
Dual tuning of nodes and functional guests of polyoxometalate@metal–organic frameworks for enhanced photocatalytic oxidative coupling†
Zhiqiang
Jiang‡
a,
Qixin
Zhao‡
a,
Yang
Zeng
a,
Tengfei
Gong
b,
Zhenxuan
Huang
a,
Zeyu
Chen
a,
Jiefeng
Shen
*a and
Weimin
Xuan
 *a
*a
aState Key Laboratory of Advanced Fiber Materials & College of Chemistry and Chemical Engineering, Donghua University, Shanghai 201620, P. R. China. E-mail: jiefeng@dhu.edu.cn; weiminxuan@dhu.edu.cn
bJiaxing Jiayuan Inspection Technology Service Co., Ltd, Building 2, No. 1403, Hongbo Road, Economic and Technological Development Zone, Jiaxing City, Zhejiang Province, P. R. China. E-mail: tengfeigong@126.com
First published on 8th May 2025
Abstract
Metal–organic frameworks are versatile platforms for photocatalysis owing to their facile capacity to incorporate active sites within organic linkers, nodes and open channels. However, the dual tuning of both nodes and functional guests for enhanced photocatalytic performance remains largely unexplored. Herein, we report the incorporation of functional polyoxometalate guests, {PM10V2} (M = Mo or W), into isoreticular 2D MOFs that are constructed from an anthraquinone-derived ligand and dinuclear nodes of {Cd2X(H2O)4} (X = Br or I), yielding isostructural polyoxometalate@metal–organic frameworks, denoted as POMOF 1–POMOF 3. By systematically varying the node from {Cd2Br(H2O)4} to {Cd2I(H2O)4} and the polyoxometalate guest from {PMo10V2} to {PW10V2}, we achieve a structural evolution from POMOF 1 to POMOF 3. Notably, POMOF 1 shows the highest activity towards photocatalytic oxidative coupling of benzylamine and its derivatives due to the synergistic effect of {PMo10V2} and {Cd2Br(H2O)4}, which facilitates the highest photocurrent and an optimal band gap distribution for reactive oxygen species formation and substrate activation. This study demonstrates a promising strategy for designing photocatalytically active materials with enhanced performance via dual regulation.
Introduction
Polyoxometalates (POMs), a class of functional metal–oxygen clusters, are widely used in photocatalysis due to their facile photoredox properties, tunable band gap structures and high efficiency in substrate activation.1–6 Upon photoirradiation, POMs can undergo reversible redox reactions, triggering a series of photocatalytic transformations, from oxidation to unactivated C–H functionalization.7–10 A prominent example is the direct activation of CH4via decatungstate.7,11 The photocatalytic activities of POMs can be tailored by metal substitution and photosensitizer/organic functionalization, allowing for facile tunability.12–17 For example, introducing heterometal sites on the pristine Keggin-type POM skeleton or switching between isostructural {PW12}, {PW6Mo6}, and {PMo12} with varying metal compositions can afford significantly distinct photocatalytic performances.18–20 These unique features enable POM clusters to act as photoactive building blocks for constructing multifunctional photocatalysts.21–24As an emerging type of crystalline porous material, metal–organic frameworks (MOFs) show great potential as photocatalytic platforms, as their active sites can be facilely integrated into organic linkers, metal nodes and the pore structures of MOFs.25–32 Consequently, linker engineering, metal node modulation and the incorporation of multifunctional nanostructured guests have been developed as effective approaches for optimizing MOF photocatalytic activity.33–37 In this context, POM-encapsulated MOFs (POM@MOFs) have recently received substantial research interest. The rational combination of POMs with MOFs not only ensures the uniform dispersity of POMs within the pore structure and synergistic photocatalytic properties between MOFs and POMs, but also facilitates fast diffusion of reactants and products.38–41 As such, a variety of POM@MOFs have been constructed and applied for photocatalytic water splitting, CO2 reduction, degradation of organic pollutants and selective oxidation of organics.42–46 Despite the great progress in applying these materials to photocatalysis, structural regulation typically focuses on either MOFs or POMs; dual tuning of both sides for enhanced performance is rarely explored.34,35,47–51 Considering the structural diversity and tunability of MOFs and POMs, a dual tuning approach will drive the further development of novel, high-performance photocatalytic systems while providing deeper insight into the structure–activity relationship.
Herein, we report the dual tuning of nodes and POM guests within a series of isostructural {PM10V2}@MOFs (M = Mo or W), POMOF 1–POMOF 3, for enhanced photocatalytic oxidative coupling of amines under green LED irradiation. POMOF 1–POMOF 3 feature a 2D square-like layered framework constructed from a photosensitive 2,6-di-(4H-1,2,4-triazol-4-yl) anthracene-9,10-dione (AQ) ligand and dinuclear {Cd2X(H2O)4} (X = Br or I) nodes, with {PM10V2} clusters intercalated between 2D metal–organic layers, leading to 1D open channels along the a-axis (Scheme 1). By dual tuning of the nodes from {Cd2I(H2O)4} to {Cd2Br(H2O)4} and POM guests from {PW10V2} to {PMo10V2}, POMOF 1 demonstrates the best performance in the photocatalytic oxidative coupling of benzylamine under green LED irradiation, and a series of benzylamines and their derivatives can be facilely converted into the corresponding imines with yields up to 99%. Photochemical studies revealed that POMOF 1 exhibited the highest photocurrent and appropriate band gap distribution for the generation of reactive oxygen species and substrate activation, attributed to the synergistic combination of {Cd2Br(H2O)4} and {PMo10V2}.
