
Functional biomimetics for copper oxidases: interesting catalytic promiscuity of novel monocopper(II) complexes†
Vigneswara Chellam
Ravisankar
a,
Balasubramaniam
Selvakumaran
 a,
Tamilarasan
Ajaykamal
b and
Mariappan
Murali
*a
a,
Tamilarasan
Ajaykamal
b and
Mariappan
Murali
*a
aCoordination and Bioinorganic Chemistry Research Laboratory, Department of Chemistry, National College (Autonomous) affiliated to Bharathidasan University, Tiruchirappalli 620 001, Tamil Nadu, India. E-mail: murali@nct.ac.in; ma66mu@yahoo.co.in
bSchool of Chemistry, Bharathidasan University, Tiruchirappalli 620 024, Tamil Nadu, India
First published on 10th April 2025
Abstract
Employing H(L1) [2-((pyridine-2-ylmethyl)imino)methylphenol] or H(L2) [2-((pyridine-2-ylethyl)imino)methylphenol] and phen (1,10-phenanthroline), two novel monocopper(II) complexes, [Cu(L1/L2)(phen)](ClO4) (1 or 2), have been produced and studied. The single-crystal structure of the complex ion in 2, as determined by X-ray structure analysis, shows a trigonal bipyramidal geometry with distortion (τ, 0.65). DFT calculations were used to investigate the molecular geometry of copper(II) complexes in solution as well as their electronic characteristics. The electronic and EPR spectra in the solid-state of 1 and 2 reveal a trigonal bipyramidal geometry, whereas the geometry in solution is square pyramidal. The positive and reversible nature of the redox pair (CuII/CuI) makes redox states easily interconvertible. The catalysts in methanol and/or the buffer induced three separate chemical changes: (i) ascorbic acid → dehydroascorbic acid, (ii) benzylamine → benzaldehyde, and (iii) 3,5-di-tert-butylcatechol → 3,5-di-tert-butylquinone. Their kcat results show higher activities of amine oxidase (105 h−1). Ascorbate oxidase (107 h−1) and catechol oxidase (106 h−1) activity in the buffer yields kcat values that are closer to those of the natural enzyme. This is due to the presence of ligand flexibility, structural distortion, an appropriate chelate ring size, a labile donor, a positive redox potential, and a persistent catalyst–substrate interaction. Therefore, the two monocopper(II) complexes serve as the most efficient promiscuous catalysts, acting as complementary agents to the activity of copper oxidase enzymes and superior models for oxidation processes.
Introduction
Organic synthesis relies heavily on oxidation processes, which are crucial for endowing valuable molecules with the functionality they require, like drugs, agricultural chemical products, and other valuable substances.1–3 The perfect oxidant is molecular oxygen, which can be taken simply from the air and is both affordable and safe for the environment. Stoichiometric proportions of inorganic oxidants used in traditional oxidation processes are hazardous and contribute to contamination in the environment. For this reason, oxidations involving a catalytic dose of an activator possess the potential for activating molecular oxygen with the least amount of chemical loss. Since molecular oxygen is kinetically inert, it is difficult to make it active for applications involving oxidation processes. One of the main issues with employing dioxygen in chemical change happens to be that it is difficult to manage its reactivity, which frequently results in poor selectivity and over-oxidation.4 Through the use of transition metals integrated into proteins, known as “metalloenzymes”, nature discovered a sophisticated way to get over the kinetic hurdle of dioxygen being activated.5,6 By developing oxygen activation catalysts, which function as miniature molecule mimics of metalloenzymes and aid in the understanding of mechanistic processes, inorganic chemists have largely capitalized on the idea of nature. The donor sites of enzymes are modeled using ligands, which are tiny molecules that are mixed with metals to generate complexes that are examined as structural and functional models. This is done by applying the understanding of coordination chemistry, electronic parameters, and redox potential.5,7,8 The catalytic activity of the majority of metal complexes is generally quite promising. Several of these complexes would not be considered extremely effective catalysts since they cannot efficiently perform that particular organic oxidative reaction.9 Despite their seeming inefficiency, several of the complexes are unquestionably very helpful in giving us invaluable knowledge into the significant mechanistic elements of the metalloenzymes. Over the past few decades, bioinorganic chemists have focused on developing catalytic reactions with the goal of comprehending and simulating the enzymatic functions of metalloenzymes.Copper ions are essential for enzymatic catalysis of redox processes and are used by the majority of enzymes that employ molecular dioxygen. Members of copper proteins (‘Type-2’ and ‘Type-3’) that have received less attention are ascorbate oxidase (AOase), amine oxidase (AmOase), and catechol oxidase (COase). The copper site in AOase oxidizes ascorbic acid to dehydroascorbate using dioxygen. One of its three copper center types is ‘Type-2’ (A∥ > 140 × 10−4 cm−1), which is part of the functional trinuclear cluster,7,10 being coordinated to two histidine molecules and a water molecule. Electron transfer is an essential action of the ‘Type-2’ copper center, and ascorbate functions as an electron donor under physiological conditions of the reaction. Dioxygen is activated by the reduced copper(I) sites and transformed into water by AOase11 using four electrons and four protons. An important two electron enzymatic process that undergoes conversion of amine to aldehyde (RCH2NH2 → RCHO + NH3) is the deamination reaction, which includes the ‘Type-2’ copper centers in AmOase.8,12 It manifests as homodimers and exhibits a square pyramidal shape at the active site. A N3O2 chromophore comprising two O-donor sites13,14 and being labile is formed by the presence of three imidazole and two water molecules at the copper(II) site. A frequently occurring enzyme in plants, insects, and crustaceans15 is COase, a ‘Type-3’ copper protein. It consists of a hydroxo bridged dicopper(II) center, where each copper(II) center is coordinated by three histidine residues (two nitrogens equatorially and one nitrogen axially) and takes on a nearly trigonal pyramidal structure.16 In the presence of dioxygen, it catalyzes the oxidation of catechol to o-quinone (highly reactive).17–19 The auto-polymerization of o-quinone results in the synthesis of melanin. Among its various defensive functions, it protects the injured tissues from insects and pathogens.20
The importance of metals in a variety of biochemical reactions is the main reason for the intense interest in the production of unique metal complexes with creative structures and reactivity in diverse biological scenarios. Consequently, the structural characteristics of the metalloenzyme active sites serve as an inspiration for the molecular synthesis of several new tiny inorganic metal complexes. The fundamental objective of these complexes, which are known to be biomimetic, is to convert structural similarities with biological originals into functional ones.21 A variety of biological activities, such as electron transport or the oxidation of different organic substrates, employ the copper ions at the active site of proteins as redox catalysts. This modeling investigation led to an important comprehension of the structural and functional characteristics of copper oxidases.17 Even while many dicopper(II) complexes made from N/O donor dinucleating ligands are successful at replicating the structure and function of the enzyme,18,19,22 the copper oxidase activity of numerous monocopper(II) complexes is likewise well known. The established structure-property relationships are only suitable for a limited number of molecules. In order for ‘Type-2’ and ‘Type-3’ copper oxidases to exhibit their functional characteristics during the catalytic process, it was noted that the Cu⋯Cu separation in dicopper(II) complexes must be smaller than 5 Å. In contrast, it is crucial to examine the oxygen transfer mechanism in AOase and AmOase using mixed-ligand monocopper(II) complexes, which provide a 4 + 1 square-pyramidal geometry that includes labile binding site(s).20,23,24 However, for monocopper(II) complexes to become functional in COase activity, a non-planar shape about copper(II) is required.25 The turnover numbers (kcat) of the majority of the replicas are rather small in comparison with the enzymes, which is an important disadvantage of the model complexes. In light of this information, it is important to investigate whether novel varieties of monocopper(II) compounds can demonstrate copper oxidase activity in relation to the ligand flexibility, exogenous ligand type, and coordination structure surrounding the metal ion.
Enzymatic catalysis tends to be very efficient when the active site of an enzyme is accurately optimized towards a particular reaction. The ability of an enzyme to do numerous operations or enzymatic promiscuity is currently being sought after as a desirable quality since it facilitates use in biocatalysis and biotechnology. Enzymatic promiscuity encompasses three main aspects: (i) substrate: ‘enzymes with a broad or relaxed specificity for substrates’, (ii) condition: ‘catalytic activity under different reaction conditions than those found in nature, such as high temperatures, pH levels, or anhydrous media’, and (iii) catalytic promiscuity: ‘enzymes that catalyze unique chemical changes with various transition states’. The two further categories of catalytic promiscuity26 are (a) induced: ‘a novel reaction arising from one or more mutations that diverge the reaction in which the wild-type enzyme was catalyzing’, and (b) accidental: ‘an additional reaction that the wild-type enzyme catalyzes’. However, using native enzymes can occasionally be quite costly; using functionally mimicking substances could be frequently a superior cost-effective option. As models, complexes consist of a simpler structure than enzymes and offer the potential to exhibit greater catalytic promiscuity, or the ability to facilitate a wider range of reactions through catalysis.24,27
In the current study, we have produced mixed-ligand, green-colored monocopper(II) complexes of the general formula [Cu(L1/L2)(phen)](ClO4) (1 or 2) {where H(L1) and H(L2), the Schiff bases [condensation of salicylaldehyde and 2-picolylamine, H(L1) or (2-(2-aminoethyl)pyridine), H(L2)]; phen, 1,10-phenanthroline} (Scheme 1). They vary in their degree of flexibility due to changes within the chelate ring members that occur between imine and pyridine nitrogens of coordinated Schiff base ligands. Using X-ray investigations of a single crystal, we have identified the solid-state structure of 2. The spectroscopic and electrochemical characteristics were assessed when the chelate ring members were modified from methylene (–CH2–) to ethylene (–CH2–CH2–). The solution geometry of 1 or 2 was established in order to comprehend the structure–reactivity correlation. Their potential promiscuous catalytic activities have been tested through spectroscopic and electrochemical methods using a model substrate, ascorbic acid (H2A) or benzylamine (Ph–CH2–NH2) or 3,5-di-tert-butylcatechol (3,5-DTBC), with the aim of mimicking the functional sites of AOase, AmOase and COase.
 | ||
| Scheme 1 Structure of ligands (tridentate NNO and bidentate NN donors). | ||
Experimental
Materials
Salicylaldehyde, 2-picolylamine, 2-(2-aminoethyl)pyridine, 1,10-phenanthroline, sodium perchlorate, copper(II) acetate monohydrate, 3,5-di-tert-butylcatechol, tetra-N-butylammonium perchlorate (Aldrich), potassium iodide, ammonium molybdate, hydrogen peroxide (30% w/v) solution, silica gel (Merck), L-ascorbic acid (Fisher Scientific), and benzylamine (Avra) were used as received. Disodium hydrogen phosphate (Merck), sodium dihydrogen phosphate (Merck), and NaOH (Merck) were used for the preparation of phosphate buffer solutions (pH, 5.0–8.0). Phthalate (pH, 4.0), borate (pH, 9.0) and ammonia (pH, 10.0) were obtained from SRL. Solvents like dichloromethane, methanol (HPLC), hexane, N,N-dimethylformamide, acetonitrile (HPLC), diethylether (AR), and ethyl acetate were purchased from Merck.Buffer solutions
Phthalate (pH 4.0), phosphate (pH 5.0–8.0), borate (pH 9.0), and ammonia (pH 10.0) were used as buffer solutions to examine the pH effect. The concentration of the buffer solution was 0.1 M despite the ionic strength. Sodium phosphate monobasic and sodium phosphate dibasic were dissolved in deionized water, and the pH was then adjusted between 5 and 8 using NaOH (1 M). The final volume (250 mL) was reached by using deionized water. The buffer solution was filtered using a 0.22 μm membrane filter and kept at 4 °C before use. After being prepared and standardized, the buffer solution was used for three days. The buffer solutions used in this investigation were measured for pH using a Thermo Scientific EUTECH pH meter with an ATC electrode and a pH resolution of 0.01 for the pH range of 0 to 14. Before each set of readings, the device was calibrated using standard pH buffers (pH 4.0, 7.0, and 10.0) to guarantee accuracy. With automatic temperature adjustment enabled, all pH measurements were obtained at room temperature (25 ± 1 °C). The purchased buffer solutions were prepared using potassium hydrogen phthalate (phthalate, pH 4.0), boric acid, sodium tetraborate and sodium chloride (borate, pH 9.0), and ammonia chloride and ammonia (ammonia, pH 10.0).Physical methods
Elements C, H, and N were analyzed using a Vario EL III elemental analyzer. Using an EQUIPTRONICS EQDCMP bridge, molar conductivity measurements in methanol were performed at 25 °C (solute concentration: 1 × 10−3 M). A JMS-T100LC spectrometer was used to record the ESI-MS spectra. An HRMS Exactive Plus EMR spectrometer was used to perform high-resolution mass spectrometry. With a PerkinElmer Spectrum Two FTIR spectrophotometer, infrared spectra were recorded in the 4000–400 cm−1 region using KBr pellets. A PerkinElmer Lambda 365 was used to record diffuse reflectance spectra, and a 365+ UV-VIS spectrophotometer attached to a refrigerated circulating water bath (25 ± 0.2 °C) using cuvettes (path length, 1 cm) to capture solution electronic absorption spectra in the 200–1100 nm range. The X-band EPR spectra of the solid at ambient temperature and the solution at 77 K were acquired using a JEOL JES-FA200 ESR spectrometer at an X-band frequency (9–10 GHz) with 100 kHz field modulation. A Bruker 400 MHz AVANCE III HD NMR spectrometer was used to record the 1H NMR spectral data (CDCl3) for the ligands and products separated from catalytic reactions. A CHI 620C electrochemical analyzer operating at 25 ± 0.2 °C was used to evaluate the redox potentials of copper(II) complexes 1 and 2 (0.001 M) in methanol with TBAP (0.1 M) as the supporting electrolyte. Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were employed. The standard configuration of three electrodes, such as the working electrode (glassy carbon; A, 0.0707 cm2), counter electrode (platinum wire), and reference electrode (saturated calomel), was employed. The potential varied between −1 and +1 V at scan speeds of 0.01–0.1 V s−1. Before measurements, N2 was quickly bubbled into the solution to eliminate oxygen. Using the formula E1/2 = (Epc + Epa)/2 and ΔEp = Epa − Epc, redox data have been calculated.Synthesis of Schiff base ligands
A solution of salicylaldehyde (3.1 g, 0.025 mol) in methanol (10 mL) was added, under constant stirring, to a solution of 2-picolylamine (2.7 g, 0.025 mol) or 2-(2-aminoethyl)pyridine (3.1 g, 0.025 mol) in methanol (30 mL). The reaction mixture was refluxed at 70 °C for 2 h and cooled to room temperature. The solution was rotaevaporated to dryness, and the yellow residue was extracted with dichloromethane. The organic layer was dried over magnesium sulfate, filtered and dried in a vacuum. The residue was chromatographed through silica gel (dichloromethane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane = 1:3 v/v) to give a yellow oil of H(L1) or H(L2).
