
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold bis(dithiolene) radical with fused pyrazine and dithiine rings on dithiolene ligand turns metallic under pressure†
Haia
Kharraz
a,
Pere
Alemany
 b,
Enric
Canadell
*cd,
Thierry
Roisnel
a,
Hengbo
Cui
e,
Kee Hoon
Kim
e,
Marc
Fourmigué
*a and
Dominique
Lorcy
*a
b,
Enric
Canadell
*cd,
Thierry
Roisnel
a,
Hengbo
Cui
e,
Kee Hoon
Kim
e,
Marc
Fourmigué
*a and
Dominique
Lorcy
*a
aUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes), 35042 Rennes, France. E-mail: marc.fourmigue@univ-rennes.fr; dominique.lorcy@univ-rennes.fr
bDepartament de Ciència de Materials i Química Física and Institut de Química Teòrica i Computacional (IQTCUB), Universitat de Barcelona, Martí i Franquès 1, 08028 Barcelona, Spain
cInstitut de Ciencia de Materials de Barcelona, ICMAB-CSIC, Campus de la UAB, 08193 Bellaterra, Spain. E-mail: canadell@icmab.es
dRoyal Academy of Sciences and Arts of Barcelona, Chemistry Section, La Rambla 115, 08002 Barcelona, Spain
eInstitute of Applied Physics, Department of Physics and Astronomy, Seoul National University, Seoul 08826, Korea
First published on 1st April 2025
Abstract
Conducting molecular materials built on dithiolene complexes, as mixed-valence salts or single component materials, are most often based on planar structures which efficiently stack on top of each other. Herein, we report an original dithiolene ligand, namely, [1,4]dithiino[2,3-b]pyrazine-2,3-bis(thiolate) (hereafter noted as pzdtdt), which combines an electron-withdrawing pyrazine ring and a folded (by 40–50°) dithiine ring. Until now, such strong distortions from planarity have been hindering the isolation of highly conducting materials from dithiine-containing dithiolene complexes. However, in this study, we showed that the gold complex radical [Au(pzdtdt)2]˙ obtained via electrocrystallization from the 1e− oxidation of [Ph4P][Au(pzdtdt)2] organized into regular, non-dimerized chains in the solid state. Interestingly, [Au(pzdtdt)2]˙ exhibited a semi-conducting behaviour at ambient pressure and turned metallic upon application of pressures above 4 GPa. The electronic structure of [Au(pzdtdt)2]˙ was investigated in terms of electron localization effects through spin-polarized band-structure calculations.
Introduction
Bis(dithiolene) metal complexes are now more than sixty years old, and among their various applications, their utility as precursors for many highly conducting molecular materials,1 such as the prototypical [Ni(dmit)2] mixed valence salts2 or their Pd analogs with quantum spin liquid (QSL) behavior, has garnered significant attention.3 Among the most recently studied complexes, the open-shell, neutral gold radical complexes [Au(dt)2]˙ are being investigated as single component molecular conductors.4 Such radical complexes often exhibit a tendency to dimerize in the solid state,5 leading to a semiconducting behaviour, but recent examples, based on the original substitution pattern on the dithiolene ligands,6 enabled the isolation of non-dimerized, regular stack structures with strong intermolecular overlap and associated metallic behaviour, either under high pressure or ambient pressure.7 The properties of such radical species are strongly dependent on the nature of the dithiolene ligands, often involving fused heterocycles incorporating nitrogen atoms (pyridine and pyrazine) or sulfur atoms (thiophene, dithiole, and dithiine) or both (thiazoline and thiazole). Among these, complexes with fused N-aromatic dithiolene ligands (Scheme 1a), such as pyrazine moieties [Au(pzdt)2]8,9 and [M(qdt)2] (qdt = quinoxaline-2,3-dithiolate),10 have been reported for their ability to coordinate with other metals for the generation of MOF11 and for their proton-dependent solution luminescence properties.12 However, owing to the strong electron-withdrawing effect of the fused pyrazine, only the reduced monoanionic gold complexes can be isolated,13 and no single component molecular conductors based on the neutral species have been reported so far. Alternatively, the introduction of sulfur heterocycles at the periphery of the metal bis(dithiolene) complexes is extensively explored to favour intermolecular interactions in the solid state, as illustrated in numerous dmit and dddt complexes.14 Besides, complexes of the dithiine dithiolate ligand (ddt, Scheme 1b) have also been studied, but only poorly conducting materials were obtained from M(ddt)2 derivatives so far owing to a lack of sizeable intermolecular interactions,15,16 which is a consequence of the non-planar structure of the dithiine ring that is strongly folded along the S⋯S axis. Consequently, no conducting mixed-valence nickel salts or conducting single-component gold complexes have been reported to date from such systems. Only one complex combining the pyrazine and dithiine heterocycles has been reported (Scheme 1c), with two dithiino[2,3-b]quinoxaline-2,3-dithiolates surrounding either the Au3+ or Ni2+ in M(qdtdt)2 complexes; however, no conducting materials have been prepared from them so far.17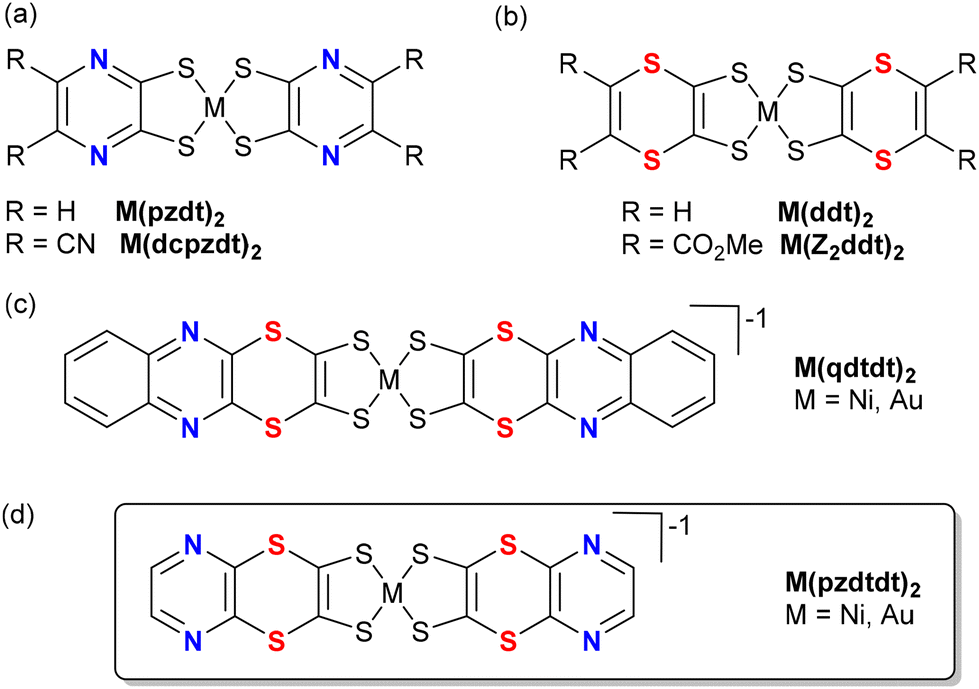 | ||
| Scheme 1 Molecular structures of dithiolene complexes involving pyrazine, dithiine or both the heterocycles. | ||
Therefore, we decided to investigate the synthesis of another dithiolene ligand with only the pyrazine and dithiine moieties and concentrated our efforts on the [1,4]dithiino[2,3-b]pyrazine-2,3-bis(thiolate) ligand (hereafter pyrazinedithiinedithiolate is denoted as pzdtdt) (Scheme 1d). Herein, we report the preparation of the ligand precursor, its Ni and Au monoanionic complexes, and the successful isolation of the neutral gold complex radical [Au(pzdtdt)2]˙, behaving as a single-component molecular conductor, which is a remarkable behaviour when considering its non-planar geometry.
