
Fluorescent zinc(II) thione and selone complexes for light-emitting applications†
Suman
Mandal‡
a,
Bikash
Lahkar‡
a,
Gopendra
Muduli‡
 a,
Arushi
Rawat
b,
Abhilash
Sahu
b,
Osamu
Tsutsumi
b and
Ganesan
Prabusankar
*a
a,
Arushi
Rawat
b,
Abhilash
Sahu
b,
Osamu
Tsutsumi
b and
Ganesan
Prabusankar
*a
aOrganometallics and Materials Chemistry Lab, Department of Chemistry, Indian Institute of Technology Hyderabad, Kandi, Sangareddy, Telangana 502285, India. E-mail: prabu@chy.iith.ac.in
bDepartment of Applied Chemistry, Ritsumeikan University, Kusatsu 525-8577, Japan. E-mail: tsutsumi@sk.ritsumei.ac.jp
First published on 9th December 2024
Abstract
Three 1-(anthracene-9-ylmethyl)-3-isopropyl-imidazol-2-thione Zn(II) halide complexes (1–3) and one 1-(anthracene-9-ylmethyl)-3-isopropyl-imidazol-2-selone Zn(II) dichloride complex (4) were synthesized and characterized. Complexes 2, 3, and 4 exhibited distorted tetrahedral geometries, while complex 1 adopted a regular tetrahedral geometry. All these complexes displayed emission in the crystalline state, with complex 3 emitting in the yellow region and complex 1 and 4 in the blue region, while complex 2 gave a bluish-green emission. The ligands L1 and L2, however, showed no emission in the solution and crystalline state. The photophysical properties of these four complexes were studied, and their quantum yields in the crystalline states were determined. Complex 1 exhibited the highest quantum yield of 7.72%, and complexes 2 and 3 demonstrated 5.95% and 5.07% yield, respectively. Complex 4 exhibited a relatively lower quantum yield of 3.87%. The crystalline state quantum yields for the complexes were found to vary with the variation of the halide ion coordinated to the metal center, following the trend Cl− > Br− > I−. The quantum yield for the thione complexes 1–3 was found to be superior to that of the selone complex 4. Density functional theory calculations were performed to study their structural properties and emissive nature. TD-DFT natural transition orbital calculations revealed that the observed emission behaviour is primarily driven by intra-ligand charge transfer (1ILCT) mediated through the metal center.
Introduction
Light-emitting diodes (LEDs) have become indispensable in addressing high energy consumption and increasing the durability of electronic display devices.1 Since the discovery of the first organic molecules used in light-emitting devices by Tang in 1987 and Burroughes in 1990, there has been tremendous advancement in organic light-emitting devices.2–6 However, a major drawback of such organic molecules in these devices is their lower stability, which affects their longevity.7,8 In contrast, transition metal coordination complexes provide longer durability and various photophysical mechanisms that maximize quantum efficiency, making luminescent transition metal complexes a useful tool for LED fabrication.9–11A wide range of transition metal complexes has been synthesized using different metals in various oxidation states, demonstrating significant emission properties for use in light-emitting diodes.12–14 Recently, interest in Zn(II) luminescent complexes has grown considerably due to their high reaction yield, stability, and natural abundance, offering a natural alternative to precious metals such as iridium and platinum.15–17 In closed-shell d10 metal complexes, the stable shell configuration prevents metal-to-ligand charge transfer, making luminescence in these complexes challenging.18 While luminescent polynuclear d10 metal complexes having metal–metal interactions are well known, ligands such as thiolates or halides can significantly enhance their photophysical behavior.19 In 2021, Koshevoy and co-workers reported Zn(II) halide luminescent complexes using phenanthrene-based pyridyl-imidazole chromophores, achieving a quantum yield of up to 28%.20 Later, in 2024, Tadokoro and co-workers reported Zn(II) halide-based tetranuclear clusters [Zn4La4(μ3-OMe)2X2] (HLa ethyl-5-iode-3-methoxysalicylate; X = I, Br, Cl), which exhibited phosphorescence in the crystalline state. In these complexes, the iodine-substituted ligand played a vital role in the luminescent behaviour.21
In contrast to green- and red-emitting LED materials, the efficiency of blue light-emitting LEDs is particularly critical.22 Most blue-emitting complexes exhibit phosphorescence or thermally-activated delayed fluorescence behaviour along with high quantum yield and long lifetimes, but these complexes typically require high-cost and low-abundance metals such as Rh, Ir, and Pt.23 Notably, such complexes must be doped with other host materials having wider band gaps and higher triplet energy levels to avoid concentration-caused emission quenching and exciton annihilation. For such complications, the use of blue phosphorescent and TADF materials in practical day-to-day life is very complex, opening the field of blue-emitting fluorescent molecules using cost-effective metals with higher natural abundance such as zinc and copper.24,25
Herewith, we report four fluorescent mononuclear Zn(II) halide imidazol-2-chalcogenone complexes. 1H NMR, 13C NMR, FT-IR, PXRD, and SCXRD techniques were used to characterize the complexes [(L1)2ZnCl2] (1), [(L1)2ZnBr2] (2), [(L1)2ZnI2] (3) and [(L2)2ZnCl2] (4); L1 = 1-(anthracene-9-ylmethyl)-3-isopropyl-imidazol-2-thione and L2 = 1-(anthracene-9-ylmethyl)-3-isopropyl-imidazol-2-selone. DFT, TD-DFT, and NTO analyses were carried out on all the complexes to understand their emissive characteristics.
