
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand-assisted nickel catalysis enabling N,N-dialkylation and cyclization of acyl hydrazides using aliphatic alcohols†
Ayanangshu Biswas ,
Sourav Mandal,
Supriya Halder,
Bikramaditya Mandal and
Debashis Adhikari*
,
Sourav Mandal,
Supriya Halder,
Bikramaditya Mandal and
Debashis Adhikari*
Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Mohali, Sector-81, Knowledge City, Manauli-140306, India. E-mail: adhikari@iisermohali.ac.in
First published on 17th June 2025
Abstract
Herein, we describe a nickel-catalyzed N,N-dilakylation protocol for acyl hydrazides. A series of aliphatic alcohols and diols were successfully dehydrogenated and used for this challenging dialkylation as well as some cyclization reactions. The reaction is chemoselective, as many of the N,N-dialkylated products contain an olefinic motif, with the imine linkage selectively reduced while the olefinic one remains intact. The azo–hydrazo redox couple plays a crucial role in the hydrogenation of imines. The reaction proceeds via a radical pathway and in striking contrast to previous reports that involve metal hydrides.
The construction of a C–N bond via alkylation of an amine has garnered tremendous attention in recent years, owing to the widespread occurrence of substituted amines in natural products, agrochemicals and pharmaceuticals.1 The conventional methods for C–N bond formation largely conducted the cross-coupling of amines and alkyl halide in the presence of a base, leaving a large amount of waste.2–5 Replacing the mutagenic alkyl halides by their precursor alcohols might be very effective, although the lower reactivity of alcohols can be a bottleneck. To this end, the conversion of alcohols to carbonyls via in situ dehydrogenation, followed by hydrogenation of the resulting imine bond using the borrowed hydrogen from alcohol, has emerged as an appealing method towards C–N bond formation.6–10 Thus, N-alkylation using alcohols as the starting substrates has become a burgeoning field, and a plethora of precious as well as base metal catalysts have been discovered for this purpose.11–19
N,N-Dialkylated acyl hydrazides can serve as key building blocks in the synthesis of biologically active compounds20–23 appealing as PG12 agonists,24 antibacterial,25 and antifungal agents.26 Their conventional preparation includes the reaction of acyl halides with substituted hydrazine or the reaction of acyl hydrazides with a stoichiometric amount of strong base, subsequently treating with toxic alkyl halides.21 All these methods have pitfalls, including harsh reaction conditions, usage of toxic acyl halides, and generation of copious amounts of waste. The use of simple alcohols to dialkylate acyl hydrazides can be very practical, yet such protocols are scarce. Gunanathan recently established a Ru-Macho catalyst capable of N,N-dialkylation and cyclization of acyl hydrazides starting from alcohols.27 Subsequently, a diaminocyclopentadienone ruthenium tricarbonyl complex and an iridium amidato complex derived from N-phenylpicolinamide have also been shown to successfully dialkylate acyl hydrazides using alcohols.28,29 Notably, there is a severe dearth of base metal catalysts that can perform this challenging reaction, and the sole representative in this direction features a manganese catalyst reported by Balaraman.30 Nevertheless, the Mn(I) precursor for the catalyst preparation is expensive, which leaves plenty of scope to develop other inexpensive base metal catalysts. Herein, we report the first example of base metal nickel that N,N-dialkylates acyl hydrazides under relatively mild reaction conditions (Scheme 1). The use of a redox-active azo-phenolate ligand backbone31 also brings a new mechanistic paradigm that is remarkably different from all prior catalysts used for this purpose. Both dehydrogenation and hydrogenation steps are mediated by a radical pathway that is unprecedented in N,N-dialkylation reactions.
 | ||
| Scheme 1 Selective catalytic N,N-dialkylation of acyl hydrazides using aliphatic alcohols. | ||
We recently established that a nickel azo-phenolate catalyst, 1 (Scheme 1), is very effective in N-alkylation reaction.32 In addition, it can expand the scope of borrowing hydrogen reactions in C-alkylation,33–35 heterocycle formation36 or diol cyclization.37 Our preliminary investigation commenced with the reaction of benzoyl hydrazide with hexanol as the alkylating partner in KOtBu base (Table 1). When the catalytic reaction was performed using 5 mol% of nickel catalyst 1 with 2.2 equiv. of the alcohol in the presence of 0.25 equiv. of KOtBu in toluene, the desired dialkylation product 4b was isolated in 29% yield, along with a large amount of monoalkylation product 4bb (42% yield, Table 1). This condition also generated some amount of hydrazone compound 4bc in 15% yield. However, increasing the base loading to 0.5 equiv. led to the predominant formation of the expected N,N-dialkylated product 4b in 53% yield (entry 2). A further increase in base loading to 1 equiv. improved the formation of 4b to a commendable 72%, along with a tiny amount of 4bb and 4bc. Subsequently, increasing the catalyst loading to 7 mol% significantly augmented the yield of the desired N,N-dihexylbenzohydrazide product to 87%. A blank reaction performed in the absence of a catalyst, keeping other reaction conditions identical, did not afford any N′,N′-dialkylated product, although some hydrazone formed. Another control reaction reveals that the presence of base is critical for the reaction. A solvent screening revealed that xylene afforded 4b in a 46% yield, while THF did not give any product (entries 7–8). Thus, solvent screening clearly established toluene as the ideal solvent for the reaction.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst loading | Alcohol (equiv.) | Solvent | Base (mmol) | Yield | ||
| 4b | 4bb | 4bc | |||||
| Reaction conditions: 1 (x mol %, with respect to benzoyl hydrazide), benzoyl hydrazide (1 mmol), alcohol (2.2 mmol), KOtBu (x mmol), toluene (2 mL), 130 °C, 24 h (isolated yield). | |||||||
| 1. | 1 (5 mol%) | 2.2 | Toluene | KOtBu (0.25) | 29 | 42 | 15 |
| 2. | 1 (5 mol%) | 2.2 | Toluene | KOtBu (0.5) | 53 | 25 | 17 |
| 3. | 1 (5 mol%) | 2.2 | Toluene | KOtBu (1) | 72 | 10 | 8 |
| 4. | 1 (7 mol%) | 2.2 | Toluene | KOtBu (1) | 87 | 10 | n.r. |
| 5. | — | 2.2 | Toluene | KOtBu (1) | n.r. | n.r. | 11 |
| 6. | 1 (7 mol%) | 2.2 | Toluene | — | n.r. | n.r. | n.r. |
| 7. | 1 (7 mol%) | 2.2 | Xylene | KOtBu (1) | 46 | 21 | 16 |
| 8. | 1 (7 mol%) | 2.2 | THF | KOtBu (1) | n.r. | n.r. | 9 |
| 9. | 1 (7 mol%) | 2.2 | Toluene | KOH (1) | n.r. | n.r. | 12 |
| 10. | 1 (7 mol%) | 2.2 | Toluene | K2CO3 (1) | n.r. | n.r. | 9 |
| 11. | 1 (7 mol%) | 2.2 | Toluene | KH (1) | n.r. | n.r. | 14 |
| 12. | 1 (7 mol%) | 2.2 | Toluene | NaOtBu (1) | 23 | 36 | 19 |
| 13. | 1 (7 mol%) | 2.2 | Toluene | LiHMDS (1) | n.r. | n.r. | 9 |
| 14. | 1 (7 mol%) | 2.2 | Toluene | LDA (1) | n.r. | n.r. | 13 |
| 15. | 1 (7 mol%) | 2.2 | Toluene | DABCO (1) | n.r. | n.r. | n.r. |
| 16. | 1 (7 mol%) | 2.2 | Toluene | DBU (1) | n.r. | n.r. | n.r. |

Other bases, such as KOH and K2CO3 were examined and gave only trace amounts of product, highlighting the efficiency of KOtBu, likely due to its mild reducing character in addition to its basicity (entries 9–10). Bases, such as LDA, LiHMDS, and DBU did not afford any product. Further temperature screening also establishes that the reaction performs well at 130 °C.
Upon achieving the optimized reaction conditions, we next progressed towards exploring the scope of this Ni-catalyzed dialkylation reaction by utilizing an assortment of acyl hydrazide derivatives with an array of aliphatic alcohols as the alkylating partners. Primarily, short-chain aliphatic alcohols, such as pentanol and hexanol were reacted with benzoyl hydrazide to deliver the corresponding dialkylated products 4a and 4b in 82–87% yields (Table 2). When the same reaction was performed on a gram scale, 4b was isolated in 71% yield. Similarly, acyl hydrazides bearing heteroarenes, such as N-containing pyrazinoic acid hydrazide as well as thiophene-2-carbohydrazide, worked well under the current dialkylation protocol, furnishing products N,N-dihexyl pyrazinyl hydrazide 4c and N,N-dihexyl thiophenyl hydrazide 4d in 91% and 69% yields, respectively. Pleasingly, long-chain aliphatic alcohol such as tetradecanol smoothly dialkylated benzoyl hydrazide, synthesizing 4e in 90% yield. In comparison, 3-phenylpropanol furnished the N,N′-dialkylated product 4f in 83% yield.
| Reaction conditions: acylhydrazide (1 mmol), primary alcohol (2.2 mmol), KOtBu (1 mmol), 1 (7 mol%), toluene (2 mL), 130 °C, 24 h. When diol was the substrate, 1.2 mmol was used. |
|---|
 |
Subsequently, a series of branched-chain alcohols were examined, which reacted smoothly with acyl hydrazides containing electron-donating groups (−Me and –tBu) at the para position of the acyl ring to afford products 4h–4i in 88–93% yields. The furanoyl hydrazide was N,N-dialkylated by the proelectrophile 3,5,5-trimethylhexan-1-ol to furnish 4j in 81% yield, where the Ru-Macho complex provided only the monoalkylated product.27 Remarkably, this dialkylation protocol hinges on the chemoselective reduction of the imine group, leaving a terminal olefin completely untouched. Accordingly, when hex-5-en-ol and 9-decen-ol were reacted with substituted benzoyl hydrazide and 1-naphthyl hydrazide, the dialkylated products 4k–4p were isolated in 72–84% yields, preserving the terminal double bond. Other long-chain enol substrates responded well to the reaction conditions to furnish dialkylated products 4n–4p in 80–87% yields (Table 2). Likewise, citronellol, a sterically hindered unsaturated alcohol, underwent smooth dehydrogenation with various substituted acyl hydrazides, affording products 4q–4y. As will be discussed in the mechanistic sketch, the involvement of a radical intermediate in both the dehydrogenation and hydrogenation steps plays a key role in governing the observed chemoselectivity. Encouraged by the excellent efficiency of our catalyst towards dialkylation using acyclic aliphatic alcohol partners, we were intrigued to investigate diols as the alkylating agents, which may lead to intramolecular cyclization. Of note, such cyclic hydrazide motifs are valuable building blocks in drug synthesis.38 Gratifyingly, 1,4-butandiol was oxidized and successfully cyclized to provide product 6a in 71% yield. Furthermore, a diverse set of acyl hydrazides bearing electron-donating groups such as –tBu, −phenyl, and –OMe, as well as electron-withdrawing –CF3 groups at the p-position of the phenyl ring, reacted with the aforementioned diol to afford the corresponding acyl hydrazide derivatives 6b–6e in 81–88% yields.
Further, the same alcohol upon treatment with 1-naphthyl hydrazide resulted in the formation of 6f in 79% yield. An analogous reaction of 1,5 pentanediol with different benzoyl hydrazide substrates yielded the corresponding 6-membered cyclic products (6g–6i) in good yields. Analogously, 1,6 hexanediol smoothly prepared the seven-membered cyclized product 6j in 83% yield. Next, we interrogated whether substituted diols can also be an efficient substrate for such cyclization reactions. Interestingly, 2,5-hexanediol and 3-methylpentane-1,5-diol smoothly reacted with benzoyl hydrazide, furnishing the corresponding cyclized products 6k and 6l in 77 and 78% yields, respectively.
Upon establishing a successful dialkylation protocol, we were interested to delineate the mechanistic sketch of the reaction. It is very intriguing that the reaction involves radical intermediates, and the dehydrogenation steps are strikingly different from prior catalysts, where metal hydride formation was imminent.27,29,39 Earlier we described the explicit details of alcohol dehydrogenation and showed how a radical centralized in the azo motif can facilitate the critical hydrogen atom transfer (HAT) step to conduct smooth dehydrogenation reactions.31,32 We have also shown clearly that upon dehydrogenation, the azo motif is converted to a hydrazo species, 1H,H, so that two-electron oxidation of a substrate alcohol is concomitant with two-electron reduction of the azo motif present in the catalyst molecule. Aliphatic alcohols are also dehydrogenated by the same pathway that we have proved recently via a Bell–Evans–Polanyi analysis.31 In the current mechanistic investigation, we will use 1-hexanol as the model aliphatic alcohol and delineate how two alkyl units are introduced to alkylate the terminal nitrogen of the benzoyl hydrazide. Initially, we attempted to prove the homogeneous nature of the catalytic reaction by performing a mercury drop test, which did not have any negative impact on the reaction (Scheme 2a). In strong support of our radical mechanism, where an azo-promoted HAT generates a ketyl radical, we intercepted such a radical intermediate. The TEMPO-adduct of the ketyl radical from 1-hexanol was detected by ESI high-resolution mass spectrometry (Scheme 2b).
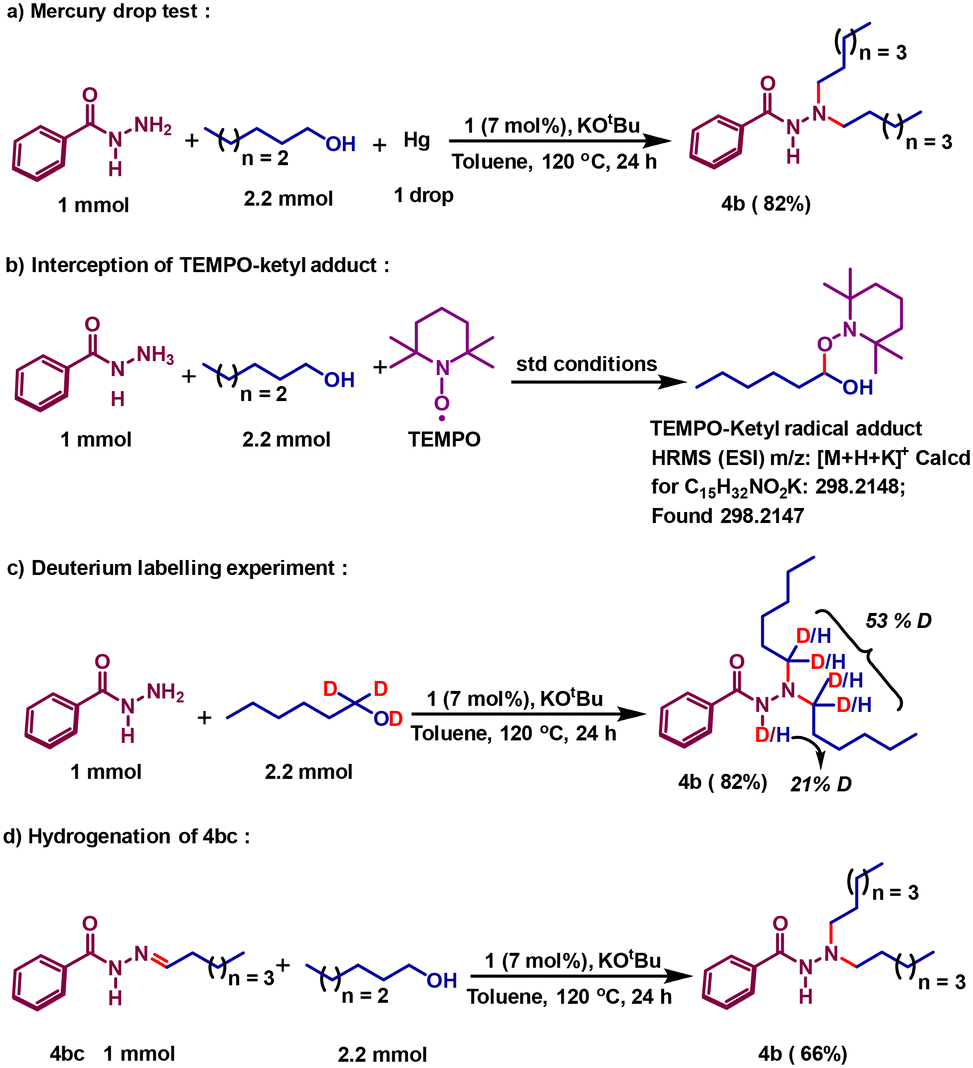 | ||
| Scheme 2 Control reactions to mechanistically probe the N,N-dialkylation protocol. | ||
To prove that the process undergoes a borrowing hydrogen method, we further conducted a deuterium labelling experiment. Starting with α-dideuterated 1-hexanol, the deuterium incorporation at the designated carbons in product 4b was proved (Scheme 2c). Upon dehydrogenation, a hexanal forms, which condenses with benzoyl hydrazide to afford the imine intermediate 4bc. To prove the involvement of this intermediate, it was synthesized separately and hydrogenated using 1-hexanol in the presence of catalyst 1. Such a control reaction affords the desired dialkylated product 4b in 66% yield (Scheme 2d) along with the isolation of dehydrogenated byproduct hexanal in 57% yield. For the hydrogenation of the imine bond, the hydrogen stored at the hydrazo motif is used, adopting a radical mechanism. The details of radical-promoted imine hydrogenation during the monoalkylation was proved earlier.40 In this current work, we will describe how that second alkylation proceeds, notably via the involvement of the radical intermediate. Intuitively, the mono-alkylated product further condenses with hexanal to give 4bd as the intermediate (Scheme 3). The nickel catalyst, upon conducting dehydrogenation, remains as 1H,H˙−, where one of the azo-arms is mono-reduced. During C-alkylation reactions, we have shown that this electron is passed to the enone substrate and reduces it, and such a reduced intermediate conducts further HAT.37 Along the same line, the iminium intermediate 4bd can be mono-reduced, and this reduction is even more plausible given the cationic nature of the substrate.
 | ||
| Scheme 3 Plausible radical-mediated mechanistic cycle highlighting iminium hydrogenation. | ||
In the context of chemoselectivity, it is clear that the iminium cation is reduced much faster than the isolated double bond, ensuring selective reduction of the imine over olefin.
The easy generation of the radical on the imine or iminium cation by single electron reduction facilitates the hydrogenation, leaving the terminal olefin untouched. Once reduced by a single electron, the resulting carbon-centered radical facilitates a HAT step from the hydrazo motif. The dialkylated product is released upon HAT, while the catalyst N–H is deprotonated by KOtBu so that the mono-reduced form of the catalyst is regenerated (Scheme 3). This catalytic intermediate, 1˙− starts the next round of alcohol dehydrogenation. Notably, all prior literature describing transition metal-catalyzed hydrogen autotransfer invoked a metal hydride.41–43 Usually, a β-hydride elimination from a metal-bound alkoxide generates the metal hydride. These findings clearly demonstrate the utility of the redox-responsive ligand backbone, which can store the reducing equivalent.44–46 This way, it provides an orthogonal avenue for conducting smooth (de)hydrogenation reactions to yield the final N,N-dialkylation product.
Conclusions
In summary, we describe an efficient N,N-dialkylation protocol using a nickel catalyst where the redox-active azo ligand plays an extremely important role in steering both dehydrogenation and hydrogenation steps. The efficiency of the catalyst in dehydrogenating aliphatic diols was obvious, as the long aliphatic chains were installed on amine nitrogen to achieve dialkylation. A series of products comprising a long aliphatic straight chain, a branched chain, and an olefinic motif at the terminal position of the chain have been successfully synthesized in high yields using this simple catalyst. The catalyst also showcases excellent chemoselectivity by preferentially reducing the imine bonds over the olefinic bonds. Both critical steps for the reaction—the dehydrogenation of alcohol and the hydrogenation of iminium cation—are driven by a radical mechanism. During imine hydrogenation, a radical housed in the azo ligand is transported to monoreduce such substrate, which subsequently performs HAT from the hydrazo of the ligand framework. Thus, two azophenolates present in the ligand scaffold behave as a redox reservoir, switch between azo–hydrazo redox shuttle, and operate in a very cooperative fashion to conduct this reaction successfully. We hope the catalytic efficiency of the present system shows promise for future BH catalyst design, where redox-active ligands can play an increasingly important role.Experimental section
All starting materials utilized in this study were procured from commercial suppliers. Potassium tert-butoxide, potassium hydroxide, potassium carbonate, and sodium hydroxide were purchased from Avra Synthesis Pvt. Ltd., India. Primary and secondary alcohols were purchased from TCI (India) and BLD Pharma. Fluorene was purchased from BLD Pharma. All these chemicals were used without further purification. Glassware was dried overnight at 160 °C. Solvents such as acetonitrile, ethanol, and dichloromethane were used as received (Finar Chemicals). Toluene was dried by heating over sodium with benzophenone as an indicator. For thin-layer chromatography (TLC), aluminum foil coated with silica and a fluorescent indicator @254 nm (from Merck) was used. Column chromatography was performed using SD Fine silica gel (100–200 mesh) using a gradient of hexane and diethyl ether as the mobile phase.Analytical information
All synthesized products were isolated and characterized by 1H and 13C NMR spectroscopy and high-resolution mass spectrometry. IR spectra were recorded on a Perkin-Elmer FTIR spectrometer using KBr pellets.1H NMR and 13C NMR spectra were recorded on a 400 MHz Bruker Biospin Advance III FT-NMR spectrometer. NMR shifts were reported as delta (δ) units in parts per million (ppm), and coupling constants (J) were reported in hertz (Hz). Chemical shifts (δ) were quoted to the nearest 0.01 ppm relative to the residual protons in CDCl3 (δ 7.26 ppm). Carbon chemical shifts were internally referenced to the deuterated solvent signal of CDCl3 (δ 77.1 ppm). 13C NMR data were proton decoupled. High-resolution mass spectra (HRMS) were recorded on a Waters QTOF mass spectrometer. For chlorine-containing molecules, the most abundant 35Cl was considered for the monoisotopic mass calculation.Synthesis of 1
The azo-phenolate ligand was synthesized using a previously reported procedure.32 In a 100 mL round-bottom flask, a methanolic solution of azo-phenolate ligand (0.1 mmol) and an equimolar amount of KOH were added, and the mixture was stirred at room temperature for 30 min. Next, Ni(OAc)2 (0.05 mmol) was added, and the reaction mixture was further refluxed for another 30 min, during which precipitation was observed. After completion of the reaction, the solution was filtered, affording a dark brown-colored compound 1 in 82% yield. The desired product was fully characterized by 1H and 13C NMR and IR spectroscopy.Synthesis of 1H,H
In a 25 mL Schlenk flask, equimolar quantities of 1, benzyl alcohol and KOtBu (1 mmol each) were added, and the flask was connected to a condenser under a nitrogen flow. The solution was heated to reflux for 4 h, after which the color changed to maroon. The reaction mixture was dried in vacuo, and the crude mixture was washed with dry hexane (5 mL × 3). The precipitate was dried to collect 1H,H in 68% yield. The desired product was fully characterized by 1H, 13C NMR and IR spectroscopy.36General procedure for N,N-dialkylation of acyl hydrazides
In a typical reaction, a 15 mL pressure tube was charged with 1 (7 mol%), KOtBu (1 mmol), alcohols (2.2 mmol), and substituted acylhydrazide (1 mmol), along with 2 mL toluene. The reaction flask was purged with an inert gas for a few minutes before closing the flask tightly. The reaction mixture was stirred at 130 °C for 24 h. The reaction mixture was cooled to room temperature upon completion of the reaction and concentrated in vacuo. The residue was purified by column chromatography using hexane/diethyl ether as an eluent to afford pure products. The desired products were fully characterized by 1H and 13C NMR spectroscopy.Data availability
All data required for the report are available in the form of ESI.†Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
We thank STARS-2 (MHRD), India (Grant No. STARS-2/2023/474), and IISER Mohali for financial support. The authors further thank the IISER Mohali central facility for analytical data.References
- J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177–2250 CrossRef CAS PubMed.
- S. Lee, M. Jørgensen and J. F. Hartwig, Org. Lett., 2001, 3, 2729–2732 CrossRef CAS PubMed.
- X. Huang and S. L. Buchwald, Org. Lett., 2001, 3, 3417–3419 CrossRef CAS PubMed.
- B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 15914–15917 CrossRef CAS PubMed.
- X.-B. Lan, Y. Li, Y.-F. Li, D.-S. Shen, Z. Ke and F.-S. Liu, J. Org. Chem., 2017, 82, 2914–2925 CrossRef CAS PubMed.
- M. H. S. A. Hamid, C. L. Allen, G. W. Lamb, A. C. Maxwell, H. C. Maytum, A. J. A. Watson and J. M. J. Williams, J. Am. Chem. Soc., 2009, 131, 1766–1774 CrossRef CAS PubMed.
- P. Piehl, M. Peña-López, A. Frey, H. Neumann and M. Beller, Chem. Commun., 2017, 53, 3265–3268 RSC.
- G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Science, 2013, 341, 249–260 CrossRef CAS PubMed.
- M. Siddique, B. Boity and A. Rit, Catal. Sci. Technol., 2023, 13, 7172–7180 RSC.
- R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit and N. Tongpenyai, J. Chem. Soc., Chem. Commun., 1981, 611–612, 10.1039/C39810000611.
- G. Guillena, D. J. Ramón and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS PubMed.
- T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed.
- B. G. Reed-Berendt, D. E. Latham, M. B. Dambatta and L. C. Morrill, ACS Cent. Sci., 2021, 7, 570–585 CrossRef CAS PubMed.
- K. Das, A. Mondal, D. Pal and D. Srimani, Org. Lett., 2019, 21, 3223–3227 CrossRef CAS PubMed.
- D. Bhattacharyya, P. Adhikari, K. Deori and A. Das, Catal. Sci. Technol., 2022, 12, 5695–5702 RSC.
- P. Daw, A. Kumar, N. A. Espinosa-Jalapa, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2019, 141, 12202–12206 CrossRef CAS PubMed.
- S. Elangovan, J. Neumann, J.-B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 12641 CrossRef PubMed.
- K. Das, A. Mondal, D. Pal, H. K. Srivastava and D. Srimani, Organometallics, 2019, 38, 1815–1825 CrossRef CAS.
- P. Majumdar, A. Pati, M. Patra, R. K. Behera and A. K. Behera, Chem. Rev., 2014, 114, 2942–2977 CrossRef CAS PubMed.
- U. Ragnarsson, Chem. Soc. Rev., 2001, 30, 205–213 RSC.
- S. N. Mali, B. R. Thorat, D. R. Gupta and A. Pandey, Eng. Proc., 2021, 11, 21 Search PubMed.
- A. Shamsabadi and V. Chudasama, Org. Biomol. Chem., 2017, 15, 17–33 RSC.
- N. Hamanaka, K. Takahashi, Y. Nagao, K. Torisu, S. Shigeoka, S. Hamada, H. Kato, H. Tokumoto and K. Kondo, Bioorg. Med. Chem. Lett., 1995, 5, 1087–1090 CrossRef CAS.
- H. M. Eisa, A. S. Tantawy and M. M. el-Kerdawy, Pharmazie, 1991, 46, 182–184 CAS.
- N. T. Thu-Cuc, N. P. Buu-Hoï and N. D. Xuong, J. Med. Pharm. Chem., 1961, 3, 361–367 CrossRef CAS PubMed.
- S. Thiyagarajan and C. Gunanathan, Org. Lett., 2020, 22, 6617–6622 CrossRef CAS PubMed.
- W.-H. Wang, W.-Y. Shao, J.-Y. Sang, X. Li, X. Yu, Y. Yamamoto and M. Bao, Organometallics, 2023, 42, 2623–2631 CrossRef CAS.
- N. Joly, L. Bettoni, S. Gaillard, A. Poater and J.-L. Renaud, J. Org. Chem., 2021, 86, 6813–6825 CrossRef CAS PubMed.
- R. Babu, S. S. Padhy, G. Sivakumar and E. Balaraman, Catal. Sci. Technol., 2023, 13, 2763–2771 RSC.
- R. Singh, A. K. Bains, A. Kundu, H. Jain, S. Yadav, D. Dey and D. Adhikari, Organometallics, 2023, 42, 1759–1765 CrossRef CAS.
- A. K. Bains, A. Kundu, S. Yadav and D. Adhikari, ACS Catal., 2019, 9, 9051–9059 CrossRef CAS.
- A. Biswas, A. K. Bains and D. Adhikari, Catal. Sci. Technol., 2022, 12, 4211–4216 RSC.
- A. K. Bains, A. Biswas, A. Kundu and D. Adhikari, Adv. Synth. Catal., 2022, 364, 2815–2821 CrossRef CAS.
- A. K. Bains, A. Biswas and D. Adhikari, Adv. Synth. Catal., 2022, 364, 47–52 CrossRef CAS.
- A. K. Bains, V. Singh and D. Adhikari, J. Org. Chem., 2020, 85, 14971–14979 CrossRef CAS PubMed.
- A. K. Bains, A. Kundu, D. Maiti and D. Adhikari, Chem. Sci., 2021, 12, 14217–14223 RSC.
- M. Ślusarczyk, W. M. De Borggraeve, G. Hoornaert, F. Deroose and J. T. M. Linders, Eur. J. Org. Chem., 2008, 2008, 1350–1357 CrossRef.
- A. Kaithal, B. Chatterjee, C. Werlé and W. Leitner, Angew. Chem., Int. Ed., 2021, 60, 26500–26505 CrossRef CAS PubMed.
- A. K. Bains, A. Kundu, R. Singh, H. Jain and D. Adhikari, Eur. J. Inorg. Chem., 2024, 27, e202400558 CrossRef.
- S. Fu, Z. Shao, Y. Wang and Q. Liu, J. Am. Chem. Soc., 2017, 139, 11941–11948 CrossRef CAS PubMed.
- K. Sarkar, K. Das, A. Kundu, D. Adhikari and B. Maji, ACS Catal., 2021, 11, 2786–2794 CrossRef CAS.
- M. Bertoli, A. Choualeb, A. J. Lough, B. Moore, D. Spasyuk and D. G. Gusev, Organometallics, 2011, 30, 3479–3482 CrossRef CAS.
- V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- K. Singh, A. Kundu and D. Adhikari, ACS Catal., 2022, 12, 13075–13107 CrossRef CAS.
- J. I. van der Vlugt, Chem. – Eur. J., 2019, 25, 2651–2662 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedure, control experiments, characterization details, NMR spectra. See DOI: https://doi.org/10.1039/d5cy00433k |
| This journal is © The Royal Society of Chemistry 2025 |