Results and discussion
Synthesis of the proligands
For the synthesis of the dithiolene ligand (Scheme 2), we focused on the synthesis of the corresponding proligands 1 and 2, as 1,3-dithiole-2-thione (1) or 1,3-dithiole-2-one (2) derivatives, via a copper-catalysed coupling reaction between aryl iodides and zinc–thiolate complexes.18 Thus, reacting 2,3-diiodopyrazine19 and [Et4N]2[Zn(dmit)2] in the presence of Cu2O afforded dithiole-2-thione 1 in 63% yield. Trans-chalcogenation with Hg(OAc)2 afforded the desired proligand 2 in 94% yield. | ||
| Scheme 2 Synthetic scheme of 1, 2 and [Au(pzdtdt)2]˙. | ||
In order to evaluate the possibly different reactivity of 1 and 2 as precursors of the dithiolate ligand, both were separately engaged with sodium methanolate followed by successive additions of KAuCl4 and Ph4PBr, to afford the monoanionic complex [Ph4P][Au(pzdtdt)2] in 10 and 40% yields, respectively, demonstrating the usefulness of the transformation of dithiole-2-thione 1 into dithiole-2-one 2 before treatment with sodium methanolate. Note that the corresponding nickel complex has also been prepared, as Et4N+ salt, by the same method (see ESI for experimental details and properties†).
Both 1 and 2 were crystallized from CH2Cl2, and their molecular structures were obtained from X-ray diffraction (Fig. 1). The dithiine ring in both of them strongly deviates from planarity, with a folding along the S⋯S axis of 49.3(2)° and 50.0(3)° in 1 and 2, respectively, a recurrent feature of such dithiine derivatives (43–53°).20,21
 | ||
| Fig. 1 Molecular structures of (a) 1 and (b) 2, showing the distortion of the central dithiine ring from planarity. | ||
The redox properties of [Au(pzdtdt)2]− were investigated by cyclic voltammetry in CH3CN and the oxidation potentials are collected in Table 1, together with those of the related complexes. At variance with the pyrazinodithiolate complex [Au(pzdt)2]− which oxidizes irreversibly at high potential (+1.12 V vs. SCE), the insertion of the dithiine ring between the pyrazine and the dithiolate moiety suppresses here to a large extent the electron-withdrawing nature of the pyrazine ring on the redox-active dithiolene and provides a −1/0 oxidation potential for [Au(pzdtdt)2]− around +0.47 V vs. SCE (Fig. S2 in ESI†). Such an effect was already observed, in a more limited way, from the introduction of a benzo ring (rather than a dithiine one) in a Ni complex with two quinoxaline-2,3-dithiolate ligands22 compared with the complex surrounded by two quinoxaline-6,7-dithiolate (Scheme 3) which oxidizes at lower potential (by 300 mV).23 This effect opens the possibility to isolate the radical [Au(pzdtdt)2]˙ species within an accessible potential window. Besides, spectroelectrochemical experiments performed on [Ph4P][Au(pzdtdt)2] show, upon oxidation, the appearance of a low-energy NIR absorption band for the neutral complex centred at 1410 nm, characteristic of such radical complexes.24,25
 | ||
| Scheme 3 Molecular structures of quinoxaline-2,3-dithiolate and quinoxaline-6,7-dithiolate complexes. | ||
| E 1 −2/–1 | E 2 −1/0 | E 3 0/+1 | ΔEpa | Ref. | |
|---|---|---|---|---|---|
| E = (Epa + Epc)/2.a Epa/Epc: anodic and cathodic peak potentials.b As Bu4N+ salt.c As Et4N+ salt.d A correction of 0.39 V has been applied to the given redox potentials (in italics) versus Fc+/Fc.e Performed in CH2Cl2.f Irreversible. | |||||
| [Au(pzdtdt)2] | –1.01f | +0.54/0.40a | +0.96f | 0.42 | This work |
| [Au(pzdt)2] | −1.27d | +1.12d,f | +1.89d,f | 0.77 | 8 |
| (−1.66) | (+0.73) | (+1.5) | |||
| [Au(qdtdt)2] | −0.89b | +0.49b | — | — | 17 |
| −1.02c | +0.44c | — | |||
| [Au(Z2ddt)2] | — | +0.86 | +1.2 | 0.34 | 16b |
| [Au(Py-ddt)2] | −0.93 | +0.74 | — | — | 16c |
| [Au(Et-thiazdt)2]e | –0.90f | +0.55/+0.49 | +0.71/+0.61 | 0.16 | 6a |
Accordingly, electrocrystallization experiments of a solution of [Ph4P][Au(pzdtdt)2] were conducted with [Bu4N][PF6] in THF as the electrolyte and crystals of the neutral radical species [Au(pzdtdt)2]˙ were indeed successfully obtained applying a constant current intensity of 1 μA for 3 days. The molecular structure of the radical [Au(pzdtdt)2]˙ can be now compared with that of the monoanionic [Au(pzdtdt)2]− complex, as detailed below, while its solid-state structure will be analysed in a second step, in relation with its conducting properties.
