
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA tridentate and dianionic N-heterocyclic olefin (NHO) with two unsymmetrically-tethered aryloxide sidearms on bismuth: a fortuitous followed by systematic discovery†
Santu
Goswami
a,
Subham
Sarkar
ab,
Anisha
Guha Roy
a,
Dibyendu
Mallick
 *b and
Debabrata
Mukherjee
*a
*b and
Debabrata
Mukherjee
*a
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur, 741246, West Bengal, India. E-mail: d.mukherjee@iiserkol.ac.in
bDepartment of Chemistry, Presidency University, 86/1 College Street, Kolkata, 700073, West Bengal, India. E-mail: dibyendu.chem@presiuniv.ac.in
First published on 25th March 2025
Abstract
Herein, we report our previously developed bifunctional imidaozlium-phenol LH2Br ([HO-4,6-tBu2-C6H2-2-CH2{CH(NCH = CHNAr)}]Br; Ar = Dipp = 2,6-iPr2-C6H3), leading to three distinct outcomes with bismuth by simply altering the relative ratios of the reacting components. Treating LH2Br and Bi(HMDS)3 (HMDS = N(SiMe3)2) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio gives the NHC-Bi complex [(L)BiBr(HMDS)] (1). In contrast, LH2Br, Bi(HMDS)3, and KHMDS in a 2:1:1 ratio serendipitously result in an unprecedented NHO-Bi complex [(DippNHO2ArO)BiBr] (2), featuring a novel dianoinic and tridentate NHO ligand with two unsymmetric aryloxide sidearms. A tri-acidic imidazolium salt L′H3I ([HO-4,6-tBu2-C6H2-2-CH2{C(HO-4,6-tBu2-C6H2-2-CH2)(NCH = CHNMe)}]I) is synthesized independently as a potential precursor to such an NHO framework, and the corresponding NHO-BiI complex [(MeNHO2ArO)Bi(μ-I)]2 (32) is made as proof of concept. Lastly, treating LH2Br and Bi(HMDS)3 in a 2:1 ratio also unexpectedly leads to an ‘abnormal’ NHC-Bi complex [(aL)BiBr2(DippImd)] (4; DippImd = Dipp-imidazole). The multi-component and essentially multi-step reactions leading to 2 and 4 are challenging to fully elucidate mechanistically. Still, control experiments indicate 1 as a possible intermediate in both cases. Based on these results and prior insights into LH2Br and an intermediate LH, plausible routes for both 2 and 4 are hypothesized. DFT calculations are also performed to analyze the bonding in 2 and 32 and to justify an NHC to aNHC isomerization towards the formation of 4.
:1 ratio gives the NHC-Bi complex [(L)BiBr(HMDS)] (1). In contrast, LH2Br, Bi(HMDS)3, and KHMDS in a 2:1:1 ratio serendipitously result in an unprecedented NHO-Bi complex [(DippNHO2ArO)BiBr] (2), featuring a novel dianoinic and tridentate NHO ligand with two unsymmetric aryloxide sidearms. A tri-acidic imidazolium salt L′H3I ([HO-4,6-tBu2-C6H2-2-CH2{C(HO-4,6-tBu2-C6H2-2-CH2)(NCH = CHNMe)}]I) is synthesized independently as a potential precursor to such an NHO framework, and the corresponding NHO-BiI complex [(MeNHO2ArO)Bi(μ-I)]2 (32) is made as proof of concept. Lastly, treating LH2Br and Bi(HMDS)3 in a 2:1 ratio also unexpectedly leads to an ‘abnormal’ NHC-Bi complex [(aL)BiBr2(DippImd)] (4; DippImd = Dipp-imidazole). The multi-component and essentially multi-step reactions leading to 2 and 4 are challenging to fully elucidate mechanistically. Still, control experiments indicate 1 as a possible intermediate in both cases. Based on these results and prior insights into LH2Br and an intermediate LH, plausible routes for both 2 and 4 are hypothesized. DFT calculations are also performed to analyze the bonding in 2 and 32 and to justify an NHC to aNHC isomerization towards the formation of 4.
Introduction
N-Heterocyclic carbenes (NHCs; Fig. 1) have truly ushered in a new era in chemistry.1 More importantly, their easy tunability by functionalizing with extra donors has significantly enhanced their scope and versatility as a ligand class.2 Alkylidene-appended NHCs, better known as N-heterocyclic olefins (NHOs; Fig. 1), have distinct properties such as high nucleophilicity and strong basicity but nearly no π-acidity.3 Although reported even before the first stable NHC,4 NHOs remained mostly dormant for a fairly long time. However, they have gained significant attention over the past decade, making major contributions in organocatalysis, CO2 fixation, small molecule activation, polymerization, frustrated Lewis pairs, and, importantly, as neutral 2e− C-donor ligands.5 In fact, NHOs have often matched or even outperformed NHCs in various aspects.5b NHO to metal bonds can adapt to varying stereoelectronic demands, which is considered beneficial for catalytic cycles.6 | ||
| Fig. 1 (left): NHC and NHO frameworks with possible sites for donor functionalization highlighted in green (wingtips) and pink (NHO-Cβ). (right): selective examples of donor-functionalized NHO complexes priorly known. | ||
Curiously, despite their potential, tailoring NHOs with extra donors has been explored only sporadically, unlike the numerous cases of NHCs.5c A systematic push in this direction is thus essential to further advance NHO chemistry. Iglesias and Oro reported a ‘PCP’ pincer featuring an NHO with two flexible phosphine arms (Fig. 1).6,7 While its cationic Ir complexes A and B catalyze transfer hydrogenation and solvent-free dehydrogenation of HCO2H, respectively, the chloride complex C isomerizes into its ‘abnormal’ NHC (aNHC) version at only 40 °C.6b,7 The ‘OCO’ pincer in D is non-chelating (Fig. 1) and undergoes a similar NHO to aNHC isomerization at 70 °C.6b This isomerization is an inherent issue as aNHCs are seemingly better donors.8 In some cases, even the synthetic conditions and the ligand substituents or metal precursors dictate the choice between NHO and aNHC or can give mixtures of both. Examples (Fig. 1) from Li, Zhao, and Zhang (E, F),9 Bernhard and Albrecht (G),10 and Bera (H)11 with pyridine or phosphine sidearms and metals like Ru, Rh, Ir, and Pd also face the isomerization issue. Bera's pyridyl-based ‘ONC’ pincer has an NHO on the side (I, J).
The additional donor function(s) in all of these cases are invariably appended to the N-wingtip(s) of imidazoline. However, an NHO can also afford an additional donor at its exocyclic Cβ position. The resulting chelation might be more effective in resisting unwanted isomerization. Rivard made NHOs with an amine or a phosphine donor, but those are directly bonded to the Cβ and are non-chelating (K, L; Fig. 1).12 The NHO-phosphine binds to two AuCl molecules, one through the phosphorus and the other through the NHO-Cβ (L).12b Besides, Clot and Peris reported a –CH2-linked NHO-aNHC combo and its cationic Ir complex.13 A few bis-NHOs are also known, but they are not ligands.14 A few chiral NHOs are also there.15
We recently explored a bidentate NHC-aryloxide hybrid ligand [DippNHC-CH2-ArO]− (L−; Ar = Dipp = 2,6-iPr2-C6H3; LH2Br = [HO-4,6-tBu2-C6H2-2-CH2{CH(NCH = CHNAr)}]Br), with a flexible –CH2– linker, on d0 metals such as TiIV and AlIII (Fig. 2).16LH2Br's deprotonation by MHMDS (M = Li, K; HMDS = N(SiMe3)2) is also investigated in details,17 in which the zwitterionic intermediate LH undergoes an intriguing fragmentation pattern (Fig. 2).17 Considering the recent surge in bismuth organometallics18 but the paucity of its NHC chemistry,19 we started probing L− on BiIII and the present report is on the findings thereof. With Bi, LH2Br interestingly leads to three distinct cases, giving an NHC-BiIII, an NHO-BiIII, and an aNHC-BiIII complexes with aryloxide sidearms by simply varying the ratios of Bi(HMDS)3 and KHMDS (Fig. 2). The dianionic and tridentate NHO ligand framework is especially appealing because of its novelty and unsymmetric nature. Both the NHO-Bi and aNHC-Bi complexes seem to involve perplexing and essentially multi-step formative routes, which we attempt to postulate based on control experiments and prior insights on LH2Br and the intermediate LH. Furthermore, the same NHO framework with a substitutional change is made independently through a systematic design as proof of concept.
