | −30, −15 °C, 0.1–2.4 MPa | MCM-41 vs. Beta and FAU: Ni site deactivation, working at low T | 17 |
| Ni-AlMCM-41 | AlMCM-41 | 120 °C, 3.5 MPa | Beta vs. MCM-41 and SIRAL active sites, effect of the support | 12 |
| Ni-AlMCM-41 | AlMCM-41 | 120 °C, 0.1 MPa | Active sites | 48 |
| Ni-AlMCM-41 | AlMCM-41 | 375 °C, 1 bar | ETP | 49 |
| Ni-AlMCM-41 | AlMCM-41 | 75–475 °C, 0.1 MPa | ETP | 50 |
| Ni-AlMCM-41 | AlMCM-41 | Active sites, mechanism, DFT | 51 | |
| Ni-AlMCM-41 | AlMCM-41 | −30 °C, 1.5 MPa | Active sites, low T, CO poison | 52 |
| Ni-AlMCM-41 | AlMCM-41 | −30, −20 °C, 2.6–3.5 MPa | Mechanism | 53 |
| Ni-AlMCM-41 | AlMCM-41 | 180–300 °C, 1–4 MPa | Distribution of the active sites | 54 |
| Ni-AlMCM-41 | AlMCM-41 | DFT, ethylene adsorption on Ni sites | 55 | |
| Ni-AlMCM-41 | AlMCM-41 | Theory, mechanism | 56 | |
| Ni-non-ordered mesoporous catalysts | ||||
|---|---|---|---|---|
| Ni-ASA | ASA | 150 °C, 3.5 MPa | ETP, metathesis catalyst: MoOx/(Al)SiO2 | 57 |
| Ni-ASA | ASA | 170–230 °C, 1.5–3.5 MPa | Kinetic study | 58 |
| Ni-ASA | ASA | 150–350 °C, 3 MPa | Nature of the Ni species | 59 |
| Ni-ASA | ASA | 300 °C, 0.1 MPa | Nature of the Ni species, effect of the catalyst properties | 60 |
| Ni-ASA | ASA | 300 °C, 3–4 MPa | Effect of the catalyst properties | 61 |
| Ni-ASA | ASA | 50–350 °C, 0.35 MPa | Nature of the Ni species | 62 |
| Ni-ASA | ASA | 375 °C, 0.1 MPa | ETP | 49 |
| Ni-ASA | ASA | 80–360 °C, 3.5 MPa | Fuel production | 63 |
| Ni-ASA | ASA | 200 °C, 1 MPa | Nature of the Ni species | 64 |
| Ni-ASA | ASA | 300 °C, 0.1 MPa | Effect of the catalyst preparation | 65 |
| Ni-ASA | ASA | 60 °C, 3 MPa | Nature of the Ni species | 66 |
| Ni-ASA | ASA | 60 °C, 3 MPa | Structure of Ni active sites | 67 |
| Ni-ASA | ASA | 275 °C, 4 MPa | Fuel production | 68 |
| Ni-ASA | ASA | 350 °C, 0.1 MPa | ETP, mechanism | 69 |
| Ni-AlSiO2 | AlSiO2 | 250 °C, 1.5 MPa | Ni-AlSiO2 vs. Ni-AlSBA-15 diluted stream | 39 |
| Ni-SiAlOx | SiAlOx | 375 °C, 0.1 MPa | ETP | 70 |
| Ni-ASA | SIRAL-30 | 200 °C, 1 MPa | Fuel production | 71 |
| Ni-ASA | SIRAL-30 | 50–200 °C, 4, 6.5 MPa | Subcritical and supercritical ethylene | 72 |
| Ni/Siralox-30 | SIRALOX-30 | 120 °C, 3.5 MPa | Active sites, Beta vs. MCM-41 and SIRAL: effect of the support | 12 |
| Ni/Siralox-30 | SIRALOX 40 HPV | 120 °C, 5 MPa | Fuel production | 73 |
| Ni/Siralox-30 | SIRALOX 40 | 120 °C, 4 MPa | Fuel production | 74 |
| Ni-ASA | SIRAL-30 | 200–350 °C, 1 MPa | Fuel production, cascade reactions | 75 |
| H-ZSM-5 | ||||
| Ni–Siral-70 | SIRAL-70 | 375 °C, 0.1 MPa | ETP | 76 |
| Ni-ASA composite | ZSM-5, MCM-41 | 250 °C, 2 MPa | Synergetic effect of Ni-ions and acid sites | 77 |
| 450 °C, 0.15 MPa | ||||
| Other catalytic systems | ||||
|---|---|---|---|---|
| Ni-clay | Montmorillonite | 150–350 °C, 3 MPa | New catalysts | 78 |
| NiSO4/Al2O3 | Al2O3 | 70 °C, 3.5 MPa | Mechanism | 79 |
| NiSO4–ReOx/Al2O3 | Al2O3 | 70 °C, 0.1 MPa | ETP | 80 |
| Ni-POM-WD (NiK10P2W17O61) | POM-WD | 200 °C, 2 MPa | Ni2+ single sites | 81 |
| Ni-POM-WD (NiK10P2W17O61) | POM-WD | 200 °C, 2 MPa | Kinetic study | 82 |
Ni-containing zeolites
Zeolites are ordered crystalline microporous aluminosilicates, which are widely used as catalysts for many industrial applications. Thanks to their properties, such as high thermal, mechanical and chemical stability, these materials were frequently used as carriers for preparing Ni-containing catalysts, which showed real abilities in ethylene oligomerization.The major drawback exhibited by Ni-based zeolites was their low stability against deactivation.3,17 Indeed, they often suffered severe deactivation, mainly due to the blocking of micropores with heavy products. As shown in Table 1, Ni-Beta was the most used catalyst among the Ni-zeolites. Beta is a 3D zeolite, with pores of 12 MR (0.66 × 0.67; 0.56 × 0.56 nm). This topology is favorable for the transfer of bulky molecules, which are responsible for catalyst deactivation.
Usually, the nickel species were incorporated into the zeolite support using two post-synthesis procedures, namely ionic exchange9–11,13,15,16,23,29 and incipient wet impregnation.6,7,11,12,14,19,22,25,27 The general protocol applied for preparing Ni-zeolites by ionic exchange consists of three steps: (i) exchange of the as-synthesized Na-zeolite with aqueous NH4NO3 which results in NH4-zeolite; (ii) exchange with aqueous Ni(NO3)2 and (iii) thermal treatment. Fig. 1 shows such a protocol, used by McCaig and Lamb14 for preparing the Ni-Beta catalyst. The authors found that [NiOH]+ and H+ were the primary charge-compensating cations in the uncalcined catalyst, as evidenced by TPR (Fig. 2). More generally, it is accepted that in the uncalcined Ni-exchanged catalyst the nickel ions are in a hydrated state, similar to isolated Ni hexaaqua ions.83 To remove the water ligands and thus release the Ni ions, heat treatment at temperatures above 500 °C is mandatory prior to catalytic application.84
 | ||
| Fig. 1 General protocol for preparing the Ni-Beta catalyst. This figure has been adapted from ref. 14. | ||
 | ||
| Fig. 2 H2 TPR profiles of Ni-Beta catalysts after in situ pretreatment at 300 °C (solid lines) and 500 °C (dashed lines). This figure has been reproduced from ref. 14. | ||
The impregnation approach was also used for incorporating the Ni species in zeolites. This is a simpler method, but it often produces a large amount of NiO, which is not active in the oligomerization of ethylene.3
Ni-containing mesoporous catalysts
Ni-mesoporous aluminosilicates, which possess large pore diameters, were also widely used in ethylene oligomerization. These catalysts have taken advantage of the increasing knowledge in materials science during the last decades. It allowed the design of tailored mesoporous materials, which are able to improve the catalytic activity and stability.Catalysts with different topologies including Ni-AlMCM-41,12,17,48–54 Ni-AlSBA-15,36–42 and Ni-KIT-6 (ref. 43–47) revealed excellent behavior in terms of catalytic activity and deactivation stability. For example, productivities up to 175 g of oligomers per gram of catalyst per hour and high conversions during 80 h on stream were obtained over Ni-AlSBA-15, in both batch and flow modes by Andrei et al.40 This result, obtained at 150 °C and 3.5 MPa, was superior to those exhibited by other Ni-based heterogeneous catalysts, without using alkylaluminum cocatalysts. The outstanding behavior exhibited by the Ni-mesoporous materials has been attributed to their pores, which are large enough to allow free diffusion of large molecules, resulting in a lower deactivation rate.17
The general protocol applied for preparing these catalysts starts either from Al-containing mesostructured silica (in the case of Ni-AlMCM-41) or from mesostructured silica (in the case of Ni-AlSBA-15 and Ni-AlKIT-6). Such a protocol is given in Scheme 1. The Al-containing sample was obtained from SBA-15 silica by grafting with sodium aluminate. The nickel ions were incorporated into aluminosilicates using an ionic exchange procedure, and after the thermal treatment at 550 °C, a bifunctional catalyst with both Ni and acid sites was obtained.
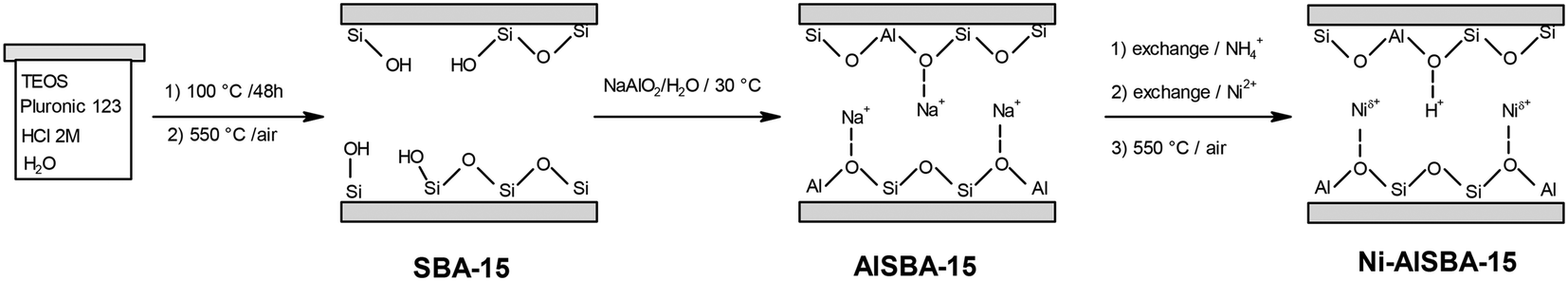 | ||
| Scheme 1 General protocol for preparing the Ni-AlSBA-15 catalyst. This scheme has been adapted from ref. 40. | ||
Although Ni-mesoporous aluminosilicates exhibit good catalytic activity, their preparation cost is high enough, which makes them difficult to apply on a commercial scale.