Results and discussion
Imidazol-2-chalcogenones owing to their versatile electronic and steric properties, have established themselves as a potential neutral donor ligand. However, not much has been explored for applications of d10 imidazol-2-chalcogenone complexes and most of these complexes have been demonstrated for catalysis.26 The interesting structural features of Zn(II) chalcogenones have been well established.27–29 There has been only one report of luminescent Zn(II) imidazol-2-chalcogenone complexes, and the quantum yield of that complex was also comparatively very low.30
L1 and L2 were prepared as per the previously reported procedures.31 Zn(II) complexes 1–3 were synthesized from the reaction between L1 and corresponding zinc(II) halide salts (Scheme 1). Complex 4 was isolated from the reaction between L2 and ZnCl2. These complexes were characterized by 1H NMR, 13C NMR, FT-IR, PXRD, and SCXRD. The formation of complexes 1–4 can be confirmed by the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S stretching frequencies at 1220 cm−1 (1), 1228 cm−1 (2), 1225 cm−1 (3), and for CSe stretching frequency at 1195 cm−1 (4). The change in chemical shift values was observed for the CS/C=Se carbon in the 13C NMR spectra of 1–4 as compared to that of corresponding ligands L1 and L2 confirming the formation of the respective metal halide complexes (see ESI, Table S1†). All the complexes were stable at room temperature and soluble in CHCl3, DCM, methanol, acetonitrile, and DMSO. The phase purity of the bulk samples 1–4 was confirmed by comparing the PXRD of bulk samples with simulated PXRD from a single crystal X-ray data.
S stretching frequencies at 1220 cm−1 (1), 1228 cm−1 (2), 1225 cm−1 (3), and for CSe stretching frequency at 1195 cm−1 (4). The change in chemical shift values was observed for the CS/C=Se carbon in the 13C NMR spectra of 1–4 as compared to that of corresponding ligands L1 and L2 confirming the formation of the respective metal halide complexes (see ESI, Table S1†). All the complexes were stable at room temperature and soluble in CHCl3, DCM, methanol, acetonitrile, and DMSO. The phase purity of the bulk samples 1–4 was confirmed by comparing the PXRD of bulk samples with simulated PXRD from a single crystal X-ray data.
 | ||
| Scheme 1 Synthesis of 1–4. | ||
The solid-state structures of 1–4 were characterized by single crystal X-ray diffraction technique. The desired single crystals of 1–4 were obtained from a mixture of methanol and acetonitrile solutions. Complexes 1 and 3 crystallizes in a monoclinic system with space groups C2/c and P21/c, respectively, while 2 and 4 crystallize in a triclinic system with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Fig. 1). The coordination environment of complexes 1–4 is comparable. However, the structural parameters of 1–4 are not comparable. Two ligands and two halides fulfill the coordination environment of Zn(II) (see ESI, Fig. S13–S16†).
(Fig. 1). The coordination environment of complexes 1–4 is comparable. However, the structural parameters of 1–4 are not comparable. Two ligands and two halides fulfill the coordination environment of Zn(II) (see ESI, Fig. S13–S16†).
 | ||
| Fig. 1 (i) Solid-state structure of 1–4 (ii) A polyhedral view around Zn(II) and (iii) space-filling model of 1–4. | ||
The Zn1–S1 bond distance in 2 is [2.368(13) Å] slightly shorter than 1 (2.388(6) Å) and 3 (2.385(13) Å). The Zn–S bond distances in 1–3 are comparable with that of the previously reported tetra coordinated [{1-(2-hydroxyethyl)-3-isopropyl-benzimidazol-2-thione}ZnBr2] complex (2.3390(15) Å).26 The average CS bond distance in 1–3 is (1.715 Å), which is slightly shorter than that of CSe in 4 [1.876(3) Å]. The bond distance (2.504(10) Å) between Zn1 and Se1 in 4 is comparable with that in [{1-methyl 3-naphthylmethylimidazol-2-selone}ZnCl2] (2.496(5) Å).32 The Se1–Zn1–Se2 bond angle of 4 is found to be (104.26(17)°) nearly comparable with that of 102.67(16)°.32 The S1–Zn1–S2 bond angle supports that 1 is in tetrahedral geometry while 2 and 3 are in a distorted tetrahedral geometry.
The solid-state packing of 1–4 is distinctly different (Fig. 2 and see ESI†). Molecules 1–3 show rare intramolecular S⋯H hydrogen bonding intersections, while molecule 4 depicts the Se⋯H interaction. In complex 1, four such intramolecular S⋯H interactions are present, varying from 2.708 to 3.630 Å. Also, complexes 2 and 3 have two interactions ranging from 2.709 to 2.764 Å. In the case of complex 4, two similar Se⋯H interactions are also present with 2.797 and 2.863 Å.
