
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revisiting the structure and properties of mid-valent monopentamethylcyclopentadienylchromium complexes†
Adrián
Calvo-Molina
 a,
Jesús
Jover
*b,
Adrián
Pérez-Redondo
*a and
Carlos
Yélamos
*a
a,
Jesús
Jover
*b,
Adrián
Pérez-Redondo
*a and
Carlos
Yélamos
*a
aDepartamento de Química Orgánica y Química Inorgánica, Instituto de Investigación Química “Andrés M. del Río” (IQAR), Universidad de Alcalá, 28805 Alcalá de Henares-Madrid, Spain. E-mail: adrian.perez@uah.es; carlos.yelamos@uah.es
bSecció de Química Inorgànica, Departament de Química Inorgànica i Orgànica, Institut de Química Teòrica i Computacional (IQTC-UB), Universitat de Barcelona, Martí i Franquès 1-11, 08028 Barcelona, Spain. E-mail: jjovermo@ub.edu
First published on 31st March 2025
Abstract
The structure and properties of half-sandwich chromium complexes derived from the dinuclear chloride compound [{CrCp*(μ-Cl)}2] (1) (Cp* = η5-C5Me5) are revisited. Complex 1 does not react with H2 and N2 but cleaves the nitrogen–nitrogen bonds of azobenzene and 1,2-diphenylhydrazine at room temperature to give dinuclear chromium(IV) bis(imido) [{CrCp*Cl(μ-NPh)}2] (2) and chromium(III) bis(amido) [{CrCp*Cl(μ-NHPh)}2] (3) derivatives, respectively. Reactions of 1 with cyclopentyllithium [Li(C5H9)] in hexane or toluene under reflux conditions afford the previously reported tetranuclear chromium(II) hydride complex [{CrCp*(μ3-H)}4] (4) and the unsymmetrical chromium(I) sandwich compound [CrCp*(η6-C6H5Me)] (5) as crystals suitable for X-ray diffraction studies. While the treatment of 1 with excess LiBH4 leads to an analogous dinuclear complex [{CrCp*(μ-κ3-BH4)}2] (6), the reaction of the chromium(III) compound [{CrCp*Cl(μ-Cl)}2] with LiBH4 gives the mononuclear species [CrCp*(κ2-BH4)2] (7). Complex 6 cleanly reacts with the 2,6-lutidinium salt (LutH)(BPh4) to form the zwitterionic sandwich derivative [CrCp*(η6-C6H5-BPh3)] (8). Compounds 1 and 6 react with LiAlH4 to give a diamagnetic tetrachromium aggregate [(Al{(μ-H)4CrCp*})4] (9), which can be described as low-spin chromate(II) {CrCp*H4}3− units stabilizing the Al3+ ions primarily through Cr–H → Al interactions and weaker Cr → Al donation according to density functional theory (DFT) calculations. The thermal decomposition of 9 in benzene at 90 °C affords a mixed-valence CrII/CrI hexachromium species [(Al2{(μ-H)4CrCp*}{(μ3-H)4Cr2Cp*2})2] (10) with analogous interactions of {CrCp*H4}3− and {Cr2Cp*2H4}3− units with Al3+ ions.
Introduction
Open-shell mid-valent chromium organometallic complexes are relatively underdeveloped in comparison with their low-valent counterparts due to the paramagnetic nature and air-sensitivity associated with the electron-deficient configurations of most of these combinations.1,2 The use of cyclopentadienyl (Cp = η5-C5H5) and especially pentamethylcyclopentadienyl (Cp* = η5-C5Me5) ligands has been highly successful in stabilizing these reactive species and improving the crystallinity of the solids, thus allowing their structural characterization by X-ray diffraction methods. Noteworthy are the contributions of the Theopold group on monopentamethylcyclopentadienyl complexes of chromium(III)3 and chromium(II)4–6 in the final decades of the 20th century. In particular, these authors reported a series of thermally stable alkyl and hydride species derived from dinuclear chromium(II) chloride complexes [{Cr(η5-C5Me4R)(μ-Cl)}2] (R = Me, Et). For example, they isolated tetranuclear polyhydride complexes [{Cr(η5-C5Me4R)(μ3-H)}4] (R = Me, Et)4,5 and [{Cr(η5-C5Me4Et)}4(μ-H)5(μ3-H)2]6 by hydrogenolysis of the dimethyl compounds [{Cr(η5-C5Me4R)(μ-Me)}2]. By using a similar methodology with analogous methyl chromium precursors bearing the bulkier η5-C5Me4SiMe3 ligands, Hou and co-workers prepared di- and trinuclear polyhydride complexes [{Cr(η5-C5Me4SiMe3)(μ-H)}2] and [{Cr(η5-C5Me4SiMe3)(μ-H)}3(μ3-H)] that enable N2 cleavage and partial hydrogenation under mild conditions.7 Analogous hydride-bridged dinuclear complexes [{Cr(η5-C5H2tBu3-1,2,4)(μ-H)}2] and [{Cr(η5-C5iPr5)(μ-H)}2] have been isolated by Sitzmann and co-workers through the reaction of the corresponding alkylcyclopentadienylchromium(II) bromide precursors with isopropylmagnesium chloride via β-hydride elimination.8 Remarkably, while the chemistry of chromium–dinitrogen complexes has been much less studied in comparison with that of the heavier Mo and W analogs,9 recent studies of the Wei and Xi group on monocyclopentadienylchromium(I,0) systems have developed novel reactivity patterns in the functionalization of dinitrogen.10As part of a research program devoted to the study of the structure and reactivity of mid- and low-valent monocyclopentadienyl complexes of early transition metals, we initially reported a series of titanium(III) dihalide aggregates [{TiCp*X2}n] (X = Cl, Br, I).11 Further reduction of [TiCp*Cl2] with magnesium led to the mixed-valence titanium(III)/titanium(II) trinuclear complex [{TiCp*(μ-Cl)}3(μ3-Cl)], which reacts with dinitrogen under ambient conditions to give a stable derivative with a μ3-η1:η2:η2-N2 ligand.12 We have also prepared several titanium(II) and titanium(III) species stabilized with aluminum/boron hydride fragments by the reaction of [TiCp*Cl3] (X = Cl, Br) with LiEH3R (E = Al, R = H; E = B, R = H, Me).13 The resulting heterometallic hydride-bridged Ti–Al compounds exhibited unprecedented Ti–Ti → Al interactions according to crystallographic and theoretical studies. Well-characterized hydride complexes pairing aluminum with transition metals are rare despite current interest in the preparation and applications of this type of heterometallic compound.14,15 We reasoned that the easier access to low oxidation states of other Earth-abundant 3d metals, such as vanadium and chromium, could be beneficial for generating polymetallic species more suitable for cooperative small-molecule activation processes. While we have communicated our preliminary results on vanadium compounds,16 here we report the use of the readily available complex [{CrCp*(μ-Cl)}2] (1)4,5 for the synthesis of mid-valent monopentamethylcyclopentadienylchromium derivatives. Thus, we first evaluated the capability of 1 to activate the nitrogen–nitrogen bonds of N2, azobenzene, and 1,2-diphenylhydrazine. In addition, we report the syntheses and crystal structures of several chromium hydride species prepared by the reaction of 1 with LiEH4 (E = B, Al). The electronic structure of the heterometallic Cr–Al polyhydride complexes have been elucidated by theoretical calculations.
Results and discussion
The chromium(II) complex [{CrCp*(μ-Cl)}2] (1) readily reacted with one equivalent of azobenzene and 1,2-diphenylhydrazine in toluene at room temperature to give the dinuclear chromium(IV) imido [{CrCp*Cl(μ-NPh)}2] (2) and chromium(III) amido [{CrCp*Cl(μ-NHPh)}2] (3) derivatives, respectively (Scheme 1). In contrast to the facile cleavage of the double N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and single N–N bonds, compound 1 did not show any reaction with dinitrogen (up 8 atm) in benzene-d6 solution at room temperature. Upon heating this solution at 90 °C, the 1H NMR spectrum showed resonance signals assignable to the chromium(III) chloride compound [{CrCp*Cl(μ-Cl)}2] (δ = −47.6, Δν1/2 = 462 Hz)17 and decamethylchromocene [CrCp*2] (δ = −6.5, Δν1/2 = 120 Hz). These resonances are identical to those observed in the thermal treatment of 1 in benzene-d6 solution under an argon atmosphere, indicating decomposition of compound 1 at this temperature. In addition, unfortunately, reduction of 1 with magnesium in tetrahydrofuran under a nitrogen atmosphere did not allow the isolation of any chromium–dinitrogen complex.
N and single N–N bonds, compound 1 did not show any reaction with dinitrogen (up 8 atm) in benzene-d6 solution at room temperature. Upon heating this solution at 90 °C, the 1H NMR spectrum showed resonance signals assignable to the chromium(III) chloride compound [{CrCp*Cl(μ-Cl)}2] (δ = −47.6, Δν1/2 = 462 Hz)17 and decamethylchromocene [CrCp*2] (δ = −6.5, Δν1/2 = 120 Hz). These resonances are identical to those observed in the thermal treatment of 1 in benzene-d6 solution under an argon atmosphere, indicating decomposition of compound 1 at this temperature. In addition, unfortunately, reduction of 1 with magnesium in tetrahydrofuran under a nitrogen atmosphere did not allow the isolation of any chromium–dinitrogen complex.
 | ||
| Scheme 1 Reactions of 1 with azobenzene and 1,2-diphenylhidrazine. | ||
Compounds 2 and 3 were isolated in 53 and 66% yields as green and purple solids, respectively, which exhibit a good solubility in aromatic hydrocarbon solvents and are poorly soluble in hexane. A weak band at 3241 cm−1 in the IR (ATR) spectrum of 3 was assigned to an N–H stretching vibration. While the 1H NMR spectrum of 3 in benzene-d6 at room temperature was silent, a broad resonance at δ = −10.0 (Δν1/2 = 569 Hz) attributed to the η5-C5Me5 groups was observed in the spectrum of 2. The paramagnetic nature of compounds 2 and 3 was confirmed by an Evans method determination of their magnetic susceptibility (μeff = 2.2 and 3.6μB, respectively) in benzene-d6 solutions at room temperature.18 These effective magnetic moments are significantly lower than those expected for two magnetically isolated chromium(IV) (S = 1, μeff = 4.0μB) and chromium(III) (S = 3/2, μeff = 5.48μB) ions and suggest antiferromagnetic coupling of the chromium ions in compounds 2 and 3. The 1H NMR spectra of complexes 2 and 3 in benzene-d6 did not show any apparent change after heating the solutions at 100 °C for several days, and therefore, the complexes appear to be stable up to that temperature.