:hexane = 1:3 v/v) to give a yellow oil of H(L1) or H(L2).
Synthesis of copper(II) complexes
1,10-Phenanthroline (0.18 g, 1 mmol) dissolved in 10 mL of methanol was added drop by drop to a 10 mL methanol solution of copper(II) acetate monohydrate (0.20 g, 1 mmol) and stirred for 1 h at 25 °C. After the appearance of a blue solution, a 10 mL methanol solution of H(L1) (0.21 g, 1 mmol) or H(L2) (0.23 g, 1 mmol) was added, and the resultant green solution was refluxed for 3 h. A green crystalline precipitate formed upon the addition of 3 mL methanol solution of sodium perchlorate (0.12 g, 1 mmol). The greenish crystalline powder was collected by filtration and dried in a vacuum under pressure to give 0.32 g or 0.38 g of compound [Cu(L1)(phen)](ClO4) (1) or [Cu(L2)(phen)](ClO4) (2).![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1586 νpy(CN), 1228 ν(Cph–O), 1090, 623 ν(ClO4−) 475 ν(Cu–O), 513 ν(Cu–N). Electronic spectrum in solid/MeOH [λmax/nm (εmax/dm3 mol−1 cm−1)]: 709/688 (80), 932 (40), 456 sh (250), 342 (6325), 326 (10495), 295 (17870), 267 (24430). Electronic spectrum in MeOH:H2O (4:1 v/v) [λmax/nm (εmax/dm3 mol−1 cm−1)]: 684 (70), 931 (40), 456 sh (250), 343 (5810), 326 (10020), 321 (11500), 295 (20460), 270 (33400). Polycrystalline EPR spectrum (RT): g⊥ = 2.054, g∥ = 1.967. EPR spectrum in solution (77 K): g∥ = 2.239, g⊥ = 2.051, A∥ = 176 × 10−4 cm−1, g∥/A∥ = 127 cm, G = 4.7, α2 = 0.79, β2 = 0.66, γ2 = 0.65, K∥ = 0.72, K⊥ = 0.65 (DMF, 1 × 10−2 M); giso = 2.044 (MeOH, 1 × 10−2 M); giso = 2.027 (MeCN, 1 × 10−2 M); g∥ = 2.255, g⊥ = 2.056, A∥ = 169 × 10−4 cm−1, g∥/A∥ = 133 cm, G = 4.5 (MeOH, 2.9 × 10−5 M); g∥ = 2.264, g⊥ = 2.052, A∥ = 165 × 10−4 cm−1, g∥/A∥ = 137 cm, G = 5.1 (buffer solution, 2.9 × 10−5 M). Redox behavior in MeOH (0.1 M TBAP): CV, E1/2 = 0.045 V, ΔEp = 90 mV, ipa/ipc = 1.1, D = 8.4 × 10−6 cm2 s−1; DPV, E1/2 = 0.044 V. Redox behavior in aqueous MeOH (0.1 M TBAP): CV, E1/2 = −0.028 V, ΔEp = 83 mV, ipa/ipc = 1.0, D = 7.8 × 10−6 cm2 s−1; DPV, E1/2 = −0.032 V.
N), 1588 νpy(CN), 1231 ν(Cph–O), 1090, 623 ν(ClO4−), 463 ν(Cu–O), 515 ν(Cu–N). Electronic spectrum in solid/MeOH [λmax/nm (εmax/dm3 mol−1 cm−1)]: 670/670 (100), 915 (60), 469 sh (230), 372 (6230), 295 sh, 266 (22200). Electronic spectrum in MeOH:H2O (4:1 v/v) [λmax/nm (εmax/dm3 mol−1 cm−1)]: 670 (90), 912 (62), 467 sh (240), 368 (6160), 294 sh, 266 (21810). Polycrystalline EPR spectrum (RT): g⊥ = 2.034, g∥ = 1.956. EPR spectrum in solution (77 K): g∥ = 2.238, g⊥ = 2.050, A∥ = 173 × 10−4 cm−1, g∥/A∥ = 129 cm, G = 4.7, α2 = 0.78, β2 = 0.66, γ2 = 0.65, K∥ = 0.72, K⊥ = 0.65 (DMF, 1 × 10−2 M); giso = 2.020 (MeOH, 1 × 10−2 M); giso = 2.031 (MeCN, 1 × 10−2 M); g∥ = 2.253, g⊥ = 2.048, A∥ = 172 × 10−4 cm−1, g∥/A∥ = 131 cm, G = 5.3 (MeOH, 2.9 × 10−5 M); g∥ = 2.253, g⊥ = 2.048, A∥ = 168 × 10−4 cm−1, g∥/A∥ = 134 cm, G = 5.3 (buffer solution, 2.9 × 10−5 M). Redox behavior in MeOH (0.1 M TBAP): CV, E1/2 = 0.044 V, ΔEp = 88 mV, ipa/ipc = 1.1, D = 7.6 × 10−6 cm2 s−1; DPV, E1/2 = 0.035 V. Redox behavior in aqueous MeOH (0.1 M TBAP): CV, E1/2 = −0.073 V, ΔEp = 106 mV, ipa/ipc = 1.1, D = 6.8 × 10−6 cm2 s−1; DPV, E1/2 = −0.060 V.
N), 1586 νpy(CN), 1228 ν(Cph–O), 1090, 623 ν(ClO4−) 475 ν(Cu–O), 513 ν(Cu–N). Electronic spectrum in solid/MeOH [λmax/nm (εmax/dm3 mol−1 cm−1)]: 709/688 (80), 932 (40), 456 sh (250), 342 (6325), 326 (10495), 295 (17870), 267 (24430). Electronic spectrum in MeOH:H2O (4:1 v/v) [λmax/nm (εmax/dm3 mol−1 cm−1)]: 684 (70), 931 (40), 456 sh (250), 343 (5810), 326 (10020), 321 (11500), 295 (20460), 270 (33400). Polycrystalline EPR spectrum (RT): g⊥ = 2.054, g∥ = 1.967. EPR spectrum in solution (77 K): g∥ = 2.239, g⊥ = 2.051, A∥ = 176 × 10−4 cm−1, g∥/A∥ = 127 cm, G = 4.7, α2 = 0.79, β2 = 0.66, γ2 = 0.65, K∥ = 0.72, K⊥ = 0.65 (DMF, 1 × 10−2 M); giso = 2.044 (MeOH, 1 × 10−2 M); giso = 2.027 (MeCN, 1 × 10−2 M); g∥ = 2.255, g⊥ = 2.056, A∥ = 169 × 10−4 cm−1, g∥/A∥ = 133 cm, G = 4.5 (MeOH, 2.9 × 10−5 M); g∥ = 2.264, g⊥ = 2.052, A∥ = 165 × 10−4 cm−1, g∥/A∥ = 137 cm, G = 5.1 (buffer solution, 2.9 × 10−5 M). Redox behavior in MeOH (0.1 M TBAP): CV, E1/2 = 0.045 V, ΔEp = 90 mV, ipa/ipc = 1.1, D = 8.4 × 10−6 cm2 s−1; DPV, E1/2 = 0.044 V. Redox behavior in aqueous MeOH (0.1 M TBAP): CV, E1/2 = −0.028 V, ΔEp = 83 mV, ipa/ipc = 1.0, D = 7.8 × 10−6 cm2 s−1; DPV, E1/2 = −0.032 V.
N), 1588 νpy(CN), 1231 ν(Cph–O), 1090, 623 ν(ClO4−), 463 ν(Cu–O), 515 ν(Cu–N). Electronic spectrum in solid/MeOH [λmax/nm (εmax/dm3 mol−1 cm−1)]: 670/670 (100), 915 (60), 469 sh (230), 372 (6230), 295 sh, 266 (22200). Electronic spectrum in MeOH:H2O (4:1 v/v) [λmax/nm (εmax/dm3 mol−1 cm−1)]: 670 (90), 912 (62), 467 sh (240), 368 (6160), 294 sh, 266 (21810). Polycrystalline EPR spectrum (RT): g⊥ = 2.034, g∥ = 1.956. EPR spectrum in solution (77 K): g∥ = 2.238, g⊥ = 2.050, A∥ = 173 × 10−4 cm−1, g∥/A∥ = 129 cm, G = 4.7, α2 = 0.78, β2 = 0.66, γ2 = 0.65, K∥ = 0.72, K⊥ = 0.65 (DMF, 1 × 10−2 M); giso = 2.020 (MeOH, 1 × 10−2 M); giso = 2.031 (MeCN, 1 × 10−2 M); g∥ = 2.253, g⊥ = 2.048, A∥ = 172 × 10−4 cm−1, g∥/A∥ = 131 cm, G = 5.3 (MeOH, 2.9 × 10−5 M); g∥ = 2.253, g⊥ = 2.048, A∥ = 168 × 10−4 cm−1, g∥/A∥ = 134 cm, G = 5.3 (buffer solution, 2.9 × 10−5 M). Redox behavior in MeOH (0.1 M TBAP): CV, E1/2 = 0.044 V, ΔEp = 88 mV, ipa/ipc = 1.1, D = 7.6 × 10−6 cm2 s−1; DPV, E1/2 = 0.035 V. Redox behavior in aqueous MeOH (0.1 M TBAP): CV, E1/2 = −0.073 V, ΔEp = 106 mV, ipa/ipc = 1.1, D = 6.8 × 10−6 cm2 s−1; DPV, E1/2 = −0.060 V.
Single-crystal X-ray structure determination
The green crystalline powder (2) was again dissolved in a methanol:acetonitrile mixture (1 mM solution) and taken in a test tube with diethyl ether (1:1, v/v) for slow evaporation at 5 °C. Suitable crystals for X-ray analysis were obtained from the solution after 8 days. A single-crystal of 2 with dimensions of 0.29 × 0.26 × 0.22 (2) mm3 was selected for viewing under a polarizing microscope and mounted on glass fiber. X-ray data were collected on a Rigaku Saturn 724 CCD diffractometer with Mo-Kα radiation (λ = 0.71075 Å) at 150 K under the continuous flow of cooled nitrogen gas. The Rigaku Crystalclear software was used for data collection and integration. Absorption corrections were carried out using a multi-scan/numerical method. The crystal structure was solved by direct methods using SIR-9228 and refined by full-matrix least-squares calculation using SHELXL version 2016/4.29 All the calculations were carried out using the programs in the WinGX module.30 All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were refined isotropically as rigid atoms in their idealized locations. ORTEP3 software for Windows30 was used to describe graphically the molecule using 50% probability displacement ellipsoids. The details of the crystallographic data are presented in Table 1. Crystallographic data in CIF format were deposited with the Cambridge Crystallographic Data Centre (CCDC-2390596).
| 2 | |
|---|---|
| Formula | C26H21N4O5ClCu |
| Formula weight | 568.46 |
| Temperature (K) | 150(2) |
| Wavelength (Å) | 0.71075 |
| Crystal system | Monoclinic |
| Space group | P21/n |
| a (Å) | 9.547(2) |
| b (Å) | 17.906(3) |
| c (Å) | 14.077(3) |
| α (°) | 90 |
| β (°) | 107.131(5) |
| γ (°) | 90 |
| V (Å)3, Z | 2299.7(8), 4 |
| D calc (g cm−3) | 1.642 |
| μ (mm−1) | 1.115 |
| F(000) | 1164 |
| Crystal size (mm) | 0.29 × 0.26 × 0.22 |
| θ (°) | 2.733–24.996 |
| Index ranges | −11 ≤ h ≤ 11, −21 ≤ k ≤ 20, −11 ≤ l ≤ 16 |
| Reflections collected | 16803 |
| Independent reflections | 4034 |
| Reflections observed [I > 2σ(I)] | 3897 |
| R int | 0.0233 |
| GOOF | 1.007 |
| R 1 [I > 2σ(I)] | 0.0272 |
| wR2 [I > 2σ(I)] | 0.0850 |
| R 1/wR2 all data | 0.0281/0.0859 |
Experimental procedure for ascorbate oxidase (AOase) activity
By reacting the copper(II) complexes (1 or 2; 3 × 10−3 M) and varying concentrations of ascorbic acid (H2A), the ascorbate oxidase activity of the complexes was measured. The studies carried out under an aerobic atmosphere with a MeOH:H2O (4:1 v/v) solution (MS) or buffer solution (BS)31 were monitored using UV-Vis spectroscopy. To ascertain the ascorbate oxidase activity of complexes, 60 equivalents of ascorbic acid (H2A) were added to a 1.667 × 10−6 M complex (1 or 2) solution in MS (pH 7.3) or BS (pH 7.6) in an aerobic environment at 25 °C. The UV spectra of the solution were recorded at regular intervals of 1 min after the addition. Over the first 30 min, the decrease in the absorption value at ∼268 nm was observed as a function of time. In an aerobic environment, the MS or BS of copper complexes (1.667 × 10−6 M) were treated with 20–140 equivalents of H2A to ascertain the dependence of the rates on the concentration of the substrate and other kinetic parameters.32
Experimental procedure for amine oxidase (AmOase) activity
The conversion of benzylamine (Ph–CH2–NH2) to benzaldehyde (Ph–CHO) occurs as a consequence of the catalytic reaction between 1 or 2 (1 × 10−3 M) and Ph–CH2–NH2 (1 × 10−1 M) in 10 mL of MeOH:H2O (4:1 v/v)33 in a H2O2 (1 mL of 30% w/v) driven deamination process. In reactions involving Ph–CH2–NH2 and H2O2 alone or in the absence of the complex, no appreciable Ph-CHO was produced. After the methanol was removed by evaporation, diethyl ether was used as a solvent to extract organic substances from the reaction mixture. A rotary evaporator was used to remove the diethyl ether, and the residue was dissolved respectively in CDCl3 and MeOH for 1H NMR and HRMS analyses. Using spectrophotometry, the aqueous methanolic solution (3.3 × 10−7 M) of complexes was subjected to sequential additions of 2–12 equivalents of Ph–CH2–NH2 and 50 equivalents of H2O2. The reaction was monitored for a 60 second interval. Because of the increasing concentration of benzaldehyde (Ph–CHO), there is a progressive increase in the absorbance band at ∼252 nm as the reaction proceeds. The rate of the reaction was determined using the initial rate method, and the kinetic parameters were estimated using the Michaelis–Menten approach.