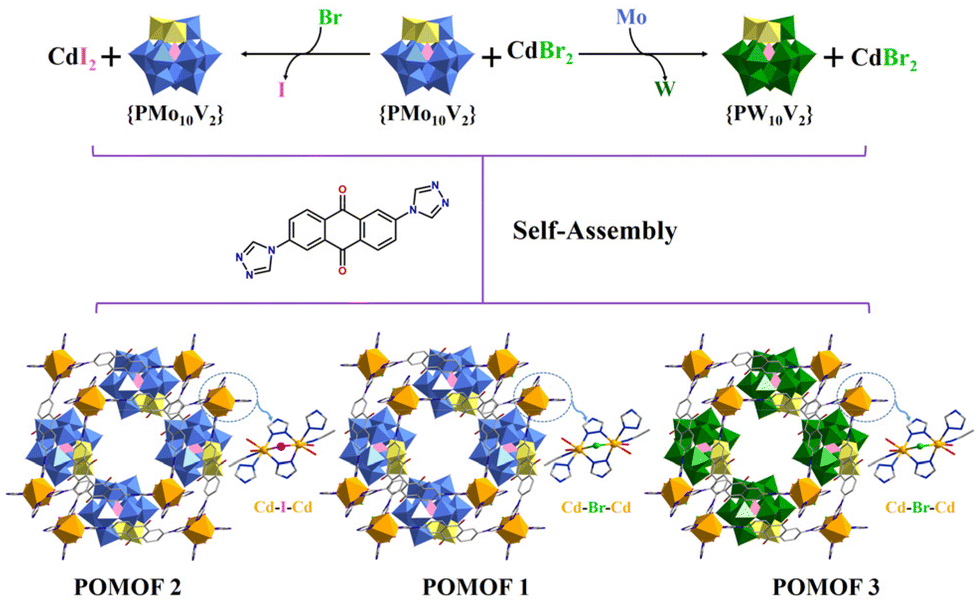 | ||
| Scheme 1 Schematic diagram showing the assembly of POMOF 1, POMOF 2 and POMOF 3, which share the isostructural networks but with different nodes and POM guests (color code: Cd, orange; C, grey; N, deep blue; Br, green; I, purple; O, red; Mo, blue; W, dark green; V, yellow; P, pink. H atoms are omitted for clarity). | ||
Results and discussion
Structural description
POMOF 1, POMOF 2, and POMOF 3 were synthesized via the self-assembly of classical Keggin-type POMs {PMo10V2}/{PW10V2} with cadmium halides and the AQ ligand under hydrothermal conditions. Single-crystal X-ray diffraction analysis reveals that the three compounds are isostructural, differing only in their metal nodes and POM guests. Therefore, POMOF 1 is selected to elucidate the detailed structural features. POMOF 1 crystallizes in the monoclinic space group I2/a, and its asymmetric unit comprises half the formula, i.e., one {PMo10V2} cluster, one {Cd2Br(H2O)4} unit and two AQ ligands (Fig. S5a† and Fig. 1a). Replacing {Cd2Br(H2O)4} with {Cd2I(H2O)4} affords POMOF 2, while substituting {PMo10V2} with {PW10V2} generates POMOF 3 (Fig. S5 and S6†). The Cd center adopts a six-coordinate geometry, coordinated by three nitrogen atoms from three AQ ligands, one bromine atom, and two oxygen atoms from two water molecules (Fig. 1a). One triazole moiety of AQ employs two N atoms to bridge two Cd sites within {Cd2Br(H2O)4}, while the other adopts a monodentate coordination mode to connect with Cd atoms. Consequently, {Cd2Br(H2O)4} behaves as a 4-connected node, and the AQ ligand acts as a 2-connected linker, resulting in a 2D square-like metal–organic layer with an aperture of 19.2 Å × 15.4 Å (Fig. 1b). Regulated by the a-glide plane, the adjacent layers display a staggered packing mode along the a-axis (Fig. 1c). To balance the charge, {PMo10V2} clusters are evenly encapsulated between different layers, yielding a 3D supramolecular POM@MOF (Fig. 1d). Accordingly, the dimensions of 1D microporous channels along the a-axis are reduced to 8.3 Å × 8.0 Å (Fig. 1d). Besides electrostatic interaction, a network of hydrogen bonds is identified between the surface oxygen atoms of {PMo10V2} and the aromatic hydrogen atom of 2D layers (C–H⋯O distances of 2.2791–2.6203 Å) (Fig. S7b†). Additionally, the shortest distance between the terminal oxygen atom of the {PMo10V2} cluster and the central quinone unit of the AQ structure is 2.8686 Å, indicative of a typical anion–π interaction (Fig. S7c†).52–54 The presence of these interactions is believed to further enhance the structural stability (Fig. 1d). | ||
| Fig. 1 (a) Molecular structure of POMOF 1, (b) view of the single layer of the 2D framework, (c) view of the staggered packing of 2D layers along the a-axis, and (d) 3D supramolecular structure of POMOF 1 along the a-axis, built from intercalation of {PMo10V2} clusters between adjacent layers. | ||
Characterization of POMOFs 1–3
Initially, the phase purity of the three complexes was assessed using powder X-ray diffraction (PXRD) analysis. The close correspondence between the experimentally obtained PXRD peaks and the simulated peaks validated the high phase purity of POMOF 1, POMOF 2, and POMOF 3 (Fig. 2a and Fig. S8, S9†). Furthermore, the thermal stability of the three complexes was evaluated via thermogravimetric analysis (TGA) under a nitrogen atmosphere over a temperature range of 30–800 °C. As depicted in the TGA curves, POMOF 1, POMOF 2, and POMOF 3 exhibited analogous weight loss profiles (Fig. 2b and Fig. S10, S11†). Taking POMOF 1 as an example, the initial weight loss stage occurs between 30 °C and 335 °C, attributed to the removal of lattice water and coordinated water molecules, followed by the gradual loss of halogen atoms. Subsequently, the system enters the second weight loss stage, during which the AQ ligand undergoes decomposition. Beyond 400 °C, complete structural degradation converts the framework into related metal oxides, including CdO, MoO3, and V2O5. | ||
| Fig. 2 (a) Experimental and simulated PXRD patterns of POMOF 1, (b) TGA curve of POMOF 1, (c) PXRD patterns of POMOF 1 after being soaked in different organic solvents and aqueous solutions of pH = 1 and 12 for 24 hours, and (d and e) resolved Br 3d, Cd 3d, Mo 3d, V 2p, and XPS core-level spectra of POMOF 1. | ||
Fourier transform infrared spectroscopy (FT-IR) was employed to elucidate the chemical structures of POMOF 1, POMOF 2, and POMOF 3. As depicted in Fig. S12–S14,† the FT-IR spectra of these compounds clearly exhibit characteristic peaks associated with PMo10V2 or PW10V2. This observation confirms the intact incorporation of POM units within the framework of the compounds. In POMOF 1 and POMOF 2, the vibrational modes of P–Oa (a: tetrahedral oxygen atom), Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Od (d: terminal oxygen atom), Mo–Ob–Mo (b: corner-sharing oxygen atom), and Mo–Oc–Mo (c: edge-sharing oxygen atom) are observed at 1051 cm−1, 943 cm−1, 871 cm−1, and 777 cm−1, respectively. These frequencies are consistent with those of pristine {PMo10V2}, indicating similar local structural environments.55 In POMOF 3, the vibrational peaks at 1070 cm−1, 957 cm−1, 885 cm−1, and 782 cm−1 are assigned to the P–Oa (tetrahedral oxygen), WOd, W–Ob–W, and W–Oc–W (edge-sharing oxygen) groups, respectively.56 Additionally, the CO stretching vibration peak, observed at 1675 cm−1, is attributed to the symmetrical ketone groups within the anthraquinone core in all three compounds.57