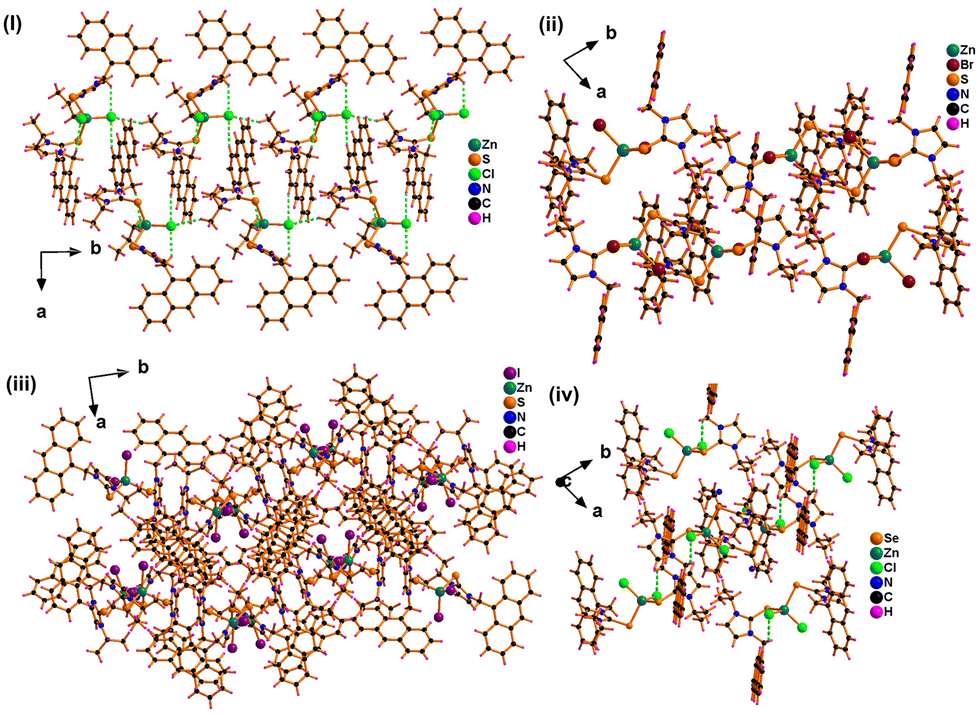 | ||
| Fig. 2 Solid-state packing of (i) 1, (ii) 2, (iii) 3, and (iv) 4. Viewed along the “c” axis. | ||
The density functional theory (DFT) calculations were carried out using CIF files 1–4 to understand the structural and bonding aspects of the complexes (Tables S5–S9†). The optimized structural parameters are comparable with the experimental structural parameters. The HOMO and LUMO energies of 1–4 are comparable (Fig. 3). The topological analysis was carried out using the ground state structure optimization. The bond critical points for S⋯H hydrogen bonding interactions of 1–3 and Se⋯H hydrogen bonding interactions of 4 are comparable with the experimental observations (see ESI, Fig. S25–S28†). The total electron density ρ(r) and Laplacian of electron density ∇2ρ(r) are convincing with the existence of S⋯H hydrogen bonding interactions of 1–3 and Se⋯H hydrogen bonding interactions of 4.
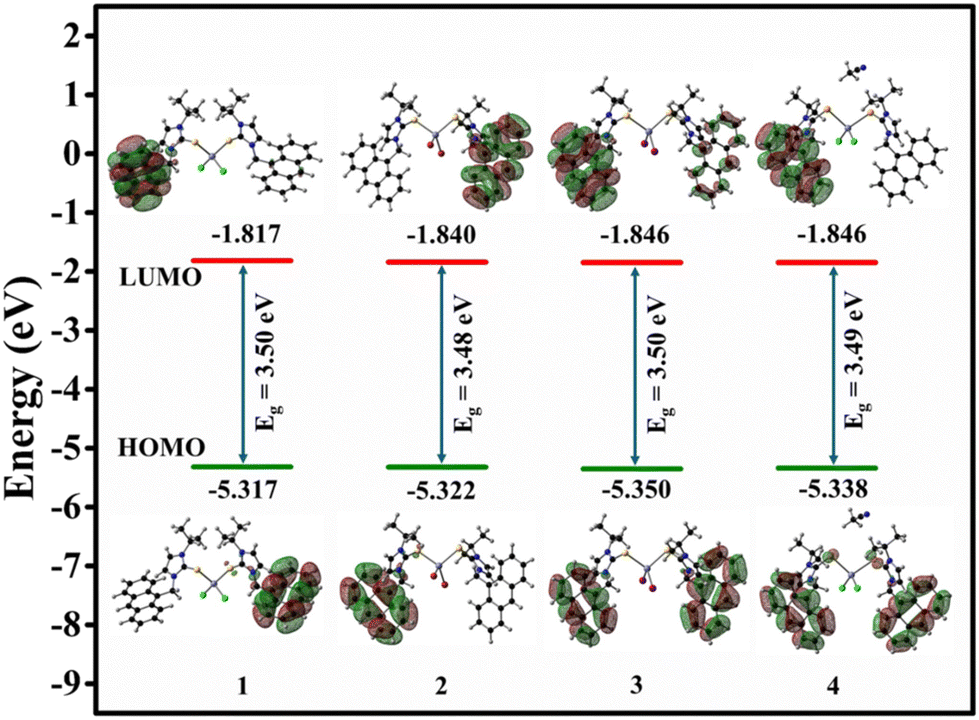 | ||
| Fig. 3 Frontier molecular orbital (HOMO–LUMO) levels of 1–4. | ||
In addition, the packing diagram of 1–4 shows various intermolecular interactions that are not similar in all the complexes (see ESI, Fig. S21–S24†). In the packing diagram of 1, the number of interactions such as Cl⋯H–C(sp3) (2.974 Å), shortest S⋯H–C(sp2) (2.996 Å), π⋯H–C(sp3) (2.852 Å), and weak π⋯π interaction (3.609 Å) are observed. In case of 2, only S⋯H–C(sp2) (3.067 Å), π⋯H–C(sp3) (3.059 Å) and π⋯H–C(sp2) (3.248 Å) were present. There were no Br⋯H interactions present in the packing diagram of 2. Complex 3 demonstrated a weak I⋯H–C(sp3) interaction (3.493 Å) and I⋯H–C(sp2) (3.092 Å) along with a weak π⋯π interaction (3.143 Å) without any presence of S⋯H interaction. In the case of complex 4, only Se⋯H–C(sp3) with 3.362 Å was present with Cl⋯H interaction of 2.992 Å.