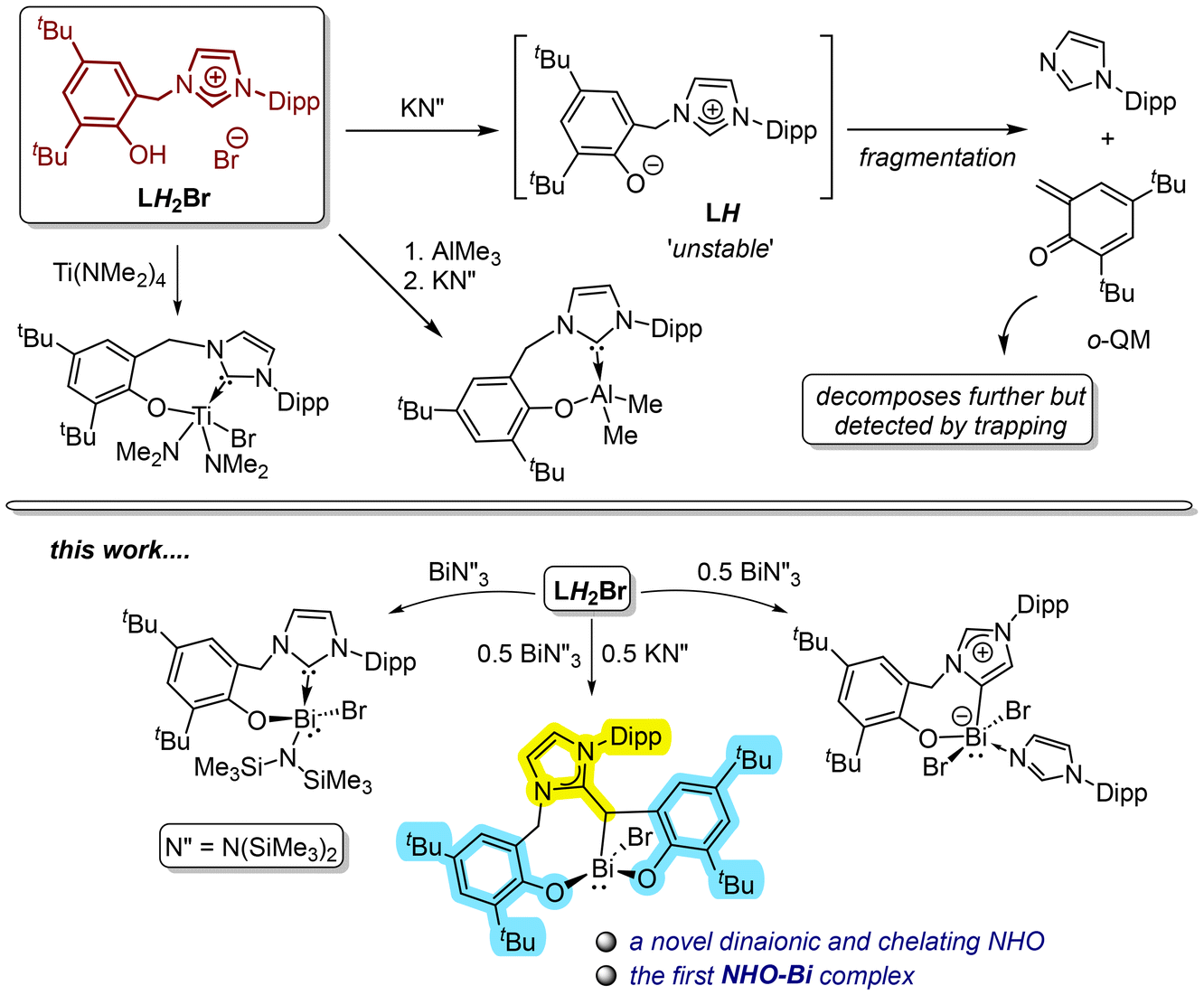 | ||
| Fig. 2 (top): Bifunctional imidazolium-phenol ligand precursor LH2Br, the corresponding Ti and Al complexes, and the fragmentation of the zwitterionic LH; (bottom): a summary of the present work where the same LH2Br is shown to give a NHC-Bi, an aNHC-Bi, and a NHO-Bi complex simply by varying the relative ratios of Bi(HMDS)3 and KHMDS. | ||
Results and discussion
The phenolic O–H (pKa ∼ 12) of LH2Br is more acidic than its imidazolium-2-H (pKa ∼ 20) as seen in Al-chemistry.16bLH2Br and Bi(HMDS)3 in a 1:1 ratio react slowly over 48 h in toluene to give the NHC-Bi complex [(L)BiBr(HMDS)] (1; Scheme 1) as a yellow solid. The relatively long reaction time was likely due to the slower deprotonation of imidazolium-2-H by moderately basic Bi-HMDS. While monitoring the reaction at the NMR scale, the peaks attributed to complex 1 were visible only from roughly the 24 h mark. This result also supports the stepwise double deprotonation of LH2Br by Bi(HMDS)3. The Bi center in 1 is ‘stereogenic’ with four different substituents along with a stereoactive lone pair trans to the NHC (Fig. 3). However, without chiral resolution, it exists as a racemic mixture. X-ray diffraction analysis shows the presence of both enantiomers in the asymmetric unit. However, the poor data quality prevented us from further discussing the metric parameters. 1's 1H NMR spectrum in C6D6 unusually splits the Bi–N(SiMe3)2 signal into two slightly broad singlets (9 H each), most likely due to structural rigidity in the complex that seems to make the two SiMe3 groups distinguishable on the NMR timescale. The linker –CH2 protons are diastereotopic as well.
 | ||
| Scheme 1 Bifunctional LH2Br leading to NHC-Bi (1), NHO-Bi (2), and aNHC-Bi (4) complexes by reacting with varying ratios of Bi(HMDS)3 and KHMDS. | ||
 | ||
| Fig. 3 DIAMOND20-rendered molecular structures of NHC-Bi complex 1. Relevant ellipsoids are set at a 50% probability level, while the rest of the skeleton is shown as sticks for better viewing. The H atoms and Co-crystallized toluene molecules are omitted for clarity. | ||
While studying the coordination of the bulky L− on Ti(IV), trying to fit two L− on a single TiIV center failed and instead led to fragmentation of the ligand framework.16a Since Bi(III) [rionic (C.N. = 6; C.N. = coordination number): 1.03 Å] is larger than Ti(IV) [rionic (C.N. = 6): 0.605 Å], we reconceived the idea of placing two L− on a single metal, this time on a Bi(III). As two LH2Br requires four HMDS units to be fully deprotonated, a 2:1:1 reaction of LH2Br, Bi(HMDS)3, and KHMDS is thus formulated to expect a complex like [(L)2BiBr]. However, surprisingly, the reaction gives an unexpected Bi-complex [(DippNHO2ArO)BiBr] (2; Scheme 1), with a dianionic and tridentate NHO ligand having two distinct aryloxide sidearms. 2 is the only isolable Bi-containing product obtained in a 40% yield as an orange solid. In addition, Dipp-imidazole (DippImd) was identified as a major byproduct in the NMR spectrum analysis of the crude product. The X-ray-determined solid-state structure of 2 (Fig. 4) again shows stereogenic Bi with a four-coordinate distorted trigonal pyramidal geometry, where the stereoactive lone pair is trans to the NHO-Cβ. While one ArO− is tethered to an N-wingtip through a –CH2– linker, the other is directly connected to the NHO-Cβ. The tridentate and flexible NHO framework leads to rare 8,5-metallacycles upon chelation in facial mode. The Cβ–Bi bond is 2.310(4) Å long. Notably, 2 is the first NHO-Bi complex. The 1H NMR spectrum of C6D6 shows the Cβ–H at 4.14 ppm, which overlaps with one of the two diastereotopic –CH2– protons. The four inequivalent tBu groups gave four individual singlets (9 H each) within 1.40–1.67 ppm. The Dipp-CHMe2 protons gave four doublets within 0.87–1.39 ppm and the two methine protons as two septets at 2.53 and 2.82 ppm, respectively. The olefinic backbone of NHO gives two overlapping peaks at 5.75 ppm (2 H).