Solid state structures and properties
[Ph4P][Au(pzdtdt)2] crystallizes in the triclinic system, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with two crystallographically independent complexes, both located on inversion centres. The molecular structures of these two independent anions (Mol1 and Mol2) are shown in Fig. 2a, and the bond lengths and angles are shown in Table 2. As observed for the proligands 1 and 2, the 1,4-dithiine rings deviate from planarity with a distortion angle along the S⋯S hinge of 46.4(1)° and 49.5(4)° in Mol1 and Mol2, respectively. The AuS2C2 metallacycles of these gold complexes are also distorted from planarity along the S⋯S axis with a much less pronounced effect (20.2(1)° and 10.4(2)°). The overall shape of this complex is similar to that of the corresponding monoanionic Au complex, with an additional fused benzene ring at each end, [Au(qdtdt)2]−1 (cf.Scheme 1c), where the dithiine folding angle was found to be 50.6(6)°.17
, with two crystallographically independent complexes, both located on inversion centres. The molecular structures of these two independent anions (Mol1 and Mol2) are shown in Fig. 2a, and the bond lengths and angles are shown in Table 2. As observed for the proligands 1 and 2, the 1,4-dithiine rings deviate from planarity with a distortion angle along the S⋯S hinge of 46.4(1)° and 49.5(4)° in Mol1 and Mol2, respectively. The AuS2C2 metallacycles of these gold complexes are also distorted from planarity along the S⋯S axis with a much less pronounced effect (20.2(1)° and 10.4(2)°). The overall shape of this complex is similar to that of the corresponding monoanionic Au complex, with an additional fused benzene ring at each end, [Au(qdtdt)2]−1 (cf.Scheme 1c), where the dithiine folding angle was found to be 50.6(6)°.17
 | ||
| Fig. 2 Side views of (a) two crystallographically independent monoanionic [Au(pzdtdt)2]− complexes and (b) neutral complex [Au(pzdtdt)2]˙, showing the recurrent distortion of the central dithiine ring from planarity. | ||
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Complex | a | b | c | d | e | f | θ 1 | θ 2 | Ref. | |
| [Au(pzdtdt)2]−1 | Mol1 | 2.3116(9) | 1.750(3) | 1.334(4) | 1.754(3) | 1.768(3) | 1.394(5) | 20.2(1) | 46.4(1) | This work |
| 2.3085(8) | 1.751(3) | 1.766(3) | 1.766(3) | |||||||
| Mol2 | 2.3170(7) | 1.751(3) | 1.339(4) | 1.766(3) | 1.764(3) | 1.404(4) | 10.4(2) | 49.5(4) | ||
| 2.3114(8) | 1.754(3) | 1.768(3) | 1.768(3) | |||||||
| [Au(qdtdt)2]−1 | 2.300(4) | 1.738(12) | 1.397(19) | 1.731(12) | 1.733(16) | 1.470(25) | 3.6(4) | 53.03(6) | 17 | |
| 2.282(4) | 1.738(12) | 1.746(12) | 1.757(14) | |||||||
| [Au(pzdtdt)2]˙ | 2.306(2) | 1.725(10) | 1.354(13) | 1.764(9) | 1.760(10) | 1.407(13) | 7.5(2) | 41.4(10) | This work | |
| 2.291(2) | 1.723(9) | 1.758(9) | 1.768(9) | |||||||

The neutral [Au(pzdtdt)2]˙ complex crystallizes in the monoclinic system, space group P21/n, with one crystallographically independent complex located on an inversion centre. The neutral gold complex is slightly less distorted than its monoanionic counterpart: for the dithiine ring, the distortion angle decreases from 46–49° to 41.4°, while for the metallacycle, it decreases from 10–20° to 7.5°. Comparison of the intramolecular bond lengths within the monoanionic and neutral states shows that the main modifications concentrate on the AuS2C2 metallacycles, with a shortening of the C–S bonds (bond b) and a lengthening of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds (bond c) upon oxidation, which is typical of the evolution of such dithiolene complexes upon oxidation. The absence of notable structural effects on the dithiine and pyrazine rings, together with the recurrent strong distortion of the dithiine ring from planarity, clearly indicates that the oxidation is strongly concentrated on the two metallacycles, with limited effects on the outer rings.
C bonds (bond c) upon oxidation, which is typical of the evolution of such dithiolene complexes upon oxidation. The absence of notable structural effects on the dithiine and pyrazine rings, together with the recurrent strong distortion of the dithiine ring from planarity, clearly indicates that the oxidation is strongly concentrated on the two metallacycles, with limited effects on the outer rings.
As shown in Fig. 3a, the radical complexes organize into layers in the (ac) plane, with very limited interactions between layers. Despite their non-planar structure, these radical complexes form regular stacks along the a axis with a gold-to-gold atomic distance of a = 3.70 Å (Fig. 3b). The shortest S⋯S contacts within the stacks amount to the same distance of a = 3.70 Å, while shorter lateral S⋯S intermolecular contacts are found between stacks within layers (Fig. 3a), down to 3.49–3.52 Å.
 | ||
| Fig. 3 Structure of [Au(pzdtdt)2]˙ with (a) projection view of the unit cell along the a axis and (b) view of the stack running along a. | ||
The electrical resistivity and its temperature dependence were measured on single crystals of the neutral complex [Au(pzdtdt)2]˙ by the four-point technique at pressures between 1 bar and 19.6 GPa, using a diamond anvil cell (DAC). At room temperature and ambient pressure, the resistivity amounts to 489.5 Ω cm (σRT = 2.04 10−3 S cm−1), and the compound is a semiconductor with an activation energy of 227 meV (Fig. 4a). Further application of pressure strongly increases the room temperature conductivity up to σRT(4.2 GPa) = 150 S cm−1 and decreases the activation energy (Fig. 4b). This exponential pressure dependence is observed up to 4.2 GPa to reach a quasi-metallic behaviour. At 4.8 GPa, the complex is metallic in the whole temperature range. Moreover, above 6.2 GPa (Fig. S5 in ESI†), a slight increase in both the resistivity and the activation energy is observed up to 19.6 GPa, with σRT(19.6 GPa) = 2.33 S cm−1 (ρRT(4.2 GPa) = 0.43 Ω cm) and Eact (19.6 GPa) = 22 meV.
 | ||
| Fig. 4 (a) Temperature dependence of resistivity of [Au(pzdtdt)2]˙ at different applied pressures; (b) pressure dependence of the conductivity of [Au(pzdtdt)2]˙, and the activation energy at room temperature. | ||
Electronic structure
The unit cell of [Au(pzdtdt)2]˙ contains two symmetry equivalent ac layers built from parallel chains of the [Au(pzdtdt)2]˙ complex along the a-direction (Fig. 3a). Although there are numerous short S⋯S contacts within the ac layers, there are no S⋯S inter-layer contacts along b, so this solid is expected to be at most a 2D conductor. As shown in Fig. 5a, where all intermolecular S⋯S contacts shorter than 3.9 Å are shown as red dashed lines, there are three different types of intermolecular interactions, one (I) is intra-chain and two (II and III) are inter-chain interactions. | ||
| Fig. 5 (a) Layers of the [Au(pzdtdt)2]˙ solid, where the different intermolecular interactions are noted. The S⋯S bonds shorter than 3.9 Å are shown as red dashed lines. (b) Three different intermolecular interactions. | ||
In principle the transport properties of [Au(pzdtdt)2]˙ should be determined by the SOMO⋯SOMO (Singly Occupied Molecular Orbital) interactions. However, previous work on such radical gold bis(dithiolene)7 complexes has shown that both the SOMO⋯SOMO and SOMO-1⋯SOMO-1 interactions should be considered when the complexes interact strongly along one of the two directions of the layer. In that case, there is indeed the possibility of a two-band behaviour,26i.e. some electron transfer can occur between the SOMO-1 and SOMO levels. The simplest way to get a hint on the relative strength of the SOMO⋯SOMO and SOMO-1⋯SOMO-1 interactions is by evaluating the so-called intermolecular interaction energies (β).27 The absolute values of the interaction energies |βSOMO⋯SOMO|/|βSOMO-1⋯SOMO-1| calculated for this system are 0.1993/0.1960 (I), 0.0039/0.0375 (II), and 0.0043/0.0030 (III) eV: the inter-chain interactions are either one or two orders of magnitude weaker than the intra-chain interactions. Thus, it is clear that the intra-chain interactions completely control the electronic properties of the solid. These interactions are of similar strength for the SOMO and SOMO-1. The strength of interaction I is easy to understand since, as shown in Fig. 5b, such interaction is associated with four pairs of S⋯S interactions at 3.703 Å distance. Despite the sliding of adjacent complexes, the interaction has a substantial σ-type character and does not almost differentiate between the SOMO and SOMO-1 interactions. More surprising is the very weak character of the inter-chain interactions II and III despite quite short S⋯S contacts (3.495 and 3.766 (×2) Å for II and 3.528 (×2), 3.640 (×2) and 3.716 Å for III). These interactions are of the usually weak lateral π-type. However, except for the βSOMO-1⋯SOMO-1 of interaction II, the calculated values are around one order of magnitude too small when compared with the usual values for these interactions. As far as the interactions of the SOMO are concerned, since this orbital is antisymmetric with respect to the plane perpendicular to the inner core of the complex (see Fig. 6) separating the two ligands, several of the S⋯S interactions almost cancel and the final interaction is very small. However, the SOMO-1 orbital is symmetric with respect to the same plane (see Fig. 6) and the previous effect cannot explain the weakness of the SOMO-1⋯SOMO-1 interaction III, which can only be due to the poor orientation of the S pz orbitals. Thus, both poor S orbital interactions and the topology of SOMO orbitals are responsible for very weak inter-chain interactions. In summary, [Au(pzdtdt)2]˙ is predicted to be a strongly 1D system from the electronic viewpoint with SOMO and SOMO-1 bands of similar width.
 | ||
| Fig. 6 Band structure calculated for the [Au(pzdtdt)2]˙ solid, assuming the double occupation of levels. The dashed line refers to the Fermi level and Γ = (0, 0, 0), X = (a*/2, 0, 0), Y = (0, b*/2, 0), Z = (0, 0, c*/2) and M = (a*/2, 0, c*/2). | ||
The calculated DFT band structure when double occupation of the levels is assumed is shown in Fig. 6, where every band is really the superposition of two bands because the unit cell contains two practically non-interacting layers. The SOMO band is strongly 1D, whereas the SOMO-1 has a small but non-negligible dispersion along the inter-chain direction (see lines Γ to Z and X to M in Fig. 6). As expected, the band dispersion along the inter-layer direction (Γ to Y) is nil. The two bands have a strong and similar dispersion and exhibit an important overlap. However, the SOMO-1 band does not cross the Fermi level so it is completely full and the SOMO band is half-filled. In other words, the possible two-band behaviour exhibited by other gold bis(dithiolene) solids does not develop at 150 K and ambient pressure for this solid and the SOMO⋯SOMO interactions should govern the transport and magnetic properties. The band structure in Fig. 6 is associated with a Fermi surface with a nesting vector having an a*/2 component (see Fig. S6 in ESI†). Consequently, a dimerization along the chain direction with the consequent opening of a band gap and a semiconducting behaviour is predicted, in contradiction with the observed uniform stack structure. However, strongly 1D gold bis(dithiolene) with such half-filled uniform chains almost invariably exhibits electronic localization.28 It is thus compulsory to look at possible electronic states with electron localization (i.e. uniform chains of [Au(pzdtdt)2]˙ radicals) before concluding anything about the nature of the ground state of this solid.
Initial evaluation of the magnetic coupling constants through dimer calculations showed that only the intra-layer ones corresponding to interactions I, II and II were larger than 1 K, with the first being almost two orders of magnitude larger. All inter-layer coupling constants were found to be as weak as 0.05 K so inter-layer interaction can be neglected. We have thus carried out spin-polarized calculations for four different localized states of the [Au(pzdtdt)2] layers with either ferromagnetic or antiferromagnetic interactions along (i) the intra-chain a direction, and (ii) the inter-chain c direction (see Fig. 7a).
 | ||
| Fig. 7 (a) Four different localized states considered in this study. DFT band structure for the metallic (b) and antiferromagnetic AF12 (c) and AF14 (d) states calculated for a single-layer of [Au(pzdtdt)2]˙ obtained using a 2 × 1 × 2 supercell and by deleting the four molecules of one of the layers. The dashed line refers to the highest occupied level, and Γ = (0, 0, 0), X = (a*/2, 0, 0), Y = (0, b*/2, 0), Z = (0, 0, c*/2) and M = (a*/2, 0, c*/2). The bands in (b) are doubly occupied, and the two sets of four bands noted with green/blue asterisks originate from SOMO/SOMO-1 level. Spin-up and spin-down bands in (c) and (d) are shown in blue and red, respectively. The spin-up and spin-down bands are identical. They are located in spatially different but equivalent sites, so only the blue bands are visible. | ||
Two of the localized states, AF12 and AF14, having antiferromagnetic interactions along the chains are more stable than the metallic state by 53.3 and 52.7 K, respectively. The two states with ferromagnetic interactions along the chains are found to be even less stable than the metallic state. The AF12 state is found to be the lowest energy state although the energy difference between the AF12 and AF14 states is very small (0.6 K) so we conclude that the ground state of this system should exhibit antiferromagnetic interactions along the chains but no magnetic order along the inter-chain and inter-layer directions. The calculated magnetic coupling constants are JI = −427.5 K, JII = +16.7 K and JIII = +24.2 K, respectively (a negative sign means antiferromagnetic interactions). The calculated band structures for the AF12 and AF14 states are shown in Fig. 7c and d, respectively. For comparison, the band structure of the metallic state calculated with the same cell is shown in Fig. 7b. The set of four bands noted with green asterisks in Fig. 6b is the folded version (because of the larger unit cell) of the SOMO bands in Fig. 6 which are globally half-filled. As shown in Fig. 7c and d, the development of antiferromagnetic spin interactions along the direction results with the opening of a large energy gap, which is the reason for the higher stability of the AF12 and AF14 states. The two upper doubly filled SOMO bands (green asterisks) in Fig. 7b are strongly destabilized around the X point of the Brillouin zone and become two SOMO spin up and two SOMO spin down bands of higher energy. The opposite occurs for the lower doubly filled SOMO bands (green asterisks) in Fig. 7b, which become two SOMO spin up and two SOMO spin down bands of lower energy in the region around Γ for the AF12 and AF14 states. Since these bands are filled, they provide a stabilization to the system. The opening of this band gap in the spin polarized band structure is the equivalent of the development of the well-known Peierls gap for a half-filled band in the metallic state when electronic repulsions are important. The small differences between Fig. 7c and d decode the different inter-chain magnetic interactions between antiferromagnetic chains. Since JII and JIII are very similar, the differences are small and the two states are of similar energy, leading most likely to an absence of magnetic ordering along the inter-chain direction.