To understand the thermal stability of complexes 1–4, thermogravimetric analysis was carried out under an inert environment in the range of 40–790 °C at a heating range of 10 °C per minute (see ESI, Fig. S29†). Notably, complexes 1–4 are thermally stable till 270 °C. There is no significant weight loss up to 270 °C for 1–3 and 250 °C for 4. The very minimal weight loss observed till 270 °C (less than 3.00% for 1–3 and till 250 °C) (less than 4.00%) for 4 can be explained by the loss of moisture or methanol molecules from the sample. A gradual loss of organic moieties from the corresponding complex from 270 to 790 °C range was observed for 1 (75.00%), 2 (84.20%) 3 (74.20%), and 4 (69.70%). The final residue weight of 22.00% (observed 17.00%) for the corresponding ZnCl2 of 1, 12.80% (observed 11.00%) for the corresponding ZnS of 2, 22.80% (observed 21.00%) for the corresponding mixture of ZnI2 and ZnS (0.5 equiv. ZnI2 + 0.5 equiv. ZnS) of 3 and in the case of 4, 26.30% (observed 26.30%) for ZnCl2 along with Se.
Photophysical studies
The UV-vis absorption and emission studies of complexes 1–4 were carried out in acetonitrile solution at room temperature (Fig. 4 and Table 1). The complexes show a strong absorption band at around λmax = 253 nm and weak bands at around λmax = 347, 366, and 386 nm. The strong absorption band around λmax = 253 nm can be attributed to the π → π* and the shoulder bands around 347 nm to 386 nm can be ascribed to the n → π* transition. The UV-vis absorption spectra of 1–4 resemble the absorption spectrum of their corresponding ligands i.e.L1 and L2 (see ESI, Fig. S30†). The solution state emission of complexes 1–4 were measured using the same acetonitrile solution at room temperature and all the complexes gave emission at λmax = 418 nm. Since complexes 1–4 possess the same electronic structures, the emission behavior in the solution state is unaffected. | ||
| Fig. 4 Top: UV-vis absorption spectra of 1–4 (C = 1 × 10−5 M) in acetonitrile, bottom: normalized emission and excitation spectra of the same in acetonitrile solution (excited wavelength is 253 nm for all four complexes). | ||
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| Solution state | ||||
| λ ab (nm) | 253, 347, 366, 386 | 253, 347, 366, 386 | 253, 347, 366, 386 | 253, 347, 366, 386 |
| λ ex (nm) | 253 | 253 | 253 | 253 |
| λ em (nm) | 418 | 418 | 418 | 418 |
| CIE coordinates | x = 0.1580 | x = 0.1580 | x = 0.1580 | x = 0.1580 |
| y = 0.0347 | y = 0.0347 | y = 0.0347 | y = 0.0347 | |
| Crystalline state | ||||
| λ ex (nm) | 396 | 395 | 410 | 395 |
| λ em (nm) | 455 | 475 | 535 | 425 |
| τ (ns) | 4.58 | 8.50 | 18.08 | 0.95 |
| Φ F (%) | 7.72 | 5.95 | 5.07 | 3.87 |
| k r (106 s−1) | 16.8 | 7.0 | 2.8 | 40 |
| CIE coordinates | x = 0.1760 | x = 0.2187 | x = 0.3343 | x = 0.1731 |
| y = 0.1964 | y = 0.3417 | y = 0.5292 | y = 0.1025 | |
The crystalline state photoluminescence of the complexes was studied using single crystals of the complexes (Fig. 5 and 6). The crystalline state emission spectra of 1–4 were distinctly different. The emission spectra of 1–4 were considerably sharp. The emission of 3 was slightly broader and red-shifted than those of 1, 2 and 4. Complex 1 emits blue emission (λmax = 455 nm) and 2 was found to be bluish-green emission (λmax = 475 nm). 3 showed emission at λmax = 535 nm, which falls in the yellow region. The emission behavior of complexes 1–3 in the crystalline state is influenced by the molecular aggregation due to the halide ligand. The luminescence in these complexes can be ascribed to the intra-ligand π → π* and n → π* transition and the observed redshift from 1 to 3 can be explained by the molecular aggregation (due to electronic effects of halides, the coordination environment of Zn(II) metal center and intermolecular interactions) present in the crystal packing. Thus, the spectral analysis leaves a significant role in the halide ligand coordinated to the zinc core of the complexes. For the Zn(II) complexes, spectral luminescence emission spectra display the following trend: Cl− > Br− > I−, thus Cl− is the most efficient. This can be explained from the effective increase in the bond rigidity and lower loss of emission through non-radiative processes.33 For the Zn(II) selone complex 4, the emission was found to be at λem = 425 nm, which also emits in the blue region. The Commission Internationale de l'Eclairage (CIE) chromaticity coordinates of the complexes are listed in Table 1 along with all photophysical parameters.