Single crystals suitable for X-ray crystal structure determinations of complexes 2 and 3·2C6H5F were obtained from toluene and fluorobenzene solutions at −35 °C, respectively. The molecular structures are shown in Fig. 1, and selected distances and angles of both complexes are compared in Table 1. The crystal structures of complexes 2 and 3 show dimers with two {CrCp*Cl} moieties held together by two bridging arylimido μ-NPh or arylamido μ-NHPh groups. Molecules of 2 and 3 lie on a crystallographic inversion center at the midpoint between the two chromium atoms, which are separated by 2.663(2) and 2.978(1) Å, respectively. The chromium–nitrogen bond lengths associated with the imido linkages of 2 (1.869(5) and 1.888(5) Å) are clearly shorter than those of the bridging amido groups of 3 (2.069(3) and 2.077(3) Å). The sum of angles Cr–N–Cr and Cr–N–C(1) (C(1) is the carbon atom of the phenyl ring bound to nitrogen) about the imido nitrogens N(1) in 2 is 360.0°, as expected for a trigonal planar geometry, while the analogous sum for the amido nitrogens N(1) in 3 is smaller (346.4°) but still greater than that expected for the ideal tetrahedral arrangement (328.5°). The structural parameters of the imido and amido groups of 2 and 3 are comparable to those of the analogous monocyclopentadienyl bis(imido) chromium(IV) complex [{CrCp(OCMe3)(μ-NPh)}2]19 and the amido/imido chromium(III) derivative [(CrCp*)2(μ-Br)(μ-NHR)(μ-NR)]20 (R = 2,6-iPr2C6H3).
 | ||
| Fig. 1 Perspective views of (a) 2 and (b) 3·2C6H5F with thermal ellipsoids at the 50% probability level. Hydrogen atoms of the η5-C5Me5 ligands and phenyl groups are omitted for clarity. The two fluorobenzene solvent molecules in 3 are also omitted. Symmetry code: (i) 1 − x, 1 − y, 1 − z. | ||
| 2 | 3 | |
|---|---|---|
| a Cm = Centroid of the η5-C5Me5 ring. b C(1) is the carbon atom of the phenyl ring bound to nitrogen. Symmetry code: (i) 1 − x, 1 − y, 1 − z. | ||
| Cr(1)–Cl(1) | 2.309(2) | 2.345(1) |
| Cr(1)–N(1) | 1.869(5) | 2.069(3) |
| Cr(1)–N(1)i | 1.888(5) | 2.077(3) |
| Cr(1)–Cm(1)a | 1.927 | 1.902 |
| Cr(1)⋯Cr(1)i | 2.663(2) | 2.978(1) |
| N(1)⋯N(1)i | 2.651(7) | 2.885(5) |
| Cl(1)–Cr(1)–N(1) | 100.5(2) | 100.3(1) |
| Cl(1)–Cr(1)–N(1)i | 99.8(2) | 87.5(1) |
| N(1)–Cr(1)–N(1)i | 89.7(2) | 88.2(1) |
| Cr(1)–N(1)–Cr(1)i | 90.3(2) | 91.8(1) |
| Cr(1)–N(1)–C(1)b | 135.9(4) | 128.3(3) |
| Cr(1)i–N(1)–C(1)b | 133.8(4) | 126.3(2) |
Since compound 1 in benzene-d6 solution did not react with H2 even at 100 °C, the reaction of 1 with cyclopentyllithium [Li(C5H9)] was examined as a potential route to the preparation of chromium hydride derivatives, given the presence of β-hydrogens in the alkyl group. The treatment of 1 with 2 equiv. of [Li(C5H9)] in hexane or toluene immediately gave brown solutions of presumably the alkyl species [{CrCp*(μ-C5H9)}2] (Scheme 2). Most likely, this complex adopts a butterfly structure similar to those of 1 and analogous alkyl derivatives [{CrCp*(μ-R)}2] (R = Me, Et, nBu, Ph, CH2SiMe3) reported by Theopold and co-workers.4,5 This compound was obtained as a dark brown solid, which is highly soluble in hydrocarbon solvents such as hexane and pentane and could not be isolated in a crystalline form, precluding structural characterization. In accord with its paramagnetic nature, a benzene-d6 solution of the product gave a silent 1H NMR spectrum. Heating this benzene-d6 solution at 80 °C in an NMR tube and then slowly cooling it to room temperature gave single crystals of the tetranuclear hydride complex [{CrCp*(μ3-H)}4] (4) suitable for X-ray crystal structure determination. Compound 4 has been previously prepared in 68% yield by Theopold through the hydrogenolysis of [{CrCp*(μ-Me)}2] in pentane at room temperature.4,5 However, an accurate determination of its crystal structure by X-ray diffraction was not possible, and the authors isolated and structurally characterized the analogous compound [{Cr(η5-C5Me4Et)(μ3-H)}4].5
 | ||
| Scheme 2 Reactions of 1 with cyclopentyllithium. | ||
The X-ray crystal structure of 4 shows a distorted {Cr4(μ3-H)4} cube-type core with no crystallographically imposed symmetry (Fig. 2). Therefore, the molecular structure displays six different Cr–Cr distances ranging from 2.509(1) to 2.753(1) Å. This range is wider than those observed in related tetrachromium hydride complexes [{Cr(η5-C5Me4Et)(μ3-H)}4] (2.612(2)–2.681(2) Å)5 and [{Cr(η5-C5Me4Et)}4(μ-H)5(μ3-H)2] (2.759(3)–2.797(2) Å).6 These data and the Cr–Cr–Cr angles ranging from 55.9(1) to 65.3(1)° indicate a high distortion of the tetrahedron of chromium atoms. Furthermore, Cr–H bond lengths and Cr–H–Cr and H–Cr–H angles show broad ranges of values.
 | ||
| Fig. 2 Perspective view of 4 with thermal ellipsoids at the 50% probability level. Hydrogen atoms of the η5-C5Me5 ligands are omitted for clarity. Selected lengths (Å) and angles (°): Cr⋯Cr 2.509(1)–2.753(1), Cr–H 1.72(6)–2.07(5), Cr–Cm av. 1.920(3), Cr–Cr–Cr 55.9(1)–65.3(1), H–Cr–H 79(2)–96(3), Cr–H–Cr 82(2)–103(3). | ||
In a preparative-scale reaction, the hexane solution resulting from a mixture of 1 and [Li(C5H9)] was heated at 70 °C for 4 days to give a dark solution and the precipitation of black crystals of 4. Compound 4 was isolated in poor yield (12%), but subsequent workup of the dark solution did not give any isolable compound. In contrast, heating a toluene solution of the putative intermediate [{CrCp*(μ-C5H9)}2] at 110 °C for 3 days gave a few black crystals of 4 along with a green solution. After the workup of this solution, orange crystals of the chromium(I) arene complex [CrCp*(η6-C6H5Me)] (5) were isolated in 19% yield. Complex 5 has also been previously prepared by Köhler and co-workers through the reaction of [{CrCp*Cl(μ-Cl)}2] with AlEt3 in toluene under reflux conditions,21 but the crystal structure of 5 was determined in the course of the work described here. The X-ray diffraction of a single crystal revealed an unsymmetrical sandwich structure for 5 (Fig. S1 in the ESI†). The 1H NMR spectrum of 5 in benzene-d6 is silent, and the magnetic moment measurement at room temperature by the Evans method gave a μeff of 1.7μB, which is in good agreement with the spin-only magnetic moment of a mononuclear species with one unpaired electron (1.73μB).
In view of these results, it appears that the hypothetical intermediate [{CrCp*(μ-C5H9)}2] is highly stable, even though the cyclopentyl groups contain β-hydrogens, and only slowly decomposes either under argon or dinitrogen atmospheres at high temperatures to give the expected tetranuclear hydride complex 4 along with other paramagnetic chromium species. Monitoring the reaction of 1 with [Li(C5H9)] in cyclohexane-d12 at 70 °C and toluene-d8 at 100 °C by 1H NMR spectroscopy showed only resonance signals attributable to cyclopentene and cyclopentane as diamagnetic byproducts.
To obtain other chromium hydride complexes, we have also explored the reactions of 1 and the analogous chromium(III) dichloride complex [{CrCp*Cl(μ-Cl)}2]17,22 with lithium tetrahydridoborate (Scheme 3). Structurally documented tetrahydridoborato complexes of chromium are extremely rare in the extensive literature of metal complexes stabilized with these ligands.23,24 Treatment of 1 with LiBH4 (≥2 equiv.) in tetrahydrofuran at room temperature afforded the dinuclear chromium(II) derivative [{CrCp*(μ-κ3-BH4)}2] (6) in 81% isolated yield after workup. Similarly, the reaction of [{CrCp*Cl(μ-Cl)}2] with LiBH4 (≥4 equiv.) gives the mononuclear chromium(III) species [CrCp*(κ2-BH4)2] (7) in 83% yield. Compounds 6 and 7 were obtained as green solids, which are highly soluble in hydrocarbon solvents. The IR spectrum (KBr) of 6 displays one strong absorption at 2457 cm−1 and one broad band centered at 2092 cm−1 for the terminal and bridging B–H stretching vibrations of the μ-κ3-BH4 ligands, respectively. In contrast, the terminal κ2-BH4 ligands of 7 give rise to two strong absorptions at 2448 and 2396 cm−1 for the terminal B–H bonds, several bands in the range 2211–1957 cm−1 for the bridging B–H bonds, and one intense absorption at 1110 cm−1 for the BH2 deformation in the IR spectrum.23a The 1H NMR spectra of complexes 6 and 7 in benzene-d6 display one broad resonance signal for the η5-C5Me5 ligands at δ = 6.7 (Δν1/2 = 52 Hz) and 42.6 (Δν1/2 = 355 Hz), respectively. The magnetic moment measurements for compound 6 in benzene-d6 at ambient temperature using the Evans method gave a μeff of 1.9μB per dimer. This effective magnetic moment in solution is consistent with some degree of metal–metal bonding in this compound. In contrast, the magnetic moment measurement of 7 in benzene-d6 at room temperature gave a μeff of 4.1μB, in good agreement with that estimated (3.87μB) for the spin-only magnetic moment of a mononuclear species with three unpaired electrons.
 | ||
| Scheme 3 Reactions of 1 and [{CrCp*Cl(μ-Cl)}2] with LiBH4. | ||
Complex 6 has been previously prepared by Fehlner and co-workers using the same procedure, but the compound was only characterized by spectroscopic techniques.25 Fortunately, green crystals of 6 suitable for X-ray crystal structure determination were grown from a toluene solution at −35 °C. The molecular structure of 6 exhibits a pseudo C2v symmetry and shows two CrCp* units connected by two bridging tetrahydridoborato ligands (Fig. 3). The {Cr2B2} core adopts a butterfly shape similar to those found in the dimers [{CrCp*(μ-X)}2] (X = Cl (1), Me)4,5 and [(CrCp*)2(μ-Et)(μ-Ph)].5 The Cr(1)–Cr(2) distance of 2.586(1) Å in 6 is slightly shorter than that determined in 1 (2.642(2) Å), suggesting a weak bonding interaction between the chromium atoms. This is consistent with an effective magnetic moment of 6 of 1.9μB, which is slightly lower than the value reported for 1 (μeff = 2.0μB) at room temperature.4,5 For comparison, the shorter Cr–Cr distances and lower μeff values for the alkyl derivatives [{CrCp*(μ-Me)}2] (2.263(3) Å and 1.2μB) and [(CrCp*)2(μ-Et)(μ-Ph)] (2.289(4) Å and 1.4μB) are indicative of significant metal–metal bonding.5 The tridentate tetrahydridoborato ligands bridge the two metal centers in the rare coordination mode μ-κ3-BH4, which has been documented only in the structure of the dinuclear cobalt complex [{Co(μ-κ3-BH4)(Ph2P(CH2)5PPh2)}2]26 and a few compounds of group 1 and 2 metals,23b such as the dimers with bidentate ligands [{Li(μ-κ3-BH4)(ligand)}2] (ligand = TMEDA,27 {H2C(3,5-Me2pz)2}, and 4,4′-Me2bipy).28
 | ||
| Fig. 3 Perspective view of 6 with thermal ellipsoids at the 50% probability level. Hydrogen atoms of the η5-C5Me5 ligands are omitted for clarity. Selected average lengths (Å) and angles (°): Cr–Cr 2.586(1), Cr⋯B 2.369(1), Cr–H(μ) 1.94(6), Cr–H(μ3) 2.06(6), Cr–Cm 1.900(5), Cr–B–Cr 66.2(1). | ||
The X-ray diffraction of a single crystal of 7 showed a mononuclear structure with the chromium center bonded to one η5-C5Me5 group and four μ-H bridging hydrides of two BH4 ligands (Fig. 4). Thus, the chromium atom exhibits a classical four-legged piano-stool coordination geometry. The κ2-coordinated BH4 groups display Cr(1)⋯B(1) and Cr(1)⋯B(2) distances of 2.312(2) and 2.322(2) Å, which are shorter than those found for the κ2-BH4 ligands of the chromium(II) complexes [Cr(BH4)2(TMEDA)] (2.44(1) and 2.42(1) Å)29 and [Cr(POCOP-tBu)(BH4)] (2.47(2) Å),24 and slightly longer than those determined in the chromium(I) [Cr(POCOP-tBu)(NO)(BH4)] (2.292(4) Å)24 and chromium(0) [Cr(CO)4(BH4)]− (2.29(1) Å)30 complexes. To our knowledge, complex 7 is the first structurally characterized example of a chromium(III) complex with tetrahydridoborato ligands, though the analogous species [CrCp(BH4)2] was detected by mass spectrometry in the reaction mixtures of [CrCpCl2(thf)] and excess NaBH4.31
 | ||
| Fig. 4 Perspective view of 7 with thermal ellipsoids at the 50% probability level. Hydrogen atoms of the η5-C5Me5 ligands are omitted for clarity. Selected lengths (Å): Cr(1)⋯B(1) 2.312(2), Cr(1)⋯B(2) 2.322(2), Cr(1)–H 1.82(3)–1.91(3), Cr(1)–Cm 1.838. | ||
Given the good yields and facile preparation of compounds 6 and 7, we decided to explore their reactivity to prepare other chromium derivatives. The treatment of 6 with 2 equiv. of 2,6-lutidinium tetraphenylborate (LutH)(BPh4) in toluene at room temperature led to the precipitation of derivative [CrCp*(η6-C6H5-BPh3)] (8) with vigorous gas evolution (Scheme 3). The reaction presumably involves the interaction of protonic (LutH)+ ions with the hydridic BH4− ligands of 6, leading to the formation of H2 and subsequent trapping of the generated BH3 with lutidine to give the soluble acid–base adduct (Lut)BH3. Complex 8 was isolated in 71% yield as an orange solid, which is slightly soluble in toluene and benzene. The zwitterionic complex 8 has been previously prepared and structurally characterized, although the syntheses were rather peculiar and led to lower yields of the compound.3b,32 Complex 8 was also obtained in the reaction of 7 with (LutH)(BPh4) in toluene, but this procedure afforded 8 in low yield due to the presence of free BPh3 and other paramagnetic species as byproducts.
Additionally, treatment of the chromium(II) tetrahydridoborato complex 6 or the chloride precursor 1 with 2 equiv. of lithium tetrahydridoaluminate in tetrahydrofuran at room temperature gave the diamagnetic tetrachromium aggregate [(Al{(μ-H)4CrCp*})4] (9) (Scheme 4). Compound 9 was isolated in good yields (70–73%) as a brown solid, which exhibits high solubility in toluene and benzene but is poorly soluble in hexane. Complex 9 was characterized by spectroscopic and analytical methods, as well as by X-ray crystal structure determination of 9·1.5C7H8 from single crystals grown in toluene. The 1H NMR spectrum of 9 in benzene-d6 at room temperature shows two sharp singlet resonances at δ = 1.92 and −10.51 in a 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio for the η5-C5Me5 and hydride ligands, respectively. The 13C{1H} NMR spectrum also shows resonances for equivalent η5-C5Me5 ligands in the typical range of chemical shifts of a diamagnetic compound. The solid-state infrared spectrum (KBr) of 9 exhibits the characteristic bands of η5-C5Me5 ligands and two very broad and strong absorptions at 1572 and 1546 cm−1, which could be assigned to the bridging Cr–H–Al stretching modes.33,34
:4 ratio for the η5-C5Me5 and hydride ligands, respectively. The 13C{1H} NMR spectrum also shows resonances for equivalent η5-C5Me5 ligands in the typical range of chemical shifts of a diamagnetic compound. The solid-state infrared spectrum (KBr) of 9 exhibits the characteristic bands of η5-C5Me5 ligands and two very broad and strong absorptions at 1572 and 1546 cm−1, which could be assigned to the bridging Cr–H–Al stretching modes.33,34
 | ||
| Scheme 4 Reactions of [{CrCp*(μ-X)}2] (X = Cl, BH4) with LiAlH4. | ||
Complex 9 is stable in benzene-d6 at room temperature according to 1H NMR spectroscopy but slowly decomposes at 90 °C to give a dark solution. Upon cooling this solution to room temperature, single crystals of the hexachromium species [(Al2{(μ-H)4CrCp*}{(μ3-H)4Cr2Cp*2})2]·C6D6 (10·C6D6) were grown. In a preparative-scale reaction, black crystals of 10·0.5C6H6 were isolated in poor yield (20%) upon heating a benzene solution of 9 at 90 °C. Compound 10 is not soluble in common organic solvents, precluding its characterization by NMR spectroscopy and the determination of its magnetic moment in solution. Similarly to 9, the IR spectrum (ATR) of 10 shows two very broad and strong bands at 1615 and 1576 cm−1 attributable to the Cr–H–Al stretching modes.
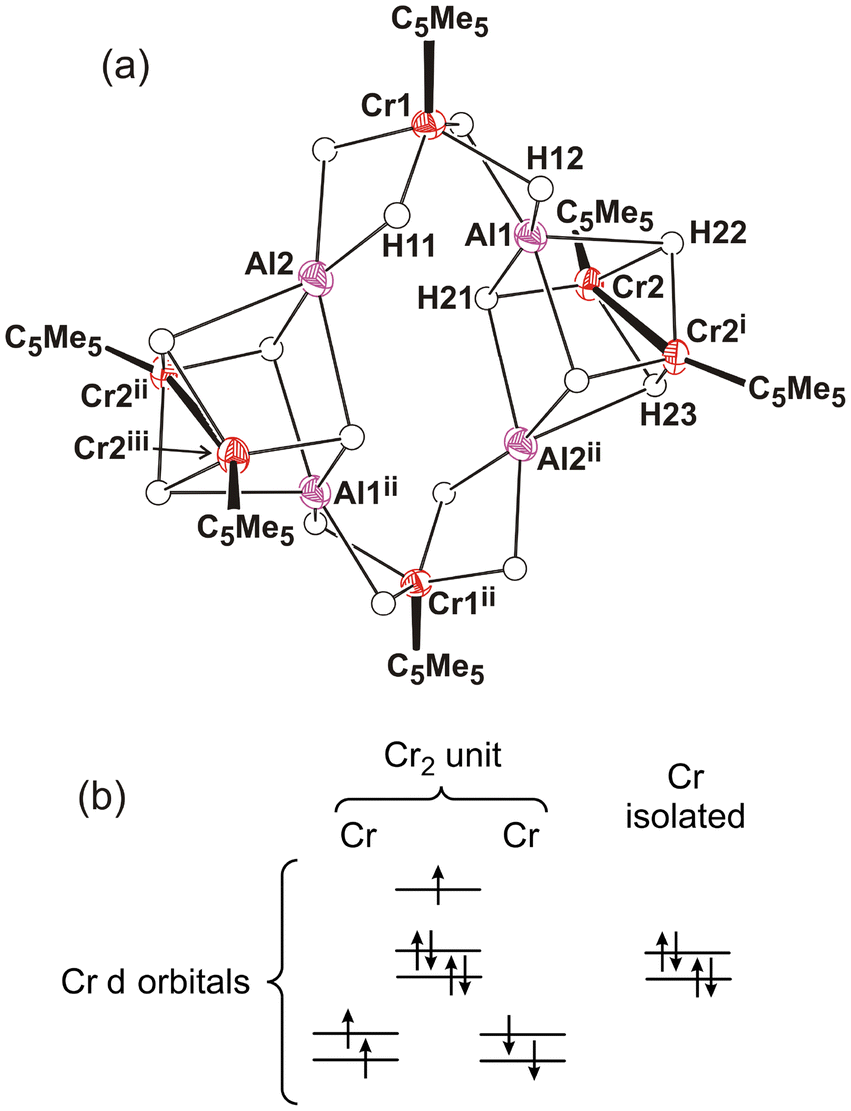
The X-ray crystal structure of compound 9 shows four {CrCp*H4} units connected to four aluminum centers by hydride ligands (Fig. 5 and S2†). The molecules do not show any crystallographic symmetry in the solid state, but the structure of 9 is nearly symmetric, with a twofold rotation axis perpendicular to the midpoint of the Cr(2)–Cr(4) segment, a vertical mirror plane bearing the rotation axis and Cr(1) and Cr(3) atoms, and another mirror plane also bearing the rotation axis and the Cr(2) and Cr(4) atoms. This pseudo C2v symmetry makes all the chromium and aluminum atoms in the structure essentially equivalent and is consistent with the single peak for the η5-C5Me5 ligands observed in the 1H NMR spectrum of 9 in solution. The whole molecule takes the form of a butterfly structure with both wings consisting of three CrCp* units forming an isosceles triangle (Cr(1)/Cr(3)⋯Cr(2) av. 4.748(2), Cr(1)/Cr(3)⋯Cr(4) av. 4.750(4), and Cr(2)⋯Cr(4) 6.119(1) Å). The aluminum atoms bridge the equal edges of the chromium triangles with Cr–Al–Cr angles of average 176(2)° and are connected to each chromium center by two bridging hydride ligands. The Cr–Al distances of average 2.376(2) Å are less than the sum of covalent radii for the two atoms (2.60 Å)35 and, along with the butterfly shape of the {Al(μ-H)2Cr} fragments with Cr–H–Al angles of average 90(2)°, could be indicative of some degree of metal–metal interaction (see below). Each chromium atom exhibits a classical four-legged piano-stool arrangement with four hydrides at the legs and H–Cr–H angles ranging from 72(2)° to 88(2)° for cis positions and 131(2) to 136(2)° for trans positions. Each pair of hydride ligands is also coordinated to an aluminum center, which shows a four-coordinate environment with three distinct H–Al–H angles ranging 79(2)–102(2), 121(2)–132(2), and 172(2)–178(2)°. Thus, the arrangement of hydride ligands around each aluminum atom could be described as a sawhorse geometry (τ4 = 0.45, 0.43, 0.35, 0.35; τδ = 0.32, 0.29, 0.26, 0.26).36
 | ||
| Fig. 5 Perspective view of 9·1.5C7H8 with thermal ellipsoids at the 50% probability level. Methyl groups of the η5-C5Me5 ligands and toluene solvent molecules are omitted for clarity. Selected averaged lengths (Å) and angles (°): Cr–H 1.63(5), Al–H 1.72(2), Cr–Cm 1.808(2), Cr–Al 2.376(2), Cr(1)/Cr(3)⋯Cr(2) 4.748(2), Cr(1)/Cr(3)⋯Cr(4) 4.750(4), Cr(2)⋯Cr(4) 6.119(1), Cr–Al–Cr 176(2), Al–Cr–Al 78.2(3), Cr–H–Al 90(2), H–Cr–H 72(2)–88(2) and 131(2)–136(2), H–Al–H 79(2)–102(2), 121(2)–132(2) and 172(2)–178(2). | ||
The X-ray crystal structure of 9 is essentially identical to that determined by Sitzmann and co-workers for the tetrachromium complex [(Al{(μ-H)4Cr(η5-C5H2tBu3-1,2,4)})4].8 The authors suggested that this tetramer could be described as four {Cr(η5-C5H2tBu3-1,2,4)H4}3− chromate(II) anions coordinating four Al3+ cations. The unusual structure of the compound and the unexpected distribution of the four hydride ligands around aluminum were attributed to the acquisition of full valence electron configurations for the chromium(II) and aluminum(III) centers. Interestingly, Camp and co-workers have described the structure and bonding of several heterometallic polyhydride complexes containing {IrCp*Hx}n− iridate units stabilizing Al3+ ions through Ir–H → Al and Ir → Al interactions.15 More recently, the Arnold and Camp groups have extended these studies to a series of heterometallic actinide-transition metal An-TM polyhydride species (An = Th, U; TM = Ir, Os, Re) with analogous interactions.37 To elucidate the electronic structure of complex 9 and to explain its diamagnetic nature, we have carried out density functional theory (DFT) calculations (BP86/TZVP + NBO analysis, see the Computational details section). In principle, we expect the aluminum atoms to be in the +3 oxidation state and, given the total negative charges (20−) of the η5-C5Me5 and μ-H ligands, all the chromium atoms must be in the +2 oxidation state. Several different spin distributions for these Cr2+ ions were tried. Initially, we started assuming that the chromium atoms were in a relatively weak field to generate different high-spin Cr2+ distributions that would be canceled in antiferromagnetic arrangements. All the calculations done in this way ended up providing the same closed-shell structure in which the chromium atoms are found to be in the low-spin configuration, i.e. the 4 electrons in each chromium center are paired in two d orbitals. This seems to be the best option for this complex and, in addition, it matches the diamagnetic nature observed experimentally. Furthermore, the computed structure reproduces very well all the geometric parameters of the crystallographic structure of 9 (e.g., averaged Cr–Al–Cr angles of 177.0(1)°, Al–Cr–Al angles of 77.6(1)°, and Cr–Al distances of 2.386(3) Å, see Table S8†). The computed IR spectrum for 9 reveals that the maximum absorption bands corresponding to Cr–H–Al vibrations appear at 1636 and 1590 cm−1 and the whole range for these vibrations is between 1648 and 1556 cm−1. These values agree with the two very strong and broad bands centered at 1572 and 1546 cm−1, which are assigned to these vibrations in the experimental solid-state IR spectrum of complex 9.
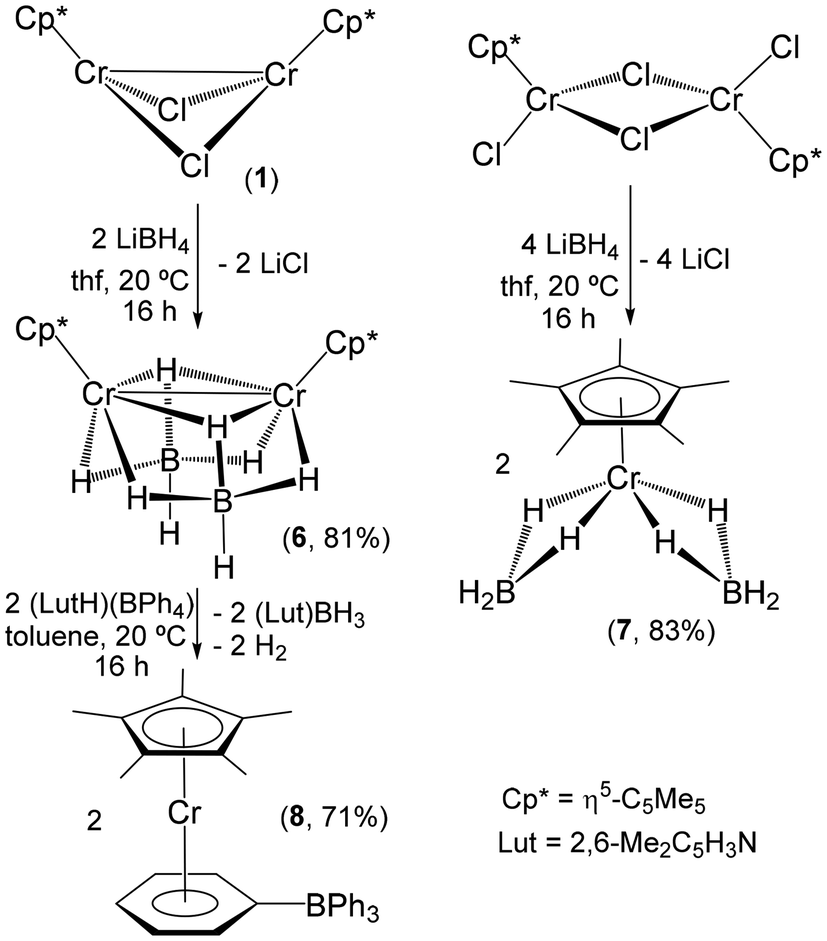
This electronic distribution and subsequent Natural Bond Orbital (NBO) analysis facilitate the interpretation of the bonding of complex 9. Thus, in the NBO analysis, we can find the two full d orbitals of each chromium atom and the corresponding four Cr–H bonds (Fig. S4†). The analysis also shows the donation from these occupied orbitals to the empty orbitals of aluminum (Fig. 6), which clearly stabilize the structure. The most remarkable donor/acceptor interactions in complex 9 correspond to the Cr–H donations to the empty orbitals of the Al atoms (Fig. 6a–c). However, some weaker donation from the full d orbitals of Cr to Al can also be identified (Fig. 6d and e). Since all the chromium and aluminum centers are equivalent in the molecule, the same orbitals and donor/acceptor interactions are found in the remaining parts of the complex. Interestingly, the peculiar geometric arrangements of the bridging hydride ligands around each aluminum are maintained upon geometry optimization. Therefore, each {Al(μ-H)2Cr} core adopts a butterfly geometry, with Al–H–Cr angles of average 87(1)° in the computed structure, in line with the X-ray crystallographic structure. The positioning of both bridging hydrogen atoms to one side of the Cr–Al vector allows the displacement of the chromium and aluminum centers towards each other, making possible the existence of the (weak) bonding interaction between the two metal atoms described above.
 | ||
| Fig. 6 Donor/acceptor (a–c) Cr–H → Al and (d and e) Cr → Al interactions in compound 9. Solid and transparent orbitals represent electron donor (full) and acceptor (empty) orbitals, respectively. The number near each representation corresponds to the NBO donor/acceptor stabilization energy in kcal mol−1. Hydrogen atoms of the η5-C5Me5 ligands are omitted for clarity. | ||
The X-ray diffraction of single crystals of 10·C6D6 revealed a molecular structure for the complex, with two alternating mononuclear {CrCp*H4} and dinuclear {(CrCp*H2)2} units connected to four aluminum centers by hydride ligands (Fig. 7a and S3†). Molecules of 10 have a twofold rotation axis perpendicular to the midpoint of the Cr(1)–Cr(1)ii segment and a mirror plane that bears the Cr(1), Cr(1)ii, Al(1), Al(1)ii, Al(2), and Al(2)ii core atoms. This mirror plane is perpendicular to the twofold rotation axis previously mentioned and also perpendicular to the midpoint of the Cr(2)–Cr(2)i segment of the Cr2 units. The whole structure has a D2h symmetry, which makes the two mononuclear {CrCp*H4} and the two dinuclear {(CrCp*H2)2} units of the complex equivalent, with the latter moieties also showing equivalent Cr(2) atoms. The mononuclear {CrCp*H4} fragments have a four-legged piano-stool geometry with four bridging hydrides at the legs. Each pair of these hydride ligands is also coordinated to an aluminum atom with H–Cr(1)–H and H–Al–H angles of average 82(5) and 78(2)°, which compare well with those found in the structure of 9. In addition, the Cr(1)–Al length of average 2.390(5) Å and the butterfly shape of the {Cr(μ-H)2Al} cores are consistent with the existence of a (weak) bonding interaction between the two metal atoms, as described above for 9. The dinuclear {(CrCp*H2)2} units contain two CrCp* moieties held together by two bridging hydride ligands also bonded to Al atoms. The {Cr(μ-H)2Cr} fragments exhibit a butterfly structure with average Cr(2)–H bond lengths of 1.66(2) Å and Cr(2)–H–Cr(2)i angles of 89(1)°. The short Cr(2)–Cr(2)i distance of 2.324(1) Å is indicative of significant metal–metal bonding (see below). In addition, each Cr(2) atom is bonded to one hydride ligand, which is also bridging two aluminum centers. Thus, the dinuclear {(CrCp*H2)2} units are connected to each aluminum atom through three μ3-H bridging hydride ligands with Cr(2)–Al distances of average 2.532(2) Å, which are longer than the Cr(1)–Al distances of 2.390(5) Å. The five-coordinate geometry about the aluminum atoms is best described as basally distorted square pyramidal (τ5 = 0 for Al(1) and Al(2))38, with the Al(1) and Al(2) atoms sitting 0.24 and 0.30 Å respectively above the mean plane of the four hydrogen atoms that form the basal plane of the pyramid and the apical hydrogen atom bent towards the Cr2 unit.
 | ||
| Fig. 7 (a) Perspective view of complex 10·C6D6 with thermal ellipsoids at the 50% probability level. η5-C5Me5 ligands and benzene solvent molecules are omitted for clarity. Selected lengths (Å) and angles (°): Cr(2)–Cr(2)i 2.324(1), Cr(1)–H(11) 1.54(3), Cr(1)–H(12) 1.62(3), Cr(2)–H(21) 1.55(3), Cr(2)–H(22) 1.65(3), Cr(2)–H(23) 1.68(3), Al(1)–H(12) 1.71(3), Al(1)–H(21) 1.97(3), Al(1)–H(22) 2.24(4), Al(2)–H(11) 1.72(3), Al(2)–H(21)ii 2.05(3), Al(2)–H(23)ii 2.12(4), Cr(1)–Cm 1.812, Cr(2)–Cm 1.837, Cr(1)–Al(1) 2.387(1), Cr(1)–Al(2) 2.394(1), Cr(2)–Al(1) 2.531(1), Cr(2)–Al(2)ii 2.534(1), Al(1)–Cr(1)–Al(2) 79.4(1), Cr(1)–H(11)–Al(2) 94(1), Cr(1)–H(12)–Al(1) 92(1), H(11)–Cr(1)–H(11)i 87(2), H(12)–Cr(1)–H(12)i 85(1), H(12)–Al(1)–H(12)i 79(1), H(11)–Al(2)–H(11)i 76(1). Symmetry code: (i) x, y, 1 − z; (ii) 1 − x, 1 − y, z; (iii) 1 − x, 1 − y, 1 − z. (b) Most stable computed triplet electronic structure for compound 10. The full structure comprises two equivalent Cr2 dinuclear units and two equivalent isolated Cr centers, not shown here. | ||
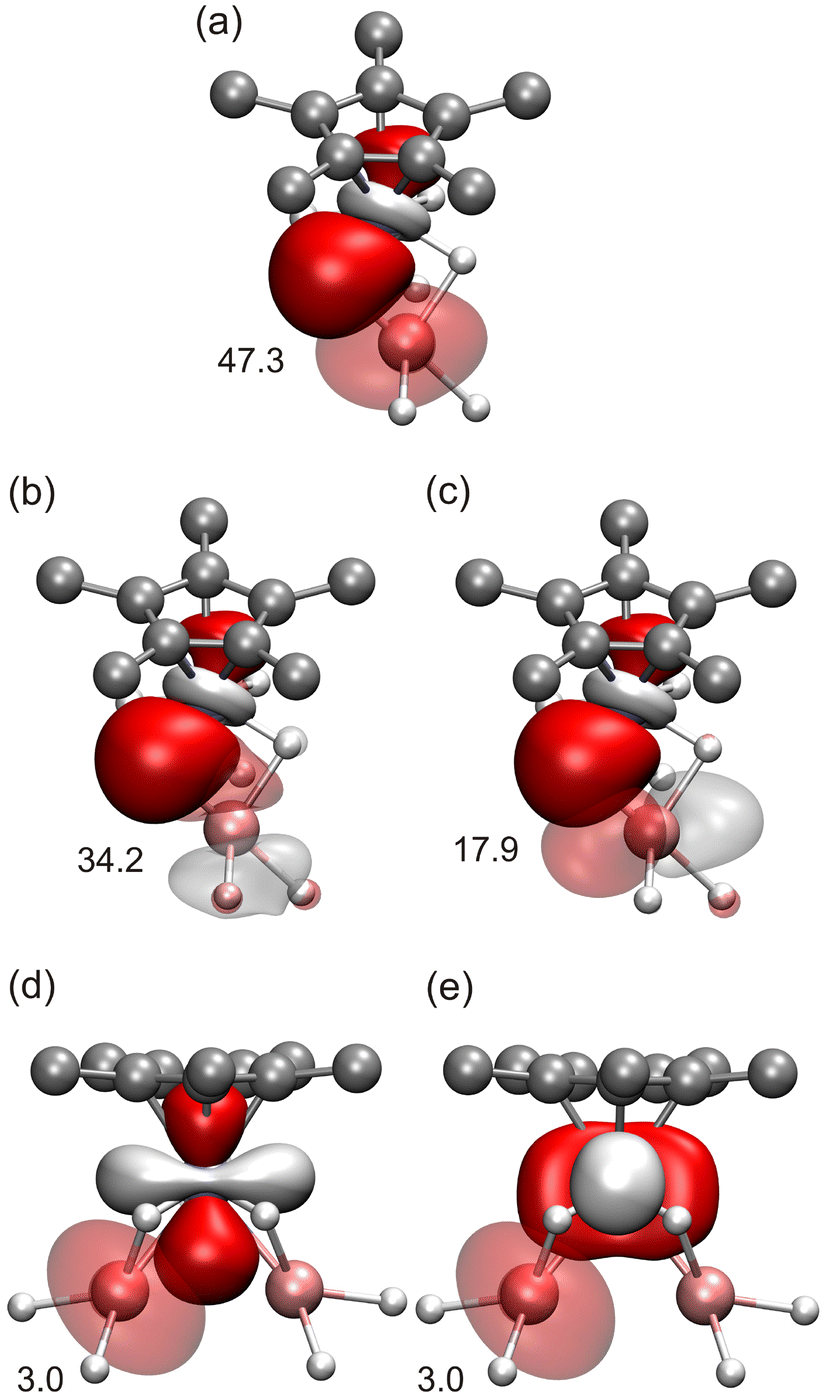
DFT calculations have been carried out to understand the electronic structure of complex 10. In this case, the sum of the negatively charged fragments is 22− (hydrides and η5-C5Me5) and, since all the aluminum atoms are in the +3 oxidation state, the sum of the chromium charges must be +10, indicating a mixture of oxidation states among these ions. At first sight, the two likely combinations should be: 4Cr+ + 2Cr3+ and 4Cr2+ + 2Cr+. Therefore, different initial spin distributions accounting for these options were computed. However, the final results indicate that none of the initial options considered seemed to be correct; eventually, all the calculations carried out ended up converging to a unique configuration, whose electronic structure can be described as shown in Fig. 7b. The standalone Cr atoms, following the behavior observed in complex 9, seem to be low-spin chromium(II) ions. In the case of these isolated chromium atoms, the NBO analysis of the electronic structure shows two full d orbitals (Fig. S5†), which account for the low-spin nature of these atoms. This indicates that each dinuclear unit contains two low-spin chromium atoms in a +1.5 oxidation state, in which one unpaired electron is shared between both metals. The specific arrangement of d electrons found, as generated from NBO analysis, indicates that there are two Cr–Cr doubly occupied bonds (Fig. 8a), one Cr–Cr orbital containing the unpaired alpha electron (Fig. 8b), and two one-electron lone pairs on each chromium center pointing in either the alpha or beta orientation (Fig. S6†). Since the relative orientations of the latter electrons are opposite, two additional Cr–Cr bonds might be expected where these orbitals overlap. However, the NBO calculation does not consider the existence of those bonds as such and provides independent NBOs. The overall complex is a paramagnetic triplet, in which the unpaired electrons are arranged in the same orientation (alpha), one on each dinuclear unit. The spin density of the triplet complex shows that the unpaired electrons are mainly located within the dinuclear units of the structure (Fig. 8c).
 | ||
| Fig. 8 Computed NBOs of the dinuclear Cr unit in compound 10: (a) for the doubly occupied Cr–Cr orbitals and (b) for the alpha semioccupied Cr–Cr orbital. (c) Computed spin density for compound 10. Color code: C = gray, H = white, Cr = iceblue, Al = pink. Hydrogen atoms of the η5-C5Me5 ligands are omitted for clarity. | ||
As observed in complex 9, the optimization leading to this electronic structure does not entail a large reorganization of the tridimensional arrangement of the compound (Table S10†). The computed IR spectrum for 10 reveals that the maximum absorption bands corresponding to Cr–H–Al vibrations appear at 1648, 1632 and 1612 cm−1, and the whole range for these vibrations is between 1650 and 1536 cm−1. These values agree with the two very strong and broad bands centered at 1615 and 1576 cm−1 assigned to these vibrations in the experimental solid-state IR spectrum of complex 10.
Additionally, higher multiplicity complexes were computed, but we were not able to converge to any satisfactory electronic structure. We also computed the open-shell singlet, in which both unpaired electrons (one on each dinuclear unit) are arranged antiferromagnetically. The energy of this singlet structure is quite close to that found for the triplet, but the calculation indicates a relatively high degree of spin contamination, which suggests that the structure would prefer to adopt the triplet electronic structure in the ground state.
Moreover, the theoretical analysis reveals other important NBOs, which are related to Cr–H bonds and participate in donation processes to aluminum centers (Fig. 9). In a fashion similar to that observed in complex 9, no Al–H bonds were found in the NBO calculations; all aluminum valence orbitals are almost empty and have some population due to the donation from the Cr–H orbitals.
 | ||
| Fig. 9 Cr–H bonding NBOs in complex 10: (a) one of the four Cr–H bonds of the mononuclear unit, (b) one of the two Cr–H bonds of the dinuclear unit, and (c) bridging Cr2–H bond of the dinuclear unit. Color code: C = gray, H = white, Cr = iceblue, Al = pink. Hydrogen atoms of the η5-C5Me5 ligands are omitted for clarity. | ||
Finally, relevant donor/acceptor interactions were also identified, corresponding to Cr–H donation to the empty orbitals of the aluminum atoms. Whereas the donor/acceptor Cr–H → Al and Cr–Al interactions within the mononuclear chromium units in complex 10 (Fig. S7†) are similar to those described above for compound 9, donor/acceptor interactions in the dinuclear units were observed for Cr–H and Cr2–H bonds, producing significant stabilization of the complex (Fig. 10a–e). In addition, strong interactions between the Cr–Cr bonds and the empty s orbitals of the Al atoms were observed (Fig. 10f and g) in a fashion similar to those described previously by us in heterometallic Ti–Al complexes.13a
 | ||
| Fig. 10 Donor/acceptor (a–e) Cr–H → Al and (f and g) Cr–Cr → Al interactions within the dinuclear Cr units in compound 10. Solid and transparent orbitals represent electron donor (full) and acceptor (empty) orbitals, respectively. The number near each representation corresponds to the NBO donor/acceptor stabilization energy in kcal mol−1. Hydrogen atoms of the η5-C5Me5 ligands are omitted for clarity. | ||
Conclusions
A series of half-sandwich chromium species have been prepared and structurally characterized using the easily available dinuclear chromium(II) chloride complex [{CrCp*(μ-Cl)}2]. This compound does not react with N2 but is capable of reducing the NN and N–N bonds of azobenzene and 1,2-diphenylhydrazine in four or two electrons at room temperature to give dimeric imido- and amido-bridged chromium(IV) and chromium(III) complexes, respectively. The treatment of [{CrCp*(μ-Cl)}2] with LiBH4 leads to the dinuclear chromium(II) derivative [{CrCp*(μ-κ3-BH4)}2] with rare bridging μ-κ3-tetrahydridoborato ligands. An analogous reaction with LiAlH4 affords a diamagnetic heterometallic Cr–Al species [(Al{(μ-H)4CrCp*})4] with a butterfly structure, where four chromium atoms are connected through four μ-κ2:κ2-AlH4 moieties, generating a sawhorse environment around the aluminum centers. Theoretical calculations demonstrate that the compound contains low-spin chromate(II) {CrCp*H4}3− anions, stabilizing the Al3+ ions primarily through Cr–H → Al interactions and weaker Cr → Al donation. The thermal decomposition of this tetrachromium(II) aggregate generates the mixed-valence CrII/CrI hexachromium species [(Al2{(μ-H)4CrCp*}{(μ3-H)4Cr2Cp*2})2], with analogous {CrCp*H4}3− and {Cr2Cp*2H4}3− chromate units coordinating Al3+ ions. The latter dinuclear chromate units display two Cr–Cr doubly occupied bonds that establish strong electron density donation to the empty s orbitals of the Al atoms.
Experimental section
General considerations
All manipulations were carried out under an argon atmosphere using Schlenk line or glovebox techniques. Toluene, benzene, hexane and pentane were distilled from Na/K alloy just before use. Tetrahydrofuran was distilled from purple solutions of sodium benzophenone just prior to use. Benzene-d6 and toluene-d8 were dried with Na/K alloy and distilled before use. Oven-dried glassware was repeatedly evacuated with a pumping system (ca. 1 × 10−3 Torr) and subsequently filled with an inert gas. 1,2-Diphenylhydrazine (97%) and lithium tetrahydridoaluminate (LiAlH4, 97%) were purchased from Aldrich and used as received. Lithium tetrahydridoborate (LiBH4, ≥95%, Aldrich) was ground using a mortar and pestle until a very fine powder was obtained. Azobenzene (PhNNPh) was purchased from Aldrich and sublimed under vacuum prior to use. [{CrCp*(μ-Cl)}2] (1),4,5 [{CrCp*Cl(μ-Cl)}2],17 2,6-lutidinium tetraphenylborate (LutH)(BPh4),39 and cyclopentyllithium [Li(C5H9)]40 were prepared according to published procedures.
Samples for infrared spectroscopy were prepared as KBr pellets, and the spectra were obtained using a FT-IR-Frontier PerkinElmer spectrophotometer, or the IR spectra were recorded as a powder using an attenuated total reflection (ATR) method on a Bruker FT-IR-ALPHA II spectrometer placed in an argon-filled glovebox. 1H and 13C{1H} NMR spectra were recorded on Varian Mercury-300, Varian Unity-500, or Bruker Avance Neo 400 spectrometers. Chemical shifts (δ) in the 1H and 13C{1H} NMR spectra are given relative to residual protons or to carbon of the solvent, C6D6 (1H: δ = 7.15; 13C: δ = 128.0) or C7D8 (1H: δ = 2.08; 13C: δ = 20.4). The effective magnetic moments in solution were determined by the Evans NMR method at 295 K (using a 300 MHz instrument with a field strength of 7.05 Tesla).18 Melting points were determined in sealed capillary tubes under argon and are uncorrected. Microanalyses (C, H, N) were performed using a PerkinElmer CHNS/O 2400 or Leco CHNS-932 microanalyzer.
Synthesis of [{CrCp*Cl(μ-NPh)}2] (2)
A toluene solution (5 mL) of azobenzene (0.12 g, 0.66 mmol) was slowly added to a solution of 1 (0.30 g, 0.67 mmol) in toluene (10 mL). The reaction mixture was stirred at room temperature for 16 h to give a green solution. After filtration, the volatile components of the solution were removed under reduced pressure to afford a sticky green solid. The solid was washed with hexane (20 mL) and vacuum-dried to give 2 as a green powder (0.22 g, 53%). IR (KBr, cm−1):![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) 3069 (w), 3049 (w), 2984 (w), 2957 (w), 2903 (m), 2851 (w), 1567 (w), 1471 (s), 1444 (s), 1427 (s), 1407 (m), 1376 (s), 1361 (m), 1262 (w), 1160 (m), 1102 (w), 1068 (m), 1022 (s), 806 (w), 773 (s), 729 (w), 697 (vs), 628 (m), 586 (s), 431 (w). 1H NMR (300 MHz, C6D6, 20 °C): δ −10.0 (s br., Δν1/2 = 569 Hz; C5Me5). Anal. Calcd for C32H40Cl2Cr2N2 (Mw = 627.57): C 61.24, H 6.42, N 4.46. Found: C 61.72, H 6.37, N 4.73. The effective magnetic moment of 2 was found to be 2.2μB (based on a unit formula of C32H40Cl2Cr2N2) in a C6D6 solution.
3069 (w), 3049 (w), 2984 (w), 2957 (w), 2903 (m), 2851 (w), 1567 (w), 1471 (s), 1444 (s), 1427 (s), 1407 (m), 1376 (s), 1361 (m), 1262 (w), 1160 (m), 1102 (w), 1068 (m), 1022 (s), 806 (w), 773 (s), 729 (w), 697 (vs), 628 (m), 586 (s), 431 (w). 1H NMR (300 MHz, C6D6, 20 °C): δ −10.0 (s br., Δν1/2 = 569 Hz; C5Me5). Anal. Calcd for C32H40Cl2Cr2N2 (Mw = 627.57): C 61.24, H 6.42, N 4.46. Found: C 61.72, H 6.37, N 4.73. The effective magnetic moment of 2 was found to be 2.2μB (based on a unit formula of C32H40Cl2Cr2N2) in a C6D6 solution.
Synthesis of [{CrCp*Cl(μ-NHPh)}2] (3)
In a fashion similar to the preparation of 2, complex 1 (0.20 g, 0.45 mmol) and 1,2-diphenylhydrazine (0.080 g, 0.43 mmol) were reacted in toluene (20 mL) at room temperature for 15 min to give 3 as a purple powder (0.18 g, 66%). IR (ATR, cm−1): 3241 (w) (NH), 3057 (w), 3008 (w), 2906 (m), 2855 (w), 1594 (s), 1489 (s), 1467 (s), 1427 (m), 1380 (m), 1359 (m), 1237 (s), 1222 (vs), 1073 (m), 1029 (s), 957 (m), 906 (s), 861 (s), 832 (vs), 815 (vs), 762 (vs), 697 (vs), 583 (m), 521 (m), 453 (vs), 415 (m). Anal. Calcd for C32H42Cl2Cr2N2 (Mw = 629.59): C 61.05, H 6.72, N 4.45. Found: C 60.94, H 6.63, N 4.93. The effective magnetic moment of 3 was found to be 3.6μB (based on a unit formula of C32H42Cl2Cr2N2) in a C6D6 solution.
Synthesis of [{CrCp*(μ3-H)}4] (4)4
A hexane solution (10 mL) of cyclopentyllithium (0.10 g, 1.31 mmol) was added to a solution of 1 (0.30 g, 0.67 mmol) in hexane (10 mL). The reaction mixture was stirred at room temperature for 30 min and filtered. The resulting dark brown solution was transferred into a 100 mL ampule (Teflon stopcock) and heated at 70 °C. After 4 days at this temperature, black crystals of 4 deposited on the walls of the ampule. The crystals were isolated by filtration and vacuum-dried to afford 4 (0.030 g, 12%). IR (ATR, cm−1): 2889 (vs), 2850 (s), 1483 (m), 1429 (s), 1371 (vs), 1258 (w), 1066 (w), 1021 (s), 799 (w), 717 (m), 618 (w), 528 (s), 479 (m), 437 (m), 411 (m).
Synthesis of [CrCp*(η6-C6H5Me)] (5)21
A toluene solution (10 mL) of cyclopentyllithium (0.10 g, 1.31 mmol) was added to a solution of 1 (0.30 g, 0.67 mmol) in toluene (10 mL). The reaction mixture was stirred at room temperature for 30 min and filtered. The resulting dark brown solution was transferred into a 100 mL ampule (Teflon stopcock) and heated at 110 °C. After 3 days at this temperature, the green solution was filtered and the volatile components of the solution were removed under reduced pressure. The resulting solid was dissolved in hexane (5 mL), and the hexane solution was filtered through a coarse glass frit. Crystallization at −35 °C afforded 5 (0.070 g, 19%) as orange crystals (mp 95–97 °C, lit.21 98 °C). IR (ATR, cm−1): 3057 (w), 3040 (w), 2967 (m), 2953 (m), 2902 (s), 2854 (m), 2720 (w), 1705 (w), 1642 (w), 1584 (w), 1530 (w), 1478 (m), 1454 (m), 1425 (s), 1377 (vs), 1120 (w), 1066 (w), 1038 (s), 1026 (vs), 999 (m), 969 (s), 793 (m), 761 (vs), 585 (w), 485 (s), 465 (s), 453 (vs), 410 (m). Anal. Calcd for C17H23Cr (Mw = 279.36): C 73.09, H 8.30. Found: C 73.28, H 8.40. The effective magnetic moment of 5 was found to be 1.7μB (based on a unit formula of C17H23Cr) in C6D6 or C7D8 solutions.
Synthesis of [{CrCp*(μ-κ3-BH4)}2] (6)
A suspension of LiBH4 (0.040 g, 1.74 mmol) in tetrahydrofuran (10 mL) was slowly added to a solution of 1 (0.30 g, 0.67 mol) in tetrahydrofuran (10 mL). The reaction mixture immediately turned dark blue and was stirred at room temperature for 16 h. The volatile components were removed under reduced pressure, and the resulting solid was extracted with toluene (15 mL). After filtration, the volatile components of the filtrate were removed under reduced pressure to give 6 as a green solid (0.22 g, 81%). IR (KBr, cm−1): 2967 (m), 2911 (vs), 2857 (s), 2457 (s) (BHterm), 2092 (m br.) (BHbridging), 1449 (s), 1380 (vs), 1262 (w), 1158 (w), 1062 (m), 1023 (s), 797 (m), 413 (s). 1H NMR (300 MHz, C6D6, 20 °C): δ 6.7 (s br., Δν1/2 = 52 Hz; C5Me5). Anal. Calcd for C20H38B2Cr2 (Mw = 404.13): C 59.44, H 9.48. Found: C 59.78, H 9.62. The effective magnetic moment of 6 was found to be 1.9μB (based on a unit formula of C20H38B2Cr2) in a C6D6 solution.
Synthesis of [CrCp*(κ2-BH4)2] (7)
In a fashion similar to the preparation of 6, [{CrCp*Cl(μ-Cl)}2] (0.50 g, 0.97 mmol) and LiBH4 (0.10 g, 4.36 mmol) were reacted in tetrahydrofuran (20 mL) for 16 h to afford 7 as a dark green powder (0.35 g, 83%). IR (KBr, cm−1): 2992 (w), 2966 (w), 2920 (m), 2448 (vs) (BHterm), 2396 (vs) (BHterm), 2211 (m) (BHbridging), 2058 (vs) (BHbridging), 2035 (vs) (BHbridging), 1957 (s) (BHbridging), 1474 (s), 1381 (vs), 1342 (vs), 1235 (w), 1110 (vs) (BH2), 1020 (s), 985 (w), 803 (w), 681 (w), 543 (w), 487 (s), 444 (m). 1H NMR (300 MHz, C6D6, 20 °C): δ 42.9 (s br., Δν1/2 = 355 Hz; C5Me5). Anal. Calcd for C10H23B2Cr (Mw = 216.91): C 55.37, H 10.69. Found: C 55.30, H 10.06. The effective magnetic moment of 7 was found to be 4.1μB (based on a unit formula of C10H23B2Cr) in a C6D6 solution.
Synthesis of [CrCp*(η6-C6H5-BPh3)] (8)3b,32
A suspension of (LutH)(BPh4) (0.20 g, 0.47 mmol) in toluene (10 mL) was added to a solution of 6 (0.10 g, 0.25 mmol) in toluene (15 mL). Gas evolution was immediately observed, and the initial green suspension turned red. The reaction mixture was stirred for 16 h at room temperature to give an orange solid and a red solution. The solid was isolated by filtration through a glass frit and was washed with toluene (10 mL) to afford 8 (0.17 g, 71%) as an orange powder (mp: 8 decomposes at 194–196 °C to give a dark solid, which melts at 238–240 °C; lit.3b 246–248 °C). IR (ATR, cm−1): 3064 (w), 3041 (w), 2912 (w), 1582 (w), 1483 (m), 1448 (m), 1425 (s), 1388 (m), 1260 (m), 1136 (m), 1066 (w), 1028 (m), 979 (m), 846 (m), 800 (m), 734 (vs), 705 (vs), 611 (s), 473 (m), 428 (m). 1H NMR (300 MHz, C6D6, 20 °C): δ 8.5 (s br., Δν1/2 = 70 Hz, 12H; C6H5), 6.7 (s br., Δν1/2 = 41 Hz, 8H; C6H5), −15.0 (s br., Δν1/2 = 538 Hz, 15H; C5Me5). Anal. Calcd for C34H35BCr (Mw = 506.45): C 80.63, H 6.97. Found: C 80.68, H 7.22. The effective magnetic moment of 8 was found to be 1.5μB (based on a unit formula of C34H35BCr) in a C6D6 solution.
Synthesis of [(Al{(μ-H)4CrCp*})4] (9)
Method A: A suspension of LiAlH4 (0.14 g, 3.58 mmol) in tetrahydrofuran (20 mL) was added to a solution of 1 (0.80 g, 1.80 mmol) in tetrahydrofuran (20 mL). The reaction mixture was stirred at room temperature for 20 min, and the volatile components were removed under reduced pressure to give a brown solid. This solid was extracted with toluene (30 mL), and, after filtration, the volatile components of the filtrate were removed under reduced pressure. The resulting solid was washed with hexane (3 × 5 mL) and vacuum-dried to give 9 as a brown powder (0.55 g, 70%). Method B: In a fashion similar to Method A, the reaction of 6 (0.10 g, 0.25 mmol) with LiAlH4 (0.020 g, 0.51 mmol) in tetrahydrofuran (25 mL) for 16 h afforded 9 as a brown powder (0.080 g, 73%). IR (KBr, cm−1): 2969 (s), 2952 (s), 2899 (vs), 2853 (s), 1572 (vs br.) (CrH), 1546 (vs br.) (CrH), 1483 (s), 1455 (s), 1378 (vs), 1217 (m), 1111 (m), 1069 (m), 1029 (vs), 777 (s), 730 (s), 696 (m), 579 (m), 510 (vs), 465 (w), 409 (m). 1H NMR (400 MHz, C6D6, 20 °C): δ 1.92 (s, 60H; C5Me5), −10.51 (s, 16H; AlH4). 13C{1H} NMR (100 MHz, C6D6, 20 °C): δ 97.7 (C5Me5), 14.2 (C5Me5). Anal. Calcd for C40H76Al4Cr4 (Mw = 872.94): C 55.04, H 8.78. Found: C 55.53, H 8.80.
Synthesis of [(Al2{(μ-H)4CrCp*}{(μ3-H)4Cr2Cp*2})2] (10)
A 10 mL ampule (Teflon stopcock) was charged with 9 (0.20 g, 0.23 mmol) and benzene (5 mL). The resulting brown solution was heated at 90 °C. After 10 days at this temperature, black crystals deposited on the walls of the ampule. The crystals were isolated by filtration, washed with toluene (3 × 1 mL) and vacuum-dried to afford 10·0.5C6H6 as black prismatic crystals (0.040 g, 20%). IR (ATR, cm−1): 2969 (m), 2948 (m), 2896 (s), 2847 (m), 1615 (vs br.) (CrH), 1576 (vs br.) (CrH), 1479 (s), 1450 (s), 1428 (s), 1370 (vs), 1067 (m), 1025 (vs), 763 (s), 727 (s), 693 (m), 672 (vs), 567 (m), 537 (m), 497 (vs), 478 (vs), 437 (s), 408 (m). Anal. Calcd for C63H109Al4Cr6 (Mw = 1286.44): C 58.82, H 8.54. Found: C 58.71, H 8.62.
X-ray crystal structure determinations
Brown crystals of 2, grey crystals of 6, black crystals of 7 and brown crystals of 9·1.5C7H8 were grown from toluene solutions at −35 °C. Purple crystals of 3·2C6H5F were obtained from a fluorobenzene solution at −35 °C. Black crystals of 4 and 10·C6D6 were grown by slow cooling of benzene-d6 suspensions heated at 90 °C in NMR tubes at room temperature. Orange crystals of 5 were obtained from a hexane solution at −35 °C. The crystals were removed from the Schlenk or NMR tubes and covered with a layer of a viscous perfluoropolyether (Fomblin Y). A suitable crystal was selected with the aid of a microscope, mounted on a cryoloop, and immediately placed in the low-temperature nitrogen stream of the diffractometer. The intensity data sets were collected at 150 K on a Bruker-Nonius KappaCCD diffractometer equipped with an Oxford Cryostream 700 unit. Crystallographic data for all the complexes are presented in Tables S1 and S2 of the ESI.†The structures were solved, using the WINGX package,41 by intrinsic phasing methods (SHELXT)42 and refined by least-squares against F2 (SHELXL-2018/3).43 Whereas compounds 2 and 4 crystallized as solvent-free molecules, 3 did so with two molecules of fluorobenzene. In the crystallographic studies of 2–4, all non-hydrogen atoms were anisotropically refined, while hydrogen atoms bound to carbon were included, positioned geometrically, and refined using a riding model. On the other hand, the hydrogen atoms of the imido groups in complex 3 were found in the difference Fourier map and refined isotropically. Additionally, the XHYDEX tool was employed to locate the hydride groups bound to chromium atoms in 4. Then, these hydrogen atoms, H(1), H(2), H(3) and H(4), were found in the difference Fourier map and refined isotropically. However, after the last refinement cycles, the isotropic displacement parameters for H(3) and H(4) atoms were not appropriate, so their Uiso values were forced to be 0.05.
Crystals of 5 showed disorder in the carbon atoms of the toluene ring, which was placed on a mirror plane, so these carbon atoms, C(1)–C(7), were refined in two sites with occupancy of 50%. All non-hydrogen atoms were anisotropically refined. Hydrogen atoms were positioned geometrically and refined using a riding model, but previously FREE instructions were applied to the atoms C(1)–C(7) to avoid issues with the calculated hydrogen atoms. Moreover, SADI restraints were applied to the carbon–carbon distances.
In the crystallographic studies of 6 and 7, all non-hydrogen atoms were anisotropically refined. All hydrogen atoms were placed geometrically and refined using a riding model, except those bound to boron atoms, which were located in the difference Fourier map and refined isotropically. Remarkably, it was necessary to search up to two hundred peaks in the Fourier map in order to locate all the hydrogen atoms bound to boron in complex 6.
Compound 9 crystallized with one and a half molecules of toluene, which were found in the difference Fourier map. However, it was not possible to obtain a chemically sensible model for them, so the PLATON44 squeeze procedure was used to remove their contribution to the structure factors. Additionally, this crystal presented disorder for the carbon atoms C(31)–C(40) of the pentamethylcyclopentadienyl ligand linked to Cr(3), but it was not possible to properly model it in two (or more) positions. All non-hydrogen atoms were anisotropically refined. Whereas hydrogen atoms bound to carbon were geometrically positioned and refined using a riding model, hydrogen atoms bound to chromium and aluminum were located in the Fourier map and refined isotropically. Furthermore, DELU and SIMU restraints were also applied to the carbon atoms of the disordered C5Me5 group.
Complex 10 crystallized with a molecule of benzene. All non-hydrogen atoms were anisotropically refined. Hydrogen atoms bound to carbon were geometrically positioned and refined employing a riding model, while hydrogen atoms bound to chromium or aluminum were found in the difference Fourier map and isotropically refined. Furthermore, DFIX constraints were applied to the carbon–carbon distances of the benzene molecule.
Computational details
All the structures have been fully optimized in gas phase employing the Gaussian09 suite of programs45 and the unrestricted formalism of the BP86 functional along with the TZVP basis sets for all the atoms.46 NBO analysis has been carried out at the same level of theory as above using the NBO 7.0 program.47Author contributions
Conceptualization: C. Y. and A. P.-R.; investigation: A. C.-M., J. J. and A. P.-R.; validation: A. C.-M. and C. Y.; supervision: C. Y. and A. P.-R.; visualization: A. C.-M., J. J., A. P.-R. and C. Y.; writing – original draft: C. Y.; writing – review & editing: A. C.-M., J. J., A. P.-R. and C. Y.; funding acquisition and project administration: C. Y. and A. P.-R. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data that support the findings of this study are available in the ESI† of this article.Acknowledgements
The authors acknowledge the financial support from the Ministerio de Ciencia, Innovación y Universidades of Spain MICIU/AEI/10.13039/501100011033/ERDF/EU (grant PID2023-146287NB-I00) and Universidad de Alcalá (PIUAH23/CC-004). A. C.-M. thanks Spanish Ministerio de Universidades and Universidad de Alcalá for doctoral fellowships. J. J. acknowledges the Spanish Structures of Excellence María de Maeztu program (CEX2021-001202-M) and the Generalitat de Catalunya for the 2021-SGR-00286 grant.References
- (a) K. H. Theopold and R. R. Kucharczyk, in Encyclopedia of Inorganic Chemistry, ed. R. B. King, Wiley, 2nd edn, 2006, vol. 2, pp. 907–924 Search PubMed; (b) M. J. Carney, in Comprehensive Organometallic Chemistry III, ed. M. P. Mingos and R. H. Crabtree, Elsevier, 2007, vol. 5, pp. 291–390 Search PubMed; (c) T. Song and Y. Mu, in Comprehensive Organometallic Chemistry IV, ed. G. Parkin, K. Meyer and D. O'Hare, Elsevier, 2022, vol. 5, pp. 81–173 Search PubMed.
- R. Poli, Chem. Rev., 1996, 96, 2135–2204 CrossRef CAS PubMed.
- (a) K. H. Theopold, Acc. Chem. Res., 1990, 23, 263–270 CrossRef CAS; (b) B. J. Thomas, S. K. Noh, G. K. Schulte, S. C. Sendlinger and K. H. Theopold, J. Am. Chem. Soc., 1991, 113, 893–902 CrossRef CAS; (c) S. K. Noh, R. A. Heintz, B. S. Haggerty, A. L. Rheingold and K. H. Theopold, J. Am. Chem. Soc., 1992, 114, 1892–1893 Search PubMed; (d) G. Bhandari, Y. Kim, J. M. McFarland, A. L. Rheingold and K. H. Theopold, Organometallics, 1995, 14, 738–745 CrossRef CAS; (e) R. A. Heintz, S. Leelasubcharoen, L. M. Liable-Sands, A. L. Rheingold and K. H. Theopold, Organometallics, 1998, 17, 5477–5485 CrossRef CAS ; and references therein.
- R. A. Heintz, B. S. Haggerty, H. Wan, A. L. Rheingold and K. H. Theopold, Angew. Chem., Int. Ed. Engl., 1992, 31, 1077–1079 CrossRef.
- R. A. Heintz, R. L. Ostrander, A. L. Rheingold and K. H. Theopold, J. Am. Chem. Soc., 1994, 116, 11387–11396 CrossRef CAS.
- R. A. Heintz, T. F. Koetzle, R. L. Ostrander, A. L. Rheingold, K. H. Theopold and P. Wu, Nature, 1995, 378, 359–362 CrossRef CAS.
- T. Shima, J. Yang, G. Luo, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2020, 142, 9007–9016 Search PubMed.
- I. Smytschkow, W. Gidt, Y. Sun, J. Langer, T. Böttcher, L. Kubíčková, O. Kaman and H. Sitzmann, Organometallics, 2021, 40, 2951–2969 Search PubMed.
- (a) N. Mézailles, in Transition Metals-Dinitrogen Complexes, ed. Y. Nishibayashi, Wiley-VCH, 2019, ch. 4 Search PubMed; (b) A. J. Kendall and M. T. Mock, Eur. J. Inorg. Chem., 2020, 1358–1375 CrossRef CAS ; and references therein.
- (a) G.-X. Wang, Z.-B. Yin, J. Wei and Z. Xi, Acc. Chem. Res., 2023, 56, 3211–3222 Search PubMed; (b) Z.-B. Yin, G.-X. Wang, X. Yan, J. Wei and Z. Xi, Nat. Commun., 2025, 16, 674 CrossRef CAS.
- (a) M. García-Castro, C. García-Iriepa, E. del Horno, A. Martín, M. Mena, A. Pérez-Redondo, M. Temprado and C. Yélamos, Inorg. Chem., 2019, 58, 5314–5324 Search PubMed; (b) E. del Horno, R. Jiménez-Aparicio, M. Mena, A. Pérez-Redondo, J. L. Priego and C. Yélamos, Inorg. Chem., 2020, 59, 3740–3752 CrossRef CAS PubMed.
- E. del Horno, J. Jover, M. Mena, A. Pérez-Redondo and C. Yélamos, Angew. Chem., Int. Ed., 2022, 61, e202204544 CrossRef CAS.
- (a) E. del Horno, J. Jover, M. Mena, A. Pérez-Redondo and C. Yélamos, Chem. – Eur. J., 2022, 28, e202103085 Search PubMed; (b) A. Calvo-Molina, E. del Horno, J. Jover, A. Pérez-Redondo, C. Yélamos and R. Zapata, Organometallics, 2023, 42, 1360–1372 CrossRef CAS.
- For reviews, see: (a) M. J. Butler and M. R. Crimmin, Chem. Commun., 2017, 53, 1348–1365 RSC; (b) S. Fernández, S. Fernando and O. Planas, Dalton Trans., 2023, 52, 14259–14286 Search PubMed.
- (a) L. Escomel, I. Del Rosal, L. Maron, E. Jeanneau, L. Veyre, C. Thieuleux and C. Camp, J. Am. Chem. Soc., 2021, 143, 4844–4856 CrossRef CAS PubMed; (b) L. Escomel, N. Soulé, E. Robin, I. Del Rosal, L. Maron, E. Jeanneau, C. Thieuleux and C. Camp, Inorg. Chem., 2022, 61, 5715–5730 CrossRef CAS PubMed.
- A. Calvo-Molina, A. Pérez-Redondo and C. Yélamos, Organometallics, 2024, 43, 1780–1784 CrossRef CAS.
- D. S. Richeson, J. F. Mitchell and K. H. Theopold, Organometallics, 1989, 8, 2570–2577 CrossRef CAS.
- (a) D. F. Evans, J. Chem. Soc., 1959, 2003–2005 Search PubMed; (b) S. K. Sur, J. Magn. Reson., 1989, 82, 169–173 Search PubMed; (c) G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
- I. L. Eremenko, A. A. Pasynskii, E. A. Vas'utinskaya, A. S. Katugin, S. E. Nefedov, O. G. Ellert, V. M. Novotortsev, A. F. Shestakov, A. I. Yanovsky and Y. T. Struchkov, J. Organomet. Chem., 1991, 411, 193–205 CrossRef CAS.
- A. A. Danopoulos, G. Wilkinson, T. K. N. Sweet and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1996, 271–281 RSC.
- F. H. Köhler, B. Metz and W. Strauss, Inorg. Chem., 1995, 34, 4402–4413 CrossRef.
- F. H. Köhler, J. Lachmann, G. Müller, H. Zeh, H. Brunner, J. Pfauntsch and J. Wachter, J. Organomet. Chem., 1989, 365, C15–C18 CrossRef.
- For reviews on metal tetrahydridoborato complexes, see: (a) T. J. Marks and J. R. Kolb, Chem. Rev., 1977, 77, 263–293 CrossRef CAS; (b) V. D. Makhaev, Russ. Chem. Rev., 2000, 69, 727–746 CrossRef CAS; (c) M. Besora and A. Lledós, Struct. Bonding, 2008, 130, 149–202 CrossRef CAS.
- D. Himmelbauer, B. Stöger, L. F. Veiros, M. Pignitter and K. Kirchner, Organometallics, 2019, 38, 4669–4678 CrossRef CAS.
- (a) K. J. Deck, Y. Nishihara, M. Shang and T. P. Fehlner, J. Am. Chem. Soc., 1994, 116, 8408–8409 CrossRef CAS; (b) J. Ho, K. J. Deck, Y. Nishihara, M. Shang and T. P. Fehlner, J. Am. Chem. Soc., 1995, 117, 10292–10299 CrossRef CAS.
- (a) D. G. Holah, A. N. Hughes, S. Maciaszek and V. R. Magnuson, J. Chem. Soc., Chem. Commun., 1983, 1308–1309 RSC; (b) D. G. Holah, A. N. Hughes, S. Maciaszek, V. R. Magnuson and K. O. Parker, Inorg. Chem., 1985, 24, 3956–3962 CrossRef CAS.
- D. R. Armstrong, W. Clegg, H. M. Colquhoun, J. A. Daniels, R. E. Mulvey, I. R. Stephenson and K. Wade, J. Chem. Soc., Chem. Commun., 1987, 630–632 RSC.
- D. L. Reger, J. E. Collins, M. A. Matthews, A. L. Rheingold, L. M. Liable-Sands and I. A. Guzei, Inorg. Chem., 1997, 36, 6266–6269 CrossRef CAS.
- M. Dionne, S. Hao and S. Gambarotta, Can. J. Chem., 1995, 73, 1126–1134 CrossRef CAS.
- M. Y. Darensbourg, R. Bau, M. W. Marks, R. R. Burch Jr., J. C. Deaton and S. Slater, J. Am. Chem. Soc., 1982, 104, 6961–6969 CrossRef CAS.
- J. D. Koola and H. H. Brintzinger, J. Chem. Soc., Chem. Commun., 1976, 388–389 RSC.
- C. Elschenbroich, B. Kanellakopulos, F. H. Köhler, B. Metz, R. Lescouëzec, N. W. Mitzel and W. Strauβ, Chem. – Eur. J., 2007, 13, 1191–1200 CrossRef CAS PubMed.
- (a) A. R. Barron, J. E. Salt and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1986, 1329–1332 RSC; (b) A. R. Barron and G. Wilkinson, Polyhedron, 1986, 5, 1897–1915 CrossRef CAS; (c) B. M. Bulychev, Polyhedron, 1990, 9, 387–408 CrossRef CAS.
- S. J. Bonyhady, D. Collis, N. Holzmann, A. J. Edwards, R. O. Piltz, G. Frenking, A. Stasch and C. Jones, Nat. Commun., 2018, 9, 3079 CrossRef PubMed.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Álvarez, Dalton Trans., 2008, 2832–2838 RSC.
- (a) L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC; (b) M. H. Reineke, M. D. Sampson, A. L. Rheingold and C. P. Kubiak, Inorg. Chem., 2015, 54, 3211–3217 Search PubMed.
- (a) C. Z. Ye, I. Del Rosal, M. A. Boreen, E. T. Ouellette, D. R. Russo, L. Maron, J. Arnold and C. Camp, Chem. Sci., 2023, 14, 861–868 RSC; (b) C. Z. Ye, I. Del Rosal, S. N. Kelly, I. J. Brackbill, L. Maron, C. Camp and J. Arnold, Chem. Sci., 2024, 15, 9784–9792 RSC; (c) C. Z. Ye, I. Del Rosal, E. T. Ouellette, S. Hohloch, L. Maron, C. Camp and J. Arnold, Chem. Commun., 2024, 60, 12377–12380 RSC; (d) C. Z. Ye, I. Del Rosal, S. N. Kelly, E. T. Ouellette, L. Maron, C. Camp and J. Arnold, Inorg. Chem., 2024, 63, 21167–21176 CrossRef CAS PubMed ; and references therein.
- (a) A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC; (b) A. G. Blackman, E. B. Schenk, R. E. Jelley, E. H. Krenske and L. R. Gahan, Dalton Trans., 2020, 49, 14798–14806 RSC.
- (a) J. R. Dilworth, R. A. Henderson, P. Dahlstrom, T. Nicholson and J. A. Zubieta, J. Chem. Soc., Dalton Trans., 1987, 529–540 RSC; (b) K. L. C. Grönberg, R. A. Henderson and K. E. Oglieve, J. Chem. Soc., Dalton Trans., 1998, 3093–3104 RSC.
- C. Su, R. Hopson and P. G. Williard, J. Am. Chem. Soc., 2013, 135, 12400–12406 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Speck, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (c) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2018.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental crystallographic data of compounds 2–7, 9, and 10. Perspective view of the crystal structure of compounds 5, 9, and 10. Tables for selected lengths and angles in the X-ray crystal structures of 5–7, 9, and 10. Tables for experimental vs. calculated selected lengths and angles for compounds 9 and 10. Computed NBOs and donor/acceptor interactions for complexes 9 and 10. Selected NMR and IR spectra. Cartesian coordinates of compounds 9 and 10. CCDC 2418690–2418697. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00620a |
| This journal is © The Royal Society of Chemistry 2025 |