Experimental procedure for catechol oxidase (COase) activity
By treating a copper(II) complex (1 or 2) solution (2.9 × 10−5 M) in CH3OH (MS) or a buffer solution (BS) at pH 8.0 (1) or 7.8 (2) containing 50 (MS) or 500 (BS) equivalents of 3,5-di-tert-butylcatechol (3,5-DTBC) in MS or BS, the catechol oxidase activity of the complex was determined under aerobic conditions. The electronic spectra of the reaction mixture were recorded for 3 h (MS) or 30 min (BS) at a wavelength of 400 (1) and 390 nm (2) (MS) or 404 (1) and 399 nm (2) (BS). Scaling up the reactants allowed for the same reaction to be performed, and the required 3,5-di-tert-butylquinone (3,5-DTBQ) was extracted column chromatographically using an eluent mixture of 10% ethyl acetate and hexane. Using FTIR spectroscopy and 1H NMR spectra in CDCl3,34 the isolated 3,5-DTBC was found. It became apparent that the rates and kinetic parameters of the complexes (MS, 2.9 × 10−5; BS, 2.9 × 10−6 M) under aerobic conditions depended on the substrate concentration (MS, 10–60 or BS, 100–700 equivalents).35 As buffers, ammonia (pH 10.0), borate (pH 9.0), phosphate (pH 5.0–8.0), and phthalate (pH 4.0) were used to study the pH effect. The buffer, complex, and substrate concentrations in the absence of ionic strength were [buffer] = 0.1 M, [complex] = 2.9 × 10−5 M, and [3,5-DTBC] = 1.45 × 10−3 M. The reaction mixture used to produce hydrogen peroxide catalytically was made in the same way as kinetic experiments. The pH of the reaction mixture was lowered to 2 with H2SO4 and an equivalent amount of water to cease the oxidation reaction. The formed quinone was extracted three times with dichloromethane. To the aqueous layer were added 1 mL of a 10% solution of KI and three drops of a 3% solution of ammonium molybdate. Spectrophotometric monitoring of the formation of I3− was done using the development of a typical I3− absorption band at ∼353 nm (ε, 26000 M−1 cm−1).36
Please refer to the ESI† for details of the computational protocol.
Results and discussion
Synthesis and general aspects
Copper(II) acetate monohydrate [Cu(O2CMe)2·H2O] was reacted with a Schiff base H(L1) or H(L2) (obtained via condensing 2-picolylamine or 2-(2-aminoethyl)pyridine with salicylaldehyde), phen (1,10-phenanthroline), and NaClO4 in methanol at a refluxing temperature to generate monocationic copper(II) complexes (1 or 2). At ambient temperature, the two complexes were stable in air after being separated into a crystalline substance in moderate to high yields (1, 58; 2, 67%). The molar conductivities (ΛM) of complexes 1 and 2 were determined to be 76 and 78 Ω−1 cm2 mol−1 in methanol solutions (1.0 × 10−3 M), accordingly. The predicted formulas, which show a 1:1 electrolytic nature, are completely in line with these values.37,38 They exhibited solubility in methanol (MeOH) and acetonitrile (MeCN), as evidenced by their solution molar conductivity measurements over a period of four days, which revealed no indications of degradation or solvolysis. The stability and purity of Cu(II) complexes are shown by the ESI mass spectra in MeCN solution. The molecular mass detected, with an m/z value of 454.56 (1) or 468.66 (2), is consistent with the formula of the molecular ion peak [M+ − ClO4]. Since the two copper(II) complexes exhibit stretching vibrations associated with ṽimine(CN) [1615 (1); 1624 cm−1 (2)], ṽpy(CN) [1586 (1); 1588 cm−1 (2)], and ṽ(Cph–O) [1228 (1); 1231 cm−1 (2)], which show that nitrogen (amine and pyridine) and oxygen (phenolate) donors are coordinated to copper(II), and their spectral patterns are comparable. The existence of a significant absorption for the ClO4− stretching vibrations at 1090 and 623 cm−1 indicated the emergence of a monocationic copper(II) complex. The two sharp bands were noticed (1, 475, 513; 2, 463, 515 cm−1) due to the result of ligand coordination and are caused by the ṽ(Cu–O) and ṽ(Cu–N) stretching.39,40 It becomes apparent that at 27 °C, the effective magnetic moment (μeff) value is 1.75 (1) or 1.74μB (2). This result coincides with the predicted spin-only magnetic moment for an electronic system [S = 1/2, Cu(II) d9].41 Using elemental analysis, the general form of the complexes, [Cu(Ln)(phen)](ClO4) (1 or 2), was established, which is compatible with the single-crystal X-ray structure of 2.
Single crystal X-ray structure of [Cu(L2)(phen)](ClO4) (2)
The monocationic [Cu(L2)(phen)](ClO4) (2) is shown in an ORTEP image (Fig. 1a), together with the atom numbering system. All selected bond lengths and angles are listed in Table 2. The anionic Schiff base ligand H(L2), which is tridentate in the cationic portion, facially coordinates with the central Cu(II) ion through one phenolate oxygen and one imine and one pyridine nitrogen atoms. The most accurate description of the coordination polyhedron the Cu(II) center is that it is severely distorted trigonal bipyramidal. The potential source of the perfect geometrical distortion could be the asymmetric and non-planar Schiff base, resulting in the formation of two six-membered chelate rings. Due to the sp3 and sp2 hybridizations of the carbon atoms in the chelating ligand, the six-membered chelate rings of the Schiff base [N(4)–C(17)–C(18)–C(19)–N(3) and N(3)–C(20)–C(21)–C(26)–O(1)] are puckered. The bidentate 1,10-phenanthroline (phen) and the tridentate Schiff base [H(L2)] are chelating through three pyridine [N(1), N(2), N(4)] and one imine [N(3)] nitrogens and one phenolate oxygen [O(1)] in the central metal coordination sphere. For Cu(II), this coordination is typical of either a square pyramidal (sp) or trigonal bipyramidal (tbp) geometry. There have been reports of some compounds recently where the Cu(II) ion takes on a geometry somewhere between sp and tbp.42,43 A trigonal index value (τ5), defined as τ = (β − α)/60, where β is the highest coordination angle and α is the second biggest, has been introduced by Addison et al.44 as a means of differentiating between a tbp and an sp geometry in five-coordinate metal complexes. Typically, a perfect square pyramidal geometry is denoted by τ = 0 and an ideal trigonal bipyramidal geometry by τ = 1.44 Here, the compromise between tbp and sp structures may better characterize the geometry surrounding the central copper atom (τ = 0.65 for 2). Stated otherwise, the complex ion displays a highly distorted (3 + 2) (NNO + NN) trigonal bipyramidal geometry surrounding the metal center, corresponding to the CuN4O chromophore coordination mode.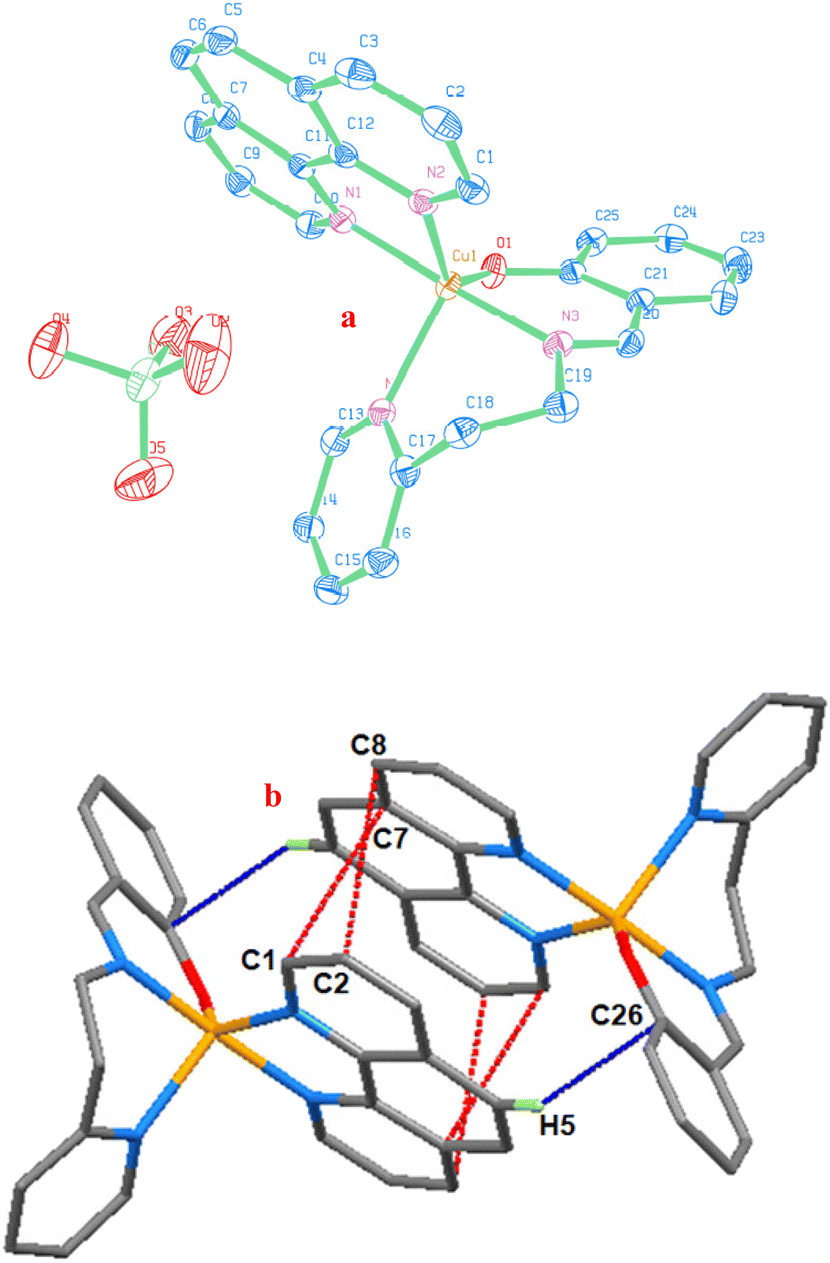 | ||
| Fig. 1 (a) ORTEP image of [Cu(L2)(phen)](ClO4) (2) showing 40% probability thermal ellipsoids with atom labeling for the metal and heteroatoms. (b) Packing diagram seen along the a-axis exhibiting intermolecular interactions of [Cu(L2)(phen)]+: (i) red, π⋯π stacking and (ii) blue, C–H⋯π (symmetry code: −x + 1/2, y + 1/2, and −z + 1/2). | ||
| X-ray | DFTa | ||
|---|---|---|---|
| 2 | 2 | 1 | |
| a Calculated from the B3LYP level of theory with mixed basis sets LANL2DZ/6-31G* using methanol as a solvent employing the CPCM method. | |||
| Cu(1)–N(1) | 2.0130 (16) | 2.096 | 2.077 |
| Cu(1)–N(2) | 2.1335 (15) | 2.143 | 2.102 |
| Cu(1)–N(3) | 1.9634 (15) | 2.011 | 1.978 |
| Cu(1)–N(4) | 2.1750 (15) | 2.372 | 2.344 |
| Cu(1)–O(1) | 1.9481 (13) | 1.946 | 1.940 |
| O(1)–Cu(1)–N(1) | 87.78 (6) | 84.947 | 81.021 |
| O(1)–Cu(1)–N(2) | 138.10 (6) | 153.457 | 161.731 |
| O(1)–Cu(1)–N(3) | 93.20 (6) | 92.398 | 92.517 |
| O(1)–Cu(1)–N(4) | 111.83 (6) | 107.800 | 108.607 |
| N(1)–Cu(1)–N(2) | 79.79 (6) | 75.398 | 76.392 |
| N(1)–Cu(1)–N(3) | 177.04 (6) | 174.889 | 174.319 |
| N(1)–Cu(1)–N(4) | 95.29 (6) | 92.172 | 95.955 |
| N(2)–Cu(1)–N(3) | 97.65 (6) | 96.889 | 95.714 |
| N(2)–Cu(1)–N(4) | 109.08 (6) | 101.768 | 102.547 |
| N(3)–Cu(1)–N(4) | 86.93 (6) | 91.971 | 88.927 |
The mean basal triangular plane is shared by one phenolate oxygen atom O(1) and one pyridine nitrogen atom N(4) of H(L2), as well as one pyridine nitrogen atom N(2) of phen. The distances between Cu(1)–N(1), Cu(1)–N(2), Cu(1)–N(3), Cu(1)–N(4) and Cu(1)–O(1) are 2.0130(16), 2.1335(15), 1.9634(15), 2.1750(15), and 1.9481(13) Å, respectively. To put it another way, axial nitrogen bond lengths are marginally smaller than equatorial nitrogen bond lengths. The bond angles are 109.08(6), 111.83(6), 138.10(6), and 177.04(6)° for the equatorial N(4)–Cu(1)–N(2), N(4)–Cu(1)–O(1), N(2)–Cu(1)–O(1), and axial N(3)–Cu(1)–N(1). The Cu(II) center deviates by 0.116(13) Å in the direction of N(1) from the mean basal plane formed by N(2), N(4), and O(1). The axial–equatorial bond angles show the deviation from the trigonal pyramidal geometry; they range from 79.79(6) to 97.65(6)° from a typical value of 90°. At the copper center, there is a little twist in the H(L2) ligand. Torsion angles of two six-membered chelate rings, which differ significantly from normal gauche and trans angles, appear to indicate strain. The values in analogous pentacoordinated complexes published elsewhere in the literature42,43,45,46 are in close agreement with the bond angles and bond lengths of 2.
Numerous non-covalent interactions (hydrogen bonding, π⋯π stacking, cation–π, anion–π, and C–H⋯π), in the crystal packing of compounds, stabilize solid state crystal structures.47,48 The cations of neighboring molecules in the crystal packing of 2 are grouped in dimeric association (Fig. 1b) as a result of attractive C–H⋯π and inter-pair π⋯π non-covalent interactions. The interaction between the phenyl ring of phen and the phenyl ring of H(L2) is the most significant intermolecular interaction, and it is represented by the C–H⋯π interaction [distance of H(5)⋯Cg(ph) is 2.640 Å; ∠C(5)–H(5)⋯Cg(ph) is 164.26°].49 Through antiparallel stacking interactions between the coordinated pyridine rings, it forms a dimer along the a axis of the unit cell. Specifically, it does so between the following coordinated pyridine rings: (i) C(2) of py and C(8) of py (distance of C(2)⋯C(8) is 3.389 Å), and (ii) C(1) of py and C(7) of py (distance of C(1)⋯C(7) is 3.297 Å). The coordinated phen ligands have a plane-to-plane distance of 3.309 Å, whereas the distance of Cg(py)⋯Cg(py) is 3.624 Å.50 These two interactions demonstrate a Cu⋯Cu distance of 7.040 Å and stabilize the structure of 2. They also show the nearby approach and orientation of the adjacent molecules.
DFT analysis of structural and electronic properties
Using the density functional theory (DFT) approach in methanol medium, geometry optimization for 1 and 2 was shown to have no negative Eigen value (Fig. 2), indicating structural stability. Quantitative examination of the geometry and ground electronic characteristics of the compounds is made possible by optimization of the molecular orbital geometry. Employing the B3LYP and 6-31G*/LANL2DZ levels and the Gaussian 09 computer program,51 the optimized energies, frontier molecular orbital (FMO), and highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) energy gaps have been determined. Bond length and bond angle calculations were made using geometry optimization, and the experimental values of 2 from single-crystal X-ray studies were compared with the calculated values (Table 2). | ||
| Fig. 2 DFT optimized structures of complexes [Cu(L1)(phen)]+1 (a) and [Cu(L2)(phen)]+2 (b). | ||
The calculated bond lengths of Cu–N3 (0.048 Å) and Cu–N4 (0.197 Å) are longer, Cu–O1 is comparable, and Cu–N1 (0.083 Å) is shorter at the basal plane while Cu–N2 (0.009 Å) is longer at the axial position. Even so, the calculated structural parameters for 2 well match those seen in its X-ray crystal structure. Although the bond angles in the crystal are 177.0° and 138.1°, the two longest calculated bond angles, N1–Cu–N3 and N2–Cu–O1, diverge from an ideal 180° by 174.9° and 153.5°, respectively. The calculated bond lengths and bond angles are nearly identical in the optimized structures of 1 and 2. As a result, the geometry of the optimized structure in 1 differed slightly from that in 2. Two longer estimated bond angles were used to determine the trigonality index (τ)of 1 (0.21) and 2 (0.36). The geometry of the copper center in 1 and 2 is distorted square pyramidal, and τ for 2 differs from the experimental result (τ, 0.65). Therefore, in contrast to the experimental geometry of 2 in the solid-state, the optimized structures of 1 and 2 are altered in the solution-state (methanol medium). The experimental electronic and EPR spectral results in methanol (cf. below) substantiate it. Moreover, the planar phen ligand coordinated axially (N2) and equatorially (N1) and L1 or L2 (N3, N4, O1) coordinated equatorially in a 4 + 1 manner with the Cu(II) cation is enforced by the lower twist angle between the O–Cu–N and N–Cu–N planes, estimated for 1 and 2, being similar (1.2) in the solution state. It leaves a positive effect on the ease of a labile oxygen donor and a continuous catalyst–substrate interaction, which results in efficient catalytic activity.
The FMOs (HOMO, HOMO+1, LUMO, LUMO−1) define the manner in which a particular molecule interacts with other species and are essential to regulating a wide range of molecular chemical reactions.52 The capacity of a compound to donate an electron is determined by its EHOMO, whereas its capacity to accept an electron is determined by its ELUMO. These MOs are significant because they use HOMO–LUMO priority orbitals to interact with other molecules. An essential foundation for assessing the stability, chemical reactivity, and selectivity of the complexes is provided by the analysis of orbital distribution, which frequently portrays the capacity of electrons to migrate from the occupied to the unoccupied orbitals.53 While the unoccupied (LUMO) orbitals were positioned on the pyridine and imine nitrogens, the phenolate oxygen of L1 or L2, and the equatorial nitrogen of phen coordinated to Cu(II) in the square plane of 1 or 2, the occupied (HOMO) orbitals were situated on the imine-phenolate unit of L1 or L2 and the axial nitrogen of phen (Fig. 3). There is a significant energy difference between ELUMO+1 and ELUMO; the LUMO has around 1.5 times the value of the LUMO+1, indicating high-energy transition from the LUMO+1 to the LUMO. On the other hand, EHOMO−1 and EHOMO values are comparable, indicating that these orbital transitions differ very little (Table 3). Research has demonstrated that, in contrast to compounds with lower EHOMO values, those with higher EHOMO values readily donate electrons.54
 | ||
| Fig. 3 Energy profile diagram of complexes [Cu(L1)(phen)]+1 (a) and [Cu(L2)(phen)]+2 (b). The doublet spin state: HOMO and LUMO in restricted spin calculations were carried out using the B3LYP level of theory with mixed basis sets LANL2DZ/6-31G* using methanol as a solvent employing the CPCM method. | ||
| Energy (eV) | 1 | 2 |
|---|---|---|
| Optimized energy (× 104) | −3.9577 | −4.0647 |
| HOMO | −5.7569 | −5.7509 |
| HOMO−1 | −5.8159 | −5.8004 |
| LUMO | −3.1971 | −3.3206 |
| LUMO+1 | −2.2831 | −2.2885 |
| Energy gap, ΔE (eV) | 2.5598 | 2.4303 |
| IP (eV) | 5.7569 | 5.7509 |
| EA (eV) | 3.1971 | 3.3206 |
| μ (eV) | −4.4770 | −4.5358 |
| η (eV) | 1.2799 | 1.2152 |
| S (eV−1) | 0.7813 | 0.8229 |
| ω (eV) | 7.8301 | 8.4651 |
| χ | 4.4770 | 4.5358 |
We anticipate a beneficial interaction since the electron donation of the complexes with another chemical system is plausible, as indicated by the relative EHOMO value of −5.8 eV found in this computation. According to these findings, more energy is involved in the electron transition in the HOMO than in the LUMO. Several characteristics of quantum chemistry including the ionization potential (IP = –EHOMO),53 electron affinity (EA = –ELUMO),53 band gap (ΔE = ELUMO − EHOMO), chemical hardness (η = ELUMO − EHOMO/2),55 global softness (S = 1/η),55 electrochemical potential (μ = –(IP + EA)/2),56 electrophilicity index (ω = μ2/2η),57 and electronegativity (χ = −μ)56 were computed. Theoretical understanding of chemical reactivity and selectivity is largely dependent on these electronic characteristics (Table 3). The electronic characteristics of 1 and 2 are comparable because they are analogous. The nucleophilic characteristics and electron-withdrawing capacity of the complexes are apparent in the quantities IP and EA, which are obtained from EHOMO and ELUMO, respectively. In particular, EA has proven to be a useful characteristic for determining the rate of electron transfer from O2˙− to Cu(II) ions and evaluating copper oxidase activity.58 There was a noticeable amount of copper oxidase activity because the complexes with lower EA values carried more electrons during the transfer. The energy difference between the EHOMO and ELUMO, or the band gap (ΔE), is one of the most crucial stability factors for assessing electron conductivity and molecular reactivity. The calculated ΔE values of 1 and 2 are relatively low, 0.91 and 1.03 eV, respectively; stability and reactivity have been attributed to low ΔE values.57,59 The computed values indicate the possible ease of reactivity in these complexes, and the most advantageous parameters are ΔE, η, and S. The complexes show electrophilic behavior57 with a higher ω index. Due to the positively charged coordinating metal (Cu), we observed a lower electronegativity (χ) power for the structures.
Spectral and electrochemical properties
One broad ligand field absorption band is visible in the solid state reflectance spectra (Fig. S1 and S2†) of complexes 1 (709 nm) and 2 (670 nm). However, in methanol solutions, two well-separated (>3000 cm−1) d–d bands with an approximate 2:1 intensity ratio are observed at 688 nm (εmax, 80 M−1 cm−1) and 932 nm (εmax, 40 M−1 cm−1) (1) or 670 (εmax, 100 M−1 cm−1) and 915 nm (εmax, 60 M−1 cm−1) (2) (Fig. S3 and S4†). This suggests that the complexes undergo a change in dissolution60–62 with the geometry changing from a trigonal bipyramidal component63 revealed in the X-ray structure of 2 to a square-derived component with significant axial interaction. Also, because of the phenolate oxygen and nitrogen coordinations to copper(II), which correspond to phenolate-to-Cu(II) and N(π)-to-Cu(II) ligand-to-metal charge-transfer (LMCT) transitions, they show an intense shoulder at 456 nm (εmax, 250 M−1 cm−1) (1) or 469 nm (εmax, 230 M−1 cm−1) (2) and a band at 342 nm (εmax, 6325 M−1 cm−1) (1) or 372 nm (εmax, 6230 M−1 cm−1) (2), respectively. This is in accordance with the coordination sphere of five-coordinate copper(II) complexes stabilizing in solution, which is more inclined toward a square pyramidal geometry than a trigonal bipyramidal geometry.64 The bands in the 266–295 nm region are caused by the intraligand transitions of phen, while the high intensity bands at 326 nm are attributed to the π–π* transitions of L1 (it is not noticeable in L2). The complexes in MeOH:H2O (4:1 v/v) solutions show a comparable spectral pattern in the UV-visible spectral measurements (Fig. S5 and S6†), indicating no change in the coordination sphere.
The polycrystalline EPR spectra of 1 and 2 are identified by two g values (Fig. S7 and S8†). Two possible alternate geometries for Cu(II)-pentacoordinated complexes are as follows: one approaches the trigonal bipyramidal, while the other approaches the limit of square pyramidal. The two conditions are distinguished by EPR spectroscopy.65 In addition to demonstrating an axial symmetry with two g values [g⊥: 2.054 (1) and 2.034 (2); g∥: 1.967 (1) and 1.956 (2)], the spectra of the complexes support the former geometry. The g factor corresponding to the higher symmetry axis (g∥) is nearly coincident with ge (2.0023). A pentacoordinate arrangement strongly shifted toward the trigonal bipyramidal60,66,67 is indicated by the relations g⊥ > g∥ and g∥ ≈ ge. While the pyridine nitrogen atom (phen) and the imine nitrogen atom [H(Ln)] occupy the two axial positions, the equatorial plane is occupied by pyridine nitrogen and phenolate oxygen atoms [H(Ln)] and a pyridine nitrogen atom (phen) (cf. above).
The axial pattern with a weakly resolved parallel feature is apparent in the frozen solution (77 K) EPR of 1 or 2 (Fig. S9 and S10†). The four parallel hyperfine patterns were well separated in DMF (conc. 1 × 10−2 M), which is ascribed to the interaction with copper nuclei in the parallel region. In contrast, they exhibit an isotropic pattern, with a perpendicular feature in MeOH (giso: 1, 2.044; 2, 2.020) and MeCN (giso: 1, 2.027; 2, 2.031) being noticeably broader (conc. 1 × 10−2 M). In diluted MeOH [conc. 2.9 × 10−5 M; g∥: 2.255 (1), 2.253 (2); g⊥: 2.056 (1), 2.048 (2)] and buffer solutions [conc. 2.9 × 10−5 M; g∥: 2.264 (1), 2.253 (2); g⊥: 2.052 (1), 2.048 (2)] the four parallel hyperfine signals were clearly discernible (Fig. S11–S14†). This implies that regardless of solution concentration, solvolysis68 breaks the intermolecular dimeric interaction in a strong coordinating DMF solvent. However, in a weak coordinating MeOH solvent, the broadening at a high concentration is caused by the near proximity of many monomers, as it was not observed at low concentrations. The derived values of g∥ (1, 2.239; 2, 2.238) and g⊥ (1, 2.051; 2, 2.050) are consistent with the EPR spectra in DMF, which show absorption characteristics for the mononuclear species and belong to an axially elongated tetragonal stereochemistry. The unpaired electron in these complexes is found in the dx2−y2 orbital of the copper(II) ion, which is situated in square-based geometries, as indicated by the trend g∥ > g⊥ > ge (2.0023).40 In both instances, the measured g∥ is less than 2.3, indicating that the copper complexes appear to be covalent in character.69 Additionally, they demonstrate that g∥ = 2.238 or 2.239, which is consistent with these chelates possessing mixed copper–oxygen and copper–nitrogen bonds.70 At a low field, A∥ (1, 176; 2, 173 × 10−4 cm−1) values ranged from 2600 to 3600 Gauss, indicating hyperfine splitting. However, the perpendicular region is not affected by the superhyperfine splitting brought on by the interaction with the nitrogen-donating 14N (I = 1) ligand. Around 2.200 and 200 × 10−4 cm−1, respectively, are the g∥ and A∥ values for the CuN4 chromophore71,72 in a square planar geometry. The g∥ value decreases and the A∥ value increases when oxygen donors are substituted for nitrogen donors. Alternatively, the g∥ and A∥ values for a CuO4 chromophore73 are about 2.420 and 145 × 10−4 cm−1, respectively. A strong axial interaction with the CuN3O chromophore based on a square is thus consistent with the observed g∥ and A∥ values. Conversely, decreased A∥ values of the complexes most likely indicate that the copper environment is distorted. Higher g∥/A∥ values (1, 127; 2, 129 cm) reflect a structural distortion in the CuN3O chromophore based on a square.74 The G value (4.7) ruled out the exchange interaction that takes place between the copper(II) centers for these complexes.75 However, these spectral characteristics along with the positioning of the d–d absorption reveal a square pyramidal structure with a CuN4O chromophore, in which the nitrogen atom engages the axial position while three nitrogens and oxygen choose the equatorial plane. Bonding characteristics of the complexes, including in-plane π-bonding (β2, covalent) and in-plane σ-bonding (α2, between ligand and 3d orbitals) have been identified.76 According to the values of β2 (1 or 2, 0.66) and α2 (1, 0.79; 2, 0.78), the π-bonding is more covalent than the σ type and σ-bonding has an ionic character. The bonding between the ligand and copper(II) ion is quite covalent, as indicated by the β2 values. Significant out-of-plane π-bonding is revealed by the orbital reduction factor,77K∥ (0.72) > K⊥ (0.65).
Using cyclic voltammetry (CV) and differential pulse voltammetry (DPV), the electrochemical behavior of 1 or 2 (0.001 M complex; 0.1 M supporting electrolyte, NBu4ClO4; working electrode, glassy carbon; and medium, CH3OH) was examined (Fig. S15–S18†). Upon scanning negatively, both complexes exhibit a reversible redox event during reduction, which was associated with the Cu(II) → Cu(I) reduction process. One reversible oxidation peak appeared as a result of the Cu(I) → Cu(II) oxidation process while scanning positively after cycling back. The copper(II) complexes that contain the CuN4O chromophore are compatible with the E1/2 values (1, 0.045; 2, 0.044 V vs. SCE). The CuII/CuI redox behavior is both chemically and electrochemically reversible, as shown by the linear plot of ipcversus ν1/2 [D, 8.4 (1), 7.6 × 10−6 cm2 s−1 (2)] and the peak current ratio [ipa/ipc, 1.1 (1); 1.1 (2)]. Under the same conditions of an experiment, the limiting peak-to-peak separation of the Fc/Fc+ couple (ΔEp, 86 mV; E1/2, 0.406 V vs. SCE) is substantially closer to the value of complex 1 or 2 [ΔEp, 90 (1) or 88 mV (2)]. In reality, a number of factors affect the molecular structure of the complexes, which in turn regulates the redox potential of the Cu(II)/Cu(I) pair. These factors include the size of the chelate ring, axial ligation, degree and distribution of unsaturation, and substitution pattern within the chelate ring.78 However, 1 and 2 are varied by the chelate ring size (1, 655; 2, 665 chelate rings), and they display similar E1/2 and ΔEp, confirming the influence of structural distortion on the redox potential. This suggests that both are more appropriate for the easy inter-conversion of the tetrahedral (CuI) to the square planar (CuII) geometry due to the structural distortion. It is suggested that during the heterogeneous electron transfer process in 1 or 2, between copper(II) and copper(I) species, the coordination sphere experiences a minimal amount of stereochemical reorganization.79 This could be attributed to the flexible equatorial phenolate oxygen coordination. These findings are in line with the structural distortion of the CuN3O chromophore based on a square in 1 or 2, which are higher values of the g∥/A∥ index (cf. above) and represent intermediate geometric features between Cu(II) and Cu(I) redox-active states.
Oxidation of ascorbic acid (H2A) to dehydroascorbic acid (DA)
By reacting with 1 or 2 (3.0 × 10−3 M) and varying concentrations of ascorbic acid (H2A) in aqueous methanol under aerobic conditions at 25 °C, the ascorbate oxidase activity of the complexes was assessed (Fig. 4 and Fig. S19†). The original green solution, attributed to the Cu(II) complexes, changed to a brown solution (Fig. 4b–e and Fig. S19b–e†) upon the immediate addition of the substrate, H2A, to the catalyst (1 or 2) solution. A new charge transfer (CT) band at 437 (1) or 434 nm (2) is observed up to 15 min (inset in Fig. 4 and Fig. S19†), which may be the result of Cu(II) converting to Cu(I) species.80,81 When exposed to air, the brown colour of the solution changes to green within 20 to 30 min of time (Fig. 4f–4h and Fig. S19f–19h†). This occurs simultaneously with the disappearance of the CT band and the reappearance of the d–d band, indicating the transformation of the reduced species, Cu(I), into the oxidized form, Cu(II), using dioxygen as an oxidant. The parent copper(II) species regenerates, resulting in an isosbestic point at either 649 (1) or 644 (2) nm. It becomes apparent that the catalytic cycle of 1 or 2 (Scheme 2) is efficient at a [H2A]:[complex] molar ratio of almost 100:1 (1) or 130:1 (2).
 | ||
| Fig. 4 Spectral trace of [CuII(L2)(phen)]+ (2, 3 × 10−3 M) in MeOH:H2O (4:1 v/v) in the visible region (spectrum a); reduction of copper(II) to copper(I) (color change from green to brown) with addition of H2A (3 × 10−3 M) up to 15 min (spectra b, c, d, and e); regeneration of copper(II) species (color change from brown to green) from 20 to 30 min (spectra f, g, and h). Inset: generation of charge transfer band in brown solution due to formation of copper(I) species up to 15 min (spectrum e). | ||
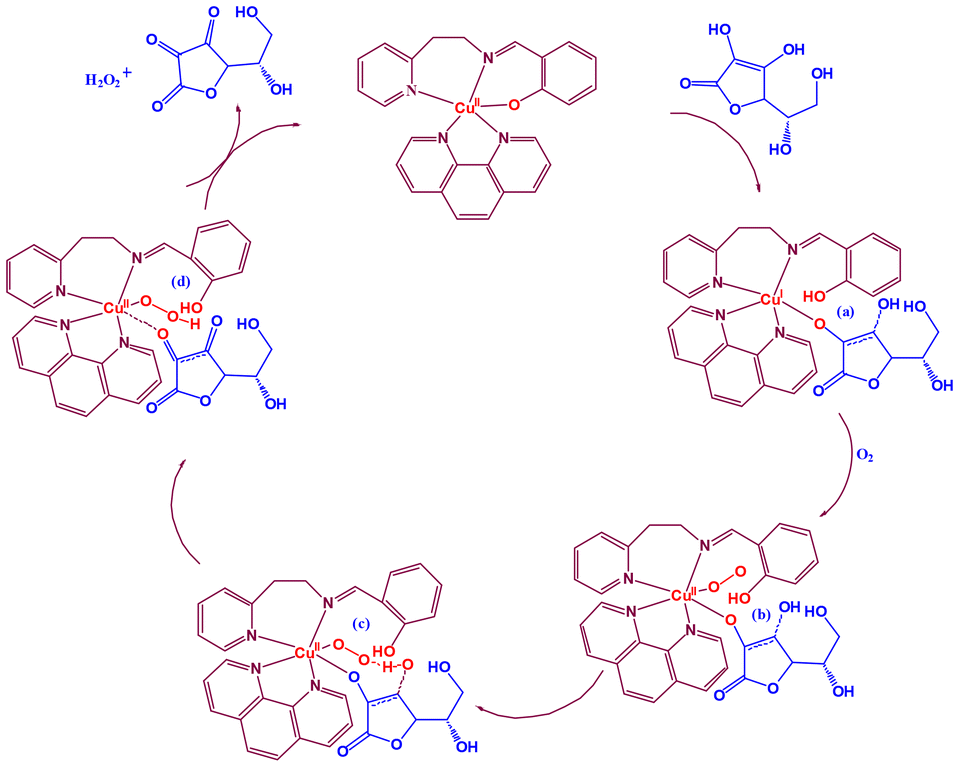 | ||
| Scheme 2 Based on experimental results, the possible catalytic cycle for the oxidation of H2A to DA {catalyst, [Cu(L2)(phen)]ClO4 (2)}. | ||
Using the initial rate approach, the kinetics of H2A oxidation in aqueous methanol solution (pH 7.3) MS or buffer solution (pH 7.6) (Fig. S20–S22†) BS was ascertained by tracking the decrease in the absorption band at ∼268 nm over the course of the first 30 minutes (Fig. 5 and Fig. S23, S24†). The decrease in the absorbance of H2A suggests that it is being oxidized in the presence of the copper(II) complex to generate DA. The rate constant data demonstrate considerable reactivity differences and ascorbate oxidase activity. Under aerobic conditions, solutions of copper complexes were treated with various concentrations of H2A (20–140 equivalent) to examine the influence of rates on the substrate concentration and other kinetic parameters. At low concentrations of H2A, first-order dependence of the substrate concentration was found, with the effect being more pronounced for 2 than 1. Both complexes displayed saturation kinetics at higher concentrations. The dependence on the substrate concentration shows that the catalyst–substrate interaction is an early step in the catalytic process.
 | ||
| Fig. 5 Oxidation of ascorbic acid by [Cu(L2)(phen)](ClO4) (2) in buffer solution (pH 7.6) monitored by UV-Vis spectroscopy. | ||
The rates of reactions measured at various H2A concentrations were fitted to the Michaelis–Menten equation and linearized employing the Lineweaver–Burk plot (Fig. 6 and Fig. S25–S27†) to derive important kinetic parameters such as the maximum rate of the reaction (Vmax) [MS, 22.2 (1) or 39.1 × 10−4 Ms−1 (2); BS, 23.1 (1) or 47.8 × 10−4 M s−1 (2)], Michaelis binding constant (KM) [MS, 1.0 (1) or 3.1 × 10−4 M (2); BS, 0.2 (1) or 0.4 × 10−4 M (2)] and turnover number (kcat) [MS, 4.8 (1) or 8.5 × 106 h−1 (2); BS, 2.5 (1) or 5.2 × 107 h−1 (2)]. The complexes are highly effective at oxidizing H2A in the presence of molecular oxygen, resulting in DA. The reaction kinetics clearly indicate that a stable copper(II) complex–substrate adduct is present. Control experiments demonstrated that the desired DA product did not develop in an inert environment. Therefore, the copper(II) complex catalyzes the synthesis of DA in the catalytic process that proceeds from H2A oxidation that involves molecular oxygen. The ascorbate oxidation feature of the Cu(II) sites in AOase is modeled by the catalytic activity of the complexes in the ascorbate oxidation by molecular oxygen involving the reduced copper(I) intermediate.
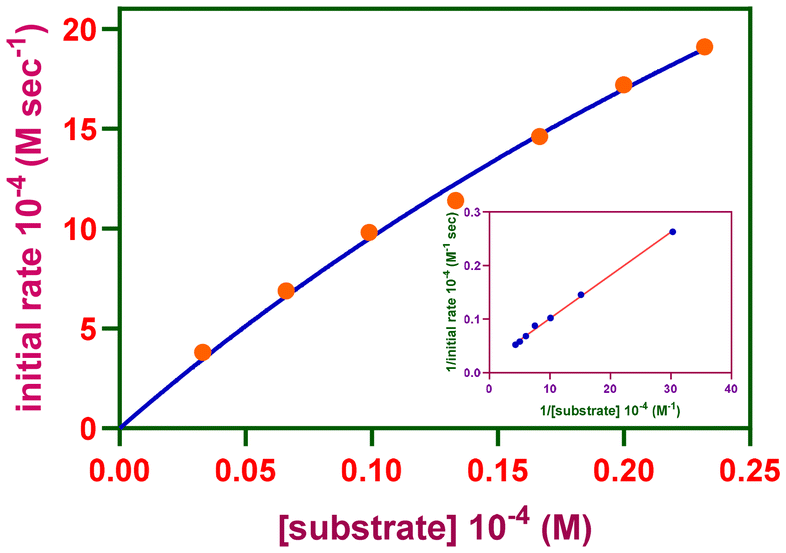 | ||
| Fig. 6 Reaction rates of the oxidation reaction catalyzed by [Cu(L2)(phen)](ClO4) (2) in buffer solution (pH 7.6) depending on the ascorbic acid content. Inset: Lineweaver–Burk plot. | ||
The chemical conversion of H2A to DA was investigated by means of a cyclic voltammetric (CV) experiment (Fig. 7) with catalyst 2 (complex 2:H2A; molar ratio, 1:5) without the purging of the nitrogen gas at ten-minute intervals. In the CV of complex 2, which was acquired at a scan rate of 50 mV s−l, one reduction wave and one matching oxidation wave were detected at −0.125 V (ipc, 6.1 μA) and −0.019 V (ipa, 4.0 μA), respectively. The obtained current functions agree with a one-electron CuII/CuI redox pair (Fig. 7a), where the E1/2 value is −0.053 V (ΔEp, 106 mV). The complex solution exhibits a reduction in E1/2 of −0.067 V (ΔEp, 133 mV) upon the addition of H2A, indicating a CuI/CuII pair redox mechanism. The addition of H2A rapidly reduces Cu(II) to Cu(I), and at the same time, it generates a five-coordinate complex-monodehydroascorbate adduct {[(phen)(HL2)-CuI-(HA)]}, which has an increased anodic peak current of 4.0 to 12.6 μA and a lowered cathodic peak current of 6.1 to 2.7 μA (Fig. 7b). A decrease in redox potential (E1/2, −0.084 V) and reversibility (ΔEp, 168 mV) are observed after 20 minutes when the uptake of dioxygen from the environment stabilizes the one-electron CuII/CuI redox pair. The anodic peak current diminishes to 6.7 μA; however, the cathodic peak current changes slightly. A six coordinate complex-monodehydroascorbate-dioxygen adduct, [(phen)(HL2)-CuII-(HA)(O2)] (Fig. 7c), is formed, as shown by these observations. A one-electron CuII/CuI redox pair is consistent with a minor increase in redox potential (E1/2, −0.076 V) and reversibility (ΔEp, 152 mV) with no change in the cathodic peak current and an additional drop in the anodic peak current to 3.9 μA after 30 minutes. The observations are indicative of a new generation of complex-dehydroascorbatehydroperoxo species, [(phen)(HL2)-CuII-(DA)(OOH)], (Scheme 2d). Eventually, the one-electron CuII/CuI pair (Fig. 7e) maintains its preferential stabilization (complex 2) with the same redox potential (E1/2, −0.051 V), reversibility (ΔEp, 102 mV), and cathodic and anodic peak currents (ipc, 6.4; ipa, 4.1 μA).
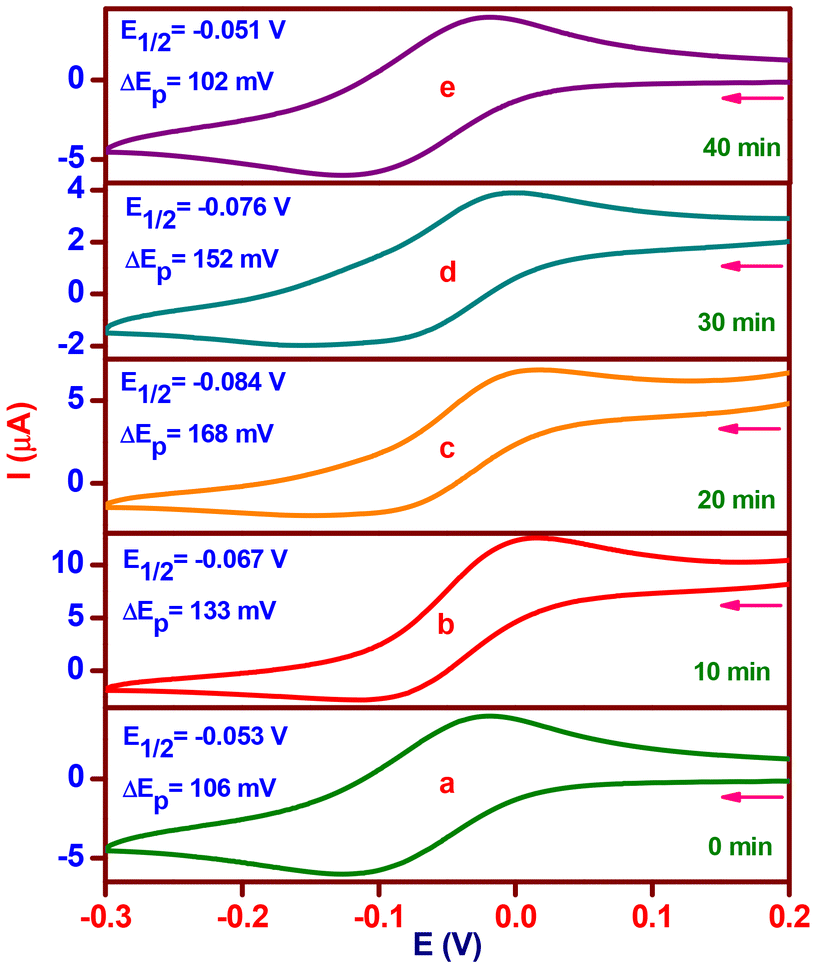 | ||
| Fig. 7 Cyclic voltammetric changes of [Cu(L2)(phen)](ClO4) (2) upon addition of H2A (molar ratio, 1:5; time interval, 10 min). | ||
The 1:50 equivalent ratio of catalyst 2 and H2A was measured using a high-resolution mass spectrum (HRMS) after mixing in methanol for 10 minutes (Fig. S28†). It shows three minor peaks at m/z = 174.08, 643.86, and 674.91 in addition to a main peak at m/z = 468.10. The former peak, which is estimated to be 469.01, corresponds to the catalyst [CuII(L2)(phen)]+. The product, DA (estimated m/z = 174.10), is indicated by the latter smaller peak at m/z = 174.08. The existence of a complex–monodehydroascorbate adduct, [(phen)(HL2)-CuI-(HA)], may be responsible for another peak at m/z = 643.86 (estimated m/z = 644.23). A complex–substrate–dioxygen aggregate (estimated m/z = 675.23) containing superoxocopper(II) species (Scheme 2c) and/or hydroperoxocopper(II) species (Scheme 2d) matches with the peak at m/z = 674.91.
We have studied the catalyst–substrate reaction using UV-Vis spectroscopy, CV, and HRMS techniques in order to deduce a likely mechanism (Scheme 2) of the AOase activity of 1 and 2. It is suggested to start with a five-coordinate Cu(I) species ‘a’ bonded to monodehydroascorbate, which is produced by catalyst 1 or 2. Subsequently, it undergoes an oxidation process that converts the Cu(I) center in ‘a’ to a Cu(II) center in ‘b’ when it reacts with atmospheric oxygen. The next stage entails a crucial intramolecular proton transfer mechanism that goes through transition state ‘c’ to produce peroxo intermediate ‘d’ from the oxygen atom in the monodehydroascorbate moiety to the oxygen atom in superoxide in the ‘b’ intermediate.
Finally, the last step includes binding yet another molecule of the H2A substrate to the copper center in intermediate ‘d’, rejuvenating the initial catalyst and releasing the expected DA product and H2O2. It has previously been demonstrated that the primary likely result of oxygen reduction in such catalytic cycle processes employing a CuII/CuI redox pair is hydrogen peroxide.82 As a result, complexes 1 and 2 replicate the ascorbate oxidase activity of the type-2 copper sites in AOase, as demonstrated by the oxidation of H2A with molecular oxygen to yield DA.
Oxidation of benzylamine (Ph–CH2–NH2) to benzaldehyde (Ph–CHO)
In a 10 mL solution of MeOH:H2O (4:1 v/v) containing Ph–CH2–NH2 as a substrate (1 × 10−1 M), the copper(II) complex (1 × 10−3 M) using H2O2 (30% w/v; 1 mL) catalyzed the chemical transformation of amine into aldehyde (RCH2NH2 → RCHO + NH3).80 There is no discernible Ph–CHO produced by the reaction involving Ph–CH2–NH2 and H2O2 or the complex (Fig. 8 and Fig. S29†). However, Ph–CHO production is aided by the complex-catalyzed deamination of Ph–CH2–NH2 in the presence of H2O2. Consequently, a significant yield of Ph–CHO (1, 0.070 g, 66%; 2, 0.077, 72%) was produced under mild reaction conditions by effectively and selectively converting Ph–CH2–NH2. Ph–CHO (δ 9.83 ppm, –CHO instead of δ 3.67 ppm, –CH2–) was observed (Fig. S30†) from 1H NMR data in CDCl3, and Ph-CHO (Fig. S31†) is associated with the substantial HRMS peak at m/z = 107.049 ((M + H)+; calcd 106.120). The novel monocopper(II) complexes have therefore been developed as biomimetic catalysts that resemble AmOase to facilitate the oxidation process of an aerobic amine to an aldehyde.
 | ||
| Fig. 8 Electronic spectra of [Cu(L2)(phen)](ClO4) (2) in methanol:water (4:1 v/v) (a), addition of benzylamine to complex (b) and addition of H2O2 to benzylamine and complex (c). | ||
The process has been monitored using UV-Vis spectrophotometry, which determines the increase in absorbance with time (60 s interval) at 252 nm to produce the Ph–CHO concentration. A room temperature aerobic reaction was carried out using both complexes (1 or 2: 3.3 × 10−7 M) with Ph–CH2–NH2 (2–12 equivalents) in an aqueous methanolic solution containing H2O2 (50 equivalents) (Fig. S32†). The kinetic features of the reaction have been explored through the use of the function of the initial rate process. The Michaelis–Menten approach was applied to assess the measured rate vs. concentration of the substrate. The linearization was performed using the Lineweaver–Burk plot, which produced the maximal initial rate, Vmax [43.4 (1) or 50.4 × 10−4 Ms−1 (2)], and Michaelis–Menten constant, KM [38.1 (1) or 39.5 × 10−4 M (2)]. The computation of the turnover number, kcat [4.7 (1) or 5.4 × 105 h−1 (2)] (Fig. S33 and S34†), involved dividing the maximal initial rate by the concentration of the related complexes. The outcomes demonstrate the significant efficacy of both complexes towards the oxidation of Ph–CH2–NH2. As expected, there is more activity in 2 than in 1. The phenolate oxygen donor of the Schiff base is readily dissociable, and the coordination number surrounding copper(II) is smaller. It is possible that these properties, which facilitate substrate attachment to the metal center, account for the high reactivity of the complexes.
The catalytic oxidation of Ph–CH2–NH2 with copper(II) complexes as catalysts was suggested as a potential mechanism (Scheme 3). First, a six-coordinate adduct between the copper(II) complex and benzylamine is formed, denoted as [CuII(Ln)(phen)(C6H5-CH2-NH2)]+ (A), which has an octahedral geometry and a CuIIN5O chromophore; (a) electronic spectra showed that an adduct formed and the visible band changed from 684 to 680 nm (1) and 670 to 628 nm (2) (hypsochromic shift); (b) electrochemical measurements demonstrated that the redox potential (E1/2) (Fig. 9 and Fig. S35†) moved to a more negative value (1, 0.044 to −0.029; 2, 0.035 to −0.037 V); and mass spectra (Fig. S36†) revealed that the HRMS peak emerged at m/z, 575.40 (calc. 575.71) (2). The second stage was the reaction of the adduct with H2O2 to cause a color change from green to brown, resulting in the vanishing of the d–d band. In parallel, the chromophore transitioned from CuIIN5O (CuII-adduct, A) to CuIN5 (CuI-adduct, B), whereby Cu(II) underwent reduction to Cu(I). In the third stage, B undergoes five simultaneous changes: (i) the Cu(I) oxidized to Cu(II) by molecular oxygen,(II) the O2 reduced to H2O, (iii) the C(α)–H bond ruptured, (iv) a hydride ion is transferred from the methylene group, and (v) the benzylimine (C6H5–CHNH) intermediate is discharged; and the catalyst, [Cu(Ln)(phen)]+, is regenerated. Lastly, in the presence of water, C6H5–CHNH underwent further hydrolysis to produce Ph–CHO and NH3.83 The oxidation of amine to aldehyde with molecular oxygen is catalyzed by complexes 1 and 2, which are similar to an enzymatic reaction84 of the type-2 copper core in AmOase.
 | ||
| Scheme 3 Based on the findings of the experiment, a catalytic cycle for the oxidation of Ph–CH2–NH2 to Ph–CHO, {catalyst, [Cu(L2)(phen)]ClO4 (2)}. | ||
 | ||
| Fig. 9 Differential pulse voltammogram of [Cu(L2)(phen)]+2 (a) and the adduct formed by the addition of benzylamine (molar ratio, 1:10) (b). | ||
Oxidation of 3,5-di-tert-butylcatechol (3,5-DTBC) to 3,5-di-tert-butylquinone (3,5-DTBQ)
The potential of copper(II) complexes to function as functional models of catechol oxidase was assessed by using them as catalysts in the oxidation of 3,5-di-tert-butylcatechol (3,5-DTBC) to 3,5-di-tert-butylquinone (3,5-DTBQ). Both monocopper(II) complexes exhibit comparable behavior in methanol solution (MS) and buffer solution (BS) at optimal pH (Fig. S37 and S38†) and the representative spectral change for 2 is displayed in Fig. 10 and Fig. S39–S41.† In order to investigate the catalytic activity of 1 or 2, 1.45 × 10−3 M of 3,5-DTBC was added to 2.9 × 10−5 M (MS) or 2.9 × 10−6 M (BS) complex solutions [catalyst:substrate ratio, 1:50 (MS) or 1:500 (BS)]. Following the addition of 3,5-DTBC to catalyst 1 or 2, the reaction was observed for 3 h (MS) or 30 min (BS). In the beginning, both complexes exhibit bands in MS at 400 (1) or 390 (2) nm and in BS at 404 (1) or 399 (2) nm. The spectral run completed in the time limit shows an increase in absorbance almost above that band upon the addition of 3,5-DTBC. The experiment clearly demonstrates the oxidation of 3,5-DTBC to 3,5-DTBQ, which is accelerated by the monocopper(II) complexes. This is because it is commonly known that in MS or BS, 3,5-DTBQ displays band maxima in the 390–404 nm range. Even so, when the autoxidation of 3,5-DTBC alone was examined in both media (MS and BS), it seemed that this process was either weak or nonexistent (Fig. S42 and S43†).
 | ||
| Fig. 10 Oxidation of 3,5-DTBC by the catalyst [Cu(L2)(phen)](ClO4) (2) in buffer solution (pH 7.8) monitored by UV-Vis spectroscopy. | ||
3,5-DTBQ was purified by column chromatography using a 10% ethyl acetate and hexane solution as the eluent. It was then extracted from the reaction mixture in MS with a significant yield (1, 70.7; 2, 71.2%). By measuring its melting point (1, 109; 2, 111 °C; and literature value, 110 °C),85 FTIR (Fig. S44†) in KBr (2: ṽ(O–H) bands absent at 3460 and 3260 cm−1; the ṽ(CO) band present at 1657 cm−1) and 1H NMR (Fig. S45†) in CDCl3 [2: –CH3 (C), s, δ 1.16 ppm, 9H; –CH3 (D), s, δ 1.20 ppm, 9H; H (B), s, δ 6.15 ppm, 1H; H (A), s, δ 6.87 ppm, 1H], the isolated 3,5-DTBQ was identified.
The rate constant for a catalyst complex was found using the conventional initial rate method in order to comprehend the kinetic component of catalysis for 1 or 2. Following that, the observed rates versus substrate concentration data were examined using the Michaelis–Menten method of enzyme kinetics. The maximum initial rate (Vmax) [MS, 20.2 (1) or 8.4 × 10−4 M min−1 (2); BS, 12.1 (1) or 14.0 × 10−4 M s−1 (2)], and the Michaelis–Menten constant, (KM) [MS, 1.2 (1) or 0.4 × 10−4 M (2); BS, 12.0 (1) or 16.9 × 10−4 M (2)], were found by linearization utilizing Lineweaver–Burk plots (Fig. 11 and Fig. S46–S50†).86 By dividing the Vmax values by the concentration of the respective complexes, the turnover number (kcat) [MS, 2.6 (1) or 1.7 × 103 h−1 (2); BS, 1.5 (1) or 1.7 × 106 h−1 (2)] values were determined. The data clearly show that both complexes 1 and 2 are quite active. The phenolate oxygen atom in complex 1 or 2 is labile, which may facilitate the catalyst–substrate interaction by forming a positive channel. This may be a necessary condition for exhibiting higher catalytic activities, which could explain the unusually high activity of the monocopper(II) complexes. The catalyst–substrate complex in BS is substantially more stable than in MS at optimum pH, as shown by the KM value in BS being 10 (1) or 42 (2) times more than that of MS. Additionally, the complexes exhibit a greater turnover number in BS at the optimum pH, while the turnover number in MS exhibits the opposite effect. It is approximately 577 (1) or 1000 (2) times greater in BS than in MS. In this instance, complex 2 performs better than complex 1, demonstrating higher catecholase activity.
 | ||
| Fig. 11 Reaction rate for the oxidation reaction catalyzed by [Cu(L2)(phen)](ClO4) (2) depending on the concentration of 3,5-DTBC in buffer solution (pH 7.8). Inset: Lineweaver–Burk plot. | ||
Based on the differences in activities, it appears that the lability of coordinated phenolate oxygen is easier in the equatorial position and that substrate substitution occurs more quickly in 2 than in 1. The flexible 665 chelate rings in 2 instead of the rigid 655 chelate rings in 1 cause high structural distortion. This enables easy access to the substrate, maximizing the steric match between the complex and the substrate, which promotes a more efficient inner-sphere electron transfer process that elevates catalytic activity.
The aerobic interaction between 100 equivalents of 3,5-DTBC and the complex (1 or 2, 3.3 × 10−3 M) in MS (Fig. 12 and Fig. S49†) was investigated using UV-Visible spectroscopy. The complex oxidizes 3,5-DTBC and converts 3,5-DTBQ, which results in the loss of the d–d band (Fig. 12b and Fig. S49b†) at 678 nm (1) or 670 nm (2) due to the Cu(II) to Cu(I) reduction produced by the addition of 3,5-DTBC and a rise in absorbance (Fig. 12d and Fig. S49d†) at 396 nm (1) or 398 nm (2). Following the completion of the reaction, the uptake of molecular oxygen by the solution displayed another significant absorbance peak (Fig. 12c and Fig. S49d†) at either 557 nm (1) or 571 nm (2). This was most probably the result of the superoxide (O2−)-to-Cu(II) LMCT transition, which suggested the formation of a superoxocopper(II) intermediate.87
 | ||
| Fig. 12 Electronic spectral changes of [Cu(L2)(phen)](ClO4) (2) (3.3 × 10−3 M) upon addition of 3,5-DTBC (molar ratio, 1:100) in MeOH. The electronic spectrum of complex (a), disappearance of the d–d band immediately after the addition of 3,5-DTBC (b), formation of a superoxide intermediate (c) and generation of 3,5-DTBQ (d). | ||
Conversely, differential pulse voltammetry (DPV) was used to study both the complex (1 or 2, 0.001 M) and 3,5-DTBC (molar ratio 1:5) in MS at intervals of five minutes throughout the reaction (Fig. 13 and Fig. S50†). When 3,5-DTBC is added to the complex solution, two peaks appear at two distinct E1/2, representing CuII/CuI and CuI/CuII one-electron redox pairs (Fig. 13b and Fig. S46b†) at +0.015 V (i, 15.9 μA) and +0.055 V (i, 15.7 μA) (1) or −0.027 V (i, 18.7 μA) and +0.054 V (i, 16.9 μA) (2), respectively. This suggests the formation of a copper(II) adduct bound to monocatecholate, which is then reduced right away to produce copper(I) species bound to monocatecholate (quick, Scheme 4b). The peak (1: E1/2, + 0.055 V; i, 15.7 μA or 2: E1/2, + 0.054 V; i, 16.9 μA) shifted to −87 mV (1) or −79 mV (2) after 10 min (Fig. 13c and Fig. S46c†), showing a one-electron CuII/CuI redox pair. Reduction becomes challenging due to the conversion of molecular oxygen bonding to copper(I) species bound to monocatecholate, as evidenced by a rise in electron density on Cu(II) and an opposite trend in E1/2. This has something to do with the development of the superoxocopper(II) intermediate (Scheme 4c). Additionally, the DPV displays a peak (Fig. 13c–13e and Fig. S46c to 46e†) at an E1/2 value of −0.308 to −0.319 V (i, 19.8–34.8 μA) (1) or −0.306 to −0.323 V (i, 22.2–35.0 μA) (2). Furthermore, an increase in current is observed (i: 1, 15.0; 2, 13.8 μA), consistent with the formation of hydroperoxocopper(II) species (Scheme 4e).
 | ||
| Fig. 13 Differential pulse voltammetric changes of [Cu(L2)(phen)](ClO4) (2) upon addition of 3,5-DTBC (molar ratio, 1:5; time interval, 5 min). | ||
 | ||
| Scheme 4 Based on observations from experiments, a plausible catalytic cycle for the oxidation of 3,5-DTBC to 3,5-DTBQ {catalyst, [Cu(L2)(phen)]ClO4 (2)}. | ||
HRMS measurements were taken in MS (Fig. S51 and S52†) and BS (Fig. S53 and S54†) after reacting the complex (1 or 2) with 3,5-DTBC (molar ratio 1:50) and allowing it to react for 10 minutes. The most common species is produced from the monomer [CuII(L1)(phen)]+ (1) or [CuII(L2)(phen)]+ (2) at m/z = 454.16 (MS)/454.23 (BS) (1) or 468.15 (MS)/468.12 (BS) (2). It is possible that a monocatecholate-bound copper(II) or monocatecholate-bound copper(I) adduct exists as a prominent peak at m/z = 676.77 (MS)/676.61 (BS) (calculated 676.89) (1) or 690.13 (MS)/690.47 (BS) (calculated 690.75) (2). According to Scheme 4a and b, they can be understood as either [(phen)(HL1/HL2)CuII-(3,5-DTBC1−)]+ or [(phen)(HL1/HL2)CuI-(3,5-DTBC1−)]. At m/z = 243.19 (MS)/243.15 (BS) (calculated 243.29) (1) or 243.16 (MS)/243.12 (BS) (calculated 243.29) (2), a well-defined peak for the product 3,5-DTBQ and Na+ was found. The complex–substrate–dioxygen aggregate is specifically responsible for the peak at m/z = 708.55 (MS)/708.46 (BS) (calculated 708.88) (1) or 722.15 (MS)/722.89 (BS) (calculated 722.97) (2). This is appropriate with the creation of intermediates, either superoxocopper(II), [(phen)(HL1/HL2)-CuII-(3,5-DTBC1−)(O2−)] (Scheme 4c), or hydroperoxocopper(II), [(phen)(HL1/HL2)-CuII-(3,5-DTBQ1−)(HOO−)] (Scheme 4e).
At ambient temperature, the reactivity of [Cu(L1)(phen)](ClO4) (1) or [Cu(L2)(phen)](ClO4) (2) with H2O2 was observed using UV-Vis spectroscopy in CH3CN (Fig. S55 and S56†). It can be observed from the results that the complex is evolving into a new species with an absorption band at λmax = 370 (1) or 368 nm (2).27 The absorption band enables an LMCT from the hydroperoxide ligand to the Cu(II) ion. These findings imply that [CuII(L1/L2)(phen)]+ transforms into [CuII(HL1/HL2)(phen)(OOH)]+. The hydroperoxocopper(II) complex develops slowly at room temperature, and it requires an hour for the resultant species to stabilize. Also, we must ascertain in the event, dioxygen undergoes reduction to H2O2 or water throughout the oxidation (H2A or 3,5-DTBC) process. A thorough work-up of the mixture of H2A or 3,5-DTBC, complex 2, and KI clearly indicates that dioxygen is reduced to H2O2, as reported by other investigators as well.88 This is supported by the oxidation of I− to I2 and subsequent production of I3−, as shown by the UV-visible spectral study of the solution (Fig. S57†). Notably, superoxide is the starting point for the formation of H2O2. Moreover, the results provided evidence for the production of the hydroperoxocopper(II) intermediate and the subsequent emergence of H2O2 during the catalytic cycle of oxidation of different substrates like H2A or 3,5-DTBC.
As a result, the following is a description of Scheme 4, which represents the most likely catalytic mechanism. In the beginning, the catalyst and 3,5-DTBC react to produce monocatecholate-bound copper(II) species (a). Following that, the copper(II) species (a) undergo fast reduction to a monocatecholate-bound copper(I) species (fast, b). Subsequently, the copper(I) center in (b) is then oxidized to a copper(II) center with aerial oxygen, resulting in the superoxocopper(II) intermediate (c). Afterwards, a proton is transferred from the unbound catecholate oxygen atom (c) to the superoxide bound to copper(II) (d) in the rate-determining step, which generates hydroperoxocopper(II) species (e). In the end, when 3,5-DTBQ is discharged from (e), H2O2 is released, returning the catalyst to its initial active state. According to the outcomes, copper(II) is reduced to copper(I) and there is a monocatecholate-bound copper(I) adduct; these events come together to form complex–substrate–dioxygen intermediates that catalytically convert catechol to o-quinone in order to resemble the functions of type-3 COase.
One benefit of the current model systems over the original enzyme is their capacity to perform in methanol solution. When comparing the values of mono- and dicopper(II) complexes in the literature, the turnover number of 1 or 2 is divided into three categories: (a) greater (3–943 h−1),89,90 (b) comparable (1.38–9.11 × 103 h−1)91,92 and (c) smaller (1.09 × 104–1.09 × 105 h−1).93,94 The complexes in buffer solution [1.5 × 106 h−1 (1) or 1.7 × 106 h−1 (2)], on the other hand, exhibit a turnover number 2.3–2.6 times smaller than the value for the dinuclear copper(II) complex, [Cu2L(NO3)2] (H2L = N,N′-bis(3-methoxysalicylidene)-1,3-propanediamine), reported by Terán et al. (3.89 × 106 h−1).95 It is one of the most potent catalytic models and is closer in value to the COase enzyme that was isolated from Ipomoea batatas (8.25 × 106 h−1).96 Interestingly, the new complexes displayed greater turnover numbers than the COase enzyme isolated from Lycopus europaeus (5.7 × 105 h−1).97 Considering that the CuII/CuI redox potential is crucial to the oxidation of catechol, both complexes work as efficient catalysts in MS and BS to convert 3,5-DTBC into 3,5-DTBQ.98 A basic limitation of the model complexes (mono- and dinuclear) is that most mimics show relatively modest catalytic activity in comparison with the enzymes. This means that there are always rooms for improvement in the design of structural and functional model systems that are more industrially potential and closely aligned with the enzyme attributable to the structure–property relationship. It follows that the monocopper(II) complex 1 or 2 is the most effective catalyst model for this oxidation process, and that the turnover number value is the highest that has been reported, complementing the quality of the enzyme assay of catechol oxidase.
Conclusions
Novel monocopper(II) complexes with mixed-ligands (tridentate NNO and bidentate NN donors) have been synthesized. The geometry of the complex ion among the active model complexes shows a distorted (3 + 2) trigonal bipyramidal (NNO + NN), which surrounds copper(II) and corresponds to the CuN4O chromophore. An attractive C–H⋯π and the inter-pair π⋯π non-covalent interactions lead to the construction of dimers, as indicated by crystal packing. Using DFT, structural optimization and electronic parameter calculations were carried out to comprehend the chemical reactivity, stability, and selectivity of both complexes in solution. The optimized structures show that the coordination geometry of the copper center is distorted square pyramidal. The solid-state electronic absorption spectra illustrate a broad band, whereas the polycrystalline EPR spectra show a considerable shift toward the trigonal bipyramidal (g⊥ > g∥; g∥ ≈ ge) geometry in addition to the axial symmetry. However, in a solution, they lean toward the axially symmetric, distorted square pyramidal shape characteristics of monocopper(II) five-coordinate complexes. The structural distortion of the positive CuII/CuI redox couple facilitates simple interconversion between CuI and CuII redox states and makes the pair chemically and electrochemically reversible. This is because the appropriate chelate ring sizes of the models provide the coordination mode with a minimal amount of stereochemical reorganization while preserving the stability of the CuI and CuII states.Biomimetic catalytic oxidative reactions can be effectively imitated by the monocopper(II) complexes. It emerged that both catalysts in MS and/or BS favored three different chemical transformations under benign conditions and functioning as a single active site, specifically, H2A → DA [kcat: AOase, MS, 4.8 (1) or 8.5 × 106 h−1 (2); BS, 2.5 (1) or 5.2 × 107 h−1 (2)], Ph–CH2–NH2 → Ph-CHO [kcat: AmOase, 4.7 (1) or 5.4 × 105 h−1 (2)], and 3,5-DTBC → 3,5-DTBQ [kcat: COase, MS, 2.6 (1) or 1.7 × 103 h−1 (2); BS, 1.5 (1) or 1.7 × 106 h−1 (2)]. Based on their kcat values, they demonstrate a high degree of AmOase activity. The kcat values for their activities in buffer solution are more similar to the turnover number of natural enzymes (AOase, 2.45 × 106 h−1 and 3.1 × 107 h−1; COase, 5.7 × 105 h−1 and 8.25 × 106 h−1). Interestingly, for these catalytic processes, these values represent the highest turnover number for monocopper(II) complexes that has been reported to date.
In summary, several characteristics, such as the labile coordinated oxygen donor, the appropriate chelate ring size, the flexibility of the coordinated planar phen, structural distortion, the ease of the substrate–catalyst association, and the positive (CuII/CuI) redox potential, can be attributed to the higher activity of catalysts for enzyme (AOase, AmOase, and COase) modeling catalysis. As a result, both monocopper(II) complexes exhibit the features of the most potent promiscuous catalysts, making them suitable models for oxidation processes and amplify the activity of copper oxidase enzymes. The comprehension of the three probable mechanistic routes has been enhanced by the catalytic promiscuity of the functional biomimetics. Gaining an understanding of functional biomimetics allows one to catalyze a number of important and industrially relevant organic syntheses
Author contributions
Vigneswara Chellam Ravisankar: formal analysis, investigation, and methodology; Balasubramaniam Selvakumaran: data curation, formal analysis, investigation, methodology, and writing – original draft; Tamilarasan Ajaykamal: methodology and software; Mariappan Murali: conceptualization, funding acquisition, investigation, methodology, project administration, resources, supervision, writing – original draft, and writing – review and editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to the ANRF-Science and Engineering Research Board (SERB), India, for the financial support (CRG/2022/002225 and EMR/2016/007756). The authors further thank Professor R. Murugavel, Indian Institute of Technology Bombay, Mumbai, for providing them access to single crystal X-ray diffraction testing that was made possible by an excellent investigator award from the DAE-SRC. We express our gratitude to the Sophisticated Analytical Instrumentation Facility (SAIF), Indian Institute of Technology Madras, Chennai for providing the EPR facility. BS is grateful to the Tamil Nadu State Council for Science and Technology (TNSCST), Chennai, for the RFRS fellowship (TNSCST/RFRS/08/VM/2021-22).References
- S. V. Ley, Comprehensive Organic Synthesis: Selectivity, Strategy and Efficiency in Modern Organic Chemistry, Oxidation, 1992, vol. 7 Search PubMed.
- R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, 1981 Search PubMed.
- B. Cornils, in Green Chemistry and Catalysis, ed. R. A. Sheldon, I. Arends and U. Hanefeld, 2007 Search PubMed.
- C. S. Foote, J. S. Valentine, A. Greenberg and J. F. Liebman, Active Oxygen in Chemistry, in Struct. Energ. React. Chem. Ser, 1995, vol. 2 Search PubMed.
- M. Rolff, J. Schottenheim, H. Decker and F. Tuczek, Chem. Soc. Rev., 2011, 40, 4077–4098 Search PubMed.
- E. I. Solomon, R. Sarangi, J. S. Woertink, A. J. Augustine, J. Yoon and S. Ghosh, Acc. Chem. Res., 2007, 40, 581–591 CrossRef CAS PubMed.
- S. Friedle, E. Reisner and S. J. Lippard, Chem. Soc. Rev., 2010, 39, 2768–2779 RSC.
- L. Que Jr and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef PubMed.
- T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2364 CrossRef CAS PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. K. Emmons, C. H. Kjaergaard, R. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS.
- H. Pracejus, Adv. Synth. Catal., 1975, 317, 350–351 Search PubMed.
- L. Casella, E. Monzani, L. Santagostini, L. D. Gioia, M. Gullotti, P. Fantucci, T. Beringhelli and A. Marchesini, Coord. Chem. Rev., 1999, 185, 619–628 CrossRef.
- A. Messerschmidt, A. Rossi, R. Ladenstein, R. Huber, M. Bolognesi, G. Gatti, A. Machesini, R. Petruzzelli and A. Finazzi-Agró, J. Mol. Biol., 1989, 206, 513–529 CrossRef CAS PubMed.
- A. Messerschmidt, R. Ladenstein and R. Huber, J. Mol. Biol., 1993, 230, 997–1014 CrossRef CAS.
- A. L. Hughes, Immunogenetics, 1999, 49, 106–114 CrossRef CAS.
- T. Klabunde, C. Eicken, J. C. Sacchettini and B. Krebs, Nat. Struct. Biol., 1998, 5, 1084–1090 CrossRef CAS PubMed.
- C. Gerdemann, C. Eicken and B. Krebs, Acc. Chem. Res., 2002, 35, 183–191 CrossRef CAS PubMed.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2606 CrossRef CAS PubMed.
- I. A. Koval, P. Gamez, C. Belle, K. Selmeczi and J. Reedijk, Chem. Soc. Rev., 2006, 35, 814–840 RSC.
- B. J. Deverall, Nature, 1961, 189, 311 CrossRef.
- M. Zhao, H. B. Eang, L. N. Ji and Z. W. Mao, Chem. Soc. Rev., 2013, 42, 8360–8375 RSC.
- M. Guell and P. E. M. Siegbahn, J. Biol. Inorg. Chem., 2007, 12, 1251–1264 CrossRef PubMed.
- B. T. O. Holt, M. A. Vance, L. M. Mirica, D. E. Heppner, T. D. P. Stack and E. I. Solomon, J. Am. Chem. Soc., 2009, 131, 6421–6438 CrossRef PubMed.
- S. K. Dey and A. Mukherjee, Coord. Chem. Rev., 2016, 310, 80–115 CrossRef.
- A. Biswas, L. K. Das, M. G. B. Drew, C. Diaz and A. Ghosh, Inorg. Chem., 2012, 51, 10111–10121 CrossRef.
- K. Hult and P. Berglund, Trends Biotechnol., 2007, 25, 231–238 CrossRef.
- T. P. Camargo, F. F. Maia, C. Chaves, B. D. Souza, A. J. Bortoluzzi, N. Castilho, T. Bortolotto, H. Terenzi, E. E. Castellano, W. Haase, Z. Tomkowicz, R. A. Peralta and A. Neves, J. Inorg. Biochem., 2015, 146, 77–88 CrossRef PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, C71, 3–8 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef.
- B. Selvakumaran, M. Murali, S. Shanmugavadivel and S. Sindhuja, J. Inorg. Biochem., 2024, 259, 112671 CrossRef CAS PubMed.
- D. Jiang, X. Li, L. Liu, G. B. Yagnik and F. Zhou, J. Phys. Chem. B, 2010, 114, 4896–4903 CrossRef CAS PubMed.
- P. A. N. Reddy, M. Nethaji and A. R. Chakravarty, Inorg. Chim. Acta, 2002, 337, 450–458 CrossRef CAS.
- A. K. Ghosh, A. Ali, Y. Singh, C. S. Purohit and R. Ghosh, Inorg. Chim. Acta, 2018, 474, 156–163 CrossRef CAS.
- A. Santra, G. Mondal, M. Acharjya, P. Bera, A. Panja, T. K. Mandal, P. Mitra and P. Bera, Polyhedron, 2016, 113, 5–15 CrossRef CAS.
- S. Mondal, M. Chakraborty, A. Mondal, B. Pakhira, A. J. Blake, E. Sinn and S. K. Chattopadhyay, New J. Chem., 2018, 42, 9588–9597 RSC.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef CAS.
- V. Sathya and M. Murali, Inorg. Chem. Commun., 2018, 92, 55–59 CrossRef CAS.
- J. B. Bates, L. M. Toth, A. S. Quist and G. E. Boyd, Spectrochim. Acta, Part A, 1973, 29, 1585–1600 CrossRef.
- S. Sangeetha, T. Ajaykamal and M. Murali, New J. Chem., 2021, 45, 7578–7593 RSC.
- I. C. Mendes, J. P. Moreira, A. S. Mangrich, S. P. Balena, B. L. Rodrigues and H. Beraldo, Polyhedron, 2007, 26, 3263–3270 CrossRef CAS.
- E. Montiel, A. J. Cruz, N. Jayanthi, S. Bernés and T. Pandiyan, Aust. J. Chem., 2010, 63, 965–977 CrossRef CAS.
- G. A. V. Albada, A. Mohamadou, I. Mutikainen, U. Turpeinen and J. Reedijk, Eur. J. Inorg. Chem., 2004, 3733–3742 CrossRef.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- K. Gudasi, R. Vadavi, R. Shenoy, M. Patil, S. A. Patil and M. Nethaji, Inorg. Chim. Acta, 2005, 358, 3799–3806 CrossRef CAS.
- J. L. Viegas and S. N. Dhuri, J. Mol. Struct., 2023, 1288, 135719 CrossRef CAS.
- M. Egli and S. Sarkhel, Acc. Chem. Res., 2007, 40, 197–205 CrossRef CAS PubMed.
- T. J. Mooibroek, P. Gamez and J. Reedijk, CrystEngComm, 2008, 10, 1501–1515 RSC.
- P. Chakraborty, S. Purkait, S. Mondal, A. Bauzá, A. Frontera, C. Masserac and D. Das, CrystEngComm, 2015, 17, 4680–4690 RSC.
- T. H. Huang, H. Yang, G. Yang, S. L. Zhu and C. L. Zhang, Inorg. Chim. Acta, 2017, 455, 1–8 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision D.01), Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- N. Cankaya, E. Tanıs, H. E. Gülbas and N. Bulut, Polym. Bull., 2019, 76, 3297–3327 CrossRef CAS.
- C.-G. Zhan, J. A. Nichols and D. A. Dixon, J. Phys. Chem. A, 2003, 107, 4184–4195 CrossRef CAS.
- J. D. Bradley and G. C. Gerrans, J. Chem. Educ., 1973, 50, 463–466 CrossRef CAS.
- W. T. Yang and R. G. Parr, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 6723–6726 CrossRef CAS PubMed.
- M. Berkowitz, J. Am. Chem. Soc., 1987, 109, 4823–4825 CrossRef CAS.
- S. B. Liu, J. Chem. Sci., 2006, 117, 477–483 CrossRef.
- L. Shen, H. Y. Zhang and H. F. Ji, J. Mol. Struct.:THEOCHEM, 2007, 817, 161–162 CrossRef CAS.
- M. M. Lawal, T. Govender, G. E. Maguire, H. G. Kruger and B. Honarparvar, Int. J. Quantum Chem., 2018, 118, e25497 CrossRef.
- I. M. Procter, B. J. Hathaway, D. E. Billing, R. Dudley and P. Nicholls, J. Chem. Soc. A, 1969, 1192–1197 RSC.
- D. E. Billing, R. Dudley, B. J. Hathaway and A. A. G. Tomlinson, J. Chem. Soc. A, 1971, 691–696 RSC.
- W. Fitzgerald and B. J. Hathaway, J. Chem. Soc., Dalton Trans., 1981, 567–574 RSC.
- B. J. Hathaway and D. E. Billing, Coord. Chem. Rev., 1970, 5, 143–207 CrossRef CAS.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, 1970 Search PubMed.
- E. Garribba and G. Micera, J. Chem. Educ., 2006, 83, 1229–1232 CrossRef CAS.
- Z. Lu, C. Duan, Y. Tian, X. You and X. Huang, Inorg. Chem., 1996, 35, 2253–2258 CrossRef CAS.
- A. M. Fathy, M. M. Hessien, M. M. Ibrahim and A. E. M. M. Ramadan, J. Mol. Struct., 2022, 1250, 131809 CrossRef CAS.
- P. S. Subramanian, E. Suresh, P. Dastidar, S. Waghmode and D. Srinivas, Inorg. Chem., 2001, 40, 4291–4301 CrossRef CAS.
- D. Kivelson and R. Neiman, J. Chem. Phys., 1961, 35, 149–155 CrossRef CAS.
- S. D. Sid, O. B. Baitich and J. P. Deloume, Polyhedron, 1997, 16, 2175–2182 CrossRef.
- A. W. Addison, M. Carpenter, L. M. K. Lau and M. Wicholas, Inorg. Chem., 1978, 17, 1545–1552 CrossRef CAS.
- P. Tamil Selvi, M. Murali, M. Palaniandavar, M. Kockerling and G. Henkel, Inorg. Chim. Acta, 2002, 340, 139–146 CrossRef.
- D. X. West and M. Palaniandavar, Inorg. Chim. Acta, 1983, 77, L97–L98 CrossRef CAS.
- U. Sakaguchi and A. W. Addison, J. Chem. Soc., Dalton Trans., 1979, 600–608 RSC.
- I. M. Procter, B. J. Hathaway and P. Nicholls, J. Chem. Soc. A, 1968, 1678–1684 RSC.
- E. Turkkan, U. Sayin, N. Erbilen, S. Pehlivanoglu, G. Erdogan, H. U. Tasdemir, A. O. Saf, L. Guler and E. G. Akgemci, J. Organomet. Chem., 2017, 831, 23–35 CrossRef CAS.
- B. J. Hathaway, Copper, in Comprehensive Coordination Chemistry, Pergamon Press, Oxford, 1987, vol. 5, p. 533 Search PubMed.
- R. P. John, A. Sreekanth, V. Rajakannan, T. A. Ajith and M. R. P. Kurup, Polyhedron, 2004, 23, 2549–2559 CrossRef.
- D. B. Rorabacher, Chem. Rev., 2004, 104, 651–697 CrossRef PubMed.
- V. K. Bhardwaj, N. A. Alcalde, M. Corbella and G. Hundal, Inorg. Chim. Acta, 2010, 363, 97–106 CrossRef.
- K. Güçlü, K. Sözgen, E. Tütem, M. Ö. Zyürek and R. Apak, Talanta, 2005, 65, 1226–1232 CrossRef.
- D. Jiang, X. Li, L. Liu, G. B. Yagnik and F. Zhou, J. Phys. Chem. B, 2010, 114, 4896–4903 CrossRef.
- R. D. Patil and S. Adimurthy, Adv. Synth. Catal., 2011, 353, 1695–1700 CrossRef.
- G. Battaini, A. Granata, E. Monzani, M. Gullotti and L. Casella, Biomimetic oxidations by dinuclear and trinuclear copper complexes, in Advances in Inorganic Chemistry, ed. R. van Eldik and J. Reedijk, Academic Press, 2006, pp. 185-233 Search PubMed.
- D. L. Reger, A. E. Pascui, M. D. Smith, J. Jezierska and A. Ozarowski, Inorg. Chem., 2012, 51, 11820–11836 CrossRef CAS.
- A. Jana, N. A. Alcalde, E. Ruiz and S. Mohanta, Inorg. Chem., 2013, 52, 7732–7746 CrossRef CAS.
- J. S. Woertink, L. Tian, D. Maiti, H. R. Lucas, R. A. Himes, K. D. Karlin, F. Neese, C. Wurtele, M. C. Holthausen, E. Bill, J. Sundermeyer, S. Schindler and E. I. Solomon, Inorg. Chem., 2010, 49, 9450–9459 CrossRef CAS.
- R. L. Peterson, J. W. Ginsbach, R. E. Cowley, M. F. Qayyum, R. A. Himes, M. A. Siegler, C. D. Moore, B. Hedman, K. O. Hodgson, S. Fukuzumi, E. I. Solomon, K. D. Karlin and J. Am, Chem. Soc., 2013, 135, 16454–16467 CrossRef CAS PubMed.
- M. K. Panda, M. M. Shaikh, R. J. Butcher and P. Ghosh, Inorg. Chim. Acta, 2011, 372, 145–152 CrossRef CAS.
- F. Sama, A. K. Dhara, M. N. Akhtar, Y. C. Chen, M. L. Tong, I. A. Ansari, M. Raizada, M. Ahmad, M. Shahid and Z. A. Siddiqi, Dalton Trans., 2017, 46, 9801–9823 RSC.
- M. Murali, V. Sathya, B. Selvakumaran and J. Biol, Inorg. Chem., 2021, 26, 67–79 CAS.
- S. Adhikari, A. Banerjee, S. Nandi, M. Fondo, J. S. Matalobos and D. Das, RSC Adv., 2015, 5, 10987–10993 RSC.
- S. Y. Shaban, A. M. Ramadan, M. M. Ibrahim, F. I. E. Shami and R. V. Eldik, Inorg. Chim. Acta, 2019, 486, 608–616 CrossRef CAS.
- F. T. Ferre, J. A. L. C. Resende, J. Schultz, A. S. Mangrich, R. B. Faria, A. B. Rocha and M. Scarpellini, Polyhedron, 2017, 123, 293–304 CrossRef CAS.
- A. Terán, A. Jaafar, A. E. S. Pelaez, M. C. Torralba, Á. Gutiérrez and J. Biol, Inorg. Chem., 2020, 25, 671–683 Search PubMed.
- C. Eicken, F. Zippel, K. B. Karentzopoulos and B. Krebs, FEBS Lett., 1998, 436, 293–299 CrossRef CAS PubMed.
- A. Rompel, H. Fischer, D. Meiwes, K. B. Karentzopoulos, A. Magrini, C. Eicken, C. Gerdemann and B. Krebs, FEBS Lett., 1999, 445, 103–110 CrossRef CAS.
- J. Mukherjee and R. Mukherjee, Inorg. Chim. Acta, 2002, 337, 429–438 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2390596. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00077g |
| This journal is © The Royal Society of Chemistry 2025 |