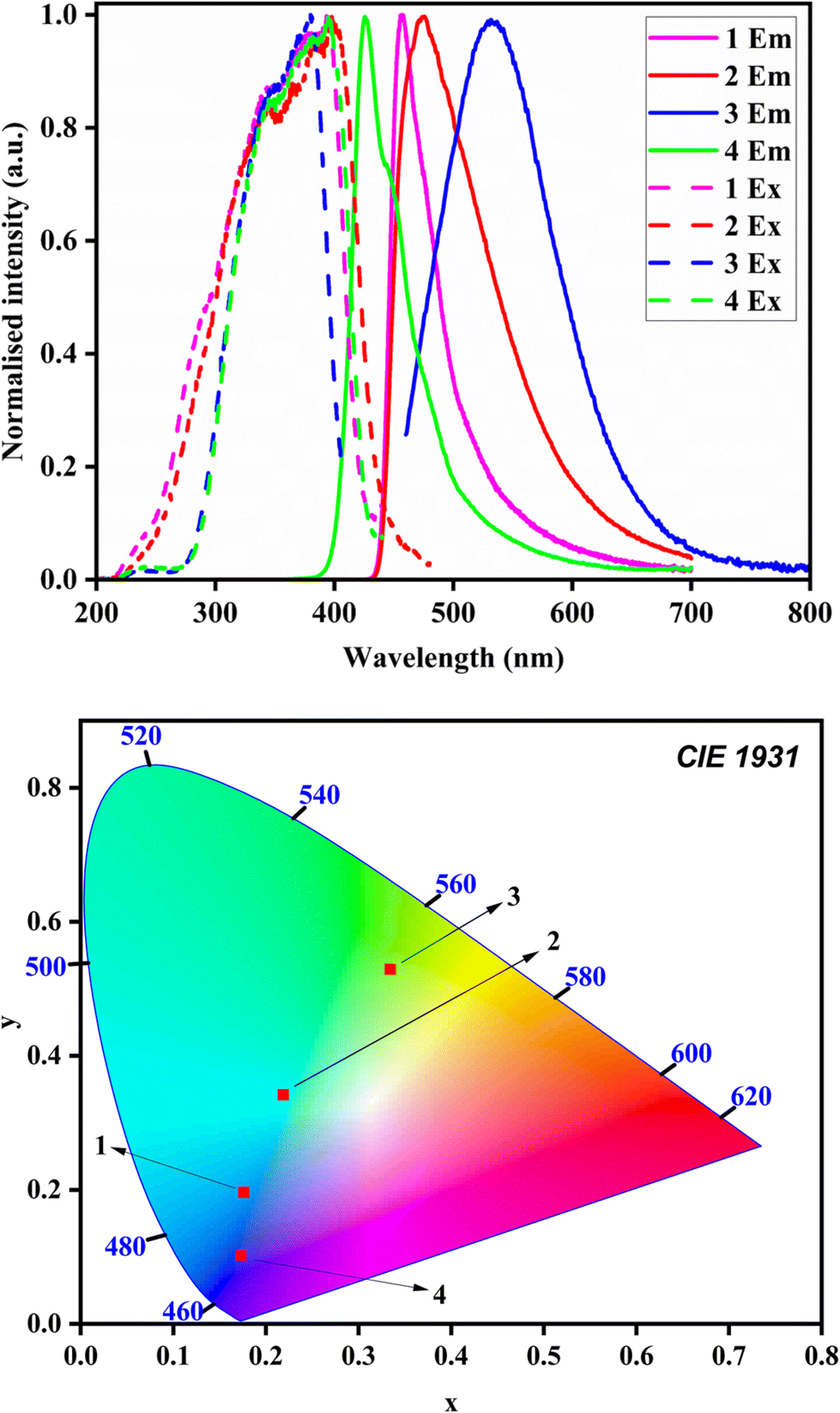 | ||
| Fig. 5 Top: Normalized emission and excitation spectra in the crystalline state, bottom: CIE diagram of emission in the crystalline state. | ||
 | ||
| Fig. 6 Crystal images under ambient light vs. UV light (395 nm). | ||
The crystalline state quantum yield (ΦF) for 1 was found to be 7.70% (λem = 455 nm) which is higher than those of 2 (5.90%, λem = 475 nm) and 3 (5.07%, λem = 535 nm). Thus, 1 displays the highest quantum yield. The quantum yield for 4 is (3.80%, λem = 425 nm) the least among all the complexes owing to the heavy atom effect of selenium and causes lesser luminescence for 4 compared to 1–3.34
However, L1 and L2 were found to be non-emissive in the solution state as well as in the crystalline state, compared to the emission of the complexes (see ESI, Fig. S31†). This confirms that the emission of the complexes arises when the metal center provides rigidity and stability to the complexes, reducing the nonradiative processes and enhancing the emission behavior. Aggregation induced emission study was performed on the complexes in DMF (c = 1 × 10−5 M), which confirmed the aggregation induced emission behaviour of molecules in the solution state when the percentage of water was increased (see ESI, Fig. S34†).
Vertical electron transitions were estimated using time-dependent density functional theory (TD-DFT). The total energy, dipole moment, energy of the HOMO–LUMO gap excitation energies, and oscillator strengths are given in the ESI (see ESI, Tables S6–S9†). The HOMO of 1–4 is predominantly contributed by anthracene moiety present in the system and a small contribution from the S/Se atom. The LUMO is predominantly distributed in the other anthracene moiety, making the local excitation from the ground state to an excited state in π → π* and n → π* transitions (Fig. 3). Most of the primary transitions happen with contributions from HOMO, HOMO−1 to LUMO and LUMO+1. The first absorption maximum is around 391 nm in complex 1 (in the gaseous phase), related to the pairwise degenerate electronic transitions. The degeneracy of S1/S2, S3/S4, S5/S6, and S7/S8 is due to an equivalent ligand on both sides connected to the metal Zn(II) atom. Therefore, the result can be seen with identical energy values between HOMO (−5.31 eV) and HOMO−1 (−5.32 eV). Similar cases happened with HOMO−2 (−5.79 eV) and HOMO−3 (−5.79 eV). A similar trend was also found in complexes 2–4, as the ligand is almost identical to 1. The most intense transition, S0 → S1/S2, can be the facilitated transition. The role of the d-orbital of the zinc atom is minimum in the transitions of HOMO, HOMO−1 to LUMO. The Zn(II) ion helps in complexation with ligands through which stability and rigidity of the whole molecule are enhanced. The enhanced emission properties of these zinc(II) complexes can be attributed to the rigidity of the molecules.
The natural transition orbital (NTO) analysis was used to find the transition density matrix. NTOs gave a simple perspective of the density change from the ground to the excited state by single particle transitions, i.e., holes as occupied and electrons as unoccupied. In the NTO analysis, holes are mainly distributed on anthracene and S/Se atoms in all complexes, whereas electrons are mainly delocalized at the anthracene group. The absorption transition with high oscillatory strength can be a combination of local excitation and 1ILCT states.