 | ||
| Fig. 4 DIAMOND-rendered molecular structure of the NHO-Bi complex 2. Relevant ellipsoids are set at a 50% probability level, while the rest of the skeleton is shown as sticks for better viewing. All H atoms except the NHO-CβH are omitted for clarity. Selected bond distances (Å): Bi–C 2.310(4), Bi–O1 2.236(3), Bi–O2 2.115(3), Bi–Br 2.8244(7). Selected bond angles (°): ∠C–Bi–O2 79.7(1), ∠C–Bi–Br 82.7(1), ∠C–Bi–O1 87.2(1), ∠O2–Bi–Br 97.3(1), ∠O1–Bi–Br 151.4(1), ∠O1–Bi–O2 107.1(1). | ||
The composition of 2 suggests a complex route of formation, seemingly with partial ligand fragmentation and a C–C bond formation. Monitoring the reaction at the NMR scale revealed a perplexed spectral pattern with broad and overlapping resonances. The reaction mixture is also partly inhomogeneous throughout because the starting LH2Br and byproduct KBr are insoluble in toluene (Fig. S7; ESI†). Resonances attributed to 2 are noted at approximately the 24 hour mark and continue to grow over the next 24 h. However, the overall spectral pattern is complex and difficult to interpret. Thus, mechanistically elucidating this multi-component and visibly multi-step reaction is challenging. As shown in Fig. 2, our previous knowledge suggests that LH2Br undergoes rapid deprotonation by KHMDS to give LH that is unstable and fragmented into DippImd and an o-quinone methide (o-QM).17 The latter is more fleeting and was only detected by trapping with maleic anhydride.17 The timeline for the formation of 2 (48 h) is roughly similar to that of the LH fragmentation. In a control experiment, reacting 1 with freshly generated LH also gives 2 within a similar time and in a similar yield of 40%. Therefore, 1 could be an intermediate towards the formation of 2. In another experiment, the addition of 1 was performed after incubating the in situ generated LH for 24 h, which decreased the yield of 2 to only 10%. This suggests that the o-QM is the coupling partner of 1 for constructing the NHO framework. The induction period allows the o-QM to degrade profoundly before it can couple with 1, which can explain the drop in 2's yield. Based on these limited insights, a plausible pathway (Scheme 2) was theorized for the formation of 2 involving two parallel 1:1 reactions of LH2Br, one with KHMDS and the other with Bi(HMDS)3. While KHMDS would produce LH and subsequently the o-QM and DippImd, Bi(HMDS)3 and the 2ndLH2Br would give 1. Given the relatively weaker nature of an NHC-Bi bond,19a,b the NHC in 1 is presumed to capture the o-QM as the C–C bond-forming step to give a zwitterionic complex 2′. Intramolecular deprotonation from 2′ by the remaining HMDS on Bi can then possibly yield 2. Notably, replacing KHMDS with other bases such as LiHMDS or KOtBu, while keeping the other reaction parameters the same gives only complex mixtures with no sign of 2. Crystallization attempts have also not afforded any identifiable species so far. This suggests the formation of 2 from LH2Br as base-specific, which is not surprising given the already observed dichotomic behavior of LH2Br towards two equiv. of KHMDS and LiHMDS.17
 | ||
| Scheme 2 A plausible mechanistic route towards the formation of the NHO-Bi complex 2. | ||
Given the complex formation of 2 with a dubious mechanistic understanding, we aimed to devise an independent and more controlled synthetic route for this novel NHO framework with options for substitutional variation. Thus, NHO can also be applied to other metals. This is achieved by starting from the bis-phenolic imidazolium salt I (Scheme 3).21 A triple deprotonation of I by NaHMDS followed by an aqueous workup gives the bis-phenolic imidazole II.21 The reaction proceeds via an NHC intermediate followed by its 1,2-benzyl migration.21 Treating II with MeI gives a new imidazolium salt L′H3I ([HO-4,6-tBu2-C6H2-2-CH2{C(HO-4,6-tBu2-C6H2-2-CH2)(NCH = CHNMe)}]I), a potential tri-acidic precursor for such an NHO framework. Reacting L′H3I and Bi(HMDS)3 in a 1:1 ratio under the same conditions as used for obtaining 1 and 2 indeed gives the NHO-BiI [(MeNHO2ArO)Bi(μ-I)]2 (32; 76%) as another orange solid. Given the relatively higher acidity of the phenols than the –CH2– linker in L′H3I, we believe the threefold deprotonation of L′H3I to give 3 is not concerted and should go through an intermediate like 3′ (Scheme 3). The similarity between 3′ and 2′ indirectly supports the route for 2via2′ proposed in Scheme 2. Solid 32 is a centrosymmetric dimer (Fig. 5), as confirmed by X-ray analysis. The dimeric nature of 32, unlike that of the monomeric 2 is likely due to the smaller Me than Dipp as the N-substituent on the NHO imidazole unit and the higher bridging aptitude of iodide than bromide. However, the DOSY analysis of THF-d8 indicated that monomeric 3 was present in solution (see ESI†). The Cβ–Bi bond is 2.298(5) Å long, similar to that in 2. The other relevant metric parameters are also in the same range.
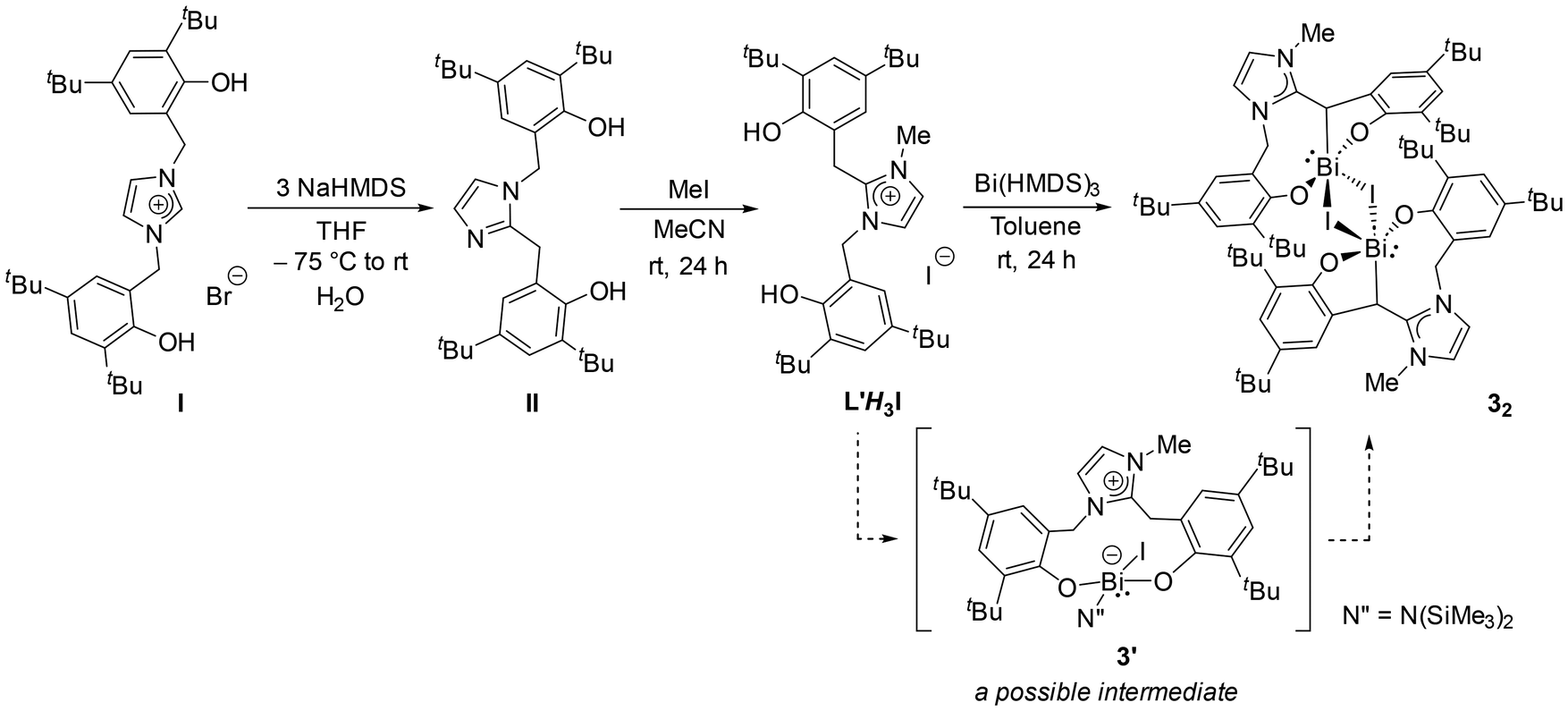 | ||
| Scheme 3 Independent synthesis of an NHO ligand precursor related to the one in 2 and its Bi complexation. | ||
 | ||
| Fig. 5 DIAMOND-rendered molecular structure of 32. Relevant ellipsoids are set at a 50% probability level, while the rest of the skeletons are shown as sticks for better viewing. The H atoms and co-crystallized THF molecules are omitted for clarity. Selected bond distances (Å): Bi1–C1 = Bi2–C2 2.298(5), Bi1–O1 = Bi2–O3 2.158(4), Bi1–O2 = Bi2–O4 2.227(3), Bi1–I1 = Bi2–I2 3.1972(4), Bi1–I2 = Bi2–I1 3.3270(4). Selected bond angles (°): [32]: ∠C2–Bi2–O4 = ∠C1–Bi1–O2 79.7(1), 79.7(1), ∠C2–Bi2–O3 = ∠C1–Bi1–O1 78.7(1), 78.7(1), ∠Bi1–I1–Bi2 = ∠Bi1–I2–Bi2 97.1(1), ∠C1–Bi1–I1 = ∠C2–Bi2–I2 79.5(1), 79.5(1), ∠C1–Bi1–I2 = ∠C2–Bi2–I1 86.0(1), 79.5(1). | ||
The bonding in 2 and 32 were probed using the NBO method at the M06-2X/def2-TZVPP level of theory on structures optimized in the gas phase at the M06-2X/6-31G** (def2-TZVP for Bi) level (see ESI†). The exocyclic Cβ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Cβ of the dianionic NHO has Wiberg Bond Indices (WBI) of 1.663 (for N-Dipp) and 1.590 (for N-Me) because the π-bond is formed by the side-on overlap of the two 2p(C) orbitals.22 The olefinic nature decreases as the same 2p orbital of Cβ forms an σ-bond with the vacant 6p of Bi (Fig. 6) as indicated by the WBIs of 1.093 and 1.078, respectively in 2 and 32.22 The Cα–Cβ is also more polarized in 2 and 32 as the Cβ is more negatively charged [−0.688 (2; Fig. 6), −0.711 (32)] than in their non-metalated NHOs (−0.373 (N-Dipp); −0.421 (N-Me)). Thus, the NHO-Bi interaction increased the ylide character as expected.
Cβ of the dianionic NHO has Wiberg Bond Indices (WBI) of 1.663 (for N-Dipp) and 1.590 (for N-Me) because the π-bond is formed by the side-on overlap of the two 2p(C) orbitals.22 The olefinic nature decreases as the same 2p orbital of Cβ forms an σ-bond with the vacant 6p of Bi (Fig. 6) as indicated by the WBIs of 1.093 and 1.078, respectively in 2 and 32.22 The Cα–Cβ is also more polarized in 2 and 32 as the Cβ is more negatively charged [−0.688 (2; Fig. 6), −0.711 (32)] than in their non-metalated NHOs (−0.373 (N-Dipp); −0.421 (N-Me)). Thus, the NHO-Bi interaction increased the ylide character as expected.
 | ||
| Fig. 6 (left): NBO plot (isovalue = 0.030) showing the NHO-Bi bond in 2; (right): natural charges on the selected atoms of 2 calculated at the M062X/B2//M062X/B1 level of theory (see ESI†). | ||
Considering the 2:1:1 stoichiometry of LH2Br, KHMDS, and Bi(HMDS)3 in the formation of 2, two more control experiments are conducted by reacting LH2Br individually with KHMDS and Bi(HMDS)3 in 2:1 ratios. The KHMDS case results the same as in the 1:1 reaction,17 giving LH and ultimately its fragmentation, while the 2nd equiv. of LH2Br remains untouched. However, Bi(HMDS)3 leads to another unexpected Bi-complex [(aL)BiBr2(DippImd)] (4 (77%); Scheme 1) as one more orange solid, this time with an aryloxide-tethered ‘abnormal’ NHC. The monomeric form and mesoionic nature of the carbene are established by X-ray diffraction analysis (Fig. 7). The Bi in 4 has a distorted square-pyramidal geometry with its stereoactive lone pair trans to the aNHC. Despite the higher coordination number of the Bi center in 4, the aNHC-Bi bond length (2.248(4) Å) is in the same range as the NHC-Bi in 1 and the NHO-Bi in 2 and 32. This is likely due to the aNHC's stronger ligating ability and lower steric bulk.23
 | ||
| Fig. 7 DIAMOND-rendered molecular structure of the aNHC-Bi complex 4. Relevant ellipsoids are set at a 50% probability level, while the rest of the skeletons are shown as sticks for better viewing. A co-crystallized toluene molecule is omitted for clarity. Only relevant H atoms are shown. Selected bond distances (Å): Bi–C 2.248(4), Bi–O 2.198(3), Bi–N4 2.608(4), Bi–Br1 2.9253(5), Bi–Br2 2.7814(5). Selected bond angles (°): [4]: ∠C–Bi–O = 88.0(1), ∠C–Bi–N4 = 76.0(1), ∠C–Bi–Br1 = 87.5(1), ∠C–Bi–Br2 = 85.9(1), ∠O–Bi–N4 = 164.0(1), ∠O–Bi–Br1 = 98.6(9), ∠O–Bi–Br2 = 96.8(8), ∠Br1–Bi–Br2 = 163.0(2), ∠Br1–Bi–N4 = 82.4(8), ∠Br2–Bi–N4 = 80.7(8). | ||
As in the case of 2, the fortuitous formation and composition of 4 also indicate a complex and multi-step synthetic route with ligand fragmentation. Monitoring it at the NMR scale again showed a complex spectral pattern, which denied the identification of any potential intermediate. Nonetheless, a control experiment by reacting LH2Br and 1 in a 1:1 ratio also gives 4 to imply that the latter can also be accessed by starting from 1, as seen in the case of 2. Based on this and prior fragmentation insights, a tentative route towards 4 is postulated in Scheme 4. We theorize that the phenolic –OH moieties of two LH2Br can be deprotonated first by Bi(HMDS)3 to give a zwitterionic species like [1HBr] and an equiv. of LH. Reacting 1 with an equiv. of LH2Br can also give the same mixture by protonating the NHC of 1, preferably over its Bi-HMDS. Intramolecular deprotonation from [1HBr] can then give a new NHC complex like [(L)BiBr2] (4′), whereas the LH can be fragmented on the side to furnish DippImd. The latter can then coordinate to the Bi of 4′ and drive NHC to aNHC isomerization to yield 4. The protonation of a metal-bound NHC in the presence of other potentially basic groups is known with the main group and early transition metal complexes, in which the NHC–metal bonds are inherently weak. For instance, Al-bound NHC can be selectively protonated by iPrOH over the Al-Me.24 Another Al-bound NHC gets selectively protonated by PhOH over Al-iBu.25 In our titanium case, the NHC-Ti bond is selectively protonated by an acidic imidazolium-2-H over two Ti-bound NMe2 moieties.16a The weak nature of an NHC-Bi bond is already noted in the spontaneous isomerization of a (NHC)-BiPhCl2 complex into its aNHC variant.19b
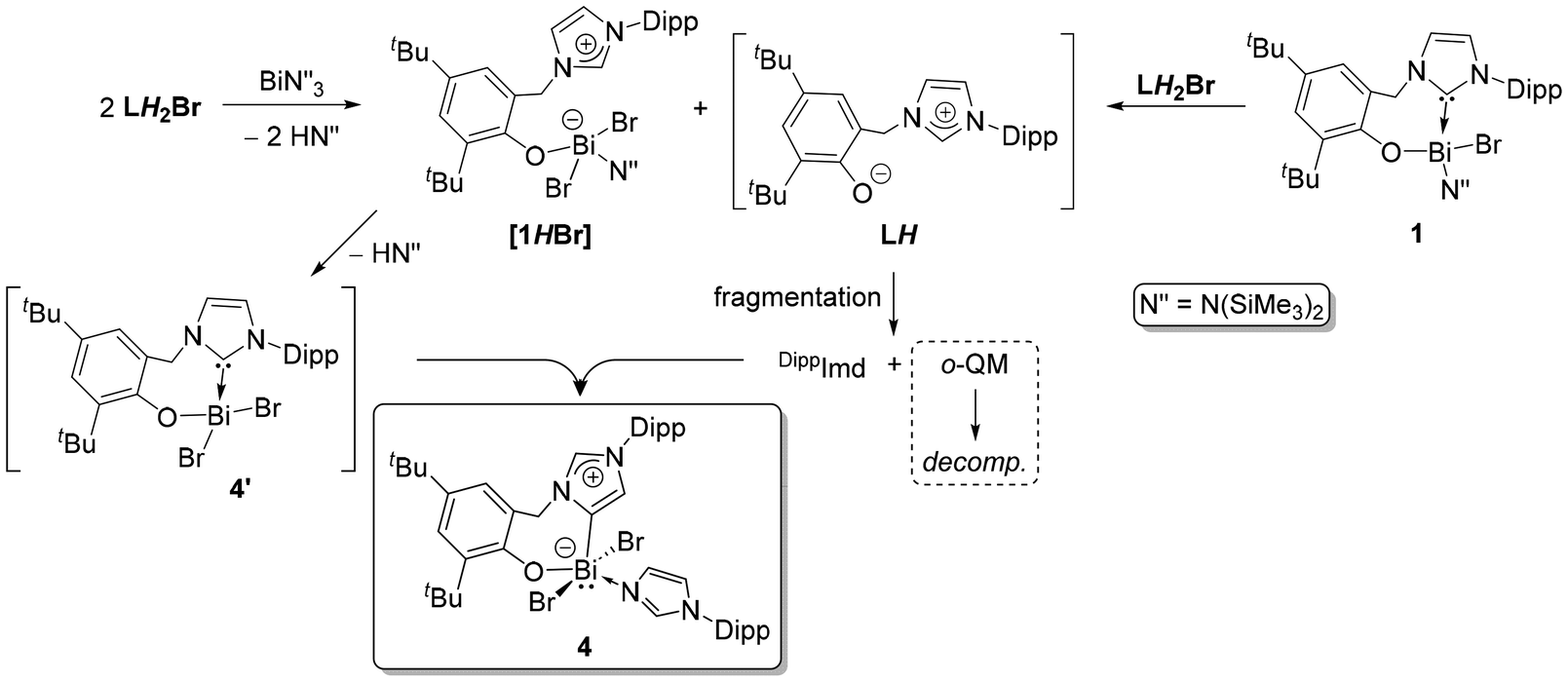 | ||
| Scheme 4 A plausible mechanistic route towards the formation of the aNHC-Bi complex 4. | ||
To further check the viability of this carbene isomerization, the postulated NHC-Bi complex 4′ and its hypothetical aNHC version are first compared by computing the free-energy change involved in replacing the NHC with its abnormal variant (Scheme 5). The same is repeated by considering DippImd as a coligand (Scheme 5). Calculations suggest that both reactions are exergonic, but their extent is greater in the presence of DippImd. This observation prompts us to probe the effect of DippImd on the difference in the Bond Dissociation Energies (BDEs) between the aNHC-Bi and NHC-Bi in concerned complexes (page S14; ESI†).
 | ||
| Scheme 5 Hypothetical ligand replacement reactions and the related free-energy changes. | ||
Here, NHC provides more effective sterics than aNHC on Bi. From the electronic energy calculations (page S14; ESI†), without the DippImd, 4′ is 3.0 kcal mol−1 lower than [(aL)BiBr2]. However, with DippImd, the order is reversed, as the aNHC-Bi complex (4) is 3.0 kcal mol−1 lower than its NHC variant [(L)BiBr2(DippImd)]. Considering only the ligands, the electronic energy of L− is 12.1 kcal mol−1 lower than that of aL−. This leads to the bond dissociation energy (BDE) gaps between Bi–CaNHC and Bi–CNHC with and without DippImd as 15.1 and 9.1 kcal mol−1, respectively (eqn (v), page S15; ESI†).26 This means, in terms of strength, aNHC-Bi is stronger than NHC-Bi for both cases. However, the difference was greater when bulky DippImd was present on Bi. It essentially weakens the NHC-Bi bond, as shown by its ∼0.05 Å elongation. In contrast, the aNHC-Bi remained virtually unchanged in the presence of DippImd. Overall, the coordination of DippImd steers the isomerization. The steric-driven isomerization of NHC to aNHC is known.27 [(IPr)BiBr3]19a and [(IPr)BiPhCl2]19b (IPr = 1,3-bis(2,6-diisopropylphenyl)-imidazol-2-ylidene) also exhibited the same properties under different conditions. Notably, mesoionic aNHCs are yet another significant and fast-evolving neutral C-based ligand class, but their donor-functionalized variants are rare, as in the case of NHOs.234 is the first such example using bismuth.
Conclusions
In summary, we made multi-layered discoveries while testing a hybrid NHC-aryloxide ligand framework on bismuth. It leads to the first ever chelating NHC-, NHO-, and aNHC-Bi complexes with aryloxide sidearms, all from the same imidazolium-phenol precursor and by just varying the relative ratios of Bi(HMDS)3 and KHMDS. The product compositions suggest that the NHC-Bi chemistry, which has been rarely explored, could be mechanistically complex. Nonetheless, an independent synthetic protocol for the precursor to the novel NHO skeleton with substitutional variation should pave the way for exploring other metals in this ligand class. Chelating variants of both NHOs and aNHCs are considerably rarer than those of NHCs. The findings of this study should improve the coordination chemistry of such ligand classes. We are also developing other donor-tethered NHOs and aNHCs.Experimental
General considerations
All experiments were performed under dry and oxygen-free nitrogen conditions using standard Schlenk techniques or in an argon-filled glovebox (MBraun), unless otherwise stated. Before use, glassware was dried overnight at 130 °C, and solvents were dried, distilled and degassed using standard methods and stored over activated 4 Å molecule sieves in the glove box. LH2Br,16a Bi(N(SiMe3)2)3,28I29 and II29 were made by following the literature procedure. BiCl3 and KN(SiMe3)2 were purchased from Sigma-Aldrich and used inside the glovebox as received. 1H and 13C{1H} NMR spectra were recorded on a Bruker Avance NEO (500 MHz) or Avance III (500 MHz) or Jeol (400 MHz) spectrometer at ambient temperature unless otherwise mentioned. Abbreviations for NMR spectra: s (singlet), d (doublet), t (triplet), q (quartet), sept (septet), br (broad). Mass spectrometric analyses were performed on a Bruker micrOTOF–Q II or Waters Xevo G2-XS QTof Spectrometers. Elemental analyses were performed on a PerkinElmer series II 2400 machine. X-ray diffraction data were collected on a Rigaku Synergy i xtalab diffractometer. The crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under the deposition numbers CCDC 2348036 (1) 2348037 (2), 2348038 (32), 2348039 (4).†
[(L)BiBr(HMDS)] (1)
A 10 mL screw-cap vial fitted with a stir bar was charged with LH2Br (0.528 g, 1.000 mmol) and suspended in 2 mL of toluene. A 3 mL toluene solution of Bi(N(SiMe3)2)3 (0.690 g, 1 mmol) was added dropwise. The reaction mixture was then stirred at room temperature for 48 h to obtain a yellowish-orange homogeneous solution. The volatiles were then removed under reduced pressure to obtain a yellow solid. Washing the solid with hexane (3 × 5 mL) and drying under vacuum gave 1 (0.750 g, 0.838 mmol, 84%) as an analytically pure yellow solid. X-ray quality single crystals were grown from its concentrated toluene solution at −30 °C.
1H NMR (C6D6, 500 MHz): δ 7.59 (s, 1 H, ArH), 7.21 (t, 3JHH = 8.0 Hz, 1 H, ArH), 7.10 (d, 3JHH = 7.5 Hz, 1 H, ArH), 7.01 (m, 2 H, ArH), 6.84 (d, 2JHH = 13 Hz, 1 H, ArCH2N), 6.34 (s, 1 H, NCHCHN), 6.29 (s, 1 H, NCHCHN), 4.13 (d, 2JHH = 13 Hz, 1 H, ArCH2N), 2.44 (br, m, 2 H, CHMe2), 1.69 (s, 9 H, CMe3), 1.55 (d, 3JHH = 6.5 Hz, 3 H, CHMe2), 1.39 (s, 9 H, CMe3), 1.29 (d, 3JHH = 6.5 Hz, 3 H, CHMe2), 0.97 (d, 3JHH = 6.5 Hz, 3 H, CHMe2), 0.54 (d, 3JHH = 6.5 Hz, 3 H, CHMe2), 0.49 (s, 9 H, NSiMe3), 0.30 (s, 9 H, NSiMe3). 13C{1H} NMR (C6D6, 126 MHz): 162.2 (N![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) N), 146.6 ((ipr), Dipp), 144.6 ((ipr), Dipp), 139.6 (N, Dipp), 135.8 (Ar–, Dipp), 133.6 (Ar–, Dipp), 131.5 (Ar–, Dipp), 128.8 (Ph–), 126.3 (Ph–), 125.6 (Ph–), 125.3 (Ph–), 124.5 (Ph–), 123.7 (Ph–), 122.8 (NHCHN), 122.6 (NCHHN), 54.5 (PhH2N), 35.9 (Me3), 34.4 (Me3), 32.5 (HMe2), 30.9 (HMe2), 29.1 (C
N), 146.6 ((ipr), Dipp), 144.6 ((ipr), Dipp), 139.6 (N, Dipp), 135.8 (Ar–, Dipp), 133.6 (Ar–, Dipp), 131.5 (Ar–, Dipp), 128.8 (Ph–), 126.3 (Ph–), 125.6 (Ph–), 125.3 (Ph–), 124.5 (Ph–), 123.7 (Ph–), 122.8 (NHCHN), 122.6 (NCHHN), 54.5 (PhH2N), 35.9 (Me3), 34.4 (Me3), 32.5 (HMe2), 30.9 (HMe2), 29.1 (C![[M with combining low line]](https://www.rsc.org/images/entities/i_char_004d_0332.gif)
![[e with combining low line]](https://www.rsc.org/images/entities/i_char_0065_0332.gif) 3), 26.7 (C3), 23.7 (CH2), 23.0 (CH2), 7.8 (NSi3), 4.4 (NSi3). Elemental analysis of C36H59BrN3OSi2Bi: Calcd C, 48.32; H, 6.65; N, 4.70; Found C, 48.07; H, 6.73; N, 4.59.
3), 26.7 (C3), 23.7 (CH2), 23.0 (CH2), 7.8 (NSi3), 4.4 (NSi3). Elemental analysis of C36H59BrN3OSi2Bi: Calcd C, 48.32; H, 6.65; N, 4.70; Found C, 48.07; H, 6.73; N, 4.59.
[(DippNHO2ArO)BiBr] (2)
A 10 mL screw-cap vial fitted with a stir bar was charged with LH2Br (0.528 g, 1.000 mmol) and suspended in 2 mL of toluene. A 3 mL toluene solution of Bi(N(SiMe3)2)3 (0.345 g, 0.500 mmol) and a 2 mL toluene solution of KHMDS (0.100 g, 0.500 mmol) were added into the reaction vial nearly at the same time. The reaction mixture was then stirred for 48 h at room temperature to obtain an orange solution along with some insoluble residues. The filtrate was then filtered through a pad of Celite and evaporated to dryness under reduced pressure to obtain an orange residue. Washing the residue with hexane (3 × 5 mL) and drying under vacuum afforded 2 (0.190 g, 0.200 mmol, 40%) as an analytically pure orange solid. X-ray quality single crystals were grown from a concentrated solution in a toluene/THF solvent mixture at −30 °C.
1H NMR (C6D6, 500 MHz, ppm): δ 7.55 (br, 1 H, ArH), 7.46 (s, 1 H, ArH) 7.12 (s, 1 H, ArH) 7.05 (s, 1 H, ArH) 6.92 (d, 3JHH = 7.5 Hz, 1 H, ArH) 6.81 (s, 1 H, ArH) 6.64 (s, 1 H, ArH), 5.75 (br, 2 H, NCHCHN), 5.49 (d, 2JHH = 14 Hz, 1 H, ArCH2N), 4.14 (s, 1 H, ArCHBi), 4.11 (d, 2JHH = 14 Hz, 1 H, ArCH2N), 2.82 (m, 1 H, CHMe2), 2.53 (m, 1 H, CHMe2), 1.67 (s, 9 H, CMe3), 1.53 (s, 9 H, CMe3), 1.41 (s, 9 H, CMe3), 1.40 (s, 9 H, CMe3), 1.39 (d, 3JHH = 6.5 Hz, 6 H, CHMe2), 1.14 (d, 3JHH = 6.5 Hz, 6 H, CHMe2), 0.90 (d, 3JHH = 6.5 Hz, 6 H, CHMe2), 0.88 (d, 3JHH = 6.5 Hz, 6 H, CHMe2). 13C{1H} NMR (126 MHz, CDCl3): δ 161.4 (NN), 147.6 ((ipr), Dipp), 145.6 ((ipr), Dipp), 140.4 (N, Dipp), 137.8 (Ar–, Dipp), 132.3 (Ar–, Dipp), 130.1 (Ar–, Dipp), 128.6 (Ph–) 125.3 (Ph–) 124.6 (Ph–), 124.2 (Ph–), 123.6 (Ph–), 121.4 (NHCHN), 120.9 (NCHHN), 35.7 (CHBi), 35.2 (ArH2N), 34.5 (Me3), 34.4 (Me3), 32.6 (Me3), 32.5 (Me3), 31.0 (HMe2), 30.5 (HMe2), 29.2 (C3), 28.3 (C3), 27.5 (C3), 26.3 (C3), 25.9 (CH2), 25.8 (CH2), 24.0 (CH2), 23.0 (CH2). Elemental analysis of C45H62BrN2O2Bi: calculated C, 56.78; H, 6.57; N, 2.94; found C, 56.29; H, 6.48; N, 2.98.
II
A 100 mL Schlenk flask fitted with a stir bar was charged with I (0.920 g, 1.570 mmol) and suspended in 10 mL of THF. It was then cooled to −78 °C before dropwise adding a 10 mL THF solution of KN(SiMe3)2 (0.940 g, 4.710 mmol) via a cannula. The reaction mixture was then allowed to come to room temperature slowly before being quenched with an excess of water. Et2O was then added, and the organic layer was separated. The aqueous layer was further washed with fresh Et2O (2 × 50 mL), and the combined organic portion was dried with MgSO4. Removing the volatiles under reduced pressure afforded II (0.450 g, 0.891 mmol, 56% yield) as a white solid.
1H NMR (CDCl3, 400 MHz): δ 7.28 (s, 1 H, ArH), 7.19 (d, 3JHH = 2.4 Hz, 1 H, ArH), 7.01 (d, 3JHH = 2.4 Hz, 1 H, ArH), 6.74 (b, 1 H, ArH), 6.73 (d, 3JHH = 2.4 Hz, 1 H, NCHCHN), 6.13 (d, 3JHH = 2.4 Hz, 1 H, NCHCHN), 5.14 (s, 2 H, ArCH2N), 3.92 (s, 2 H, ArCH2C), 1.44 (s, 9 H, CMe3), 1.43 (s, 9 H, CMe3) 1.20 (s, 9 H, CMe3), 1.06 (s, 9 H, CMe3). 13C{1H} NMR (CDCl3, 100 MHz): 153.4 (NN), 148.9 (Ar–), 148.3 (Ar–), 144.1 (Ar–), 141.3 (Ar–), 139.7 (Ar–), 138.4 (Ar–), 125.9 (Ar–), 124.6 (Ar–), 124.3 (Ar–), 123.7 (Ar–), 123.3 (Ar–), 121.9 (NHCHN), 120.0 (NCHHN), 47.6 (CH2Ar), 35.2 (ArH2N), 35.1 (Me3), 34.2 (Me3), 34.1 (Me3), 31.7 (Me3), 31.3 (C3), 30.5 (C3), 29.9 (C3). HRMS (M/Z): calculated [M + H] = 505.3800; found 505.3804.
L′H3I
A Teflon-stopped storage tube fitted with a magnetic stir bar was charged with II (0.400 g, 0.792 mmol) and dissolved in 10 mL of acetonitrile. 800 μL of iodomethane (0.226 g, 1.580 mmol) was added to this solution. The reaction mixture was then placed in an oil bath pre-heated to 90 °C and stirred for 2 h, during which a colourless solid precipitated. The reaction tube was then cooled to room temperature. The solid was then filtered and washed with Et2O (3 × 10 mL) and toluene (3 × 10 mL) successively to obtain L′H3I (0.480 g, 0.742 mmol, 94%) as a white powder.
1H NMR (DMSO-D6, 400 MHz): δ 8.47 (s, 1 H, ArOH), 8.25 (s, 1 H, ArOH), 7.71 (d, 3JHH = 2.4 Hz, 1 H, ArH), 7.55 (d, 3JHH = 2.4 Hz, 1 H, ArH), 7.18 (d, 3JHH = 2.4 Hz, 1 H, ArH), 7.13 (d, 3JHH = 2.4 Hz, 1 H, ArH), 6.85 (d, 3JHH = 2.4 Hz, 1 H, NCHCHN), 6.18 (d, 3JHH = 2.4 Hz, 1 H, NCHCHN), 5.37 (s, 2 H, ArCH2N), 4.51 (s, 2 H, ArCH2C), 3.62 (s, 3 H, NCH3), 1.36 (s, 9 H, CMe3), 1.32 (s, 9 H, CMe3) 1.11 (s, 9 H, CMe3), 1.06 (s, 9 H, CMe3). 13C NMR (DMSO-D6 126 MHz): 151.5 (NN), 150.6 (Ar–), 145.4 (Ar–), 142.5 (Ar–), 142.4 (Ar–), 138.7 (Ar–), 124.4 (Ar–), 124.1 (Ar–), 123.1 (Ar–), 122.6 (Ar–), 122.5 (Ar–), 122.4 (NCHHN), 121.7 (NCHHN), 47.7 (CH2Ar), 35.0 (ArH2N), 34.7 (N–), 33.9 (Me3), 33.7 (Me3), 31.2 (Me3), 31.1 (Me3), 29.9 (C3), 29.8 (C3), 25.8 (C3). HRMS (M/Z): calculated M = 519.3900; found 519.3959.
[(MeNHO2ArO)Bi(μ-I)]2 (32)
A 10 mL screw-cap vial fitted with a stir bar was charged with L′H3I (0.100 g, 0.155 mmol) and suspended in 2 mL of toluene. Another 2 mL of a toluene solution of Bi(N(SiMe3)2)3 (0.107 g, 0.155 mmol) was then added dropwise into the suspension. The reaction mixture was then stirred for 48 h to produce an orange suspension. The orange solid isolated by filtration was then washed with fresh toluene (3 × 5 mL) and dried under vacuum to afford 32 (0.100 g, 0.117 mmol, 76%). X-ray quality single crystals were grown from a concentrated solution of a toluene/THF mixture at −30 °C.
1H NMR (THF-D8/C6D6 (2:5), 500 MHz): δ 7.48 (s, 1 H, ArH), 7.43 (s, 1 H, ArH), 6.76 (s, 1 H, ArH), 6.36 (s, 1 H, ArH), 6.34 (s, 1 H, NCHCHN), 6.03 (s, 1 H, NCHCHN), 5.79 (d, 2JHH = 14 Hz, 1 H, ArCH2N), 4.80 (s, 1 H, ArCHBi), 4.30 (d, 2JHH = 14 Hz, 1 H, ArCH2N), 3.33 (s, 3 H, NCH3), 1.73 (s, 9 H, CMe3), 1.62 (s, 9 H, CMe3), 1.35 (s, 9 H, CMe3), 1.32 (s, 9 H, CMe3). 13C{1H} NMR (THF-D8/C6D6, (2:5) 126 MHz): 169.8 (NN), 161.1 (Ar–), 151.8 (Ar–), 143.2 (Ar–), 141.0 (Ar–), 139.9 (Ar–), 139.3 (Ar–), 129.8 (Ar–), 128.9 (Ar–), 126.0 (Ar–), 125.0 (Ar–), 124.4 (Ar–), 123.7 (Ar–), 121.2 (NHCHN), 120.3 (NCHHN), 84.0 (CHBi), 48.1 (ArH2N), 36.2 (N–), 35.9 (Me3), 35.7 (Me3), 34.5 (Me3), 34.4 (Me3), 32.6 (C3), 32.5 (C3), 31.7 (C3), 30.4 (C3). Elemental analysis of C34H48N2O2IBi: calculated C 47.89; H 5.67; N 3.29; found C 47.46; H 5.58; N 3.32.
[(aL)BiBr2(DippImd)] (4)
A 10 mL screw-cap vial fitted with a stir bar was charged with LH2Br (0.528 g, 1.000 mmol) and suspended in 2 mL of toluene. Another 3 mL solution of Bi(N(SiMe3)2)3 (0.345 g, 0.500 mmol) was added dropwise to the reaction vial. The reaction mixture was stirred for 48 h at room temperature to obtain an orange suspension. The orange solid isolated by filtration was washed with fresh toluene (3 × 5 mL) and dried under vacuum to afford 4 (0.400 g, 0.384 mmol, 77%). X-ray quality single crystals were grown from concentrated solutions of a toluene/THF mixture at room temperature.
1H NMR (CDCl3, 500 MHz): δ 8.47 (s, 1 H, NCHN), 7.93 (s, 1 H, NCHNimd), 7.61–7.15 (m, 10 H, ArH), 7.03 (s, 1 H, ArCH2N), 6.86 (s, 1 H, ArCH2N), 2.30 (br, m, 4 H, CHMe2), 1.45 (s, 9 H, CMe3), 1.25 (s, 9 H, CMe3), 1.11 (d, 3JHH = 7 Hz, 6 H, CHMe2), 1.07 (d, 3JHH = 7 Hz, 12 H, CHMe2). 13C{1H} NMR (CDCl3, 126 MHz): 160.6 (NCHN), 146.2 (NHN), 145.6 (NHN, ImdDipp), 141.9 (Ar–), 140.0 (Ar–), 139.8 (Ar–), 138.2 (Ar–), 137.9 (Ar–), 133.5 (Ar–), 132.4 (Ar–), 131.6 (Ar–), 130.7 (Ar–), 130.1 (Ar–), 129.2 (Ar–), 125.1 (Ar–), 124.7 (Ar–), 124.5 (Ar–), 123.8 (NCHN), 123.1 (AHCHN, ImdDipp), 121.6 (ACHHN, ImdDipp), 53.3 (ArH2N), 35.4 (HMe2), 33.9 (HMe2), 31.9 (Me3), 31.2 (Me3), 28.6 (C3), 28.2 (C3), 24.4 (CH2), 24.3 (CH2), 24.2 (CH2). Elemental Analysis of C45H61N4OBr2Bi: calculated C 51.83; H 5.90; N 5.37; found C 51.45; H 5.83; N 5.39.
Data availability
Synthetic descriptions of all reported compounds, spectroscopic data, computational details and coordinates, and crystallographic data files [CCDC 2348036 (1) 2348037 (2), 2348038 (32), 2348039 (4)†].Conflicts of interest
There are no conflicts to declare.Acknowledgements
D Mukherjee acknowledges SERB, India for SRG/2019/001931, CRG/2023/005396, and SB/S2/RJN-028/2018. D. Mallick thanks SERB, India for SRG/2019/001461 and additionally DST, India, for DST/NSM/R&D_HPC_Applications/2021/8 for providing computing resources of ‘PARAM Shakti’ at IIT Kharagpur and of ‘PARAM Brahma’ at IISER Pune. S. G. thanks CSIR-India, and S. S. thanks IISER Kolkata for their fellowships.References
- (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CAS; (b) D. Munz, Organometallics, 2018, 37, 275–289 CAS; (c) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed; (d) P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem., 2021, 5, 711–725 CrossRef CAS PubMed.
- (a) F. Pape and J. F. Teichert, Eur. J. Org. Chem., 2017, 2017, 4206–4229 CrossRef CAS; (b) C. Fliedel, A. Labande, E. Manoury and R. Poli, Coord. Chem. Rev., 2019, 394, 65–103 CrossRef CAS; (c) S. Hameury, P. de Frémont and P. Braunstein, Chem. Soc. Rev., 2017, 46, 632–733 RSC; (d) E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239–2246 CrossRef CAS.
- Z. Li, P. Ji and J.-P. Cheng, J. Org. Chem., 2021, 86, 2974–2985 CrossRef CAS PubMed.
- (a) U. Gruseck and M. Heuschmann, Chem. Ber., 2006, 120, 2053–2064 Search PubMed; (b) A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 Search PubMed; (c) N. Kuhn, H. Bohnen, J. Kreutzberg, D. Bläser and R. Boese, J. Chem. Soc., Chem. Commun., 1993, 1136–1137 CAS.
- (a) M. G, D. Sharma, R. Dandela and V. Dhayalan, Chem. – Eur. J., 2023, 29, e202302106 CrossRef CAS; (b) Q. Liang and D. Song, Dalton Trans., 2022, 51, 9191–9198 RSC; (c) S. Naumann, Chem. Commun., 2019, 55, 11658–11670 RSC; (d) A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed; (e) M. M. D. Roy and E. Rivard, Acc. Chem. Res., 2017, 50, 2017–2025 CrossRef CAS PubMed; (f) R. S. Ghadwal, Dalton Trans., 2016, 45, 16081–16095 RSC; (g) R. D. Crocker and T. V. Nguyen, Chem. – Eur. J., 2016, 22, 2208–2213 CrossRef CAS PubMed.
- (a) M. Iglesias, A. Iturmendi, P. J. Sanz Miguel, V. Polo, J. J. Pérez-Torrente and L. A. Oro, Chem. Commun., 2015, 51, 12431–12434 CAS; (b) A. Iturmendi, N. García, E. A. Jaseer, J. Munárriz, P. J. Sanz Miguel, V. Polo, M. Iglesias and L. A. Oro, Dalton Trans., 2016, 45, 12835–12845 CAS.
- A. Iturmendi, M. Iglesias, J. Munarriz, V. Polo, V. Passarelli, J. J. Pérez-Torrente and L. A. Oro, Green Chem., 2018, 20, 4875–4879 RSC.
- A. Schumann and C. Hering-Junghans, Eur. J. Inorg. Chem., 2018, 2018, 2584–2588 CrossRef CAS.
- G. Song, X. Li, Z. Song, J. Zhao and H. Zhang, Chem. – Eur. J., 2009, 15, 5535–5544 CrossRef CAS PubMed.
- A. Petronilho, J. A. Woods, H. Mueller-Bunz, S. Bernhard and M. Albrecht, Chem. – Eur. J., 2014, 20, 15775–15784 CrossRef CAS.
- M. Kaur, K. Patra, N. U. D. Reshi and J. K. Bera, Organometallics, 2020, 39, 189–200 CrossRef CAS.
- (a) N. R. Paisley, M. W. Lui, R. McDonald, M. J. Ferguson and E. Rivard, Dalton Trans., 2016, 45, 9860–9870 RSC; (b) M. W. Lui, O. Shynkaruk, M. S. Oakley, R. Sinelnikov, R. McDonald, M. J. Ferguson, A. Meldrum, M. Klobukowski and E. Rivard, Dalton Trans., 2017, 46, 5946–5954 RSC.
- M. Viciano, M. Feliz, R. Corberán, J. A. Mata, E. Clot and E. Peris, Organometallics, 2007, 26, 5304–5314 CrossRef CAS.
- (a) C. C. Chong, B. Rao, R. Ganguly, Y. Li and R. Kinjo, Inorg. Chem., 2017, 56, 8608–8614 CrossRef CAS PubMed; (b) R. Guthardt, J. Mellin, C. Bruhn and U. Siemeling, Eur. J. Inorg. Chem., 2021, 2022, e202100903 CrossRef.
- S. Wang, J. Yang, H. Zeng, Y. Zhou, F. Wang, X. Feng and S. Dong, Org. Lett., 2023, 25, 7247–7251 CrossRef CAS PubMed.
- (a) S. Goswami, P. Mandal, D. Mallick and D. Mukherjee, Organometallics, 2023, 42, 1232–1241 CrossRef CAS; (b) S. Goswami, P. Mandal, S. Sarkar, M. Mukherjee, S. Pal, D. Mallick and D. Mukherjee, Dalton Trans., 2024, 53, 1346–1354 RSC.
- S. Goswami, S. Sarkar, D. Mallick and D. Mukherjee, Organometallics, 2024, 43, 1308–1316 CrossRef CAS.
- (a) F. Wang, O. Planas and J. Cornella, J. Am. Chem. Soc., 2019, 141, 4235–4240 CrossRef CAS PubMed; (b) O. Planas, F. Wang, M. Leutzsch and J. Cornella, Science, 2020, 367, 313–317 CrossRef CAS PubMed; (c) J. Cornella and Y. Pang, in Comprehensive Organometallic Chemistry IV, ed. G. Parkin, K. Meyer and D. O′hare, Elsevier, Oxford, 2022, pp. 478–522 Search PubMed; (d) Y. Pang, N. Nothling, M. Leutzsch, L. Kang, E. Bill, M. van Gastel, E. Reijerse, R. Goddard, L. Wagner, D. SantaLucia, S. DeBeer, F. Neese and J. Cornella, Science, 2023, 380, 1043–1048 CrossRef CAS PubMed; (e) A. Stoy, M. Jurgensen, C. Millidoni, C. Berthold, J. Ramler, S. Martinez, M. R. Buchner and C. Lichtenberg, Angew. Chem., Int. Ed., 2023, 62, e202308293 CrossRef CAS PubMed; (f) M. Mato and J. Cornella, Angew. Chem., Int. Ed., 2024, 63, e202315046 CAS.
- (a) J. B. Waters, Q. Chen, T. A. Everitt and J. M. Goicoechea, Dalton Trans., 2017, 46, 12053–12066 RSC; (b) G. Wang, L. A. Freeman, D. A. Dickie, R. Mokrai, Z. Benkő and R. J. Gilliard, Inorg. Chem., 2018, 57, 11687–11695 CrossRef CAS PubMed; (c) G. Wang, L. A. Freeman, D. A. Dickie, R. Mokrai, Z. Benkő and R. J. Gilliard Jr., Chem. – Eur. J., 2019, 25, 4335–4339 CAS; (d) L. P. Ho and M. Tamm, Dalton Trans., 2021, 50, 1202–1205 RSC; (e) M. M. Siddiqui, S. K. Sarkar, M. Nazish, M. Morganti, C. Köhler, J. Cai, L. Zhao, R. Herbst-Irmer, D. Stalke, G. Frenking and H. W. Roesky, J. Am. Chem. Soc., 2021, 143, 1301–1306 CAS; (f) J. E. Walley, L. S. Warring, G. Wang, D. A. Dickie, S. Pan, G. Frenking and R. J. Gilliard Jr., Angew. Chem., Int. Ed., 2021, 60, 6682–6690 CrossRef CAS PubMed; (g) R. Deka and A. Orthaber, Dalton Trans., 2022, 51, 8540–8556 RSC.
- https://www.crystalimpact.com/diamond/ .
- H. Aihara, T. Matsuo and H. Kawaguchi, Chem. Commun., 2003, 2204–2205 RSC.
- H. Steffenfauseweh, Y. V. Vishnevskiy, B. Neumann, H. G. Stammler, D. M. Andrada and R. S. Ghadwal, Angew. Chem., Int. Ed., 2022, 61, e202207415 CrossRef CAS PubMed.
- (a) R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755–766 CrossRef CAS; (b) Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef PubMed; (c) S. C. Sau, P. K. Hota, S. K. Mandal, M. Soleilhavoup and G. Bertrand, Chem. Soc. Rev., 2020, 49, 1233–1252 RSC; (d) R. S. Ghadwal, Angew. Chem., Int. Ed., 2023, 62, e202304665 CrossRef CAS PubMed.
- C. Romain, C. Fliedel, S. Bellemin-Laponnaz and S. Dagorne, Organometallics, 2014, 33, 5730–5739 CAS.
- V. Dardun, L. Escomel, E. Jeanneau and C. Camp, Dalton Trans., 2018, 47, 10429–10433 RSC.
- B. M. Day, T. Pugh, D. Hendriks, C. F. Guerra, D. J. Evans, F. M. Bickelhaupt and R. A. Layfield, J. Am. Chem. Soc., 2013, 135, 13338–13341 CAS.
- (a) M. Uzelac, A. Hernán-Gómez, D. R. Armstrong, A. R. Kennedy and E. Hevia, Chem. Sci., 2015, 6, 5719–5728 RSC; (b) M. Wagner, T. Zöller, W. Hiller, M. H. Prosenc and K. Jurkschat, Chem. Commun., 2013, 49, 8925–8927 CAS.
- M. Vehkamäki, T. Hatanpää, M. Ritala and M. Leskelä, J. Mater. Chem., 2004, 14, 3191–3197 Search PubMed.
- D. Zhang, H. Aihara, T. Watanabe, T. Matsuo and H. Kawaguchi, J. Organomet. Chem., 2007, 692, 234–242 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data, computational details, and crystallographic data files. CCDC 2348036 (1) 2348037 (2), 2348038 (32), 2348039 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00706b |
| This journal is © The Royal Society of Chemistry 2025 |