Let us now briefly consider the interesting evolution of conductivity under pressure illustrated above in Fig. 4. Pressure application will certainly increase the dispersion of the SOMO bands (both because of the induced shortening of intermolecular contacts and possible decrease of the non-planarity), thus stabilizing the metallic state and increasing the conductivity, as it is observed until around 4–5 GPa. At this point, an almost metallic behaviour is observed. However, a further increase in pressure still leads to activated conductivity and one can foresee three different scenarios behind this behaviour. First, a metallic state is stabilized and a typical Peierls type instability for a half-filled strongly 1D metal develops, leading to a dimerization along the chains and the appearance of a band gap. Second, pressure leads to an increase of intra-chain interactions but also to a change in the magnetic inter-chain interactions and one of the two becomes clearly dominant, leading to the more stable state at high pressure. As a result, a different magnetic state now with magnetic order also along the inter-chain direction develops. Third, since the Fermi level for the metallic state is not that far from the top of the SOMO-1 band in Fig. 2, the increase in band dispersion due to pressure may lead to the crossing of the top of the SOMO-1 band and the Fermi level, leading to a small electron transfer between the SOMO-1 and SOMO bands (i.e. a two-band behaviour) as it occurs for instance in [Au(Me-thiazdt)2].4b In that case, the occurrence of a metallic or semiconducting behaviour subtly depends on the degree of electron transfer. However, if this was the case, a further increase of pressure should stabilize the metallic state, which is apparently not the case. Consequently, one of the first two possibilities seems to be more appropriate. Structural work under pressure is needed in order to clarify this question. Indeed, the pressure dependence of the conductivity of [Au(pzdtdt)2]˙ at room temperature (Fig. 4b) shows a very abrupt evolution between 0 and 3 GPa which might indicate a structural transition under pressure and a solid-state organization above 3–4 GPa, different from that determined here by X-ray diffraction at ambient pressure. Work is in progress to tentatively determine the crystal structure above 3–4 GPa.
Conclusions
To date, for dithiolene complexes formed by fusing of dithiine heterocycles, their crystallization into conducting materials has been hindered owing to strong distortions from planarity caused by the dithiine heterocycles. Herein, we reported an original gold bis(dithiolene) radical complex which stacked into regular non-dimerized chains, in the solid-state, with a semi-conducting behaviour at ambient pressure (σRT = 2.04 10−3 S cm−1, Eact = 227 meV). Notably, electron-localization effects of antiferromagnetic nature opened a Mott gap on the SOMO band. Remarkably, this single-component conductor turned metallic upon application of pressures above 4 GPa, which is a consequence of the associated increase in the SOMO band dispersion. A further up-turn to semiconducting behaviour at higher pressures could be the signature of a structural transition, leading to dimerization (Peierls transition) and an increase in inter-stack interactions, which were negligible in the ambient pressure conditions.Experimental section
General
Chemicals and materials from commercial sources were used without further purification. All the reactions were performed under an argon atmosphere. NMR spectra were obtained in CDCl3 unless indicated otherwise. Chemical shifts are reported in ppm, 1H NMR spectra were referenced to residual CHCl3 (7.26 ppm) and 13C NMR spectra were referenced to CHCl3 (77.2 ppm). Melting points were measured on a Kofler hot-stage apparatus and are uncorrected. Mass spectra were recorded by the Centre Régional de Mesures Physiques de l'Ouest, Rennes. Methanol, acetonitrile and dichloromethane were dried using an Inert pure solvent column device. Cyclic voltammetry was performed in a three-electrode cell equipped with a platinum disk working electrode and a glass carbon counter-electrode. CV was carried out in a 10−3 M solution of the complex in CH2Cl2 with 0.1 M [Bu4N][PF6]. Potentials were measured versus Saturated Calomel Electrode (SCE). The spectroelectrochemical study was performed in 0.2 M CH2Cl2-[NBu4][PF6] using a Pt grid as the working electrode, a Pt wire as the counter electrode and a SCE as the reference electrode. A Shimadzu 3600 Plus spectrophotometer was used to record the UV-vis-NIR spectra. The starting 3,4-diiodopyrazine was prepared according to a published procedure.19Crystallography
Suitable crystals for single crystal X-ray diffraction experiment were selected and mounted on the goniometer head of a D8 Venture (Bruker-AXS) diffractometer equipped with a CMOS-PHOTON70 detector, using Mo-Kα radiation (λ = 0.71073 Å, multilayer monochromator). Crystal structures were solved by the dual-space algorithm using the SHELXT program,29 and then refined with full-matrix least-squares methods based on F2 (SHELXL program).30 All non-hydrogen atoms were refined with anisotropic atomic displacement parameters. H atoms were finally included in their calculated positions and treated as riding on their parent atom with constrained thermal parameters. Crystallographic data on X-ray data collection and structure refinements are given in Table S1 in the ESI.†Resistivity measurements
The standard four-probe DC measurements were performed for single crystal [Au(pzdtdt)2]˙ by using a diamond anvil cell (DAC) using the same technique as described in ref. 31 A rectangular shape of single crystal sample 1 (0.14 × 0.03 × 0.01 mm3) and sample 2 (0.11 × 0.03 × 0.01 mm3) was used for ambient pressure and high-pressure measurements up to 6.2 GPa for sample 2 and 19.6 GPa for sample 3 (0.09 × 0.03 × 0.01 mm3). The four contacts were made by four gold wires (10 or 5 μm) with gold paint. The culet size of 0.7 mm (0.56 mm) diamond and tension-annealed stainless still SUS301 (Inconel 625) for sample 1, 2 (sample 3) and pressure-transmitting medium Daphne Oil 7373 was used. The pressure was determined by the shift of the ruby fluorescence R1 lines at room temperature. A Cryocooler helium compressor system (Sumitomo Heavy Industries, Ltd) was used for cooling the DAC with a cooling rate of 1.0 K min−1. A KEITHLEY 224 programmable current source and 182 sensitive digital voltmeter were used for all measurements.Computational details
The first-principles calculations for the solid were carried out using a numeric atomic orbital density functional theory (DFT) approach32,33 developed for efficient calculations in large systems and implemented in the SIESTA code.34–37 We used the generalized gradient approximation (GGA) to DFT and, in particular, the functional of Perdew, Burke, and Ernzerhof.38 To study the relative energies of states with localized electrons, spin polarized band calculations for appropriate supercells have been undertaken. All calculations included a Hubbard correction term Ueff = U − J = 6.0 eV for the S 3p states.39 In previous work,40 we have found that this U term on the chalcogen atoms is needed for appropriately describing the electronic structure of molecular conductors were accurate experimental information on the bandwidth and charge transfer is available. Only the valence electrons are considered in the calculation, with the core being replaced by norm-conserving scalar relativistic pseudopotentials41 factorized in the Kleinman–Bylander form.42 We have used a split-valence double-ζ basis set including polarization orbitals with an energy shift of 10 meV (ref. 43) for S, C, N and H atoms. For gold atoms we have used a split-valence basis set of double-ζ plus polarization quality, where the 5d electrons of Au were treated also as valence electrons. The basis functions used for Au have been optimized in order to reproduce the geometry and the bulk modulus for the ccp crystal structure of metallic gold.44 The energy cutoff of the real space integration mesh was 300 Ry. The Brillouin zone was sampled using grids45 of (10 × 3 × 10), (20 × 3 × 20) and (20 × 20 × 20) k-points. The crystal structure at 150 K was used for the computations.The intermolecular interaction energies were evaluated by means of extended Hückel type46 calculations with a modified Wolfsberg–Helmholtz formula to calculate the non-diagonal Hμν values.47 All valence electrons were considered in the calculations and the basis set consisted of Slater-type orbitals of double-ζ quality for Au 5d and of single-ζ quality for C and N 2s and 2p, S 3s and 3p and H 1s. The ionization potentials, contraction coefficients and exponents were taken from previous works.4c,48
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data have been deposited at the CCDC under CCDC 2422633–2422637 for compounds 1, 2, [Et4N][Ni(pzdtdt)2], [Au(pzdtdt)2]˙ and (Ph4P)[Au(pzdtdt)2], respectively, and can be obtained directly from https://www.ccdc.cam.ac.uk/products/csd/request/request.php4.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Work in France was supported by ANR under contract no. ANR-23-CE07-0032-01 and by Rennes University through a PhD grant (to H. K.). The work at SNU was supported by the National Research Foundation (RS-2024-00338707) and by the Ministry of Education through the core center program (2021R1A6C101B418). Work in Spain was supported by MCIN/AEI/10.13039/501100011033 and AEI through Grants PID2022-139776NB-C61 and PID2021-128217NB-I00 and by Generalitat de Catalunya (2021SGR01519 and 2021SGR00286). E. C. acknowledges MCIN/AEI/10.13039/501100011033 and AEI for support through the Severo Ochoa FUNFUTURE MaTrans42 (CEX2023-0001263-S) Excellence Centre distinction. P. A. acknowledges MCIN/AEI/10.13039/501100011033 and AEI for support through the Maria de Maeztu Units of Excellence Program (CEX-2021-001202-M).References
- R. Kato, Conducting Metal Dithiolene Complexes: Structural and Electronic Properties, Chem. Rev., 2004, 104, 5319–5346 CrossRef CAS PubMed.
- M. Bousseau, L. Valade, J.-P. Legros, P. Cassoux, M. Garbauskas and L. V. Interrante, Highly conducting charge-transfer compounds of tetrathiafulvalene and transition metal-“dmit” complexes, J. Am. Chem. Soc., 1986, 108, 1908–1916 Search PubMed.
- M. Yamashita, N. Nakata, Y. Senshu, M. Nagata, H. M. Yamamoto, R. Kato, T. Shibauchi and Y. Matsuda, Highly mobile gapless excitations in a two-dimensional candidate quantum spin liquid, Science, 2010, 328, 1246–1248 Search PubMed.
- (a) N. C. Schiødt, T. Bjørnholm, K. Bechgaard, J. J. Neumeier, C. Allgeier, C. S. Jacobsen and N. Thorup, Structural, electrical, magnetic, and optical properties of bis-benzene-1,2-dithiolato-Au(IV) crystals, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 1773–1778 Search PubMed; (b) D. Belo, H. Alves, E. B. Lopes, M. T. Duarte, V. Gama, R. T. Henriques, M. Almeida, A. Pérez-Benítez, C. Rovira and J. Veciana, Gold Complexes with Dithiothiophene Ligands: A Metal Based on a Neutral Molecule, Chem. – Eur. J., 2001, 7, 511–519 Search PubMed; (c) O. J. Dautel, M. Fourmigué, E. Canadell and P. Auban-Senzier, Fluorine Segregation Controls the Solid-State Organization and Electronic Properties of Ni and Au Dithiolene Complexes: Stabilization of a Conducting Single-Component Gold Dithiolene Complex, Adv. Funct. Mater., 2002, 12, 693–698 Search PubMed.
- (a) H. Hachem, Z. Xu, N. Bellec, O. Jeannin, P. Auban-Senzier, T. Guizouarn, M. Fourmigué and D. Lorcy, Neutral, closed-shell nickel Bis(2-alkylthio-thiazole-4,5-dithiolate) complexes as single component molecular conductors, Dalton Trans., 2018, 47, 6580–6589 RSC; (b) M. F. G. Velho, R. A. L. Silva, G. Brotas, E. B. Lopes, I. C. Santos, A. Charas, D. Belo and M. Almeida, Conducting neutral gold bisdithiolene complex [Au(dspdt)2]˙, Dalton Trans., 2020, 49, 13737–13743 RSC.
- (a) N. Tenn, N. Bellec, O. Jeannin, L. Piekara-Sady, P. Auban-Senzier, J. Íñiguez, E. Canadell and D. Lorcy, A Single-Component Molecular Metal Based on a Thiazole Dithiolate Gold Complex, J. Am. Chem. Soc., 2009, 131, 16961–16967 CrossRef CAS PubMed; (b) A. Filatre-Furcate, T. Roisnel, M. Fourmigué, N. Bellec, P. Auban-Senzier and D. Lorcy, Subtle Steric Differences Impact the Structural and Conducting Properties of Radical Gold Bis(dithiolene) Complexe, Chem. – Eur. J., 2017, 23, 16004–16013 CrossRef CAS PubMed; (c) G. Yzambart, N. Bellec, G. Nasser, O. Jeannin, M. Fourmigué, P. Auban-Senzier, J. Íñiguez, E. Canadell and D. Lorcy, Anisotropic Chemical Pressure Effects in Single-Component Molecular Metals Based on Radical Dithiolene and Diselenolene Gold Complexes, J. Am. Chem. Soc., 2012, 134, 17138–17148 CrossRef CAS PubMed.
- Y. Le Gal, T. Roisnel, P. Auban-Senzier, N. Bellec, J. Íñiguez, E. Canadell and D. Lorcy, Stable Metallic State of a Neutral-Radical Single-Component Conductor at Ambient Pressure, J. Am. Chem. Soc., 2018, 140, 6998–7004 CrossRef CAS PubMed.
- S. R. Kennedy, M. N. Kozar, H. P. Yennawar and B. J. Lear, Synthesis and characterization of the gold dithiolene monoanion, (Bu4N)[Au(pdt=2,3-pyrazinedithiol)2], Polyhedron, 2016, 103, 100–104 CrossRef CAS.
- (a) D. Belo, J. Morgado, E. B. Lopez, I. C. Santos, S. Rabaça, M. T. Duarte, V. Gama, R. T. Henriques and M. Alemeida, Synthesis and characterisation of charge transfer salts based on Au(dcdmp)2 and TTF type donors, Synth. Met., 1999, 102, 1751–1752 CrossRef CAS; (b) D. Belo, I. Santos and M. Almeida, 5,6-Dicyano-2,3-dithiopyrazine (dcdmp) chemistry: synthesis and crystal structure of Au(III)(dcdmp)2 complexes and 2,3,7,8-tetracyano-1,4,6,9-tetraazothianthrene, Polyhedron, 2004, 23, 1351–1359 CrossRef CAS.
- (a) S. Lo Schiavo, F. Nicolo, R. Scopelliti, G. Tresoldi and P. Piraino, Self-assembly of [Bu4N][M(qdt)2] [qdt=quinoxaline-2,3-dithiolate; M=Au and Cu] in a 2D network via combination of C−H⋯M and C−H⋯S interactions, Inorg. Chim. Acta, 2000, 304, 108–113 CrossRef CAS; (b) S. A. Sousa, J. H. Leitao, R. A. L. Silva, D. Belo, I. C. Santos, J. F. Guerreiro, M. Martins, D. Fontinha, M. Prudencio, M. Almeida, D. Lorcy and F. Marques, On the path to gold: Monoanionic Au bisdithiolate complexes with antimicrobial and antitumor activities, J. Inorg. Biochem., 2020, 202, 110904 CrossRef CAS PubMed.
- (a) Y. Kobayashi, B. Jacobs, M. D. Allendorf and J. R. Long, Conductivity, Doping, and Redox Chemistry of a Microporous Dithiolene-Based Metal−Organic Framework, Chem. Mater., 2010, 22, 4120–4122 CrossRef CAS; (b) M. L. Aubrey, M. T. Kapelewski, J. F. Melville, J. Oktawiec, D. Presti, L. Gagliardi and J. R. Long, Conductivity, Doping, and Redox Chemistry of a Microporous Dithiolene-Based Metal−Organic Framework, J. Am. Chem. Soc., 2019, 141, 5005–5013 CrossRef CAS PubMed.
- S. Attar, D. Espa, F. Artizzu, M. L. Mercuri, A. Serpe, E. Sessini, G. Concas, F. Congiu, L. Marchio and P. Deplano, A Platinum–Dithiolene Monoanionic Salt Exhibiting Multiproperties, Including Room-Temperature Proton-Dependent Solution Luminescence, Inorg. Chem., 2016, 55, 5118–5126 CrossRef CAS PubMed.
- R. A. L. Silva, I. C. Santos, V. Gama, E. B. Lopes, P. Auban-Senzier, M. Almeida and D. Belo, Tetrathiafulvalene and Tetramethyltetraselenafulvalene Salts with [M(dcdmp)2] Anions (M = Au, Cu, and Ni): High Conductivity and Unusual Stoichiometries, Cryst. Growth Des., 2019, 19, 6493–6502 CrossRef CAS.
- D. G. Branzea, F. Pop, P. Auban-Senzier, R. Clérac, P. Alemany, E. Canadell and N. Avarvari, Localization versus Delocalization in Chiral Single Component Conductors of Gold Bis(dithiolene) Complexes, J. Am. Chem. Soc., 2016, 138, 6838–6851 CrossRef CAS PubMed.
- (a) H. Kim, A. Kobayashi, Y. Sasaki, R. Kato, H. Kobayashi, T. Nakamura, T. Nogami and Y. Shirota, Crystal and Molecular Structure of Tetrabutylammonium Bis(1,4-dithiin-2,3-dithiolato)nickelate(III), (Bu4N)[Ni(ddt)2], Bull. Chem. Soc. Jpn., 1988, 61, 2559–2562 CrossRef CAS; (b) T. Nakamura, T. Nogami and Y. Shirota, Syntheses of New Multi-Sulfur 1,2-Dithiolene Complexes. Ni- and Pd-Complexes of 1,4-Dithiin-2,3-dithiolate, Bull. Chem. Soc. Jpn., 1987, 60, 3447–3449 CrossRef CAS; (c) H.-J. Lee and D.-Y. Noh, Syntheses, X-ray crystal structures and properties of di- and tetra-ferrocenyl nickel-bis(1,4-dithiin-5,6-dithiolate) complexes, J. Mater. Chem., 2000, 10, 2167–2172 RSC.
- (a) Q. Yu, J.-Y. Ge, Z.-P. Lv, H.-Y. Wang and J.-L. Zuo, Structure-dependent electronic transition in a new type of π-electron delocalized multi-sulfur bis(dithiolene)nickel complex, RSC Adv., 2016, 6, 100783–100789 RSC; (b) C. J. Bowlas and A. E. Underhill, The synthesis and properties of metal bis-dithiolenes based on the new DMAD ligand, Synth. Met., 1993, 55–57, 2158–2163 CrossRef; (c) A. C. Brooks, P. Day, S. I. G. Dias, S. Rabaça, I. C. Santos, R. T. Henriques, J. D. Wallis and M. Almeida, Pyridine-Functionalised (Vinylenedithio)-tetrathiafulvalene (VDT–TTF) Derivatives and Their Dithiolene Analogues, Eur. J. Inorg. Chem., 2009, 3084–3093 CrossRef CAS.
- L. Hu, J. Qin, N. Zhou, Y.-F. Meng, Y. Xu, J.-L. Zuo and X.-Z. You, Synthesis, characterization, and optical properties of new metal complexes with the multi-sulfur 1,2-dithiolene ligand, Dyes Pigm., 2012, 92, 1223–1230 CrossRef CAS.
- J. Sun, X. Lu, J. Shao, Z. Cui, Y. Shao, G. Jiang, W. Yu and X. Shao, Straightforward access to aryl-substituted/fused 1,3-dithiole-2-chalcogenones by Cu-catalyzed C–S coupling between aryl iodides and zinc–thiolate complex (TBA)2[Z n(DMIT)2], RSC Adv., 2013, 3, 10193–10196 RSC.
- Q. Ren, C. G. Reedy, E. A. Terrell, J. M. Wieting, R. W. Wagie, J. P. Asplin, L. M. Doyle, S. J. Long, M. T. Everard, J. S. Sauer, C. E. Baumgart, J. S. D'Acchioli and P. Nathan, Bowling, Evidence of Enhanced Conjugation in ortho-Arylene Ethynylenes with Transition Metal Coordination, J. Org. Chem., 2012, 77, 2571–2577 CrossRef CAS PubMed.
- Z. Lu, H. Liu, X. Lv, W. Lv, Y. Liu and K. Zhu, Molecular Hinges by Fusion of Pyrrole and Dithiin: Synthesis, Structure, Redox Chemistry and Host–Guest Complexation of Dipyrrolo-1,4-dithiins, Chem. – Eur. J., 2023, 29, e202300101 CrossRef CAS PubMed.
- (a) K. Kobayashi and C. L. Gajurel, The Chemistry of 1, 4-Dithiins, Sulfur Rep., 1986, 7, 123–148 CrossRef CAS; (b) S. I. Etkind and T. M. Swager, The Properties, Synthesis, and Materials Applications of 1,4-Dithiins and Thianthrenes, Synthesis, 2022, 4843–4863 CAS.
- X.-B. Yang, L. B. Huang, J. J. Xu, Y. Zhou, Z.-X. Xu, V. C. Y. Lau, M. H. W. Lam, W.-Y. Wong and V. A. L. Roy, Importance of alkyl chain-length on the self-assembly of new Ni(qdt)2 complexes and charge transport properties, RSC Adv., 2013, 3, 12075–12079 RSC.
- R. Bolligarla, G. Durgaprasad and S. K. Das, Synthesis, molecular structure and supramolecular chemistry of a new nickel-quinoxaline dithiolate system [Bu4N]2[Ni(6,7-qdt)2] (6,7-qdt = quinoxaline-6,7-dithiolate) and comparison of its electronic and electrochemical properties with those of [Bu4N]2[Ni(qdt)2] (qdt = quinoxaline-2,3-dithiolate), Inorg. Chem. Commun., 2011, 14, 809–813 CrossRef CAS.
- (a) S. Kokatam, K. Ray, J. Pap, E. Bill, W. E. Geiger, R. J. LeSuer, P. H. Rieger, T. Weyhermüller, F. Neese and K. Wieghardt, Inorg. Chem., 2007, 46, 1100–1111 CrossRef CAS PubMed; (b) R. Perochon, C. Poriel, O. Jeannin, L. Piekara-Sady and M. Fourmigué, Chiral, neutral and paramagnetic gold dithiolene complexes derived from camphorquinone, Eur. J. Inorg. Chem., 2009, 5413–5421 CrossRef CAS; (c) J.-B. Pluta, R. Guechaichia, A. Vacher, N. Bellec, S. Cammas-Marion and F. Camerel, Investigations of the Photothermal Properties of a Series of Molecular Gold-bis(dithiolene) Complexes Absorbing in the NIR-III Region, Chem. – Eur. J., 2023, 29, e202301789 CrossRef CAS PubMed.
- (a) R. Perochon, F. Barrière, O. Jeannin, L. Piekara-Sady and M. Fourmigué, A radical mixed-ligand gold bis(dithiolene) complex, Chem. Commun., 2021, 57, 1615–1618 RSC; (b) H. Kharraz, P. Alemany, E. Canadell, Y. Le Gal, T. Roisnel, H. Cui, K. H. Kim, M. Fourmigué and D. Lorcy, Mixed-ligand, radical, gold bis(dithiolene) complexes: from single-component conductors to controllable NIR-II absorbers, Chem. Sci., 2024, 15, 11604–11616 RSC.
- (a) E. Canadell, I. E.-I. Rachidi, S. Ravy, J.-P. Pouget, L. Brossard and J. P. Legros, On the band electronic structure of X [M (dmit)2]2 (X = TTF, (CH3)4N; M = Ni, Pd) molecular conductors and superconductors, J. Phys. (Paris), 1989, 50, 2967–2981 CrossRef CAS; (b) E. Canadell, Electronic structure of two-band molecular conductors, New J. Chem., 1997, 21, 1147–1159 CAS.
- M.-H. Whangbo, J. M. Williams, P. C. W. Leung, M. A. Beno, T. J. Emge and H. H. Wang, Role of the Intermolecular Interactions in the Two-Dimensional Ambient-Pressure Organic Superconductors β-(ET)2I3 and β-(ET)2IBr2, Inorg. Chem., 1985, 24, 3500–3502 CrossRef CAS.
- E. H. Lieb and F. Y. Wu, Absence of Mott Transition in an Exact Solution of the Short-Range, One-Band Model in One Dimension, Phys. Rev. Lett., 1968, 20, 1445–1448 CrossRef.
- G. M. Sheldrick, SHELXT - integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2015, C71, 3–8 CrossRef PubMed.
- H. Cui, J. S. Brooks, A. Kobayashi and H. Kobayashi, Single-Component Molecular Superconductor, J. Am. Chem. Soc., 2009, 131, 6358–6359 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Inhomogeneous Electron Gas, Phys. Rev., 1965, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Self-Consistent Equations Including Exchange and Correlation Effects, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal, The SIESTA method for ab initio order-N materials simulation, J. Phys.:Condens. Matter, 2002, 14, 2745–2779 CrossRef CAS.
- E. Artacho, E. Anglada, O. Diéguez, J. D. Gale, A. García, J. Junquera, R. D. Martin, P. Ordejón, M. A. Pruneda, D. Sánchez-Portal and J. M. Soler, The SIESTA method: developments and applicability, J. Phys.:Condens. Matter, 2008, 20, 064208 CrossRef PubMed.
- A. García, N. Papior, A. Akhtar, E. Artacho, V. Blum, E. Bosoni, P. Brandimarte, M. Brandbyge, J. I. Cerdá, F. Corsetti, R. Cuadrado, V. Dikan, J. Ferrer, J. D. Gale, P. García-Fernández, V. M. García-Suárez, V. M. García, G. Huhs, S. Illera, R. Korytar, P. Koval, I. Lebedeva, L. Lin, P. López-Tarifa, S. G. Mayo, S. Mohr, P. Ordejón, A. Postnikov, Y. Pouillon, M. A. Pruneda, R. Robles, D. Sánchez-Portal, J. M. Soler, R. Ullah, V. W.-z. Yu and J. Junquera, SIESTA: Recent developments and applications, J. Chem. Phys., 2020, 152, 204108 CrossRef PubMed.
- For more information on the SIESTA code visit: https://departments.icmab.es/leem/siesta/.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1966, 77, 3865–3868 CrossRef PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA + U study, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- Y. Kiyota, I.-R. Jeon, O. Jeannin, M. Beau, T. Kawamoto, P. Alemany, E. Canadell, T. Mori and M. Fourmigué, Electronic engineering of a tetrathiafulvalene charge-transfer salt via reduced symmetry induced by combined substituents, Phys. Chem. Chem. Phys., 2019, 21, 22639–22646 RSC.
- N. Troullier and J. L. Martins, Efficient Pseudopotentials for planewave calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 1993–2006 CrossRef CAS PubMed.
- L. Kleinman and D. M. Bylander, Efficacious Form for Model Pseudopotentials, Phys. Rev. Lett., 1982, 48, 1425–1428 CrossRef CAS.
- E. Artacho, D. Sánchez-Portal, P. Ordejón, A. García and J. M. Soler, Linear Scaling ab initio Calculations for Large and Complex Systems, Phys. Status Solidi B, 1999, 215, 809–817 CrossRef CAS.
- P. Alemany, M. Llunell and E. Canadell, Uniform linear chains of group 11 atoms: do they have a bias towards a Peierls distortion?, Theor. Chem. Acc., 2009, 123, 85–92 Search PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin zone integrations, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- M.-H. Whangbo and R. Hoffmann, The band structure of the tetracyanoplatinate chain, J. Am. Chem. Soc., 1978, 100, 6093–6098 CrossRef CAS.
- J. H. Ammeter, H.-B. Bürgi, J. Thibeault and R. Hoffmann, Counterintuitive orbital mixing in semiempirical and ab initio molecular orbital calculations, J. Am. Chem. Soc., 1978, 100, 3686–3692 CrossRef CAS.
- Y. Le Gal, H. Cui, P. Alemany, E. Canadell, R. Kato, T. Roisnel, V. Dorcet, M. Fourmigué and D. Lorcy, Mixed-valence gold bis(diselenolene) complex turning metallic under pressure, J. Mater. Chem. C, 2021, 9, 12291–12302 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthetic and characterization results, Fig. S1–S4, Tables S1 and S2. CCDC 2422633–2422637. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00380f |
| This journal is © The Royal Society of Chemistry 2025 |