To understand the origin of the emission of 1–4, lifetime analysis was carried out using single crystals of these complexes. The photoluminescence lifetime was measured for 1–4 in the crystal face with luminescent bands 455 nm, 475 nm, 535 nm, and 425 nm respectively. The emission decay of all the complexes was on a nanosecond scale. The decay profiles were fitted with a bi-exponential decay for 1–4 (see ESI, Table S2†). The distinct fluorescence decay can be assigned to the electronic transitions between ground levels (S2 → S0, S1 → S0). Thus, the emission of 1–4 in the crystalline state can be explained as due to fluorescence.
LED demonstration
To demonstrate the potential application of these luminescent zinc(II) halide complexes 1–4, we demonstrated the light emitting application using commercial LEDs (λmax = 410 nm) (Fig. 7). THF solution of these complexes along with PMMA (polymethyl methacrylate) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 by weight) was coated on the surface of the commercially available blue LEDs emit (emission wavelength of 410 nm). PMMA was used as a binding agent due to the low optical absorbance to coat the Zn(II) complex on the LED surface. This wavelength (410 nm) was used to excite the complex that was coated on the surface of the LEDs. The emission spectra of the coated LEDs were measured by connecting them to an external power supply (3 V Li-ion battery) and then carefully placing them at the fluorescence spectrometer using a solid-state sample holder. The emission of the coated LEDs was measured (Fig. 7). The emission of the coated LEDs is comparable with crystalline emission. A minor variation in the emission of the coated LEDs is due to the presence of the tint of the blue light emitting from the commercial LED. Complexes 1 and 4 emit in the blue region at 455 nm and 425 nm, respectively, while the blue LED (used for coating) emits at 410 nm. The CIE coordinates for coated LEDs are distinctly different from the commercial LED. The CIE coordinates for the blue LED emission are x = 0.1660, y = 0.0226 while the CIE coordinates for complexes 1 and 4 are x = 0.1635, y = 0.1808, and x = 0.1785, y = 0.1255, respectively. This clearly shows the difference in emission for the complexes as compared to the blue LED used for coating.
:10 by weight) was coated on the surface of the commercially available blue LEDs emit (emission wavelength of 410 nm). PMMA was used as a binding agent due to the low optical absorbance to coat the Zn(II) complex on the LED surface. This wavelength (410 nm) was used to excite the complex that was coated on the surface of the LEDs. The emission spectra of the coated LEDs were measured by connecting them to an external power supply (3 V Li-ion battery) and then carefully placing them at the fluorescence spectrometer using a solid-state sample holder. The emission of the coated LEDs was measured (Fig. 7). The emission of the coated LEDs is comparable with crystalline emission. A minor variation in the emission of the coated LEDs is due to the presence of the tint of the blue light emitting from the commercial LED. Complexes 1 and 4 emit in the blue region at 455 nm and 425 nm, respectively, while the blue LED (used for coating) emits at 410 nm. The CIE coordinates for coated LEDs are distinctly different from the commercial LED. The CIE coordinates for the blue LED emission are x = 0.1660, y = 0.0226 while the CIE coordinates for complexes 1 and 4 are x = 0.1635, y = 0.1808, and x = 0.1785, y = 0.1255, respectively. This clearly shows the difference in emission for the complexes as compared to the blue LED used for coating.
 | ||
| Fig. 7 (i) Emission spectra of LEDs coated with 1–4. (ii) Images of coated LEDs under power on/power off conditions. (iii) CIE diagram of the coated LED emission. | ||
Experimental section
General methods
All the reactions were carried out under argon, using standard Schlenk techniques. Solvents were dried by standard methods and distilled before use. 1-Isopropyl imidazole, anthracene methyl chloride, and ZnX2 were purchased commercially. A Bruker Ultra Shield 400 MHz spectrometer was used to record the NMR spectra at 273.15 K (for 1–3) and 298.15 K (4). A solvent resonance was marked as an internal standard for the chemical shift assessment with respect to Me4Si. FT-IR measurements were carried out using the KBr method on a JASCO FT-IR spectrometer. The UV-vis spectra were recorded on a LAB INDIA 2000U UV-vis spectrophotometer and emission spectra were recorded using a Hitachi F-4700 instrument. Crystals of 1–4 were obtained from the saturated acetonitrile and methanol mixture solution. The crystals were measured by mounting them in the Bruker D8 Venture Single Crystal X-ray diffractometer using the source MoKα (0.71073 Å) at RT. The structures were solved with the help of the Olex2.solve program and the Charge Flipping method. The structures were refined with the Olex2 refinement package using Gauss–Newton minimization.35–37 PXRD data was collected on a Rigaku Ultima IV instrument, and compared with the simulated PXRD pattern of the corresponding SCXRD structures. A thermogravimetric study was carried out for the complexes with the help of the TA-SDT-Q600 instrument.Computational details
All computational studies were carried out using Density Functional Theory (DFT) from CIF files of zinc(II) complexes 1–4 using the Gaussian16 program package.38 Ground state structure optimization and single point energy calculations were calculated with B3LYP functional having LanL2DZ basis set implementing heavier atoms such Zn, Se, Br, and I whereas 6-31G(d,p) for lighter elements such C, H, N, S and Cl.39,40 The natural transition orbital (NTO) was analyzed for various excitations.41S), 131.5 (CAn), 131.0 (CAn), 129.6 (CAn), 129.3 (CAn), 127.6 (CAn), 125.9 (CAn), 124.5 (CAn), 116.5 (ImC), 114.8 (ImC), 48.8 (N–CH2–An), 42.7 (N–CH–Me2), 21.7 (N–CH–CH3). FT-IR (cm−1, KBr): ν = 670 (s), 715 (s), 780 (s), 1005 (m), 1145 (w), 1124 (s), 1329 (m), 1440 (vs), 1475 (vs), 1595 (s), 2974 (w), 3134 (w).
S), 131.5 (CAn), 131.0 (CAn), 129.6 (CAn), 129.3 (CAn), 127.6 (CAn), 125.9 (CAn), 124.5 (CAn), 116.4 (ImC), 114.8 (ImC), 48.8 (N–CH2–An), 42.7 (N–CH–Me2), 21.7 (N–CH–CH3). FT-IR (cm−1, KBr): ν = 535 (m), 674 (s), 729 (vs), 775 (vs), 1015 (m), 1085 (w), 1140 (w), 1229 (vs), 1305 (w), 1345 (s), 1410 (vs), 1450 (vs), 1480 (vs), 1570 (m), 1604 (m), 2989 (w), 3105 (m).
S), 131.5 (CAn), 130.9 (CAn), 129.6 (CAn), 129.3 (CAn), 127.6 (CAn), 125.9 (CAn), 124.5 (CAn), 116.4 (ImC), 114.9 (ImC), 48.8 (N–CH2–An), 42.7 (N–CH–Me2), 21.7 (N–CH–CH3). FT-IR (cm−1, KBr): ν = 550 (m), 679 (m), 745 (s), 780 (s), 1015 (m), 1095 (m), 1150 (w), 1225 (vs), 1310 (m), 1350 (s), 1455 (vs), 1485 (vs), 1570 (m), 1604 (s), 2974 (w), 3099 (s), 3154 (w).
Conclusions
We successfully synthesized the first examples of high quantum yield Zn(II) halide imidazol-2-chalcogenone complexes 1–4 and structurally characterized them. Complex 1 depicted the tetrahedral geometry around the Zn(II) center, while 2, 3, and 4 showed the distorted tetrahedral geometry. Photophysical studies were carried out for all the complexes, where 1 and 4 displayed emissions in the blue region, while 2 emitted bluish-green, and the yellow emission was observed for 3. Complex 1 demonstrated the highest quantum yield (λmax = 455 nm, τ = 4.58 ns, and Φ = 7.70%), whereas complex 4 showed the least quantum yield (λmax = 425 nm, τ = 0.95 ns, and Φ = 3.80%). The efficiency of emission in 1 can be explained by the molecular rigidity. The observed redshift from 1 to 3 can be explained by the crystal packing. As predicted from TD-DFT NTO calculations, intra-ligand charge transfer was found to be the main reason for these observed fluorescent emissions. Due to their very high air and moisture stability, such complexes can be a suitable source for OLED applications.Author contributions
S.M.: conceptualization of the original idea, designing methodology, experiments, analysis of data, and writing manuscript; B.L.: conceptualization of the original idea, designing methodology, experiments, analysis of data, and writing manuscript; G.M.: conceptualization of the original idea, designing methodology, experiments, analysis of data, and writing manuscript; A.R.: experiments, and analysis of data; A.S. and A.R.: experiments, and analysis of data; O.T.: experiments, analysis of data, supervision, fund acquisition, discussion, review and writing manuscript; G.P.: conceptualization of original idea, supervision, Fund acquisition, discussion, review and writing manuscript.Data availability
The data supporting this article, including FT-IR, NMR, TGA, PXRD, PL, LT, as well as solid-state packing, crystal data, structure refinement of the reported molecules, and DFT calculations, are provided in the ESI.† Crystallographic data for compounds 1–4 have been deposited with the CCDC under accession numbers 2392305 (1), 2392306 (2), 2392307 (3), and 2392308 (4).†Conflicts of interest
There are no conflicts to declare.Acknowledgements
G.P. gratefully acknowledges (SERB-CRG P. No: CRG/2022/000714), New Delhi, India, PMRF for financial support. S.M. thanks MoE, G.M. thanks PMRF, and B.L. thanks DST, New Delhi, India, for the fellowships. O.T. thanks JST A-STEP (JPMJTR22T1), JSPS KAKENHI (24K01561, and 24K21799), and the Toshiaki Ogasawara Memorial Foundation for financial support. G.P. and O.T. thank DST-JSPS and JICA-IITH for the financial support. Dedicated to Prof. Dr Vadapalli Chandrasekhar.References
- S. Chang, J. H. Koo, J. Yoo, M. S. Kim, M. K. Choi, D. H. Kim and Y. M. Song, Chem. Rev., 2024, 124, 768–859 CrossRef CAS PubMed.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539–541 Search PubMed.
- J. Kim, J. Roh, M. Park and C. Lee, Adv. Mater., 2024, 36, 2212220 CrossRef CAS PubMed.
- J. Song, H. Lee, E. G. Jeong, K. C. Choi and S. Yoo, Adv. Mater., 2020, 32, 1907539 CrossRef CAS PubMed.
- Y.-Z. Shi, H. Wu, K. Wang, J. Yu, X.-M. Ou and X.-H. Zhang, Chem. Sci., 2022, 13, 3625–3651 RSC.
- S. Kim, H. J. Bae, S. Park, W. Kim, J. Kim, J. S. Kim, Y. Jung, S. Sul, S.-G. Ihn, C. Noh, S. Kim and Y. You, Nat. Commun., 2018, 9, 1211 CrossRef.
- H. Aziz and Z. D. Popovic, Chem. Mater., 2004, 16, 4522–4532 CrossRef CAS.
- K. Li, Y. Chen, J. Wang and C. Yang, Coord. Chem. Rev., 2021, 433, 213755 CrossRef CAS.
- X. Zhang, Y. Hou, X. Xiao, X. Chen, M. Hu, X. Geng, Z. Wang and J. Zhao, Coord. Chem. Rev., 2020, 417, 213371 Search PubMed.
- W. Christina and S. W. Oliver, JACS Au, 2021, 1, 1860–1876 CrossRef.
- H. Amouri, Chem. Rev., 2023, 123, 230–270 CrossRef CAS PubMed.
- V. Ferraro, C. Bizzarri and S. Bräse, Adv. Sci., 2024, 2404866 CrossRef CAS.
- T. Yi Li, S.-J. Zheng, P. I. Djurovich and M. E. Thompson, Chem. Rev., 2024, 124, 4332–4392 CrossRef CAS.
- (a) R. Diana and B. Panunzi, Molecules, 2020, 25, 4984 CrossRef CAS PubMed; (b) S. Sheokand, S. Sharma, M. A. Mohite, G. Rajaraman and M. S. Balakrishna, Chem. Commun., 2024, 60, 6055–6058 RSC; (c) A. Upadhyay, K. V. Saurav, E. L. Varghese, A. S. Hodage, A. Paul, M. K. Awasthi, S. K. Singh and S. Kumar, RSC Adv., 2022, 12, 3801–3808 RSC.
- K. Singh, I. Siddiqui, V. Sridharan, R. A. Kumar Yadav, J.-H. Jou and D. Adhikari, Inorg. Chem., 2021, 60, 19128–19135 CrossRef CAS PubMed.
- O. Mrózek, M. Mitra, B. Hupp, A. Belyaev, N. Lüdtke, D. Wagner, C. Wang, O. S. Wenger, C. M. Marian and A. Steffen, Chem. – Eur. J., 2023, 29, e202203980 Search PubMed.
- M. Putscher and C. M. Marian, J. Phys. Chem. A, 2023, 127, 8073–8082 Search PubMed.
- V. W.-W. Yam and K. K.-W. Lo, Chem. Soc. Rev., 1999, 28, 323–334 Search PubMed.
- D. Temerova, K. S. Kisel, T. Eskelinen, A. S. Melnikov, N. Kinnunen, P. Hirva, J. R. Shakirova, S. P. Tunik, E. V. Grachova and I. O. Koshevoy, Inorg. Chem. Front., 2021, 8, 2549–2560 Search PubMed.
- F. Kobayashi, Y. Takatsu, D. Saito, M. Yoshida, M. Kato and M. Tadokoro, Inorg. Chem., 2024, 63, 15323–15330 Search PubMed.
- A. Monkman, ACS Appl. Mater. Interfaces, 2022, 14, 20463–20467 CrossRef CAS.
- (a) J. Yang, Q. Guo, J. Wang, Z. Ren, J. Chen, Q. Peng, D. Ma and Z. Li, Adv. Opt. Mater., 2018, 6, 1800342 CrossRef; (b) A. N. Gusev, M. A. Kiskin, E. V. Braga, M. A. Kryukova, G. V. Baryshnikov, N. N. Karaush-Karmazin, V. A. Minaeva, B. F. Minaev, K. Ivaniuk, P. Stakhira, H. Ågren and W. Linert, ACS Appl. Electron. Mater., 2021, 3, 3436–3444 CrossRef CAS; (c) H. Shi, W. Yao, W. Ye, H. Ma, W. Huang and Z. An, Acc. Chem. Res., 2022, 55, 3445–3459 CrossRef CAS.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 Search PubMed.
- B. Chen, B. Liu, J. Zeng, H. Nie, Y. Xiong, J. Zou, H. Ning, Z. Wang, Z. Zhao and B. Z. Tang, Adv. Funct. Mater., 2018, 28, 1803369 Search PubMed.
- M. Mannarsamy, M. Nandeshwar, G. Muduli and G. Prabusankar, Chem. – Asian J., 2022, 17, e202200594 CrossRef CAS PubMed.
- M. Banerjee, R. Karri, K. S. Rawat, K. Muthuvel, B. Pathak and G. Roy, Angew. Chem., Int. Ed., 2015, 54, 9323–9327 Search PubMed.
- C. N. Babu, K. Srinivasa and G. Prabusankar, Dalton Trans., 2016, 45, 6456–6465 RSC.
- S. Yadav, S. T. Manjare, H. B. Singh and R. J. Butcher, Dalton Trans., 2016, 45, 12015–12027 RSC.
- G. Prabusankar, G. Raju, M. Vaddamanu, N. Muthukumaran, A. Sathyanarayana, S. Nakamura, Y. Masaya, K. Hisano, O. Tsutsumi, C. Biswas and S. S. K. Raavi, RSC Adv., 2019, 9, 14841–14848 RSC.
- S. Mandal, D. Harijan, G. Muduli, K. Subramaniyam, A. K. Rengan and G. Prabusankar, New J. Chem., 2023, 47, 15027–15035 RSC.
- M. Vaddamanu, K. Velappan and G. Prabusankar, New J. Chem., 2020, 44, 3574–3583 Search PubMed.
- H. Lee, T. H. Noh and O.-S. Jung, Dalton Trans., 2014, 43, 3842–3849 RSC.
- G. C. Hoover and D. S. Seferos, Chem. Sci., 2019, 10, 9182–9188 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3–8 Search PubMed.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 59–75 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 272–283 Search PubMed.
- R. L. Martin, Natural Transition Orbitals, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: FTIR, NMR, UV-vis, PL, LT, TGA, SCXRD, PXRD, DFT, TD-DFT. CCDC 2392305 (1), 2392306 (2), 2392307 (3) and 2392308 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02924k |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2025 |