Ni-non-ordered aluminosilicates, also known as Ni-amorphous aluminosilicates (Ni-ASA), are also considered as promising catalysts for ethylene oligomerization, thanks to their merits of simple/easy synthesis and cheapness. Additionally, their catalytic properties, such as the moderate strength acid sites and mesoporous texture, are comparable to those exhibited by the Ni-based ordered mesoporous catalysts. Note that amorphous silica–alumina was the earlier support for nickel-based oligomerization catalysts.3
Inspecting the recent literature, we can find many sources of ASA supports used for the nickel species: commercial products (e.g. SIRAL series),12,71–75 synthetic ASA and “natural” aluminosilicates (e.g. montmorillonite pillared clay).77 The synthetic ASA were prepared by various methods: homogeneous co-precipitation,60,61,65–67 alumination of the non-order silica56 and hydrolytic non-hydrolytic sol–gel synthesis.59,62
The introduction of nickel into ASA supports was performed using either methods similar to those discussed above for zeolites and ordered mesoporous aluminosilicates (i.e. ion-exchange and impregnation), or by “one-pot” co-precipitation, using tetraethyl orthosilicate, aluminium chloride/nitrate and nickel nitrate/bis(acetylacetonate) as sources of Si, Al and Ni.62,65
3. Nickel state in oligomerization catalysts
Characterization techniques, including DRIFT, XPS, EPR, and H2-TPR, allowed the identification of the different Ni species contained in the oligomerization catalysts. Ionic species, such as Ni2+, Ni+, [Ni–O–Ni]2+ and [Ni–O–H]+, but also small NiO and metallic (Ni0) particles were formed during the introduction of nickel into the carrier matrix and the heat treatment made before to the oligomerization process (Table 2). This diversity of sites is due to the fact that Ni can adopt a number of oxidation states and that it can bind to various atoms on the aluminosilicate support. The nature of the support, as well as the presence of residual Brønsted acid sites plays an important role in the formation of Ni species.| Nickel species | Catalysts | Refs. | |
|---|---|---|---|
 |
Isolated Ni2+ cations in ion exchange state | Ni-SSZ-24, Ni-Beta, Ni-ASA, Ni-MCM-41, Ni-ZSM-5 | 6, 13–15, 18, 23, 27, 34, 46, 48 |
 |
Isolated Ni2+ cations grafted on acidic silanol | Ni-Beta | 7, 48, 67 |
| Ni-ASA | |||
| Ni-MCM-41 | |||
 |
Isolated Ni+ cations in ion exchange state | Ni-SBA-15 | 40, 66, 67 |
| Ni-Beta | |||
 |
Intrazeolitic mono(μ-oxo) dinickel ions in ion exchange state | Ni-Beta | 11 |
 |
Isolated (Ni–OH)+ species in ion exchange state | Ni-MCM-41 | 51 |
 |
Isolated hydroxylated Ni2+ monomers (I), Ni2+ dimers (II) derived upon thermal treatment (Δ) of type I species, Ni2+ monomers exchanged on two H+ sites (III) and bulk nickel oxide (IV) | Ni-MCM-41 | 52 |
 |
Ni2+ in the lacunary defect (green), grey: tungsten oxide regions (grey), phosphate regions (red) | Ni-POM | 82 |
| Ni2+ and NiO nanoclusters cationic | Ni2+ and NiO nanoclusters inside the zeolitic pores | Ni-Beta | 23 |
| Ni2+, Ni+ and NiO | Ni2+ states when pretreated in air, Ni+ when pretreated in N2, NiO when pretreated H2 | Ni-ASA | 66 |
| Ni2+ and NiO | 85% framework bounded Ni2+, when prepared by in situ procedure 50–65% NiO, when prepared by impregnation | Ni-ZSM-5 | 27 |
4. Nickel active catalytic centers: nature and oligomerization mechanism
The identification of the Ni-based active centers and of the elementary steps involved in the dimerization/oligomerization mechanism was the main challenge of the studies carried out in the last decade. But, as Table 3 shows, there is still controversy around the specific nickel species responsible for ethylene oligomerization and their contribution to the initiation of the reaction over nickel aluminosilicates.| Active catalytic sites | Catalysts | Ref. | |
|---|---|---|---|
 |
Mobile [(ethene)2–Ni–alkyl]+ species | Ni-SSZ-24 | 34 |
 |
Isolated Ni+ cations | Ni-SBA-15 | 40, 66 |
| Ni-ASA | |||
 |
In situ ethene-assisted [Ni(II)–H]+ species | Ni-Beta | 6 |
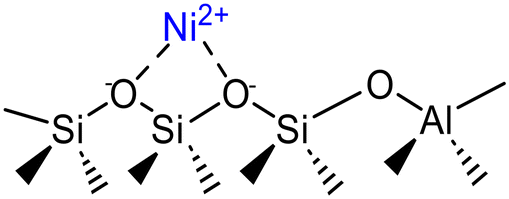 |
Isolated Ni2+ cations grafted on acidic silanols | Ni-Beta | 7, 48 |
| Ni-MCM-41 | |||
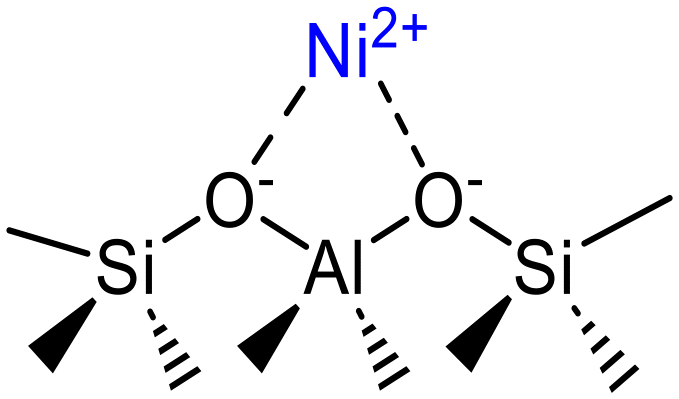
| [Ni–O–Ni]2+ | Intrazeolitic mono(μ-oxo) dinickel species | Ni-Beta | 11 |
 |
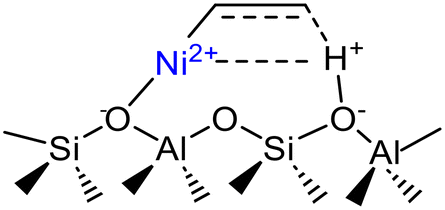
Isolated Ni2+ cations in ion exchange state | Ni-Beta | 13–15, 27 |
| Ni-ZSM-5 | |||
 |
Isolated nickel-hydride [Ni(II)–H]+ centers | Ni-Beta | 15 |
 |
Isolated hydroxylated Ni2+ | Ni-MCM-41 | 51, 52 |
The nature of the nickel species mainly depends on the preparation method of the catalysts. Generally, nickel incorporation by an aqueous ion exchange or grafting method leads to higher contents of Ni2+ cations.13–15,27,46,76 On the other hand, solid-state ion exchange mainly seems to generate intrazeolitic mono(μ-oxo) dinickel ([Ni–O–Ni]2+) species.11 The post-synthesis impregnation leads to high contents of cationic Ni2+ in ion exchange positions, but some NiO clusters are frequently formed.23,27,76 The one-pot synthesis of Ni-aluminosilicates favours the formation of a large amount of NiO particles. Xu et al.66 showed that in the case of Ni-ASA catalyst, the oxidation state of Ni can be regulated by varying the pretreatment atmosphere. Thus, the majority of the Ni species were found to exist in Ni2+, Ni+, and Ni0 states when pretreated in air, N2, and H2 respectively.
As some research groups have shown, the carrier identity had an important influence on the nature of the nickel species. Moussa et al.12 prepared bifunctional 5%Ni catalysts by impregnation of three acidic porous aluminosilicates: nanocrystalline Beta zeolite, mesoporous Al-MCM-41 and silica-doped alumina Siralox-30. According to characterization results, the authors distinguished significant differences in the nickel speciation depending on the support identity. Isolated Ni2+ cations in ion exchange positions were identified on Ni-Beta, while Ni2+ attached to weakly acidic silanol and aluminol functions and undercoordinated Ni2+ on the surface of small NiO nanoparticles prevailed on Ni-Al-MCM-41 and Ni–Siralox-30 catalysts. Comparable results were reported by Agirrezabal-Telleria and Iglesia52 for the Ni-MCM-41catalyst: isolated cations (Ni–OH)+ were identified as active sites, besides inactive NiO clusters.
Unlike homogeneous catalysis, the ethylene oligomerization performed in the presence of Ni-aluminosilicate catalysts does not require the use of activators. It is also important to note that for the oligomerization catalyzed by Ni-based complexes, the activator is involved in the formation of the first Ni–C bond. However, the oligomerization mechanism proposals for the Ni-aluminosilicate catalysts are based on organometallic chemistry.
13C NMR and FTIR spectroscopy undoubtedly proved that the conversion of ethylene over Ni-based sites starts with the formation of a π-complex.2,7 The subsequent modification of the hydrocarbon chain and its interaction with the metallic site can occur via diverse mechanisms, which can be classified according to the nature of the key intermediate species: alkyl-based, vinyl-based, allyl-based (referred to as coordination–insertion or Cossee–Arlman) and metallacycle (Scheme 2). For the coordination–insertion mechanism, largely accepted by the research groups, the main unclear aspect is the formation of the primary nickel–carbon bond. On this point there are controversies and speculation. The metallacycle mechanisms have been suggested by analogy with the homogeneous catalysis. However, some theoretical research carried out on catalysts based on Ni-aluminosilicates has discouraged the realization of this mechanism.