Od (d: terminal oxygen atom), Mo–Ob–Mo (b: corner-sharing oxygen atom), and Mo–Oc–Mo (c: edge-sharing oxygen atom) are observed at 1051 cm−1, 943 cm−1, 871 cm−1, and 777 cm−1, respectively. These frequencies are consistent with those of pristine {PMo10V2}, indicating similar local structural environments.55 In POMOF 3, the vibrational peaks at 1070 cm−1, 957 cm−1, 885 cm−1, and 782 cm−1 are assigned to the P–Oa (tetrahedral oxygen), WOd, W–Ob–W, and W–Oc–W (edge-sharing oxygen) groups, respectively.56 Additionally, the CO stretching vibration peak, observed at 1675 cm−1, is attributed to the symmetrical ketone groups within the anthraquinone core in all three compounds.57
The stability of POM@MOFs is critical for their application in heterogeneous catalysis. Therefore, POMOF 1 was chosen to assess its chemical stability in various organic solvents and aqueous solutions with different acidities. As shown in Fig. 2c, the structural integrity of POMOF 1 was well preserved after immersing in the tested organic solvents and aqueous solutions with pH values of 1 and 12, demonstrating good tolerance of POMOF 1 towards different media. Moreover, the slight change of the FT-IR signals before and after the test also further verified that POMOF 1 maintained its structure (Fig. S15†). The good thermal and chemical stabilities of POMOF 1 suggest its potential as an ideal platform for photocatalytic oxidation.
To elucidate the elemental composition and chemical oxidation states, X-ray photoelectron spectroscopy (XPS) was conducted on POMOF 1, POMOF 2, and POMOF 3. The XPS spectra unambiguously revealed the presence of essential surface elements, including C, Mo (or W), V, Cd, and Br (or I), in all three structures (Fig. S17–S19†). In the high-resolution Cd 3d spectrum, the two peaks with binding energies of 405.7 eV (Cd 3d5/2) and 412.4 eV (Cd 3d3/2) correspond to the oxidation state of Cd2+ (Fig. 2d and Fig. S20†).58 The high-resolution Mo 3d spectra of POMOF 1 and POMOF 2 showed two characteristic peaks at 235.7 eV and 232.6 eV, which were attributed to Mo6+ 3d3/2 and Mo6+ 3d5/2 of {PMo10V2} in the two structures, respectively.59 The W 4f spectra of POMOF 3 showed two characteristic peaks at 37.6 eV and 35.5 eV, corresponding to W6+ 4f5/2 and W6+ 4f7/2 of {PW10V2} species, respectively.60 In the V 2p region, the two peaks at 524.7 eV and 517.2 eV belong to V 2p1/2 and V 2p3/2, respectively, which are in accordance with the V5+ oxidation state, and the two peaks at 523.7 eV (V 2p1/2) and 516.1 eV (V 2p3/2) belong to the V4+ oxidation state, respectively (Fig. 2e and Fig. S20†).61,62
Photochemical property study
To further determine the band gaps, HOMO and LUMO levels, as well as charge separation and transfer capabilities of POMOF 1–POMOF 3, photoelectrochemical measurements were performed. First, the UV–vis diffuse reflectance spectra (UV–vis DRS) of the three complexes were recorded to determine their optical band gaps. Consistent with their structural similarities, they exhibit comparable visible-light absorption profiles, characterized by a broad band spanning from 300 to 800 nm, with the peak absorption centered at 485 nm that is approaching the edge of the green light region (Fig. 3a). From the Tauc plots, the optical band gap values (Eg) were determined to be 1.91 eV for POMOF 1, 1.90 eV for POMOF 2, and 1.95 eV for POMOF 3, respectively (Fig. S21–S23†). Despite the quite similar band gaps, the HOMO and LUMO levels of POMOF 1–POMOF 3 are dramatically distinct. The LUMO levels were derived from the flat-band potentials (Efb) obtained through Mott–Schottky analysis. As depicted in Fig. S24–S26,† the positive slopes of the fitted lines confirm that POMOF 1, POMOF 2, and POMOF 3 are n-type semiconductors.63 Their flat-band potentials were determined to be −1.42 V, −1.28 V, and −0.97 V versus the normal hydrogen electrode (NHE). The corresponding highest occupied molecular orbital (HOMO) positions were subsequently calculated using the band gap energy equation (Eg = EHOMO − ELUMO), with 0.49 V for POMOF 1, 0.62 V for POMOF 2, and 0.98 V for POMOF 3versus the NHE (Fig. 3b). Therefore, the modulation of nodes and POM guests leads to LUMO energy levels in the sequence of POMOF 1 < POMOF 2 < POMOF 3. Since the O2/O2˙− redox couple is −0.33 V vs. the NHE, the more negative the potential than this value, the more efficiency can be, in principle, achieved to convert O2 into reactive oxygen species of O2˙−.64 Moreover, as depicted in Fig. 3c, the electrochemical impedance spectroscopy (EIS) results indicate that POMOF 1 exhibits the smallest Nyquist semicircle radius, indicative of the fastest interfacial charge transfer for POMOF 1. Consequently, the photocurrent intensity of POMOF 1 also recorded the highest value among the three complexes, demonstrating enhanced electron–hole separation efficiency in this framework (Fig. 3d). Taken together, these findings suggest that the dual tuning not only enables POMOF 1 to have the most favourable LUMO level distribution, but also facilitates the highest charge separation and transfer capacity.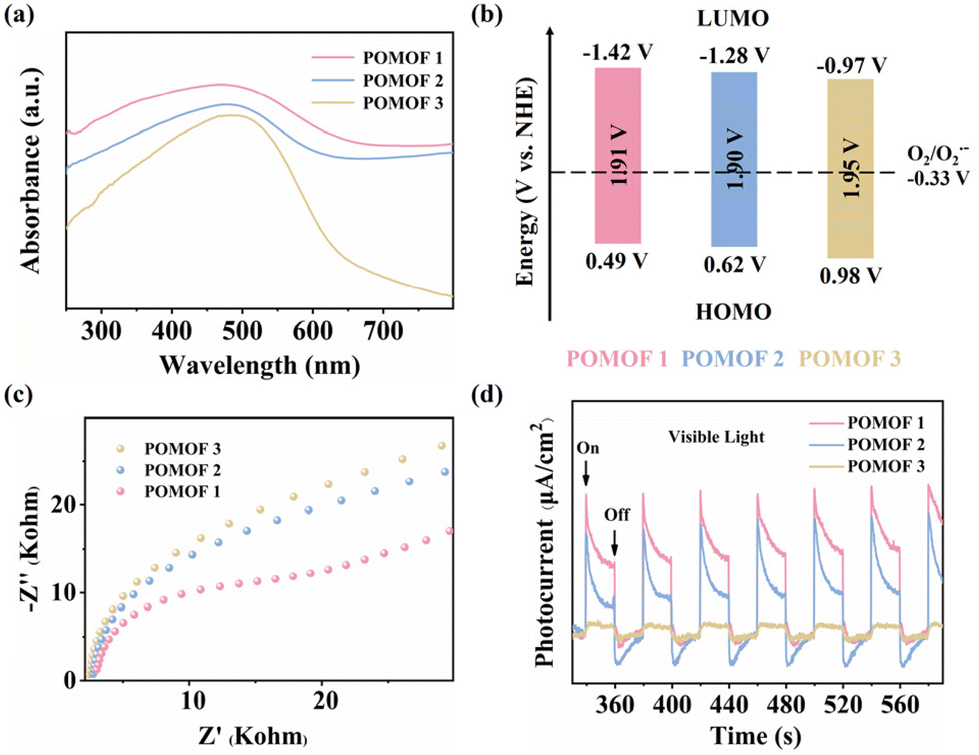 | ||
| Fig. 3 (a) UV–vis DRS of POMOFs 1–3, (b) schematic diagrams of the optical band gap of POMOFs 1–3, (c) EIS Nyquist plots of POMOFs 1–3, and (d) Photocurrent–time curves of POMOFs 1–3 under visible light irradiation. | ||
Photocatalytic oxidation of benzylamines
Imines and their derivatives are critical building blocks for the synthesis of pharmaceuticals, fine chemicals, and biologically active heterocycles.65–68 Photocatalytic oxidative coupling of amines under visible light and molecular oxygen has recently developed as a green and atom-economical approach for imine production. In view of the proper band structures and excellent chemical stability of POMOF 1–POMOF 3, we investigated their potential as heterogeneous photocatalysts in aerobic oxidation reactions. As presented in Table 1, POMOF 1 demonstrated the most superior photocatalytic performance for the oxidative coupling of benzylamine under green light irradiation and an atmosphere of 1 atm O2 in CH3CN at r.t. for 24 h, yielding the corresponding N-benzyl-1-phenylmethanimine in 98% yield (Table 1, entry 1). In stark contrast, the yields decreased significantly to 76% and 23% when POMOF 2 and POMOF 3 were employed as photocatalysts under identical conditions (Table 1, entries 2 and 3). This is consistent with the photocurrent sequence for POMOFs 1–3. Although all the three compounds can trigger the generation of O2˙−, the photocurrent intensity of POMOF 3 is only approximately one-fifth of that of POMOF 1. This significantly low electron–hole separation efficiency will thus dramatically slow down the processes of photogenerated electron/energy transfer to O2 and the activation of substrates by holes. The photocatalytic performance of POMOF 1 diminished considerably when yellow or red LEDs were employed as the light source, highlighting that the energy of green light is crucial for effective electron excitation (Table S7†). The photocatalytic aerobic oxidation of benzylamine was conducted using various solvents, including CH3OH, CH3CH2OH, cyclohexane, toluene, and CH3CN. The results demonstrate that a nonprotonic polar solvent, such as CH3CN, is advantageous for enhancing the photocatalytic performance (Table S9†).69|
|
||
|---|---|---|
| Entry | Catalyst | Yieldb (%) |
| a Reaction conditions: substrate (0.10 mmol), catalyst (2.1 mol%), CH3CN (1.0 mL), r.t., 24 h, LED (10 W), O2. b The yield was determined by GC (N-hexadecane was used as the internal standard substance, sel.: 99%). | ||
| 1 | POMOF 1 | 98 |
| 2 | POMOF 2 | 76 |
| 3 | POMOF 3 | 23 |
| 4 | PMo10V2 | 24 |
| 5 | PW10V2 | 7 |
| 6 | CdBr2 | 9 |
| 7 | CdI2 | 4 |
| 8 | AQ | 52 |
| 9 | PMo10V2 + CdBr2 + AQ | 59 |
| 10 | No catalyst | 3 |
| 11 | POMOF 1, no light | 4 |
| 12 | POMOF 1, under air | 64 |
| 13 | POMOF 1, under N2 | 3 |
Control experiments indicated that the superior photocatalytic activity of POMOF 1 is primarily attributed to the synergistic effect of its components (Table 1). The individual precursors of POMOF 1–POMOF 3, except for AQ, exhibited lower yields in the photocatalytic process (Table 1, entries 4–8). This result indicates that the photocatalytic activity is predominantly derived from the photoactive AQ ligand (Table 1, entry 8).70 Regarding the two POM precursors, {PMo10V2} demonstrated significantly higher activity than {PW10V2}, confirming the importance of regulating functional guests and the contribution of POM cluster as well (Table 1, entries 4 and 5).19,33 Similarly, the photocatalytic performance of CdBr2 was also superior to CdI2 (Table 1, entries 6 and 7), which likely accounts for the different behaviours of dinuclear {Cd2Br(H2O)4} and {Cd2I(H2O)4} nodes.71 It should be noted that a marginally enhanced yield was achieved when the mixture of PMo10V2, CdBr2, and AQ was employed as the catalyst (Table 1, entry 9). This unambiguously highlights the synergistic contribution of the integration of the dinuclear {Cd2Br(H2O)4} node, {PMo10V2} guest, and photoactive AQ into the porous framework of POMOF 1, which not only creates the optimal active sites and photochemical parameters for photocatalysis but also enhances substrate and product diffusion through the open channels. Additionally, control experiments conducted without the catalyst or light irradiation could hardly proceed (Table 1, entries 10 and 11). These results confirm that both the catalyst and light irradiation are essential for the photocatalytic oxidation process. When the reaction was performed under ambient air conditions, the imine product was harvested in a significantly reduced yield (Table 1, entry 12). Moreover, the catalytic process was nearly ineffective under a nitrogen atmosphere (Table 1, entry 13). These observations suggest that molecular oxygen is the primary source for generation of reactive oxygen species.
Under optimized conditions, we then tested the substrate scope of POMOF 1 in the photocatalytic oxidative coupling reaction (Table 2). Gratifyingly, POMOF 1 demonstrated broad applicability and efficiently converted all tested benzylamines and their derivatives into the corresponding imines with high to excellent yields. In most cases, substrates bearing both electron-donating and electron-withdrawing groups, such as methyl and halides, exhibited comparable yields (Table 2, entries 2–9). The ortho-substitution effect is observed for 2-chlorobenzylamine, resulting in a relatively low yield of imine due to steric hindrance (Table 2, entry 6). Notably, the catalytic system also showed good tolerance with a heterocyclic substrate, affording the coupling product in a yield of 97% (Table 2, entry 10). Moreover, secondary amines such as dibenzylamine, which are typically challenging to oxidize using POM-based photocatalysts, can also be facilely converted into the corresponding imines with a high yield (Table 2, entry 11).72
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yieldb (%) |
| a Reaction conditions: substrate (0.10 mmol), POMOF 1 (2.1 mol%), CH3CN (1.0 mL), r.t., 24 h, LED (10 W), O2. b The yield was determined by GC (N-hexadecane was used as the internal standard substance, sel.: 99%). c 30 h. | |||
| 1 |

|

|
98 |
| 2 |

|
|
98 |
| 3 |
|

|
99 |
| 4 |

|

|
98 |
| 5 |

|

|
97 |
| 6 |

|

|
86 |
| 7c |

|

|
93 |
| 8 |

|

|
97 |
| 9 |
|

|
94 |
| 10 |
|

|
97 |
| 11 |

|

|
82 |
| 12 |

|

|
98 |

Identifying the reactive oxygen species (ROS) generated under visible-light irradiation is essential for elucidating the mechanism of benzylamine oxidation. To this end, we probed the roles of ROS and photogenerated holes in the oxidation of benzylamine using a series of scavengers. As shown in Table S10,† the addition of the superoxide radical scavenger p-benzoquinone (BQ) resulted in a significant decrease in yield (32%), indicating the involvement of superoxide radicals (O2˙−) in the reaction (Table S10,† entry 1). Upon introducing the singlet oxygen scavenger 1,4-diazabicyclo [2.2.2] octane (DABCO), only a trace amount of product was obtained (Table S10,† entry 2), thereby confirming the significant contribution of singlet oxygen (1O2) to the oxidation process. Additionally, the addition of KI as a photogenerated hole (h+) scavenger led to a substantial reduction in the reaction yield to 19% (Table S10,† entry 3), highlighting the critical role of holes in activating the benzylamine substrate. Similarly, the introduction of AgNO3 as a photogenerated electron (e−) capture agent resulted in a yield drop to 21% (Table S10,† entry 4), indicating that electron transfer is crucial for generation of ROS. In contrast, the reaction yield remained largely unaffected when t-BuOH was used as a hydroxyl radical (˙OH) scavenger (Table S10,† entry 5), suggesting that ˙OH is not a major reactive oxidant during catalysis.
Drawing on the literature and our experimental analyses, we propose a plausible mechanism for the photocatalytic aerobic oxidation of benzylamine (Fig. 4).73–76 Upon irradiation with 500 nm LED light, the photogenerated holes of POMOF 1 initially oxidize benzylamine to form an amino radical. Concurrently, the photogenerated electrons activate adsorbed oxygen molecules on the surface of POMOF 1, generating singlet oxygen (1O2) and superoxide radicals (O2˙−). These reactive oxygen species then react with the amino radical to produce benzaldehyde, which subsequently condenses with another molecule of benzylamine to yield the corresponding imine product.
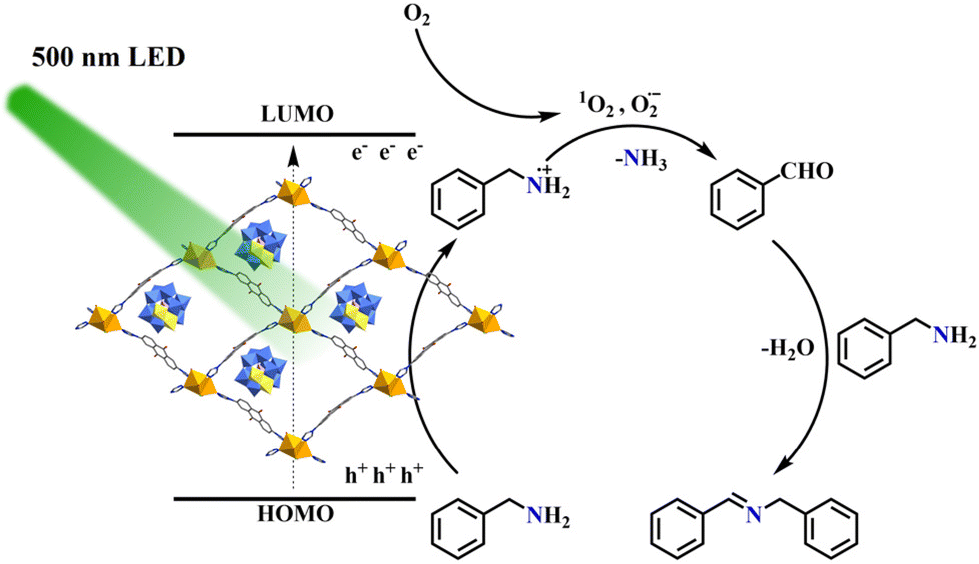 | ||
| Fig. 4 Plausible reaction mechanism of aerobic benzylamine oxidation with POMOF 1 as the photocatalyst. | ||
To evaluate the reusability of POMOF 1, recycling experiments were conducted for the benzylamine oxidation reaction. The results indicated that the photocatalytic efficiency displayed negligible loss over three cycles (Fig. S29†). PXRD and IR tests of the recycled catalyst verified that POMOF 1 retained its structural integrity after recycling (Fig. S30 and 31†). Moreover, ICP analysis of the filtered solution after catalysis indicated negligible amounts of metal ions (<0.01%), demonstrating no catalyst leakage and the heterogeneous nature during the reaction (Table S11†). Filtration tests revealed that the reaction almost completely ceased after catalyst removal, further confirming the heterogeneous nature of the catalysis (Fig. S32†).
Conclusions
In summary, we have demonstrated that the dual tuning of the nodes and POM guests in isostructural POMOF 1–POMOF 3 effectively modulates photocatalytic activity for oxidative coupling of benzylamine and its derivatives. By systematically altering the node from {Cd2Br(H2O)4} to {Cd2I(H2O)4} and POM clusters from {PMo10V2} to {PW10V2}, we achieved an optimal combination in POMOF1. This combination enables the most favorable photochemical parameters, which significantly enhance the generation of superoxide radicals and singlet oxygen, leading to the most efficient substrate oxidation. This work highlights the significant advantage of the dual tuning strategy for improving the photocatalytic performance and suggests a pathway for developing novel photocatalysts through dual or even multiple regulation approaches.Data availability
The data that support the findings of this study (synthesis and characterization data, representative spectra, and X-ray crystallography) have been included as part of the ESI.† CCDC Deposition numbers 2431828 (for POMOF 1), 2431829 (for POMOF 2) and 2431830 (for POMOF3)† contain the supplementary crystallographic data for this paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 92161111, 21901038 and 22071148), the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning, the International Cooperation Fund of Science and Technology Commission of Shanghai Municipality (No. 21130750100), and the Fundamental Research Funds for the Central Universities (No. 2232023A-07). We thank the staff from the College of Materials Science and Engineering (Donghua University) for X-ray data collection using Bruker D8 Venture and the staff from the BL17B1 beamline of the National Facility for Protein Science in Shanghai (NFPS) at the Shanghai Synchrotron Radiation Facility, for assistance during data collection.References
- M. Kamachi, K. Yonesato, T. Okazaki, D. Yanai, S. Kikkawa, S. Yamazoe, R. Ishikawa, N. Shibata, Y. Ikuhara, K. Yamaguchi and K. Suzuki, Angew. Chem., Int. Ed., 2024, 63, e202408358 CrossRef CAS PubMed.
- S. Li, N. Li, G. Li, Y. Ma, M. Huang, Q. Xia, Q. Zhao and X. Chen, Polyoxometalates, 2023, 2, 9140024 CrossRef.
- J.-M. Lin, Z.-B. Mei, C. Guo, J.-R. Li, Y. Kuang, J.-W. Shi, J.-J. Liu, X. Li, S.-L. Li, J. Liu and Y.-Q. Lan, J. Am. Chem. Soc., 2024, 146, 22797–22806 CrossRef CAS PubMed.
- P. J. Sarver, V. Bacauanu, D. M. Schultz, D. A. DiRocco, Y.-h. Lam, E. C. Sherer and D. W. C. MacMillan, Nat. Chem., 2020, 12, 459–467 CrossRef CAS PubMed.
- C. Tanielian, Coord. Chem. Rev., 1998, 178–180, 1165–1181 CrossRef CAS.
- S.-L. Xie, J. Liu, L.-Z. Dong, S.-L. Li, Y.-Q. Lan and Z.-M. Su, Chem. Sci., 2019, 10, 185–190 RSC.
- G. Laudadio, Y. Deng, K. V. D. Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92–96 CrossRef CAS PubMed.
- M. D. Tzirakis, I. N. Lykakis and M. Orfanopoulos, Chem. Soc. Rev., 2009, 38, 2609–2621 RSC.
- F. Wang, C. Jing, J. Ping, D. He, W. Shang, M. Zeng, N. Wang and Z. Jia, Polyoxometalates, 2024, 3, 9140067 CrossRef.
- Z. Yuan and R. Britton, Chem. Sci., 2023, 14, 12883–12897 RSC.
- C. B. Musgrave, K. Olsen, N. S. Liebov, J. T. Groves, W. A. Goddard and T. B. Gunnoe, ACS Catal., 2023, 13, 6382–6395 CrossRef CAS.
- Y. V. Geletii, Z. Huang, Y. Hou, D. G. Musaev, T. Lian and C. L. Hill, J. Am. Chem. Soc., 2009, 131, 7522–7523 CrossRef CAS PubMed.
- A. V. Anyushin, A. Kondinski and T. N. Parac-Vogt, Chem. Soc. Rev., 2020, 49, 382–432 RSC.
- V. Das, R. Kaushik and F. Hussain, Coord. Chem. Rev., 2020, 143, 213271 CrossRef.
- Z. Huo, I. Azcarate, R. Farha, M. Goldmann, H. Xu, B. Hasenknopf, E. Lacôte and L. Ruhlmann, J. Solid State Electrochem., 2015, 19, 2611–2621 CrossRef CAS.
- B. Matt, J. Fize, J. Moussa, H. Amouri, A. Pereira, V. Artero, G. Izzet and A. Proust, Energy Environ. Sci., 2013, 6, 1504–1508 RSC.
- K. Suzuki, N. Mizuno and K. Yamaguchi, ACS Catal., 2018, 8, 10809–10825 CrossRef CAS.
- X. Chen, A. Yang, G. Wang, M. Wei, N. Liu, B. Li and L. Wu, Chem. Eng. J., 2022, 446, 137134 CrossRef CAS.
- Y. Liu, K. Ji, J. Wang, H. Li, X. Zhu, P. Ma, J. Niu and J. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 27882–27890 CrossRef CAS PubMed.
- A. Nakajima, T. Koike, S. Yanagida, T. Isobe, Y. Kameshima and K. Okada, Appl. Catal., A, 2010, 385, 130–135 CrossRef CAS.
- Q. Gu, X.-L. Zhao, M. Meng, Z. Shao, Q. Zheng and W. Xuan, Chin. Chem. Lett., 2023, 34, 107444 CrossRef CAS.
- N. Li, J. Liu, B. X. Dong and Y. Q. Lan, Angew. Chem., Int. Ed., 2020, 59, 20779–20793 CrossRef CAS PubMed.
- X. Liu, J. Zhang, Y. Lan, Q. Zheng and W. Xuan, Inorg. Chem. Front., 2022, 9, 6534–6543 RSC.
- M. Lu, M. Zhang, J. Liu, T.-Y. Yu, J.-N. Chang, L.-J. Shang, S.-L. Li and Y.-Q. Lan, J. Am. Chem. Soc., 2022, 144, 1861–1871 CrossRef CAS PubMed.
- Y. An, L. Wang, W. Jiang, X. Lv, G. Yuan, X. Hang and H. Pang, Polyoxometalates, 2023, 2, 9140030 CrossRef.
- Z. Jiang, X. Xu, Y. Ma, H. S. Cho, D. Ding, C. Wang, J. Wu, P. Oleynikov, M. Jia, J. Cheng, Y. Zhou, O. Terasaki, T. Peng, L. Zan and H. Deng, Nature, 2020, 586, 549–554 CrossRef CAS PubMed.
- Z. W. Jiang, T. T. Zhao, S. J. Zhen, C. M. Li, Y. F. Li and C. Z. Huang, J. Mater. Chem. A, 2021, 9, 9301–9306 RSC.
- R. Li, W. Zhang and K. Zhou, Adv. Mater., 2018, 30, 1705512 CrossRef PubMed.
- X. C. Lin, Y. M. Wang, X. Chen, P. Y. You, K. M. Mo, G. H. Ning and D. Li, Angew. Chem., Int. Ed., 2023, 62, e202306497 CrossRef CAS PubMed.
- K. Muralirajan, I. S. Khan, L. Garzon-Tovar, R. Kancherla, N. Kolobov, A. Dikhtiarenko, M. Almalki, A. Shkurenko, A. Rendón-Patiño, V. Guillerm, K. N. Le, G. Shterk, H. Zhang, C. H. Hendon, M. Eddaoudi, J. Gascon and M. Rueping, Adv. Mater., 2024, 37, 2405646 CrossRef PubMed.
- S. Navalón, A. Dhakshinamoorthy, M. Álvaro, B. Ferrer and H. García, Chem. Rev., 2022, 123, 445–490 CrossRef PubMed.
- Y. Qian, F. Zhang and H. Pang, Adv. Funct. Mater., 2021, 31, 2104231 CrossRef CAS.
- H. An, H. Luo, T. Xu, S. Chang, Y. Chen, Q. Zhu, Y. Huang, H. Tan and Y.-G. Li, Inorg. Chem., 2022, 61, 10442–10453 CrossRef CAS PubMed.
- Y.-Z. Chen, Z. U. Wang, H. Wang, J. Lu, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2017, 139, 2035–2044 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- H. Liu, C. Xu, D. Li and H. L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 5379–5383 CrossRef CAS PubMed.
- P. Salcedo-Abraira, A. A. Babaryk, E. Montero-Lanzuela, O. R. Contreras-Almengor, M. Cabrero-Antonino, E. S. Grape, T. Willhammar, S. Navalón, E. Elkäim, H. García and P. Horcajada, Adv. Mater., 2021, 33, 2106627 CrossRef CAS PubMed.
- Q. Han, B. Qi, W. Ren, C. He, J. Niu and C. Duan, Nat. Commun., 2015, 6, 10007 CrossRef PubMed.
- S. Li, Y. Zheng, G.-C. Liu, X.-H. Li, Z. Zhang and X.-L. Wang, Polyoxometalates, 2024, 3, 9140061 CrossRef.
- Z. Sun, R. Wang and I. V. Kozhevnikov, Coord. Chem. Rev., 2025, 524, 216304 CrossRef CAS.
- S.-Q. You, Y.-J. Dong, B.-S. Hou, M. Dong, J.-L. Tong, L.-X. Wang, X.-L. Wang, C.-Y. Sun, W. Guan and Z.-M. Su, J. Mater. Chem. C, 2023, 11, 7389–7396 RSC.
- Y. Benseghir, A. Lemarchand, M. Duguet, P. Mialane, M. Gomez-Mingot, C. Roch-Marchal, T. Pino, M.-H. Ha-Thi, M. Haouas, M. Fontecave, A. Dolbecq, C. Sassoye and C. Mellot-Draznieks, J. Am. Chem. Soc., 2020, 142, 9428–9438 CrossRef CAS PubMed.
- L. Jiao, Y. Dong, X. Xin, L. Qin and H. Lv, Appl. Catal., B, 2021, 291, 120091 CrossRef CAS.
- Y. Liu, C. Tang, M. Cheng, M. Chen, S. Chen, L. Lei, Y. Chen, H. Yi, Y. Fu and L. Li, ACS Catal., 2021, 11, 13374–13396 CrossRef CAS.
- X. Sun, J. Zhang and Z. Fu, ACS Appl. Mater. Interfaces, 2018, 10, 35671–35675 CrossRef CAS PubMed.
- Z.-M. Zhang, T. Zhang, C. Wang, Z. Lin, L.-S. Long and W. Lin, J. Am. Chem. Soc., 2015, 137, 3197–3200 CrossRef CAS PubMed.
- J. Du, Y.-Y. Ma, W.-J. Cui, S.-M. Zhang, Z.-G. Han, R.-H. Li, X.-Q. Han, W. Guan, Y.-H. Wang, Y.-Q. Li, Y. Liu, F.-Y. Yu, K.-Q. Wei, H.-Q. Tan, Z.-H. Kang and Y.-G. Li, Appl. Catal., B, 2022, 318, 121812 CrossRef CAS.
- F. Leng, H. Liu, M. Ding, Q.-P. Lin and H.-L. Jiang, ACS Catal., 2018, 8, 4583–4590 CrossRef CAS.
- Q. Ran, B. Tao, M. Li, K. Zheng, Y. She, W. Wu, Z. Li, D. Luo and X. Xu, Coord. Chem. Rev., 2025, 525, 216324 CrossRef CAS.
- K. Wu, X.-Y. Liu, P.-W. Cheng, Y.-L. Huang, J. Zheng, M. Xie, W. Lu and D. Li, J. Am. Chem. Soc., 2023, 145, 18931–18938 CrossRef CAS PubMed.
- J.-D. Xiao and H.-L. Jiang, Acc. Chem. Res., 2019, 52, 356–366 CrossRef CAS PubMed.
- D.-X. Wang and M.-X. Wang, Acc. Chem. Res., 2020, 53, 1364–1380 CrossRef CAS PubMed.
- X.-Y. Wu, H.-L. Zhang, S.-S. Wang, W. Wu, L. Lin, X.-Y. Jiang and C.-Z. Lu, Dalton Trans., 2020, 49, 3408–3412 RSC.
- J. Zhang, L. Xiang, B. Yan and H. Zeng, J. Am. Chem. Soc., 2020, 142, 1710–1714 CrossRef CAS PubMed.
- P. Xu, L. Zhang, X. Jia, H. Wen, X. Wang, S. Yang and J. Hui, Catal. Sci. Technol., 2021, 11, 6507–6515 RSC.
- J. Y. Zhang, X. Y. Zhang, J. Wang, Y. Feng, L. L. Fan, Y. D. Cao, H. Liu, C. L. Lv and G. G. Gao, Adv. Funct. Mater., 2024, 34, 2400170 CrossRef CAS.
- X. Dong, Y. Wang, F. Huang and X. Lang, Chem. Eng. J., 2023, 469, 143934 CrossRef CAS.
- I. Brahma, S. Ray, S. Majumder, A. K. Guha and S. J. Bora, J. Mol. Struct., 2024, 1311, 138302 CrossRef CAS.
- Y.-D. Cao, W.-X. Mu, M. Gong, L.-L. Fan, J. Han, H. Liu, B. Qi and G.-G. Gao, Dalton Trans., 2023, 52, 16303–16314 RSC.
- W. Li, X. Luan, X. Zhu, J. Sun and L. Fan, J. Mater. Sci., 2022, 57, 19946–19956 CrossRef CAS.
- X. Dan, X. Yin, J. Ba, J. Li, Y. Cheng, F. Duan, Y. Wei and Y. Wang, Nano Lett., 2024, 24, 6881–6888 CrossRef CAS PubMed.
- Z. Li, J. Zhang, X. Jing, J. Dong, H. Liu, H. Lv, Y. Chi and C. Hu, J. Mater. Chem. A, 2021, 9, 6152–6159 RSC.
- K. Wu, J.-K. Jin, X.-Y. Liu, Y.-L. Huang, P.-W. Cheng, M. Xie, J. Zheng, W. Lu and D. Li, J. Mater. Chem. C, 2022, 10, 11967–11974 RSC.
- Y. Fang, Y. Zheng, T. Fang, Y. Chen, Y. Zhu, Q. Liang, H. Sheng, Z. Li, C. Chen and X. Wang, Sci. China: Chem., 2020, 63, 149–181 CrossRef CAS.
- B. Chen, L. Wang and S. Gao, ACS Catal., 2015, 5, 5851–5876 CrossRef CAS.
- S. Kobayashi and H. Ishitani, Chem. Rev., 1999, 99, 1069–1094 CrossRef CAS PubMed.
- S. I. Murahashi, Angew. Chem., Int. Ed. Engl., 1995, 34, 2443–2465 CrossRef CAS.
- T. Xia, Z. Yu, X. Wu, J. Qu and Y. Chen, ChemCatChem, 2025, 17, e202401407 CrossRef CAS.
- M. A. Bryden, F. Millward, O. S. Lee, L. Cork, M. C. Gather, A. Steffen and E. Zysman-Colman, Chem. Sci., 2024, 15, 3741–3757 RSC.
- J. Cervantes-González, D. A. Vosburg, S. E. Mora-Rodriguez, M. A. Vázquez, L. G. Zepeda, C. Villegas Gómez and S. Lagunas-Rivera, ChemCatChem, 2020, 12, 3811–3827 CrossRef.
- J. Liang, S. Huang, Y. Chang, A. Terfort, J. Gao and J. Liu, J. Mater. Chem. A, 2024, 12, 13273–13280 RSC.
- K. Suzuki, F. Tang, Y. Kikukawa, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2014, 53, 5356–5360 CrossRef CAS PubMed.
- X. Lang, H. Ji, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 3934–3937 CrossRef CAS PubMed.
- D. Sun, Y. Chen, X. Yu, Y. Yin and G. Tian, Chem. Eng. J., 2023, 462, 142084 CrossRef CAS.
- E.-T. Wu and M. H. Huang, ACS Catal., 2023, 13, 14746–14752 CrossRef CAS.
- X. Zhang, X. Pan, X. Si, L. Zhu, Q. Yao, W. Duan, X. Huang and J. Su, Inorg. Chem., 2024, 63, 19408–19417 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2431828–2431830. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00655d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |