
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-oxidative dehydrogenation of methanol to dimethoxymethane over Ag/Hβ zeolite bifunctional catalyst†
Natalia Simitsis ab,
Felix Eggerc,
Mirijam Zobelcd,
Chalachew Mebrahtuab and
Regina Palkovits*ab
ab,
Felix Eggerc,
Mirijam Zobelcd,
Chalachew Mebrahtuab and
Regina Palkovits*ab
aInstitute for a Sustainable Hydrogen Economy, Forschungszentrum Jülich GmbH, 52428 Jülich, Germany. E-mail: r.palkovits@fz-juelich.de
bChair of Heterogeneous Catalysis and Technical Chemistry, Institute for Technical and Macromolecular Chemistry, RWTH Aachen University, 52074 Aachen, Germany. E-mail: palkovits@itmc.rwth-aachen.de
cInstitute of Crystallography, RWTH Aachen University, Jägerstrasse 17–19, D-52066 Aachen, Germany
dJCNS-3: Neutron Analytics for Energy Research, Forschungszentrum Jülich GmbH, Wilhelm-Johnen-Strasse, D-52428 Jülich, Germany
First published on 23rd June 2025
Abstract
For the first time, an Ag-based catalyst is used in the gas phase non-oxidative dehydrogenation of methanol to dimethoxymethane. Auto-reductive behaviour of Ag species is observed, and the dynamic Ag oxidation state is identified as descriptive for the dynamic reaction course. Stability and regeneration tests reveal robust properties of the Ag/Hβ bifunctional catalyst.
Dimethoxymethane (DMM), the shortest of the homologous oxymethylene ethers (OMEx), with the general structure H3C–O–(CH2O)x–CH3, has recently gained interest as a sustainable diesel additive, requiring small engine modifications.1 Moreover, it bears the potential to be used as a green H2 carrier, similar to dimethyl ether (DME).2 Using methanol as an intermediate, DMM can be synthesized with carbon neutrality from CO2 and green H2.3,4 The continuous non-oxidative dehydrogenation (NOD) of methanol to DMM in the gas phase is a captivating route for technical implementation (Scheme 1).5 In this route, methanol is first dehydrogenated to formaldehyde (FA) and H2, and in the same reactor, FA directly reacts with methanol via acetalization and condensation to DMM. Since the stoichiometrically released H2 is kept as a valuable by-product, potential H2 circulation is possible and significantly improves the exergy efficiency of this process compared to established oxidative pathways.4,6
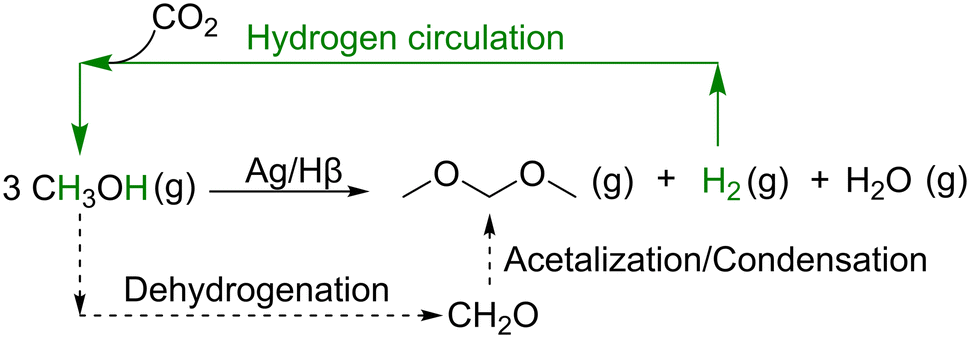 | ||
| Scheme 1 Non-oxidative dehydrogenation (NOD) of methanol to DMM over a bifunctional Ag/Hβ zeolite catalyst with potential H2 circulation. | ||
For the selective production of DMM via the NOD of methanol in one reactor, a bifunctional catalyst possessing both dehydrogenative and acidic functionality is required.5 However, both functionalities must be carefully balanced, as each can also catalyze the formation of side products, i.e., DME formation via methanol condensation over acidic sites, or methyl formate (MF) formation over dehydrogenative sites. Our group recently reported a bifunctional Cu/Hβ catalyst for the NOD of methanol to DMM,7–9 which shows a high DMM selectivity of 81.5% with a methanol conversion of 3.7% (i.e., 50% of the calculated thermodynamic equilibrium conversion) after 1500 min time on stream (TOS) at 200 °C. The high DMM selectivity and catalytic activity are attributed to the tailored dehydrogenative functionality provided by the low Cu loading (1 wt%), catalyzing the methanol dehydrogenation to FA. This is combined with a low amount of weak Lewis acid (LA) sites, provided by the post-synthetically dealuminated, Si-rich Hβ zeolite support (SiO2/Al2O3 ratio of 520), catalyzing the acetalization and condensation of FA with methanol to form DMM.8 Few other groups have also reported Cu-based catalysts for the NOD of methanol to DMM in the gas phase.10 However, besides the thermodynamic limitations inherent to the NOD pathway resulting in overall low DMM yields, challenges such as long induction times, low stability, or selectivity occur when using these Cu-based catalysts. To overcome these challenges, new catalyst systems need to be developed. Besides Cu, Ag is a well-known metal for the conversion of methanol to FA or MF, both oxidative11,12 and dehydrogenative.13,14–16 It therefore seems reasonable to transfer the existing knowledge of Ag-catalyzed methanol dehydrogenation to the NOD of methanol to DMM, employing Ag as the dehydrogenative, active metal. Very recently, the photocatalytic coupling of CO2 reduction and methanol oxidation to selectively produce DMM was published using a Ag and W modified blue TiO2 catalyst.17 However, Ag has not been used as dehydrogenative metal in the gas-phase NOD of methanol to DMM so far. Hence, to set the direction for disruptive innovation of catalyst development, in the present study, we develop a bifunctional Ag-based catalyst for the NOD of methanol to DMM in the gas-phase. Here, Ag is used as dehydrogenative metal supported on our previously optimized Si-rich Hβ zeolite.7,8 Parameters such as Ag loading, reaction temperature as well as calcination and reductive pre-treatments are varied. Based on characterization results obtained by inductively coupled plasma atomic emission spectroscopy (ICP-OES), N2-physisorption, powder X-ray diffractometry (XRD), pair distribution function (PDF), temperature programmed reduction with H2 (H2-TPR), UV/vis spectroscopy, X-ray photoelectron spectroscopy (XPS), and thermogravimetry (TG), a possible structure–activity relationship is discussed. Stability and regeneration tests reveal the good robustness of the Ag/Hβ catalyst for the NOD of methanol to DMM.
Typically, Ag was loaded by incipient wetness impregnation (IWI) on the previously optimized, dealuminated Hβ support with a SiO2/Al2O3 ratio of 520.7,8 ICP-OES results show that the targeted Ag loadings of 1, 5, and 20 wt% are obtained (Table S1, ESI†) and the catalysts are denoted as Ag1/Hβ, Ag5/Hβ and Ag20/Hβ, respectively. The N2-physisorption isotherms (Fig. S1, ESI†) of all materials exhibit a plateau at low partial pressures, characteristic of microporous materials (type I isotherm).18 Moreover, a decreasing specific surface area (SBET) and pore volume (both micro- and mesopore) are observed with increasing Ag loading, which might be due to partial pore blockage (Table S1, ESI†).
Powder XRD results (Fig. S2, ESI†) of the calcined Ag1/Hβ and Ag5/Hβ catalysts show solely reflexes attributed to the Hβ crystal structure and no visible reflexes for Ag species. In contrast to this, the Ag20/Hβ catalyst exhibits distinct reflexes attributed to Ag0, and no reflexes for oxidic Ag species,19 suggesting that the AgNO3 precursor decomposes during the calcination step in air, forming Ag0 species. This auto-reductive behavior of Ag is also known and well discussed in the literature.11,20–22
To confirm the formation of Ag0 during calcination, in situ PDF experiments were performed at the Diamond Light Source, beamline I15-1. Results give insight into a bulk AgNO3 phase present in the precursor, which starts to decompose at 160 °C in air and results in the later formation of Ag0 crystallites after ∼70 min when the temperature reaches 410 °C (Fig. S4, ESI†). Refinements of the PDF data reveal a final Ag nanoparticle diameter of 11 nm at the end of calcination. Together with the formation of these Ag nanoparticles, a structural change appears in the signal of the Hβ zeolite, indicated by the shift of two peaks from 2.63 and 3.06 Å to 2.78 and 3.21 Å, respectively. To illustrate this, the signal of the support structure was isolated by subtracting the contribution of AgNO3 and Ag for the precursor and calcined data sets, respectively (Fig. S5, ESI†). This structural change in the short-range order can be explained by the incorporation of Ag+ ions into the crystal lattice of Hβ zeolite during the calcination, as has been observed by Dzwigaj et al. based on XRD studies.23
The Ag nanoparticles undergo a sudden decrease in diameter during later reduction after 15 min to 10 nm (Fig. S4, ESI,† see the ESI† for a detailed interpretation of the PDF data). After reductive treatment (Ag20/Hβ-red) or usage in the reaction (Ag20/Hβ-spent), a broadening of the Ag0 reflexes is observed for Ag20/Hβ in the powder XRD data (Fig. S3, ESI†). PDF analysis of the total scattering data of the ex situ samples (i.e., Ag20/Hβ-red and Ag20/Hβ-spent, Fig. S6 and S7, ESI†) are not suited for a quantitative particle size analysis. However, a trend of decreasing Ag particle size can be observed for samples before and after reduction and catalytic reaction. This indicates restructuring of the Ag crystallites under reductive or catalytic conditions.
Although Ag0 crystallites are detected on the calcined Ag20/Hβ(520) catalyst in the XRD and PDF results, it is likely that oxidized Ag species are also present on this catalyst.11 Hence, Ag20/Hβ-calc was analyzed via H2-TPR, and one narrow reduction signal at 119 °C indicating the reduction of Ag+ to Ag0 species is observed (Fig. S8, ESI†).11,23,24 Thus, both Ag+ and Ag0 species coexist on the Ag20/Hβ catalyst after calcination. According to the literature, the reduction of Ag+ on zeolite can occur by the following stoichiometry:25
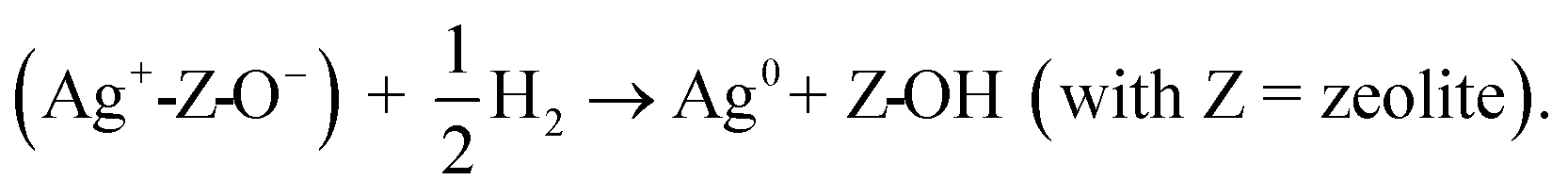
Therefore, the observed dynamics of the Ag oxidation state are expected to influence both the surface species and acidic centers of the zeolite.

To investigate the Ag species present on the Ag20/Hβ catalyst and their evolution through different treatments (calcination, reduction, reaction), UV/vis absorbance spectra of the materials were recorded (Fig. S9, ESI†). The calcined Ag20/Hβ catalyst exhibits signals at 204 nm and 245 nm, which can be attributed to the charge transfer transition between 4d10 and 4d95s1 levels of highly dispersed, isolated Ag+ species in the zeolite.23,26 The signal at 290 nm is typical for Ag clusters, which are either positively charged (Agnδ+) or metallic (Agn0).23,27 Besides, a weaker and broader signal at 400 nm is observed, which can be attributed to metallic Ag0 particles.28 After reduction, the bands at lower wavelengths attributed to isolated Ag+ and Ag clusters decrease in intensity. At the same time, the intensity of the broad signal at 380–415 nm increases significantly, indicating the formation of metallic Ag. This suggests a migration of isolated or clustered Ag species, a phenomenon also observed by Baker et al. for Ag ion-exchanged Y zeolites after reduction with H2.25 After the catalytic reaction, the intensity of the absorbances assigned to the isolated Ag+ species and Agn clusters increased slightly, which may suggest re-oxidation and re-migration of Ag species during the reaction. The reversible re-oxidation of Ag0 to Ag+ and its relocation within the zeolite lattice is also reported in the literature.25,29
Furthermore, XPS analysis of the calcined, reduced, and spent Ag20/Hβ catalyst was performed to identify the surface Ag oxidation state and its dynamic changes resulting from the respective treatments. The detailed XPS Ag 3d and Ag MNN regions are shown in Fig. S11, ESI.† Since the binding energy (BE) shifts of metallic silver vs. silver oxides are very small (i.e., within a 1.2 eV interval for the 3d5/2 BE), comparing the Auger parameters (APs) is considered a more reliable method to identify the presence of metallic or ionic Ag species.30,31 While the AP for Ag20/Hβ-calc is 724.3 eV, it increases to 726.0 eV upon reduction and then decreases to 725.5 eV for Ag20/Hβ-spent (Table S2, ESI†). Typically, an AP of 726.0 eV is found for metallic Ag species, while 724.0 eV indicates Ag ions.30 Hence, it can be assumed that Ag20/Hβ-calc possesses mainly oxidized surface Ag species, while for Ag20/Hβ-red, the surface Ag species are metallic. The decrease in AP after the reaction (725.5 eV) suggests a partial re-oxidation of surface Ag species, which is consistent with the UV/vis results. Fonseca et al. also observed a gradual shift in AP values (723.73, 724.08, and 725.26 eV) for Ag/zeolite materials, depending on the acidity of the zeolites, and attributed these changes to variations in the Ag valence states.32
Based on the characterization results, it can be assumed that the calcined Ag20/Hβ catalyst possesses both isolated Ag+ species and Agn clusters (PDF, UV/vis, H2-TPR) as well as Ag0 crystallites (XRD, PDF). The latter are presumably covered by an oxidic Ag surface layer (XPS, UV/vis). Upon reductive treatment, the oxidized surface of these Ag0 crystallites, along with the isolated Ag+ species and Agn clusters, are mostly reduced to metallic species (XPS, UV/vis). Furthermore, the observed decrease in Ag crystallite size upon reduction suggests restructuring of the Ag particles (PDF). In addition, the reduction of Ag+ to Ag0 might potentially induce changes in the surface hydroxyl species and acidic sites of the zeolite. After the catalytic reaction, a further decrease in Ag crystallite size is observed (XRD, PDF), and partial re-oxidation of the surface Ag0 particles on the Ag20/Hβ catalyst occurs (XPS, UV/vis spectra).
Finally, the Ag/Hβ catalyst was tested in the gas-phase NOD of methanol to DMM, and reaction parameters were optimized. Unless otherwise specified, the Ag/Hβ catalyst was reduced in situ prior to the reaction. Furthermore, for a fair comparison of the catalyst performance, we use the activity, which is calculated based on methanol conversion (ranging from 0.3–3.5% for the tested Ag/Hβ catalysts and reaction conditions). Preliminary testing of the Ag loading (Fig. S12, ESI†) reveals that a high loading of 20 wt% is a reasonable starting point for further optimization due to the relatively high DMM selectivity (61.5%) and activity (0.49 mmolMeOH,conv h−1 gcat−1) after 1500 min TOS. Compared to the catalysts with low Ag loading, no induction phase is observed with 20 wt% Ag. Earlier studies by Fan and coworkers14–16 and Dong et al.33 regarding the NOD of methanol to FA using Ag-based catalysts also identified a high Ag loading of 20 wt% as optimal. Hence, for further catalytic tests, 20 wt% Ag loading is selected.
Next, the reaction temperature for the NOD of methanol to DMM using the Ag20/Hβ catalyst was varied between 200–280 °C, and the results after 1500 min TOS are depicted in Table 1 (reaction courses shown in Fig. S13, ESI†). The catalytic activity increases linearly with the reaction temperature from 0.49 to 4.07 mmolMeOH,conv h−1 gcat−1. This is expected because of the endothermic nature of the first reaction step (i.e., the NOD of methanol to FA). Moreover, the DMM selectivity surpasses 70% at optimal reaction temperatures of 220–240 °C. Outside this temperature window, after 1500 min TOS, the MF formation increased significantly (>32%), while the DME formation remains relatively low, and reaches a maximum selectivity of 14.5% at 240 °C. Interestingly, no FA formation is detected even at high reaction temperatures of 280 °C, which contrasts with the established Cu/Hβ catalyst.7–9 Additionally, an increase in induction time was observed with increasing reaction temperatures, i.e., 0, 600, and >1500 min at 200, 220, and ≥240 °C. The reaction course becomes thus more dynamic with increasing temperature, suggesting an in situ change of the active sites of the Ag20/Hβ catalyst, which will be further explained below. For further catalytic tests, 240 °C is chosen as reaction temperature, offering a compromise between high DMM selectivity (73.6%) and high catalytic activity (2.04 mmolMeOH,conv h−1 gcat−1).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 549 mL h−1 gcat−1, 0.1 g of catalyst diluted with 0.9 g of SiC, n(CH3OH)/n(N2) = 0.24, N2 flow rate = 19.7 mL min−1, in situ reduction prior to reaction (450 °C, 3 h, H2 flow rate = 20 mL min−1), results given after 1500 min TOS. The equations and assumptions used for the calculations are given in the ESI†
549 mL h−1 gcat−1, 0.1 g of catalyst diluted with 0.9 g of SiC, n(CH3OH)/n(N2) = 0.24, N2 flow rate = 19.7 mL min−1, in situ reduction prior to reaction (450 °C, 3 h, H2 flow rate = 20 mL min−1), results given after 1500 min TOS. The equations and assumptions used for the calculations are given in the ESI†
| T (°C) | Activity (mmolMeOH,conv h−1 gcat−1) | S(DME) (%) | S(MF) (%) | S(DMM) (%) | Induction time (min) |
|---|---|---|---|---|---|
| 200 | 0.49 | 6.1 | 32.4 | 61.5 | 0 |
| 220 | 1.30 | 7.1 | 15.3 | 77.6 | 600 |
| 240 | 2.04 | 14.5 | 11.8 | 73.6 | >1500 |
| 280 | 4.07 | 12.1 | 37.2 | 50.3 | >1500 |
Based on the XRD, PDF, H2-TPR, XPS, and UV/vis spectroscopy results, which indicate the presence of both oxidized and metallic Ag species on the calcined catalyst as well as dynamic changes of the Ag oxidation state upon reduction and reaction, the typically performed in situ reduction prior to the reaction was skipped. Instead, the catalytic test was performed using the calcined Ag20/Hβ catalyst (Fig. S14a, ESI†). While the initial DMM selectivity over the calcined catalyst is slightly higher (49.8%) and the DME selectivity is lower (39.2%) compared to the reduced Ag20/Hβ (i.e., 40.2% and 46.1% for DMM and DME selectivity, respectively), no significant difference in the catalytic performance between the calcined and reduced catalyst can be observed after 1500 min TOS. This indicates that the Ag20/Hβ catalyst is a very robust system, as the effects of different pre-treatments, such as calcination or reduction, are balanced out during the catalytic reaction, ultimately leading to a similar catalytic performance.
The reaction course under optimized parameters reveals a 33% decrease in catalytic activity after 1500 min TOS (Fig. S13c, ESI†). A long-term experiment for 3600 min (Fig. 1) shows that further deactivation of only 6.6% occurs after the 1500 min TOS. This indicates that the catalyst is stabilized after completing the induction phase, which lasts until 2000 min TOS. Coke formation can be excluded as a possible cause of the slight catalyst deactivation, based on TG analysis showing no significant difference in mass loss between the calcined and spent catalyst (Fig. S10, ESI†). However, since dynamic changes of the Ag species and its surface oxidation state upon reduction and reaction were observed based on XRD, PDF, H2-TPR, XPS, and UV/vis spectroscopy, a regeneration experiment was performed. For this, the Ag20/Hβ catalyst was in situ re-reduced after 3600 min TOS, followed by a restart of the reaction (Fig. 1). Interestingly, after the regeneration treatment, an induction phase similar to the first reaction run is observed (i.e., decreasing DME selectivity for the benefit of DMM formation). Eventually, after an additional 1500 min TOS, a DMM selectivity of 73.0% with a catalytic activity of 1.69 mmolMeOH,conv h−1 gcat−1 is achieved, being very similar to the results obtained over the fresh catalyst. This indicates that the induction phase and the dynamic changes of the active sites are reversible.
 | ||
| Fig. 1 Long term catalytic test and regeneration of 20Ag/Hβ. Reaction conditions: 240 °C, 1 atm, GHSV = 14549 mL h−1 gcat−1, 0.1 g of catalyst diluted with 0.9 g of SiC, n(CH3OH)/n(N2) = 0.24, N2 flow rate = 19.7 mL min−1, in situ reduction prior to reaction (450 °C, H2 flow rate = 20 mL min−1, 3 h). After 3600 min TOS, the catalyst was in situ regenerated by reduction, and the reaction was restarted subsequently. | ||
The MF selectivity remains remarkably stable during the reaction course, fluctuating by less than 3%. A similar and stable MF selectivity is also observed for the calcined Ag20/Hβ (Fig. S14a, ESI†). This strongly suggests that the dehydrogenative functionality of the bifunctional Ag20/Hβ catalyst is very stable, regardless of catalyst pre-treatments or the reaction conditions, and does not contribute to the longer induction phase. In contrast, the acidic functionality, catalyzing the acetalization and condensation reactions to DMM or DME, appears to be influenced by such treatments, as evidenced by the dynamic changes of the DME and DMM formation. The dynamic changes of the Ag species and surface oxidation state might thus be descriptive for the dynamic reaction course by possibly influencing the acidity of the Ag20/Hβ catalyst. As discussed in the interpretation of the H2-TPR results, the reduction of Ag+ to Ag0 with H2 can regenerate the BA sites on the zeolite.21,22,25,34 This explains the high DME selectivity observed directly after the reduction. Interestingly, the initial DME selectivity of the reduced Ag20/Hβ is higher compared to the calcined catalyst (i.e., 46.1% vs. 39.2%, respectively), further supporting the hypothesis that reduction of Ag+ with H2 enhances the acidic functionality. During the reaction, partial re-oxidation of Ag0 to Ag+ species is observed as evidenced by XPS and UV/vis spectroscopy characterization. Jacobs et al. observed that the redox behaviour of highly dispersed Ag species in zeolites is reversible, and the oxidation of Ag0 to Ag+ can lead to the removal of BA sites due to dehydroxylation of the zeolite surface.29 This might explain the decreasing DME selectivity for the benefits of enhanced DMM selectivity during the reaction. This is in line with our previous studies, where the removal of BA sites and the presence of low amounts of weak LA sites on the Cu/Hβ catalyst are beneficial for high DMM selectivity, while DME formation is suppressed.7,8 Moreover, dynamic changes of the LA site concentration upon calcination, reduction, or after the reaction were observed for the Cu/Hβ catalyst,8 suggesting that similar dynamics may occur with the Ag/Hβ catalyst. Furthermore, isolated Ag+ ions within the zeolite framework can possess LA character35 and their mobility upon reduction or reaction conditions25,29 may, in turn, influence the overall LA properties of the Ag20/Hβ catalyst. Interestingly, these dynamic changes appear to be reversible, as the induction phase restarts after the regeneration treatment. However, further in-depth analysis of the in situ dynamics of the acid sites on the Ag20/Hβ catalyst is required and remains beyond the scope of the present study.
Conclusions
For the first time, an Ag-based bifunctional catalyst was tested in the NOD of methanol to DMM in the gas phase. During calcination, the decomposition and auto-reduction of the AgNO3 precursor to Ag0 species is observed in in situ PDF studies. After catalyst optimization (20 wt% Ag loading on a previously optimized Hβ zeolite support,7,8 240 °C reaction temperature, in situ reduction), a DMM selectivity of 73.6% and an activity of 2.04 mmolMeOH,conv h−1 gcat−1 is achieved after 1500 min TOS. A dynamic reaction course exhibiting decreasing DME selectivity for the benefit of increasing DMM selectivity is observed. Based on the characterization (XRD, PDF, H2-TPR, UV/vis-spectroscopy, XPS), the dynamic changes of the Ag oxidation state can be used as a descriptor for the dynamic changes of the product selectivity during the reaction course. Stability and regeneration tests reveal the reversibility of the induction phase and the robust performance of the Ag/Hβ bifunctional catalyst.Data availability
Raw data are available on Zenodo under the doi: https://doi.org/10.5281/zenodo.15691695. In addition, the data supporting this article have been included as part of the ESI.†Author contributions
N. Simitsis: conceptualization, investigation, data curation, visualization, project administration, and writing – original draft. F. Egger: investigation, formal analysis, visualization, writing – review & editing. M. Zobel: supervision, resources, writing – review & editing. C. Mebrahtu: conceptualization, supervision, writing – review & editing. R. Palkovits: conceptualization, supervision, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the funding by the German Federal Ministry of Research, Technology and Space (BMFTR) and the Ministry of Economic Affairs, Industry, Climate Action and Energy of the State of North Rhine-Westphalia through the project HC-H2. We acknowledge the research funding by DFG (PA 1689/22-1, Project ID: 512546329). N. S. thanks the German Federal Environmental Foundation (Deutsche Bundesstiftung Umwelt: DBU) for financial support. Part of this work was supported by the Cluster of Excellence Fuel Science Center (EXC 2186, ID: 390919832) funded by the Excellence Initiative by the German federal and state governments to promote science and research at German universities. We also acknowledge beamtime at beamline I15-1 at Diamond Light Source (Didcot, England), proposal CY29917 and local support by Phil Chater, as well as beamtime at beamline P02.1 at DESY (Hamburg, Germany), proposal I-20221101 and local support by Alexander Schoekel.Notes and references
- (a) H. Pitsch, D. Goeb, L. Cai and W. Willems, Prog. Energy Combust. Sci., 2024, 104, 101173 CrossRef; (b) L. Lautenschütz, D. Oestreich, P. Seidenspinner, U. Arnold, E. Dinjus and J. Sauer, Fuel, 2016, 173, 129 CrossRef; (c) A. Omari, B. Heuser and S. Pischinger, Fuel, 2017, 209, 232 CrossRef CAS; (d) M. Härtl, P. Seidenspinner, G. Wachtmeister and E. Jacob, MTZ Worldw., 2014, 75, 48 CrossRef; (e) A. Omari, B. Heuser, S. Pischinger and C. Rüdinger, Appl. Energy, 2019, 239, 1242 CrossRef CAS; (f) M. Härtl, P. Seidenspinner, E. Jacob and G. Wachtmeister, Fuel, 2015, 153, 328 CrossRef.
- P. Schühle, R. Stöber, M. Gierse, A. Schaadt, R. Szolak, S. Thill, M. Alders, C. Hebling, P. Wasserscheid and O. Salem, Energy Environ. Sci., 2023, 16, 3002 RSC.
- (a) J. Burre, D. Bongartz, S. Deutz, C. Mebrahtu, O. Osterthun, R. Sun, S. Völker, A. Bardow, J. Klankermayer and R. Palkovits, et al., Energy Environ. Sci., 2021, 14, 3686 RSC; (b) R. Geitner, T. Schuett, S. Zechel and U. S. Schubert, Chem. – Eur. J., 2024, e202401570 CrossRef CAS PubMed.
- S. Deutz, D. Bongartz, B. Heuser, A. Kätelhön, L. Schulze Langenhorst, A. Omari, M. Walters, J. Klankermayer, W. Leitner and A. Mitsos, et al., Energy Environ. Sci., 2018, 11, 331 RSC.
- R. Sun, I. Delidovich and R. Palkovits, ACS Catal., 2019, 9, 1298 CrossRef CAS.
- D. Bongartz, L. Doré, K. Eichler, T. Grube, B. Heuser, L. E. Hombach, M. Robinius, S. Pischinger, D. Stolten and G. Walther, et al., Appl. Energy, 2018, 231, 757 CrossRef CAS.
- N. Simitsis, C. Mebrahtu and R. Palkovits, ChemCatChem, 2024, 16, e202301704 CrossRef CAS.
- C. Mebrahtu, R. Sun, C. Gierlich and R. Palkovits, Appl. Catal., B, 2021, 287, 119964 CrossRef CAS.
- R. Sun, C. Mebrahtu, J. P. Hofmann, D. Bongartz, J. Burre, C. H. Gierlich, P. J. C. Hausoul, A. Mitsos and R. Palkovits, Sustainable Energy Fuels, 2021, 5, 117 RSC.
- (a) J. Ren, Y. Xu, C. Ye, M. Peng and F. Xin, Chem. Eng. J., 2024, 479, 147774 CrossRef CAS; (b) A. T. To, T. J. Wilke, E. Nelson, C. P. Nash, A. Bartling, E. C. Wegener, K. A. Unocic, S. E. Habas, T. D. Foust and D. A. Ruddy, ACS Sustainable Chem. Eng., 2020, 8, 12151 CrossRef CAS PubMed.
- D. Chen, Z. Qu, S. Shen, X. Li, Y. Shi, Y. Wang, Q. Fu and J. Wu, Catal. Today, 2011, 175, 338 CrossRef CAS.
- (a) A. Nagy and G. Mestl, Appl. Catal., A, 1999, 188, 337 CrossRef CAS; (b) M. Qian, M. Liauw and G. Emig, Appl. Catal., A, 2003, 238, 211 CrossRef CAS.
- (a) N. Takezawa and N. Iwasa, Catal. Today, 1997, 36, 45 CrossRef CAS; (b) J. J. Corral-Pérez, C. Copéret and A. Urakawa, J. Catal., 2019, 380, 153 CrossRef.
- J.-X. Li, L.-P. Ren, W.-L. Dai, Y. Cao and K.-N. Fan, Chin. J. Chem., 2008, 26, 1045 CrossRef CAS.
- L.-P. Ren, W.-L. Dai, Y. Cao and K.-N. Fan, Catal. Lett., 2003, 85, 81 CrossRef CAS.
- L.-P. Ren, W.-L. Dai, X.-L. Yang, Y. Cao, H. Li and K.-N. Fan, Appl. Catal., A, 2004, 273, 83 CrossRef CAS.
- Y. Wang, Y. Liu, L. Wang, S. Perumal, H. Wang, H. Ko, C.-L. Dong, P. Zhang, S. Wang and T. T. T. Nga, et al., Nat. Commun., 2024, 15, 6047 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. Sing, Pure Appl. Chem., 2015, 87, 1051 CrossRef CAS.
- (a) X. Chen, M. Chen, G. He, F. Wang, G. Xu, Y. Li, C. Zhang and H. He, J. Phys. Chem. C, 2018, 122, 27331 CrossRef CAS; (b) D. Kim, S. Jeong and J. Moon, Nanotechnology, 2006, 17, 4019 CrossRef CAS PubMed.
- (a) L. Kundakovic and M. Flytzani-Stephanopoulos, Appl. Catal., A, 1999, 183, 35 CrossRef CAS; (b) X. Zhang, Z. Qu, X. Li, M. Wen, X. Quan, D. Ma and J. Wu, Sep. Purif. Technol., 2010, 72, 395 CrossRef CAS; (c) T. Chaieb, D. Brouri, S. Casale, J.-M. Krafft, T. Da Silva, C. Thomas, L. Delannoy and C. Louis, Res. Chem. Intermed., 2019, 45, 5877 CrossRef CAS; (d) N. D. Hutson, B. A. Reisner, R. T. Yang and B. H. Toby, Chem. Mater., 2000, 12, 3020 CrossRef CAS.
- M. Chebbi, B. Azambre, L. Cantrel, M. Huvé and T. Albiol, Microporous Mesoporous Mater., 2017, 244, 137 CrossRef CAS.
- P. A. Jacobs, J. B. Uytterhoeven and H. K. Beyer, J. Chem. Soc., Faraday Trans. 1, 1979, 75, 56 RSC.
- S. Dzwigaj, Y. Millot, J.-M. Krafft, N. Popovych and P. Kyriienko, J. Phys. Chem. C, 2013, 117, 12552 CrossRef CAS.
- H. Beyer, P. A. Jacobs and J. B. Uytterhoeven, J. Chem. Soc., Faraday Trans. 1, 1976, 72, 674 RSC.
- M. D. Baker, G. A. Ozin and J. Godber, J. Phys. Chem., 1985, 89, 305 CrossRef CAS.
- N. Popovych, P. Kyriienko, S. Soloviev, R. Baran, Y. Millot and S. Dzwigaj, Phys. Chem. Chem. Phys., 2016, 18, 29458 RSC.
- J. Shibata, Y. Takada, A. Shichi, S. Satokawa, A. Satsuma and T. Hattori, Appl. Catal., B, 2004, 54, 137 CrossRef CAS.
- (a) R. Bartolomeu, R. Bértolo, S. Casale, A. Fernandes, C. Henriques, P. Da Costa and F. Ribeiro, Microporous Mesoporous Mater., 2013, 169, 137 CrossRef CAS; (b) R. Yamamoto, Y. Sawayama, H. Shibahara, Y. Ichihashi, S. Nishiyama and S. Tsuruya, J. Catal., 2005, 234, 308 CrossRef CAS.
- P. A. Jacobs, J. B. Uytterhoeven and H. K. Beyer, J. Chem. Soc., Faraday Trans. 1, 1977, 73, 1755 RSC.
- A. M. Ferraria, A. P. Carapeto and A. M. B. do Rego, Vacuum, 2012, 86, 1988 CrossRef CAS.
- (a) V. K. Kaushik, J. Electron Spectrosc. Relat. Phenom., 1991, 56, 273 CrossRef CAS; (b) G. B. Hoflund, Z. F. Hazos and G. N. Salaita, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 11126 CrossRef CAS; (c) S. G. Aspromonte, M. D. Mizrahi, F. A. Schneeberger, J. M. R. López and A. V. Boix, J. Phys. Chem. C, 2013, 117, 25433 CrossRef CAS.
- A. M. Fonseca and I. C. Neves, Microporous Mesoporous Mater., 2013, 181, 83 CrossRef CAS.
- Y. Dong, W.-L. Dai, J.-L. Li and J.-F. Deng, Chem. Lett., 2001, 30, 534 CrossRef.
- T. Baba, Catal. Surv. Asia, 2005, 9, 147 CrossRef CAS.
- (a) C. Bernardon, M. Ben Osman, G. Laugel, B. Louis and P. Pale, C. R. Chim., 2017, 20, 20 CrossRef CAS; (b) K. Góra-Marek, K. A. Tarach, Z. Piwowarska, M. Łaniecki and L. Chmielarz, Catal. Sci. Technol., 2016, 6, 1651 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cy00544b |
| This journal is © The Royal Society of Chemistry 2025 |