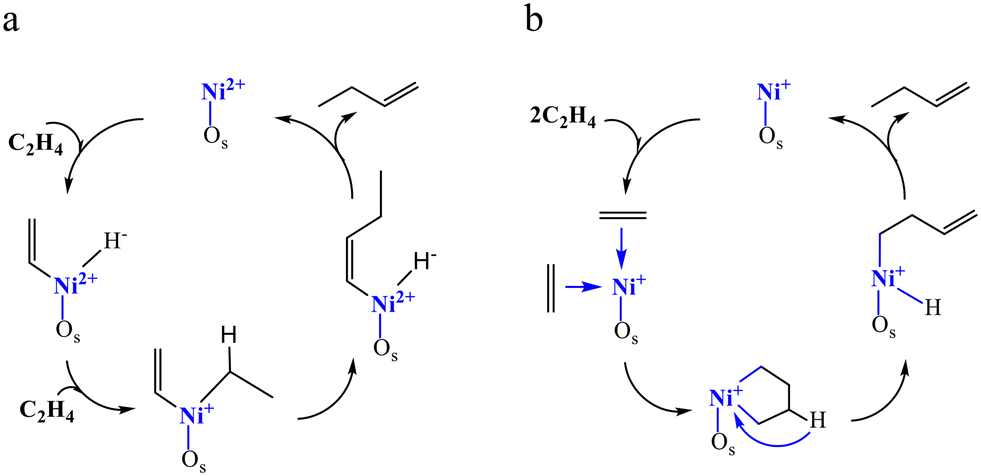 | ||
| Scheme 2 Simplified Cossee–Arlman (a) and metallacycle mechanisms (b) (Os = surface oxide ions). | ||
Cossee–Arlman mechanisms
Using H/D isotope scrambling and H2–D2 isotope exchange experiments, Joshi et al.6 evidenced the H2-assisted formation of [Ni(II)–H]+ species, which were the proposed active sites in the Cossee–Arlman mechanism (Scheme 3).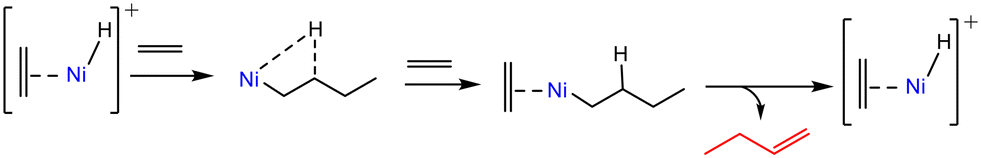 | ||
| Scheme 3 Simplified representation of the coordination–insertion mechanism for ethene dimerization at [Ni(II)–H]+ sites. | ||
Seufitelli et al.15 suggested the [Ni(II)–H]+ active centers involved in the Cossee–Arlman mechanism were generated by ethylene adsorption on a Ni2+ site, assisted by an adjacent proton (Brønsted site) (Scheme 4).
 | ||
| Scheme 4 Proposed mechanism for formation of a nickel-hydride site involving a Brønsted acid: (1) ethylene adsorption over a Ni2+ site and an adjacent Brønsted site, (2) formation of a [Ni(II)–H]+ center followed by coordination of another ethylene molecule, (3) insertion to form adsorbed butyl, (4) recovery of the Brønsted site, and (5) desorption to form a product followed by recovery of the Ni2+ site. This scheme has been adapted from ref. 15. | ||
The involvement of a neighboring Brønsted center in the Cossee–Arlman cycle was also proved by Rabeah et al.85 They used operando electron paramagnetic resonance (EPR) and in situ X-ray absorption spectroscopy (XPS) for finding the active centers in Ni-ASA catalysts during butene oligomerization. Single Ni+/Ni2+redox couples were identified as active sites and the reaction mechanism occurred as shown in Scheme 5.
 | ||
| Scheme 5 Reaction mechanism proposed for olefin oligomerization over Ni-ASA catalysts. This scheme has been adapted from ref. 85 with permission from American Chemical Society, copyright 2025. | ||
Based on the intermediates detected by FTIR-ethylene spectroscopy coupled to online MS analysis, Moussa et al.7 proposed the mechanism shown in Scheme 6. This mechanism starts with the oxidative activation of the C–H bond of ethylene at the Ni2+ site, leading to a nickel-ethenyl-hydride intermediate, which will then be involved in the coordination–insertion Cossee–Arlman cycle. According to the experimental results, the Ni2+ ions grafted on acidic silanols of the Ni-Beta catalyst appeared to be the active species, rather than the more accepted ion-exchanged nickel ions.
 | ||
| Scheme 6 Simplified catalytic cycle proposed for the activation and dimerization of ethylene on the active Ni2+ centers of Ni-Beta catalysts: (1) ethylene adsorption on Ni site; (2) oxidative adition; (3) ethylene insertion; (4) reductive elimination of butene. This scheme has been adapted from ref. 7 with permission from American Chemical Society, copyright 2025. | ||
Henry et al.13 also examined the nature of the Ni species in the Ni-Beta catalyst, using FTIR spectroscopy with CO as a probe molecule. They found that Ni2+ counterions, which were the predominant active sites, interacted with ethylene to form Cossee–Arlman oligomerization sites (Scheme 7). Moreover, the Ni ions grafted on silanol groups and NiO particles were considered as spectators in the oligomerization process.
 | ||
| Scheme 7 Formation of the active center over the Ni-Beta catalyst. This scheme has been adapted from ref. 13 with permission from Elsevier, copyright 2025. | ||
Beucher et al.46 suggested that the coordination of the Ni ions on the KIT-6 surface is similar to that observed in the coordination chemistry. Thus, the dispersed nickel ions can reversibly bind ligands (L) such as C2H4 or surface oxide ions O2−, leading to various Ni(L)n+ species. Based on these results, the following mechanism for the formation of 1-butene is proposed (Scheme 8).
 | ||
| Scheme 8 Proposed mechanism for the dimerization of ethylene by Ni-AlKIT-6. (1) Nickel complexation; (2) ethylene complexation; (3) metal–vinyl intermediate; (4) 1-butene formation. This scheme has been adapted from ref. 46. | ||
By combining advanced characterization techniques (in situ XAS, in situ FTIR, XPS, H2-TPR), DFT calculation and microkinetic simulations, Wang et al.30 demonstrated that the in-situ generated Ni–vinyl motif is the intrinsic active site and ethylene dimerization proceeds via the Cossee–Arlman mechanism (Scheme 9). The dynamic hydrogen transfer between the ethylene/vinyl ligand and zeolite framework participated in the formation of the Ni active sites.
 | ||
| Scheme 9 Proposed Cossee–Arlman pathway for ethylene dimerization to 1-butene on isolated Ni2+; Zeo = Ni-FAU zeolite. This scheme has been adapted from ref. 30 with permission from Wiley-VCH GmbH, copyright 2025. | ||
Brogaard and Olsbye33 used DFT calculation for discriminating between Cossee–Arlman and metallacycle mechanisms involved in the ethene dimerization over Ni sites (Ni2+ and Ni+) of SSZ-24 zeolite. They proved that the metallacycle mechanism was energetically unfavorable and Cossee–Arlman mechanisms prevailed for this catalyst. In order to identify the active site involved in the oligomerization mechanism, the same group34 used both experimental and computational investigations. They suggested that the active species form as shown in Scheme 10.
 | ||
| Scheme 10 Formation of the active sites involved in the Cossee–Arlman mechanism. This scheme has been adapted from ref. 34 with permission from American Chemical Society, copyright 2025. | ||
The initial [NiOH]+ ions served as a precursor for generating the Ni–alkyl species. This was followed by coordination of two ethylene molecules on Ni–alkyl species to form mobile [(ethene)2–Ni–alkyl]+ active sites. The mobilization was reversible, as ethene dynamically exchanged with oxygens of the zeolite support as a ligand on Ni during the reaction.
Metallacycle mechanisms
Andrei et al.40 showed that on the Al-SBA-15 catalyst it is possible to produce Ni+ ions and that the coordination of the Ni+ surface species is similar to that observed in the coordination chemistry.1 Accordingly, the dispersed nickel ions can reversibly bind ligands such as C2H4 or surface oxides. The growth oligomerization mechanism involves a metallacyclopentane intermediate resulting from a concerted coupling of two olefin molecules on each Ni. site (Scheme 11). | ||
| Scheme 11 Proposed catalytic cycles for linear ethylene oligomerization involving Ni+ and H+ as active sites on the Ni-AlSBA-15 catalyst (Os2− = surface oxide ions). This scheme has been adapted from ref. 40. | ||
The cyclic intermediate then liberates 1-butene by β-hydride transfer. Insertion of a third molecule of ethylene leads to a metallacycloheptane species, which can release 1-hexene, regenerating the catalytic site. When the desorbed 1-butene and 1-hexene migrate on an Brønsted acid site, they are easily converted to internal double-bond olefins.
Among the oligomerization mechanisms proposed in recent years, that proposed by Jaegers and Iglesia51 clearly stands out. Their DFT study showed that alkene dimerization cannot occur on active sites consisting of Ni+ (metallacycle mechanism) or Ni2+–H− (Cossee–Arlman cycle), which would bind ethene very strongly, in contradiction with observed kinetic trends. Instead of classical Cossee–Arlman or metallacyle pathways, they proposed a concerted Lewis acid–base pathway on (NiOH)+ sites contained on Ni-Al-MCM-41 (Scheme 12).
 | ||
| Scheme 12 Probable ethene dimerization catalytic cycle on (Ni–OH)+ moieties acting as a Lewis acid–base pair proposed from a DFT analysis of ethene reactions on Ni-Al-MCM-41. This scheme has been adapted from ref. 51 with permission from American Chemical Society, copyright 2025. | ||
Ethylene oligomerization pathways involving both nickel and acid sites
As is known, Ni-aluminosilicates are bifunctional catalysts, containing both Ni and acid sites (see Scheme 1).3 The mechanisms examined in the previous section referred to the dimerization of ethylene to butenes on Ni centers, and show that the Ni sites are indispensable for ethylene oligomerization. Some research groups considered that the Brønsted acid sites also participate in the formation of the active species involved in oligomerization.15,30 Generally, the role of catalyst acidity in the process is more complex. They catalyze a series of reactions that determine the final composition of the reaction mixture, which is usually quite complex. Typically, the products are C4, C6, C8 and C10 olefins, but under severe conditions (i.e. high temperature and high acidity of the catalyst), other hydrocarbons such as alkanes and odd-numbered alkenes are formed. To explain the formation of various molecules, several distinct reactions, involving both Ni and acid sites, have been considered (Scheme 13).3,6,18,40 | ||
| Scheme 13 Schematic representation of the ethylene oligomerization network involving Ni-ion oligomerization and acid-catalyzed alkylation, isomerization and cracking: (A) ethylene physisorption; (B) ethylene coordination on Ni-ethylene species; (C) ethylene insertion between Ni and coordinated ethylene molecule; (D) release of butene and restoration of the active species; (E) butene protonation on an acis site; (F) butene alkylation; (G) carbenium ion isomerization; (H) carbenium ion cracking. This figure has been adapted from ref. 18 with permission from Elsevier, copyright 2025. | ||
The reactions involving ethylene–ethylene and butene–ethylene couples to form linear olefins occur on nickel sites, via the Cossee–Arlman mechanism (called as true oligomerization). Once formed, these olefins are involved in further acid-catalyzed oligomerization, referred as the hetero-oligomerization pathway, including isomerization, alkylation, cracking reactions and H transfer reactions. Following an ionic mechanism, these reactions mainly lead to heavy branched olefins, aromatics and alkanes. The acid site involvement increases at high conversion, temperature, acid concentration and strenght.3,12,18
5. Kinetics on Ni-aluminosilicates
Despite the fact that a large number of studies have been devoted during the last decade to the oligomerization of ethylene over Ni-aluminosilicate catalysts, only a few of them have focused on determining the kinetic models. Among the extensive kinetic studies there is that performed by Seufitelli et al.22 with the Ni-Beta catalyst. Their micro-kinetic model obeying an Eley–Rideal-type mechanism allowed the rate expressions for the consumption of ethylene and production of butene, hexene and octene to be obtained accurately. Working at various partial pressures of ethylene, the authors found that the reaction order for butene and hexene formation was consistent with a coordination–insertion Cossee–Arlman mechanism, while the reaction order for octene formation points to a cascade co-oligomerization reaction of butene and hexene. The reaction constants for the formation of butene, hexene and octene were 1.8, 0.2 and 2.4 g gcat−1 h−1, respectively. In this model, the Ni2+ ions were the precursors for the formation of the [Ni–H]+ active sites, while the involvement of the acid sites in the process was not considered. The experimental activation energies for the formation of butene, hexene and octene were 45, 79, and 60 kJ mol−1, respectively.Combining experimental observation and single-event microkinetic modeling, Toch et al.18,58 have evaluated the kinetic parameters for ethylene dimerization on Ni-ASA and Ni-Beta catalysts. The reaction rate increased linearly with ethylene pressure (0.15 to 0.35 MPa) and the reaction was found to be first order on ethylene converted. The activation energy for the insertion and termination step was 76 and 74 kJ mol−1, respectively. The kinetic model obtained at low ethylene conversion (i.e. high selectivity to butenes) has been extrapolated to different parameters for predicting the effect of conversion, temperature, acid concentration and strength.
Agirrezabal-Telleria and Iglesia52 examined the kinetics of ethylene dimerization over the Ni-MCM-41 catalyst, at sub-ambient temperatures (−30–−15 °C) and 1.5 MPa. Under these conditions, ethylene condensed within ordered mesopores of the catalyst. A second-order dimerization rate with respect to ethylene pressure was reported. The same reaction order was observed for ethylene dimerization on Ni-SSZ-24 zeolite.34
More recently, Beucher et al.46 performed a kinetic study of ethylene oligomerization on Ni-AlKIT-6. As shown in Fig. 3, and as reported in many studies,6,10,22,25,40 1-butene is the primary ethylene dimer formed in the processes catalyzed by the Ni-aluminosilicates. The selective formation of butenes (90%) and hexenes (9%) as primary products has been explained considering a Cossee–Arlman mechanism.
 | ||
| Fig. 3 Butene selectivity (left) and 1-C4 isomer ratio (right) vs. ethylene conversion. Catalyst: 0.1 g of Ni-AlKIT-6, T = 40–80 °C, Pethylene = 0.4–2 MPa, Ptotal = 3.0 MPa, contact time = 15–98 s. This scheme has been reproduced from ref. 46. | ||
The experimental data obtained in ethylene oligomerization fitted with a first order kinetics, while the activation energy was of 15.2 kJ mol−1. The authors suggested that the kinetic-limiting step is the insertion of ethylene into the Ni–alkyl bond.
6. Effect of the reaction parameters
Effect of the temperature
Oligomerization studies of ethylene in the presence of Ni-aluminosilicates were performed in various temperature ranges, placed between −30 °C and 350 °C. Each working temperature has advantages and disadvantages, depending on the purpose of the study. Fig. 4 shows the representative behavior of a Ni-based catalyst like Ni-SBA-15, obtained at various temperatures.40 The ethylene conversion strongly increased, from 14 to 90% in the temperature range of 50 to 150 °C. After that, the ethylene conversion increases slowly up to 300 °C. | ||
| Fig. 4 Ethylene conversion and oligomer distribution at various temperatures; Ni-SBA-15 catalyst; (◊) % ethylene conversion, (□) % C4, (Δ) % C6, (x) C8; conditions: 3.0 MPa, WHSV = 10 h−1. This scheme has been reproduced from ref. 40. | ||
As shown in Fig. 4, there is a change in product distribution as a function of ethylene conversion.
Typically, at low conversion, C4 is the major oligomerization product, while at higher conversion, the oligomerization was directed toward the formation of C6 and C8 olefins.19,41,51 Among the butenes, with increasing temperature and conversion, the proportion between 1-C4 and 2-C4 decreases.40,73
Even if most recent studies have been carried out at moderate temperatures, i.e. 50–120 °C, some of them were conducted at high or very low temperatures.19,20,40,43 Jan and Resende19 worked at high temperatures, with the aim of converting ethylene into jet fuel range hydrocarbons. They found that the maximum liquid yield over Ni-Beta can be obtained at 190 °C, 52 bar, and WHSV of 2.0 h−1. Attanatho et al.41 showed that the optimum temperature for the formation of C8+ hydrocarbons was in the range of 275–300 °C.
Agirrezabal-Telleria and Iglesia51,52 studied the dimerization of ethylene over Ni-Al-MCM-41 at an ethene pressure of 1.5 MPa and sub-ambient temperatures (between −30 °C and −15 °C). The unique performances in terms of reactivity, selectivity and stability obtained under these conditions were attributed to the ethene liquid phase formed in the pores of the catalyst. The liquid phase solvates the reaction transition states, promoting the desorption of butenes and thus preventing their isomerization and their growth towards bulky oligomers which can block the Ni sites and the pores of the catalyst. A similar behavior was described by Jan et al.26 for the ethylene oligomerization carried out over Ni-Beta and Ni-SBA-15 under supercritical conditions. The increase in ethylene conversion was attributed to the high solubility of C4+ products in supercritical ethylene and the easy desorption of large molecules from the active catalytic centers. Keeping the supercritical state, the catalytic activity increased when the temperature increased from 30 to 120 °C.
Effect of the pressure
As expected, for a reaction involving a gaseous reactant, i.e. ethylene, the pressure played a major role in the process. Because the experiments carried out at low pressure led to low ethylene conversions, in most studies the reaction pressure was set between 1 and 4 MPa. The experimental data showed that the effect of the ethylene pressure in the reactor is analogous to that observed for the temperature: the ethylene conversion and the amount of higher olefins (C6+) increased with increasing ethylene pressure from 1 to 4 MPa.20,40,41,46 For example, over the Ni-SBA-15 catalyst, the specific activity increased, from 214 to 336 mmol gcat−1 h−1 in this range of pressure.40 Over Ni-ASA, the ethylene conversion was 10% at 1.0 MPa, but it increased up to 99% when the pressure increased at 3.5 MPa.73 In the presence of Ni-Beta, the steady-state ethylene conversion increased from 38 to 57% as the pressure increased from 0.85 to 2.6 MPa.20 Seufitelli and Gustafson showed that over Ni-ASA the production of liquid products reached a maximum under supercritical conditions, i.e. 6.5 MPa of ethylene.72In addition to these expected results, the recent studies revealed new aspects linked to the effect of pressure in the oligomerization reaction catalyzed by the Ni-aluminosilicates. First, it was shown that the deactivation of the catalytic centers of Ni-based mesoporous aluminosilicates during the oligomerization process can be suppressed by operating at high ethylene pressure.17,52,53 The high pressure led to capillary condensation of liquid-like ethylene within mesoporous voids, which solvates and facilitates the transport to external fluid phases of the bulky hydrocarbons that cause catalyst deactivation.
On the other hand, Rabeah et al.85 showed that the nature and the stability of the Ni active centers in the dimerization of the lower olefins depended on the ethylene pressure. Using operando EPR and in situ XAS technics, they identified the NiI/NiII couples as active sites, which were stable only at pressure higher than 1.2 MPa. In contrast, at low pressure (<0.2 MPa), NiI sites form inactive Ni0 aggregates.
Effect of the space velocity (WHSV)
Typically, over both microporous and mesoporous Ni-aluminosilicates, the ethylene conversion and the amount of the higher hydrocarbons (C6+) decreased, while the C4 fraction significantly increased with increasing WHSV (i.e. with decreasing contact time).12,20,21,40,41 Fig. 5 shows such a dependence. These results suggest that C4-hydrocarbons are the primary molecules formed in the ethylene process, while the higher olefins, i.e. C6+, are produced in a second step, by oligomerization and co-oligomerization, involving the ethylene and butenes.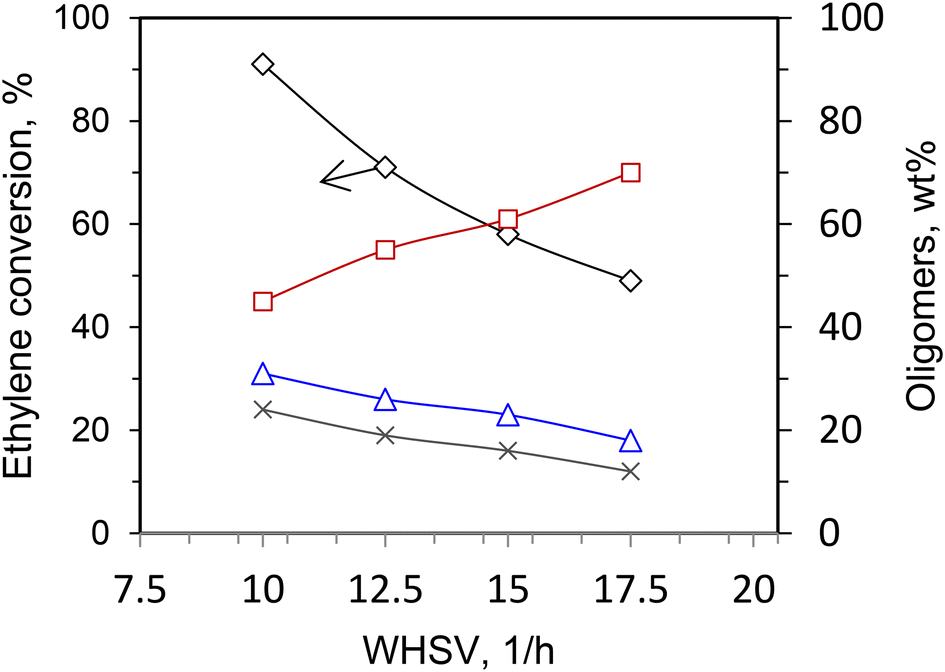 | ||
| Fig. 5 Ethylene conversion and oligomer distribution at various WHSV; Ni-SBA-15 catalyst; (◊) % ethylene conversion, (□) % C4, (Δ) % C6, (x) C8+; conditions: 3.0 MPa of ethylene, T = 150 °C, TOS = 1 h. This scheme has been reproduced from ref. 40. | ||
Jan and Resende19 evaluated the effect of WHSV on the formation of liquid fraction and coke in the presence of the HNi-Beta catalyst. At WHSV of 0.5 h−1, the yield of coke was nearly 52 wt%, along with a 3.7 wt% yield of liquid hydrocarbons. At 2.0 h−1, there was a noticeable shift in the product distribution, with the yield of the liquid product increasing three times to 10.6 wt% and a minimum coke yield of 6.2 wt%.
7. Effect of the contaminants in ethylene oligomerization
As shown above, ethylene oligomerization over Ni-containing heterogeneous catalysts follows either a Cossee–Arlman or metallacyclopentane mechanism. Whatever the mechanism, ethylene first coordinates via π-bonding to the nickel ion site. But, the nickel ions can also form complexes with other unsaturated organic compounds or with O- and S-containing compounds. During the oligomerization process, these molecules become contaminants for the catalyst, as ligands competing with ethylene.It is known that the Ni2+ cations can be easily hydrated by strong electrostatic interactions with n H2O molecules as ligands (n ≤ 6).86 Some studies showed that the Ni-aluminosilicate catalysts are extremely air- and moisture-sensitive.39,85 For example, when the Ni-ASA catalyst was prepared under aerobic conditions, no catalytic activity in the oligomerization reaction was observed, because of the blockage of active Ni sites by H2O and/or the formation of stable NiII oxide species, in which the NiII coordination sphere is saturated by O ligands and, therefore, is not prone to interact with olefin molecules.85
On the other hand, with CO, nickel cations easily form mono- and polycarbonyl complexes. For this reason, CO has been usually used in FTIR spectroscopic studies in order to characterize the Ni species.87 CO interacts with Ni+ and Ni2+ sites of low coordination.12 Exposing Ni-MCM-41 samples to 1 kPa CO at 263 K led to an infrared band at 2200 cm−1, corresponding to Ni2+–(CO).88 Fig. 6 shows dimerization rates (1.5 MPa, −30 °C) as a ratio to that before contact with CO pulses as a function of the cumulative CO uptakes.
 | ||
| Fig. 6 Ratio of rates after each CO pulse introduction (per Ni) at −30 °C, 1.5 MPa ethene. This figure has been reproduced from ref. 52 with permission from Elsevier, copyright 2025. | ||
The rate decrease as a function of cumulative CO per total Ni is similar for Ni-MCM-41 samples, leading to CO uptakes (per Ni atom) that are similar at these two Ni2+/H+ ratios.
To remove possible contaminants coordinated on Ni sites, thermal activation of the Ni-aluminosilicate catalysts at high temperature (>300 °C) before the reaction is a crucial step.84
Another way to keep the catalytic activity, even in the presence of contaminants, is to work at high temperatures. In one of the earlier studies, Kimura et al.89 reported that the dimerization of the ethylene was completely prevented when CO or H2O was pre-adsorbed over the NiO–SiO2 catalyst. The catalyst was activated by evacuation at temperatures higher than 200 °C.
In another study, Zhang et al.68 showed that at 280 °C and 4.0 MPa the oligomerization behavior of the Ni-ASA catalyst was enough high, despite the presence of other molecules, including C3H6, C4H8, CH4, CO, CO2, H2 and N2 together with the ethylene in the feed.
More recently, Andrei et al.39 evaluated the effect/role on the Ni-SBA-15 and Ni-ASA of some potential contaminants, such as H 2O, CO, and H 2, during ethylene oligomerization. The catalytic tests were carried out at 250 °C and 15 bar, while the concentrations of CO, H2O and H2 in the reactor feed were 90, 300 and 1000 ppmv, respectively. Fig. 7 compares the ethylene conversions obtained in the absence and in the presence of contaminants. While carbon monoxide has not affected the catalyst activity, water has only a minor negative effect. In contrast, in the presence of hydrogen, the ethylene conversion remained at higher values all throughout the catalytic test. Most likely, the presence of hydrogen limits the formation of large unsaturated species, which are responsible for catalyst deactivation.
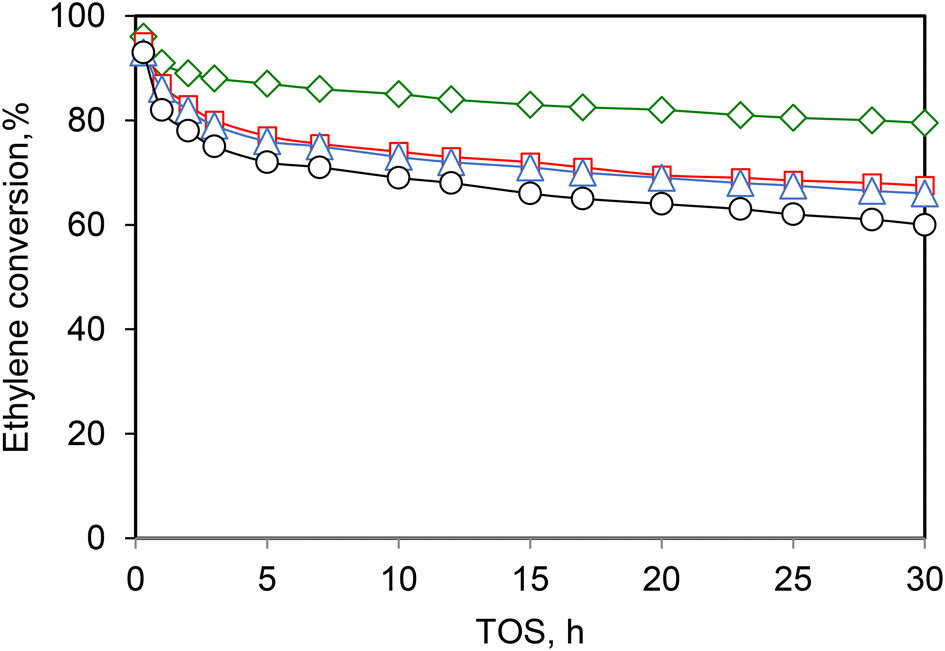 | ||
| Fig. 7 Ethylene conversion vs. time on stream for various feed compositions (□) C2H4 + N2, (◊) C2H4 + N2 + H2 and (Δ) C2H4 + N2 + CO and (○) C2H4 + N2 + H2O, over Ni-AlSBA-15 catalyst. Reaction conditions: T = 250 °C, P = 15 bar, WHSV = 1.1 h−1. This scheme has been reproduced from ref. 39. | ||
The oligomer distribution also depended on contaminants, in particular CO and H2O. In both cases, the amount of C4 was higher compared to that produced in other tests (Fig. 8).
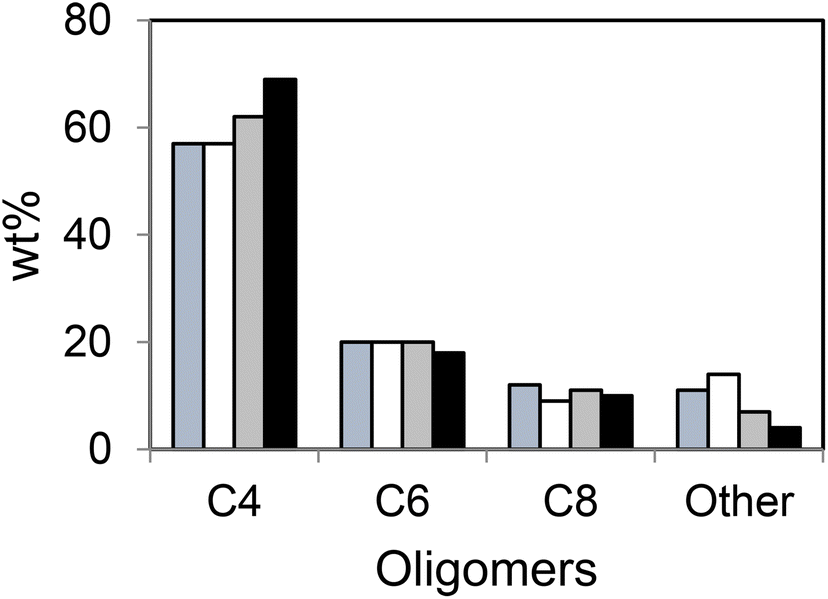 | ||
| Fig. 8 Effect of the contaminants on the product distribution; blue = C2H4 + N2, white = C2H4 + N2 + H2, grey = C2H2 + N2 + CO, black = C2H2 + N2 + H2O; TOS = 30 h, T = 250 °C, catalyst: Ni-AlSBA-15. This scheme has been reproduced from ref. 39. | ||
8. Catalyst deactivation in ethylene oligomerization
Deactivation is one of the main drawbacks of porous heterogeneous catalysts. In the case of the Ni-aluminosilicate catalysts used in the oligomerization reaction, the deactivation is mainly due to heavy hydrocarbons that can bind strongly to Ni sites and/or obstruct the porous voids. The experimental data showed that the amount of hydrocarbons trapped in the catalyst pores and the deactivation rate of the Ni centers mainly depended on the catalyst porosity, but also on its surface acidity and the reaction conditions.Generally, the Ni-based microporous zeolites deactivate more rapidly than mesoporous materials.12,13,23,29 Moussa et al.12 examined the influence of the support identity in the oligomerization of ethylene on three acidic Ni-aluminosilicates: Ni-Beta zeolite, mesostructured Ni-AlMCM-4 and Ni–Siralox-30. As shown in Fig. 9, both mesoporous catalysts were significantly more active than zeolite Ni-Beta.
 | ||
| Fig. 9 Ethylene conversion over Ni-Beta (5Ni/NB), Ni-MCM-41 (5Ni/AlM41) and Ni-ASA (5Ni/S30) catalysts. Reaction conditions: T = 120 °C, Ptot = 3.5 MPa, PC2 = 2.6 MPa. This figure has been reproduced from ref. 12 with permission from Elsevier, copyright 2025. | ||
The authors considered that the heavy oligomers formed during the reaction decreased the catalytic activity by hindering the access of the reactant to the internal Ni sites in Ni-Beta.
Similar differences in terms of activity and deactivation rate between the mesoporous Ni-ASA and microporous Ni-Beta catalysts were identified by Henry et al.13 Additionally, the authors showed that the crystal size of the Ni-Beta catalysts also had an impact on deactivation. The larger micro-crystallites (which present a longer diffusion path) exhibited a lower activity and higher deactivation rate than the nano-crystallites because of diffusion limitations inside the pores of the long chain products.
Martínez Gómez-Aldaraví et al.23 compared the behavior of Ni-ZSM-5 and Ni-Beta, two zeolite type catalysts with medium and large pores, respectively. The higher deactivation rate of Ni-ZSM-5 has been attributed to rapid blocking by the heavy oligomers of the Ni active sites placed in the small pores.
In contrast to Ni-zeolites, Ni-mesoporous catalysts such as Ni-ASBA-15,40,41 Ni-AlKIT-6 (ref. 42) and Ni-AlMCM-41,17,52 are very stable catalysts against deactivation in oligomerization processes. For example, in an experiment performed during 80 h on stream, Andrei et al.40 showed that the catalytic activity of Ni-AlSBA-15 (pore size of 7.9 nm) declined only smoothly (deactivation rate of 1.6 × 10−3 h−1). This behavior has been related to the SBA-15 topology, with large interconnected mesopores, facilitating the diffusion of the large molecules. For a Ni-KIT-6 catalyst (pore size of 5.4 nm), Hwang et al.47 found an apparent 2nd order deactivation rate constant of 2 h−1.
On the other hand, the mesoporous supports have another advantage over the microporous ones. At high pressure and low temperature, they allow the formation of a liquid phase (consisting in ethylene and oligomers) within mesoporous voids, which solvates, desorbs and facilitates the transport to external fluid phases of the bulky intermediates that are precursors to deactivation.51–53 By contrast, the capillary condensation of ethylene within the spatial constraints of microporous voids of Ni-zeolites is not possible.17
Andrei et al.39 proved that the initial deactivation rate and the lifetime of the Ni-AlSBA-15 and Ni-ASA catalysts can be improved by adding hydrogen in the reaction feed (Fig. 7 and 10). The presence of hydrogen limits the formation of large unsaturated species, which are responsible for catalyst deactivation.
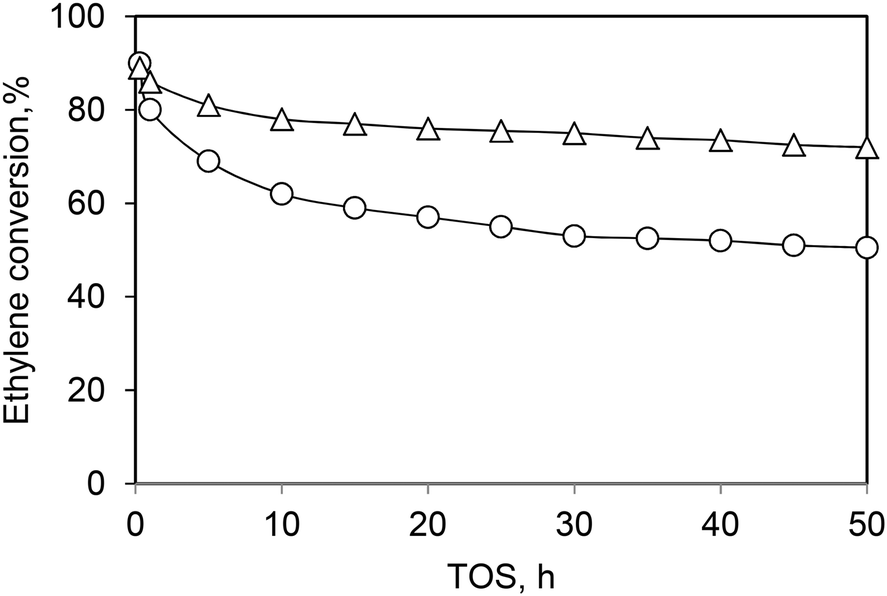 | ||
| Fig. 10 Ethylene conversion vs. time on stream in the presence of (○) C2H4 + N2 and (Δ) C2H4 + N2 + H2 over the Ni-ASA catalyst. Reaction conditions: T = 250 °C, P = 1.5 MPa, WHSV = 1.1 h−1. This scheme has been reproduced from ref. 39. | ||
Some recent studies have focused on the kinetics of catalyst deactivation. They showed that the kinetic parameters depend on the reaction parameters or the density of the Ni sites in the catalyst. Agirrezabal-Telleria and Iglesia52 modeled the deactivation of the Ni-AlMCM-41 catalyst using a first-order deactivation constants (kd), defined as follows:
| rt/r0 = exp[−kd(t − t0)] |
Fig. 11 shows the dependence between this constant and the ethene pressure, at −30 °C and −20 °C. An abrupt increase of kd as the ethene pressure decreases below 1.2 MPa at −30 °C and below 1.8 MPa at 253 K was observed.
 | ||
| Fig. 11 First-order ethylene dimerization deactivation constant (kd) as a function of ethylene pressure. This figure has been reproduced from ref. 52 with permission from Elsevier, copyright 2025. | ||
Caulkins et al.17 studied the effect of ethylene pressure on the deactivation of Ni-FAU, Ni-Beta and Ni-AlMCM-41 catalysts at sub-ambient temperature (−15 °C). The apparent deactivation constants have been calculated using the following model:

Based on the experimental results and DFR calculations, Saxena et al.8 showed that the deactivation mechanisms and kinetics depended on the Ni site density in the Ni-Beta catalyst. An exponential deactivation (i.e. single-site and deactivation rates of first-order in Ni) was observed for a low-site density sample (Ni/Al = 0.06), while a hyperbolic deactivation (i.e. dual-site and deactivation rates of second-order in Ni) was found for the high-site density catalyst (Ni/Al = 0.25).
In the first case, the deactivation was due to inhibition and poisoning by strongly bound alkyl groups formed from heavy oligomers, while in the second case the deactivation is due to the formation of bridging alkyl groups between two Ni species to form unreactive centers.
Catalyst regeneration
It is well known that the regeneration ability of solid catalysts is of vital importance for commercialization. Typically, regeneration involves thermal treatment to remove surface coatings and/or absorbed species. In most cases, the spent catalyst is treated at 500–550 °C under air, then treated under an inert gas. Using this regeneration way, Andrei et al.39 examined the behavior of the Ni-AlSiO2 catalyst in two reaction cycles, at 250 °C. The ethylene conversion versus time on stream of 75 h is presented in Fig. 12. The conversion profiles of the fresh and regenerated samples were almost the same. | ||
| Fig. 12 Ethylene conversion vs. time on stream, over (○) fresh and (●) regenerated Ni-AlSiO2 catalyst; reaction conditions: C2H4/N2 = 1/5 (vol), T = 250 °C, P = 15 bar, WHSV = 1.1 h−1. This scheme has been reproduced from ref. 39. | ||
The efficiency of the regeneration process was also evaluated for the used Ni-AlSBA-15 (ref. 40) and Ni–SIRAL-30.71 The regenerated catalysts exhibited catalytic properties similar to those of the original catalysts. Another regeneration method is to treat the used catalyst in an inert gas. Thus, Agirrezabal-Telleria and Iglesia52 demonstrated that the activity of the Ni-Al-MCM-41 catalyst can be fully restored by treating sample in He at 550 °C.
These experimental results show that nickel aluminosilicates used in ethylene oligomerization can be efficiently regenerated using recognized methods.
9. Potential applications based on Ni-aluminosilicate catalysed oligomerization
The previous sections have dealt with aspects closely related to the oligomerization reaction catalyzed by nickel aluminosilicates. However, in order to obtain products of high commercial interest, some research groups have developed in recent years' applications that involve ethylene oligomerization. Two examples, concerning the production of propylene and fuels, will be analyzed below. In both cases, multi-reaction catalytic processes, in which oligomerization is the key step, have been used.Ethylene to propylene process
Starting from the selective dimerization of ethylene over Ni-aluminosilicates, a number of recent studies have focused on the direct conversion of ethylene to propylene (ETP). This process has been carried out using only ethylene as the reagent, over heterogeneous catalysts which were able to work without activators or co-catalysts. The ETP process, carried out in flow mode, consisted in 3 cascade reactions, i.e. dimerization–isomerization–metathesis (Scheme 14). | ||
| Scheme 14 Ethylene to propylene in a catalytic cascade process (2-butenes mean cis- and trans-2-butenes). | ||
Each step requires a specific catalytic site. Dimerization and isomerization are catalysed by nickel and acid (H+) sites, respectively. Both types of catalytic sites are provided by the Ni-aluminosilicates. The metathesis reaction is catalysed by Mo, W or Re oxides. Experimentally, to produce propylene from ethylene according to Scheme 13, two catalyst beds consisting of Ni- and W/Re-based materials were placed in one36,45,57,80 or two consecutive reactors43,44 (Fig. 13). Table 5 summarizes data from relevant recent studies.
 | ||
| Fig. 13 ETP processes with one or two reactors. | ||
| Dimer–izomer catalysta | Metathesis catalyst | Reactor (s) | Conditions | C3b | C2 conv.c | C3 in prod.d | Ref |
|---|---|---|---|---|---|---|---|
| a Dimerization–isomerization.b C3 productivity (mmol g catal−1 h−1).c Ethylene conversion (%).d Propylene concentration in products. | |||||||
| Ni-AlSBA-15 | MoO3–SiO2–Al2O3 | One | 80 °C, 3 MPa | 48 | 40 | 73 | 36 |
| Ni-AlSBA-15 | MoOx/SBA-15 | One | 80 °C, 3 MPa | 54 | 43 | 71 | 57 |
| Ni-ASA | MoOx/ASA | One | 80 °C, 3 MPa | 28 | 41 | 38 | 57 |
| Ni-AlKIT-6 | ReOx/Al2O3 | One | 60–120 °C | 29 | 52 | 50 | 45 |
| 0.1–3 MPa | |||||||
| Ni-AlKIT-6 | ReOx/Al2O3 | One | 60 °C, 3 MPa | 26 | 73.5 | 61 | 43 |
| Ni-AlKIT-6 | WOx/KIT-6 | Two | 1. 60–120 °C | 39 | 48.8 | 59.4 | 44 |
| 2. 450 °C | |||||||
| NiSO4–ReOx/γ-Al2O3 | One | 50 °C, 0.1 MPa | 13 | 60 | 80 | ||
Andrei et al.36 explored for the first time the direct conversion of ethylene into propylene using two catalysts, in a single flow reactor. Under identical conditions, ethylene was first selectively dimerized/isomerized over the Ni-AlSBA-15 catalyst to form 2-butene, which reacted then with the excess of ethylene over MoO3–SiO2–Al2O3 to produce propylene. At 80 °C and 3 MPa, specific activities up to 48 mmol of propylene per gram of catalyst per hour were obtained. The authors extended the research on other catalyst couples: Ni-AlSBA-15/MoOx–SBA-15 and Ni-ASA/MoOx-ASA.57
In a similar experimental mode, but working at a lower temperature (60 °C) and using Ni-AlKIT-6 and ReOx/Al2O3 as catalysts, Beucher et al.43 showed that ethylene can be simultaneously and selectively converted into propylene and 1-butene (Fig. 14).
 | ||
| Fig. 14 Selective production of propylene and 1-butene from ethylene by catalytic cascade reactions. This scheme has been reproduced from ref. 43. | ||
The global selectivity to C3 and 1-C4 olefins was about 86%, while the yield of C3 and 1-C4 was 26 and 13 mmol gcatal−1 h−1, respectively. Despite their notable initial activity, the supported MoOx and ReOx metathesis catalysts used in these studies suffered significant deactivation in the process. In order to circumvent this inconvenience, Beucher et al.44 combined the dimerization/isomerization catalyst (Ni-AlKIT-6) with a highly efficient metathesis catalyst, i.e., WOx, placed in two reactors (Fig. 13). In the first one, at 60 °C and 3 MPa, ethylene has been partially converted into 2-butene, which reacted with the unconverted ethylene in the second reactor, operated at 450 °C and 0.1 MPa. Under these conditions, an ethylene conversion of 85% and a selectivity to propylene of 55% were maintained during 24 on reaction stream.
In a recent study, Li et al.80 performed the ETP process at 50 °C and 0.1 MPa, over a single multi-site catalyst, i.e. NiSO4/ReOx/γ-Al2O3. The two catalyst functions worked independently for dimerization/isomerization (NiSO4) and metathesis (ReOx).
Ethylene to fuels
Most experimental studies showed that, under optimized reaction conditions, ethylene oligomerization is very selective to linear C4 and C6 olefins. To produce hydrocarbons in the distillate range (>C10 carbon atoms), the Ni-catalysed ethylene oligomerization must be assisted by an acid catalysis.90 In other words, to produce jet fuels from ethylene, a multi-stage reaction is required. Thus, the dimerization/trimerization step is followed by the co-oligomerization of the C4–C6 primary molecules (Scheme 15).
 | ||
| Scheme 15 Main reaction pathways of ethylene oligomerization into C10+ olefins by catalytic cascade reactions. | ||
Practically, the process can be performed using either a dual-bed catalyst (in a continuous fixed-bed reactor),31,75 two catalysts in batch mode (autoclave),31 or by combining a fixed bed reaction system with an autoclave reactor.37 Babu et al.37 proposed an integrated process for the production of jet-fuel range olefins using Ni-AlSBA-15 and Amberlyst-35 catalysts. In a fixed bed reaction system, the Ni-based catalyst led to C4–C8 olefins, at 99% ethylene conversion, during 60 h on stream, at 200 °C and 1 MPa. Co-oligomerization reactions of the as-synthesized oligomers over the Amberlyst-35 catalyst (at 100 °C, 3 MPa N2, 24 h reaction time, batch mode) produced 98% liquid hydrocarbons, with about 42% C10+.
Kwon et al.75 demonstrated that the Ni–Siral-30 + H-ZSM-5 one-pot cascade catalysis in a continuous flow fixed-bed reactor can be an efficient method for producing jet-fuel range hydrocarbons from ethylene. Compared to the results obtained over a single catalyst (Ni–Siral-30), the cascade process with two catalysts exhibited higher ethylene conversion (close to 100%) and a completely reversed Schulz–Flory type product distribution C10 > C8 > C6 > C4, Fig. 15).
 | ||
| Fig. 15 Carbon distribution of products obtained after ethylene reaction for 16 h on stream over Ni/Siral-30 (black) and Ni/Siral-30 + H-ZSM-5 (green) in the continuous fixed-bed reactor. This figure has been reproduced from ref. 75 with permission from Elsevier, copyright 2025. | ||
In a more recent study, Mohamed et al.31 used Ni-Y and H-ZSM-5 as catalysts for oligomerization and co-oligomerization, respectively. Under optimized dual-bed conditions (300 °C, 3.5 MPa, 0.68 gcat h gC2−1), the process produced 64 wt% of jet-fuel at the beginning of the reaction and maintained a 50 wt% selectivity to this fraction for 20 h on stream.
Conclusion and future outlook
On the basis of an important quantity of research results, this contribution clearly shows that ethylene dimerization/oligomerization catalyzed by Ni-aluminosilicates is a dynamic topic, of great fundamental and practical interest. The scientific progress achieved in the last decade can be summarized as follows:1. Ni-containing zeolites, due to their highly ordered crystalline structure, have served as excellent model supports for fundamental studies in terms of mechanism, active sites and DFT simulation. On the other hand, the well-tailored Ni-mesoporous aluminosilicates, due to their high catalytic stability, were widely used for developing oligomerization processes, under optimized reaction conditions.
2. Concerning the dimerization/oligomerization mechanism, the experimental and theoretical arguments clearly favor the Cossee–Arlman mechanism over the metallacyclic pathway.
3. Unfortunately, there is still controversy surrounding the nature of the nickel active site, how it forms, its location on the aluminosilicate matrix and its contribution to the initiation of the Cossee–Arlman mechanism.
4. Studies specifically dedicated to the effects of the contaminants in ethylene oligomerization have been conducted for the first time during the last decade. It has been shown that the adsorption competition on the Ni centers between ethylene and contaminants, i.e. H2O, CO, olefins, can be circumvented by working at temperatures higher than 200 °C.
5. The conversion of ethylene in the liquid phase, i.e. at sub-ambient temperature or under supercritical conditions, highlighted the favorable effect of intrapore solvation on the catalyst activity and stability, as well as the selectivity to 1-butene.
6. Based on the remarkable results obtained in the oligomerization of ethylene, some research groups enriched the potential of the Ni-mesoporous aluminosilicates by developing multi-reaction/catalyst processes in which oligomerization is the key step. The direct ethylene-to-propene conversion and the ethylene conversion in jet fuels are the most representative applications investigated.
7. Thanks to their robust structures, Ni-aluminosilicates allow their complete regeneration by thermal treatments in an inert or oxidizing environment when deactivation occurs.
In my opinion, the main challenges regarding this topic are the following:
1. Unlike the nickel-based organometallic catalysts, Ni-aluminosilicates do not require activators or cocatalysts. Further research is needed to understand this behavior.
2. The interaction between Ni and matrix atoms and the role of the support in the formation of active species require more investigation.
3. The selective catalytic conversion of ethylene into higher hydrocarbons could be incorporated as a stage into an integrated system that includes bioethanol production from biomass and bioethanol dehydration to form ethylene (biomass → bioethanol → ethylene → higher hydrocarbons). Although promising progress has been obtained for each way, further research is still needed in order to produce the knowledge necessary to design the “ideal” catalysts and large-scale processes.
4. Developing large-scale processes capable of producing propylene and fuels from ethylene remains a challenge. Research is also needed to increase the selectivity to 1-C4 or long-chain linear hydrocarbons.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.References
- H. Olivier-Bourbigou, P. A. R. Breuil, L. Magna, T. Michel, M. F. Espada Pastor and D. Delcroix, Chem. Rev., 2020, 120, 7919–7983 CrossRef CAS PubMed.
- Z. N. Lashchinskaya, A. A. Gabrienko and A. G. Stepanov, ACS Catal., 2024, 14, 4984–4998 CrossRef CAS.
- A. Finiels, F. Fajula and V. Hulea, Catal. Sci. Technol., 2014, 4, 2412–2426 RSC.
- S. Nishimura, Handbook of heterogeneous catalytic hydrogenation for organic synthesis, John Wiley & Sons, Inc., New York, 2001 Search PubMed.
- Hydrogen production by steam reforming of natural gas and other nonrenewable feedstocks, in Compendium of hydrogen energy, Hydrogen production and purification, ed. L. Garcia, Woodhead Publishing, Kidlington, 2015, vol. 1, pp. 83–107 Search PubMed.
- R. Joshi, G. Zhang, J. T. Miller and R. Gounder, ACS Catal., 2018, 8, 11407–11422 CrossRef CAS.
- S. Moussa, P. Concepción, M. A. Arribas and A. Martínez, ACS Catal., 2018, 8, 3903–3912 CrossRef CAS.
- A. Saxena, R. Joshi, R. R. Seemakurthi, E. Koninckx, L. J. Broadbelt, J. Greeley and R. Gounder, ACS Eng. Au, 2022, 2, 12–16 CrossRef CAS.
- S. Moon, H. J. Chae and M. B. Park, Appl. Catal., A, 2018, 553, 15–23 CrossRef CAS.
- G. V. S. Seufitelli and F. L. P. Resende, Appl. Catal., A, 2019, 576, 96–107 CrossRef CAS.
- K. Lee and S. B. Hong, Appl. Catal., A, 2021, 615, 118059 CrossRef CAS.
- S. Moussa, M. A. Arribas, P. Concepción and A. Martínez, Catal. Today, 2016, 277, 78–88 CrossRef CAS.
- R. Henry, M. Komurcu, Y. Ganjkhanlou, R. Y. Brogaard, L. Lu, K.-J. Jens, G. Berlier and U. Olsbye, Catal. Today, 2018, 299, 154–163 CrossRef CAS.
- J. McCaig and H. H. Lamb, Catalysts, 2022, 12, 824 CrossRef CAS.
- G. V. S. Seufitelli, J. J. W. Park, P. N. Tran, A. Dichiara, F. L. P. Resende and R. Gustafson, Catalysts, 2022, 12, 565 CrossRef CAS.
- O. Abed, H. O. Mohamed, I. Hita, V. Velisoju, N. Morlanés, O. El Tall and P. Castaño, ChemCatChem, 2024, 16, e202301220 CrossRef CAS.
- R. Caulkins, R. Joshi, R. Gounder and F. H. Ribeiro, ChemCatChem, 2022, 14, e202101478 CrossRef CAS.
- K. Toch, J. W. Thybaut, M. A. Arribas, A. Martínez and G. B. Marin, Chem. Eng. Sci., 2017, 173, 49–59 CrossRef CAS.
- O. Jan and F. L. P. Resende, Fuel Process. Technol., 2018, 179, 269–276 CrossRef CAS.
- O. Jan, K. Song, A. Dichiara and F. Resende, Ind. Eng. Chem. Res., 2018, 57, 10241–1025021 CrossRef CAS.
- E. Koninckx, R. Gounder, J. W. Thybaut and L. J. Broadbelt, Ind. Eng. Chem. Res., 2022, 61, 3860–3876 CrossRef CAS.
- G. V. S. Seufitelli, J. J. W. Park, P. N. Tran, A. Dichiara, F. L. P. Resende and R. Gustafson, J. Catal., 2021, 401, 40–53 CrossRef CAS.
- A. Martínez Gómez-Aldaraví, C. Paris, M. Moliner and C. Martínez, J. Catal., 2023, 426, 140–152 CrossRef.
- M. Meloni and R. C. Runnebaum, Catal. Sci. Technol., 2021, 11, 3393–3401 RSC.
- Y. Bai, T. Cordero-Lanzac, A. Nova, U. Olsbye, E. Taarning and J. S. Martinez-Espin, Catal. Sci. Technol., 2024, 14, 1991–2002 RSC.
- O. Jan, K. Song, A. Dichiara and F. L. P. Resende, Chem. Eng. Sci., 2019, 197, 212–222 CrossRef CAS.
- H. O. Mohamed, V. K. Velisoju, I. Hita, O. Abed, R. K. Parsapur, N. Zambrano, M. Ben Hassine, N. Morlanes, A. H. Emwas, K. W. Huang and P. Castano, Chem. Eng. J., 2023, 475, 146077 CrossRef CAS.
- C. Wang, L. Wang, G. Wu, F. Jin, X. Zhan and Y. Ding, Catal. Lett., 2019, 150, 429–437 CrossRef.
- Y. Ganjkhanlou, G. Berlier, E. Groppo, E. Borfecchia and S. Bordiga, Top. Catal., 2017, 60, 1664–1672 CrossRef CAS.
- L. Wang, J. Ke, Y. Chai, G. Wu, C. Wang and L. Li, Angew. Chem., Int. Ed., 2025, e202502563 CAS.
- H. O. Mohamed, O. Abed, N. Zambrano, P. Castaño and I. Hita, Ind. Eng. Chem. Res., 2022, 61, 15880–15892 CAS.
- K. Lu, F. Jin, G. Wu and Y. Ding, Sustain. Energy Fuels, 2019, 3, 3569–3581 RSC.
- R. Y. Brogaard and U. Olsbye, ACS Catal., 2016, 6, 1205–1214 CrossRef CAS.
- R. Y. Brogaard, M. Kømurcu, M. M. Dyballa, A. Botan, V. Van Speybroeck, U. Olsbye and K. De Wispelaere, ACS Catal., 2019, 9, 5645–5650 CrossRef CAS PubMed.
- J. Thakkar, X. Yin and X. Zhang, ChemCatChem, 2018, 10, 4234–4237 CrossRef CAS.
- R. D. Andrei, M. I. Popa, F. Fajula, C. Cammarano, A. Al Khudhair, K. Bouchmella, P. H. Mutin and V. Hulea, ACS Catal., 2015, 5, 2774–2777 CrossRef CAS.
- B. H. Babu, M. Lee, D. W. Hwang, Y. Kim and H. Chae, Appl. Catal., A, 2017, 530, 48–55 CrossRef CAS.
- R. D. Andrei, M. I. Popa, F. Fajula and V. Hulea, J. Catal., 2015, 323, 76–84 CrossRef CAS.
- R. D. Andrei, E. Borodina, D. Minoux, N. Nesterenko, J. P. Dath, C. Cammarano and V. Hulea, Ind. Eng. Chem. Res., 2020, 59, 1746–1752 CrossRef CAS.
- R. D. Andrei, M. I. Popa, F. Fajula and V. Hulea, J. Catal., 2015, 323, 76–84 CrossRef CAS.
- L. Attanatho, S. Lao-ubol, A. Suemanotham, N. Prasongthum, P. Khowattana, T. Laosombut, N. Duangwongsa, S. Larpkiattaworn and Y. Thanmongkhon, SN Appl. Sci., 2020, 2, 971 CrossRef CAS.
- T. W. Kim, J. W. Jun, S. I. Hong and C. U. Kim, J. Nanosci. Nanotechnol., 2018, 18, 2026–2031 CrossRef CAS PubMed.
- R. Beucher, R. D. Andrei, C. Cammarano, A. Galarneau, F. Fajula and V. Hulea, ACS Catal., 2018, 8, 3636–3640 CrossRef CAS.
- R. Beucher, C. Cammarano, E. Rodríguez-Castelló and V. Hulea, Catal. Commun., 2020, 144, 10609 CrossRef.
- R. Beucher, C. Cammarano, E. Rodríguez-Castellón and V. Hulea, Ind. Eng. Chem. Res., 2020, 59, 7438–7446 CrossRef CAS.
- R. Beucher, C. Cammarano and V. Hulea, React. Chem. Eng., 2022, 7, 133–141 RSC.
- A. Hwang, S. Kim, G. Kwak, S. K. Kim, H. Park, S. C. Kang, K. Jun and Y. T. Kim, Catal. Lett., 2017, 147, 1303–1314 CrossRef.
- S. Moussa, P. Concepción, M. A. Arribas and A. Martínez, Appl. Catal., A, 2020, 608, 117831 CrossRef CAS.
- M. Stoyanova, M. Schneider, M. M. Pohl and U. Rodemerck, Catal. Commun., 2017, 92, 65–69 CrossRef CAS.
- L. A. Perea, M. Felischak, T. Wolff, C. Hamel and A. Seidel-Morgenstern, Chem. Ing. Tech., 2017, 89, 903–914 CrossRef CAS.
- N. R. Jaegers and E. Iglesia, J. Am. Chem. Soc., 2023, 145, 6349–6361 CrossRef CAS PubMed.
- I. Agirrezabal-Telleria and E. Iglesia, J. Catal., 2017, 352, 505–514 CrossRef CAS.
- I. Agirrezabal-Telleria and E. Iglesia, J. Catal., 2020, 389, 690–705 CrossRef CAS.
- W. Li, C. Zhou, W. Li, L. Ge, G. Yu, M. Qiu and X. Chen, New J. Chem., 2022, 46, 9461–9469 RSC.
- M. Ghambarian, M. Ghashghaee, Z. Azizi and M. Balar, Phys. Chem. Res., 2019, 7, 235–243 CAS.
- M. Ghambarian, M. Ghashghaee, Z. Azizi and M. Balar, Struct. Chem., 2019, 30, 137–150 CrossRef CAS.
- R. D. Andrei, M. I. Popa, C. Cammarano and V. Hulea, New J. Chem., 2016, 40, 4146–4152 RSC.
- K. Toch, J. W. Thybaut and G. B. Marin, Appl. Catal., A, 2015, 489, 292–304 CrossRef CAS.
- A. A. Khudhair, K. Bouchmella, R. D. Andrei, A. Mehdi, P. H. Mutin and V. Hulea, Microporous Mesoporous Mater., 2021, 322, 111165 CrossRef.
- K. Shimura, S. Yoshida, H. Oikawa and T. Fujitani, Microporous Mesoporous Mater., 2022, 338, 111955 CrossRef CAS.
- K. Shimura, S. Yoshida, H. Oikawa and T. Fujitani, Mol. Catal., 2022, 528, 112478 CAS.
- A. Al Khudhair, K. Bouchmella, R. D. Andrei, V. Hulea and A. Mehdi, Molecules, 2024, 29, 4172 CrossRef CAS PubMed.
- L. Chen, G. Li, Z. Wang, S. Li, M. Zhang and X. Li, Catalysts, 2020, 10, 180 CrossRef CAS.
- J. S. Yoon, M. B. Park, Y. Kim, D. W. Hwang and H. Chae, Catalysts, 2019, 9, 933 CrossRef CAS.
- K. Shimura, S. Yoshida, H. Oikawa and T. Fujitani, Catalysts, 2023, 13, 1303 CrossRef CAS.
- J. Xu, R. Wang, L. Zheng, J. Ma, W. Yan, X. Yang, J. Wang, X. Su and Y. Huang, Catal. Sci. Technol., 2021, 11, 1510–1518 RSC.
- J. Xu, R. Wang, Y. Zhang, L. Li, W. Yan, J. Wang, G. Liu, X. Su, Y. Huang and T. Zhang, Chin. J. Catal., 2021, 42, 2181–2188 CrossRef CAS.
- Q. Zhang, T. Wanga, Y. Lia, R. Xiaoe, T. Vitidsant, P. Reubroycharoen, C. Wang, Q. Zhang and L. Ma, Fuel Process. Technol., 2017, 167, 702–710 CrossRef CAS.
- Z. Chen, S. R. Docherty, P. Florian, A. Kierzkowska, I. B. Moroz, P. M. Abdala, C. Copéret, C. R. Müller and A. Fedorov, Catal. Sci. Technol., 2022, 12, 5861–5868 RSC.
- M. Stoyanova, M. Schneider, M. M. Pohl and U. Rodemerck, Catal. Commun., 2017, 92, 65–69 CrossRef CAS.
- M. Lee, J. Yoon, Y. Kim, J. Yoon, H. Chae, Y. Han and D. Hwang, Appl. Catal., A, 2018, 562, 87–93 CrossRef CAS.
- G. V. S. Seufitelli and R. Gustafson, Ind. Eng. Chem. Res., 2022, 61, 4286–4299 CrossRef CAS.
- M. Betz, C. Fuchs, T. A. Zevaco, U. Arnold and J. Sauer, Biomass Bioenergy, 2022, 166, 106595 CrossRef CAS.
- C. Fuchs, U. Arnold and J. Sauer, Chem. Ing. Tech., 2023, 95, 651–657 CrossRef CAS.
- M. H. Kwon, J. S. Yoon, M. Lee, D. W. Hwang, Y. Kim, M. B. Park and H. J. Chae, Appl. Catal., A, 2019, 572, 226–231 CrossRef CAS.
- M. Stoyanova, U. Bentrup, H. Atia, E. V. Kondratenko, D. Linke and U. Rodemerck, Catal. Sci. Technol., 2019, 9, 3137–3148 RSC.
- F. Jin, Y. Yan and G. Wu, Catal. Today, 2020, 355, 48–161 CrossRef.
- A. Aid, R. D. Andrei, S. Amokrane, C. Cammarano, D. Nibou and V. Hulea, Appl. Clay Sci., 2017, 146, 432–438 CrossRef CAS.
- S. Forget, H. Olivier-Bourbigou and D. Delcroix, ChemCatChem, 2017, 9, 2408–2417 CrossRef CAS.
- L. Li, S. Chavan, Y. Ganjkhanlou, E. Groppo, E. Sagstuen, S. Bordiga, U. Olsbye and K.-J. Jens, Appl. Catal., A, 2022, 637, 118598 CrossRef CAS.
- Y. Cho, J. A. Muhlenkamp, A. G. Oliver and J. C. Hicks, Chem. Commun., 2021, 57, 13772–13775 RSC.
- Y. Cho, A. G. Oliver and J. C. Hicks, Appl. Catal., A, 2023, 666, 119391 CrossRef CAS.
- F. P. Rotzinger, J. Am. Chem. Soc., 1996, 118, 6760–6766 CrossRef CAS.
- M. Lallemand, A. Finiels, F. Fajula and V. Hulea, J. Phys. Chem. C, 2009, 113, 20360–20364 CrossRef CAS.
- J. Rabeah, J. Radnik, V. Briois, D. Maschmeyer, G. Stochniol, S. Peitz, H. Reeker, C. La Fontaine and A. Brückner, ACS Catal., 2016, 6, 8224–8228 CrossRef CAS.
- E. Dooryhee, C. R. A. Catlow, J. W. Couves, P. J. Maddox, J. M. Thomas, G. N. Greaves, A. T. Steel and R. P. Townsend, J. Phys. Chem., 1991, 95, 4514–4521 CrossRef CAS.
- H. A. Aleksandrov, V. R. Zdravkova, M. Y. Mihaylov, P. S. Petkov, G. N. Vayssilov and K. I. Hadjiivanov, J. Phys. Chem. C, 2012, 116, 22823–22831 CrossRef CAS.
- K. Góra-Marek, A. Glanowska and J. Datka, Microporous Mesoporous Mater., 2012, 158, 162–169 CrossRef.
- K. Kimura, A.-I. Hideo and A. Ozaki, J. Catal., 1970, 18, 271–280 CrossRef CAS.
- A. Lacarriere, J. Robin, D. Swierczynski, A. Finiels, F. Fajula, F. Luck and V. Hulea, ChemSusChem, 2012, 5, 1787–1792 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |