
Tris(pentafluoroethyl)difluorophosphorane for fluoride abstraction and ligand exchange reactions of N-heterocyclic carbene and cyclic alkyl(amino)carbene copper(I) fluorides†
Melanie
Riethmann
a,
Steffen A.
Föhrenbacher
a,
Christian
Luz
a,
Nikolai V.
Ignat'ev
abc,
Maik
Finze
 *ab and
Udo
Radius
*a
*ab and
Udo
Radius
*a
aInstitute of Inorganic Chemistry, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: udo.radius@uni-wuerzburg.de; maik.finze@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität, Würzburg, Am Hubland, 97074 Würzburg, Germany
cConsultant, Merck KGaA, 64293 Darmstadt, Germany
First published on 15th May 2025
Abstract
The synthesis and structural characterization of a variety of N-heterocyclic carbene (NHC)- and cyclic (alkyl)(amino)carbene (cAAC)-ligated cationic copper(I) complexes, featuring the weakly coordinating tris(pentafluoroethyl)trifluorophosphate counteranion (FAP− anion, [(C2F5)3PF3]−) are reported. Starting with the complex [(IDipp)Cu(C6Me6)]+FAP− (IIa) reported previously, (S. A. Föhrenbacher, M. J. Krahfuss, L. Zapf, A. Friedrich, N. V. Ignat'ev, M. Finze and U. Radius, Chem. – Eur. J., 2021, 27, 3504-3516) a series of mononuclear complexes [(IDipp)Cu(LB)]+FAP− (IDipp = 1,3-bis(2,6-di-iso-propylphenyl)-imidazolin-2-ylidene) were obtained via ligand exchange of C6Me6 with neutral two valence electron (2 VE) donor molecules (LB = NH3, 1; C6H12N2 = DABCO, 2; C7H10N2 = DMAP, 3; C4H4N2 = pyrazine, 4; C13H9N = acridine, 5; η1-O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C13H9N = acridone, 6; C4H10S = SEt2, 7; C4H8S = THT, 8; PCy3, 9), alongside the dinuclear species [{(IDipp)Cu}2(C2N3H3)2]2+2FAP− (10) with 1,2,4-triazole. In a parallel strategy, [(cAACMe)Cu(C6Me6)]+FAP− (IIb) was employed as precursor for Cu(I) complex formation, leading to [(cAACMe)Cu(LB)]+FAP− (LB = C7H10N2, 13; C4H10S, 14) and the dinuclear complexes [{(cAACMe)Cu}2(C6H12N2)]2+2FAP− (11) and [{(cAACMe)Cu}2(C4H4N2)]2+2FAP− (12). Additionally, the reaction of [(carbene)CuF] with (C2F5)3PF2 in the presence of different 2 VE donor ligands induced fluoride replacement with a 2 VE donor ligand (LB). This strategy facilitated the isolation of a broad range of complexes of the type [(carbene)Cu(LB)]+FAP−, including [(IDipp)Cu(LB)]+FAP− (LB = (N
C13H9N = acridone, 6; C4H10S = SEt2, 7; C4H8S = THT, 8; PCy3, 9), alongside the dinuclear species [{(IDipp)Cu}2(C2N3H3)2]2+2FAP− (10) with 1,2,4-triazole. In a parallel strategy, [(cAACMe)Cu(C6Me6)]+FAP− (IIb) was employed as precursor for Cu(I) complex formation, leading to [(cAACMe)Cu(LB)]+FAP− (LB = C7H10N2, 13; C4H10S, 14) and the dinuclear complexes [{(cAACMe)Cu}2(C6H12N2)]2+2FAP− (11) and [{(cAACMe)Cu}2(C4H4N2)]2+2FAP− (12). Additionally, the reaction of [(carbene)CuF] with (C2F5)3PF2 in the presence of different 2 VE donor ligands induced fluoride replacement with a 2 VE donor ligand (LB). This strategy facilitated the isolation of a broad range of complexes of the type [(carbene)Cu(LB)]+FAP−, including [(IDipp)Cu(LB)]+FAP− (LB = (N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CMe)2, 16; NCPh, 17; NH2Ph, 18; NHPh2, 21; NC5H5, 22; NC5H3F2, 24; NC5H2F3, 25; η1-OCPh2, 27), [(SIDipp)Cu(NH2Ph)]+FAP− (19) (SIDipp = 1,3-bis(2,6-di-iso-propylphenyl)-imidazolidin-2-ylidene) and [(cAACMe)Cu(LB)]+FAP− (cAACMe = 1-(2,6-di-iso-propylphenyl)-3,3,5,5-tetramethyl-pyrrolidin-2-ylidene; LB = NCMe, 15; NH2Ph, 20; NC5H5, 23; THF, 28). Additionally, the dinuclear complex [{(IDipp)Cu(μ-ONC5H5)}2]2+2FAP− (26) was obtained upon reaction with pyridine-N-oxide. In all cases the carbene ligand stayed intact and the formation of Lewis acid/base pairs of the 2 VE ligand and (C2F5)3PF2 was never observed. As a result, mixtures of [(carbene)CuF] and (C2F5)3PF2 may serve as synthons for [(carbene)Cu]+, as demonstrated in this work.
CMe)2, 16; NCPh, 17; NH2Ph, 18; NHPh2, 21; NC5H5, 22; NC5H3F2, 24; NC5H2F3, 25; η1-OCPh2, 27), [(SIDipp)Cu(NH2Ph)]+FAP− (19) (SIDipp = 1,3-bis(2,6-di-iso-propylphenyl)-imidazolidin-2-ylidene) and [(cAACMe)Cu(LB)]+FAP− (cAACMe = 1-(2,6-di-iso-propylphenyl)-3,3,5,5-tetramethyl-pyrrolidin-2-ylidene; LB = NCMe, 15; NH2Ph, 20; NC5H5, 23; THF, 28). Additionally, the dinuclear complex [{(IDipp)Cu(μ-ONC5H5)}2]2+2FAP− (26) was obtained upon reaction with pyridine-N-oxide. In all cases the carbene ligand stayed intact and the formation of Lewis acid/base pairs of the 2 VE ligand and (C2F5)3PF2 was never observed. As a result, mixtures of [(carbene)CuF] and (C2F5)3PF2 may serve as synthons for [(carbene)Cu]+, as demonstrated in this work.
Introduction
Lewis acids (LAs) were originally defined by Gilbert N. Lewis as electron pair acceptors that exhibit a strong propensity to form bonds with electronegative electron pair donors, known as Lewis bases (LBs).2 The combination of a LA and a LB with both low to moderate steric demand typically results in the formation of classical Lewis acid/base adducts, characterized by stable covalent bonding interactions. However, when sterically more hindered LA/LB entities are combined, such as PPh3 with B(C6F5)3, intramolecular interactions are reduced, leading to the formation of weakly bound adducts. These systems often exhibit elongated bond distances, equilibria between the free acid and base, or structural rearrangement, as exemplified by the zwitterionic species Ph3PH–C6F4–BF(C6F5)2.3 Such systems are classified as Frustrated Lewis Pairs (FLPs), where steric constraints prevent complete Lewis acid/base adduct formation, resulting in highly reactive centers with unique chemical properties. This phenomenon enables the activation of otherwise inert small molecules, including H2,4 CO,5 CO2,6,7 N2O,7,8 or SO2.9 To further rationalize the formation of Lewis acid/base adducts while accounting for steric effects, we recently developed a novel and, to date, the first generalizable and experimentally accessible approach.10 The Lewis Acid/Base Repulsion (LAB-Rep) model utilizes the percent buried volume (%Vbur) as a quantitative measure of steric hindrance and relies on readily available structural data, such as crystallographic information or computationally derived structures. This model enables the estimation of steric constraints that may impede Lewis acid/base interactions, providing valuable insights, particularly in the context of FLP chemistry.The chemistry of Lewis acid/base combinations involving N-heterocyclic carbenes (NHCs) has been extensively studied, particularly in the context of classical adduct-type complexes with p-block elements. To date, numerous well-characterized examples exist, most of them with group 13 and 15 Lewis acids, such as BF3, AlCl3, PF5, AsF5, and SbF5. However, Lewis acid/base adducts of phosphoranes (PR5) with NHCs remain comparatively rare, and only a limited number exhibit FLP behavior.11 In 2021, we expanded the scope of such systems by isolating a series of NHC-phosphorane adducts (C2F5)3PF2·NHC via the straightforward reaction of free (small) NHCs with the readily available and highly Lewis-acidic tris(pentafluoroethyl)difluorophosphorane, (C2F5)3PF2, in Et2O. Furthermore, mixtures of (C2F5)3PF2 with sterically demanding NHCs, such as ItBu (ItBu = 1,3-di-tert-butylimidazolin-2-ylidene), IDipp, and SIDipp, exhibited FLP reactivity, facilitating the deprotonation of acetonitrile and other C–H acidic compounds (e.g., acetone, ethyl acetate) to form the corresponding imidazolium salts and [(C2F5)3PF2(R)]− (R = CH2CN, OC(CH2)CH3, CH2CO2Et).12
Recently, we reported on the reaction of (C2F5)3PF2 with the copper fluoride complexes [(IDipp)CuF] (Ia), [(SIDipp)CuF] (Ib) and [(cAACMe)CuF] (Ic) in the presence of various carbon-based ligands LB, such as alkynes and benzenes.13 This reaction facilitated fluoride transfer, yielding the corresponding tris(pentafluoroethyl)trifluorophosphate (FAP−, [(C2F5)3PF3]−) salts of the copper complex cations [(carbene)Cu(LB)]+. Due to the relatively weak interaction between the neutral carbon co-ligand and the metal center, these complexes serve as isolable synthons for cationic [(carbene)Cu]+. Furthermore, the hexamethylbenzene complexes [(IDipp)Cu(C6Me6)]+FAP− (IIa) and [(cAACMe)Cu(C6Me6)]+FAP− (IIb) were evaluated for their catalytic performance and demonstrated high efficacy as copper(I) catalysts in the cycloaddition reaction of benzyl azide and various terminal alkynes, yielding 1,4-disubstituted 1,2,3-triazoles.13 These findings indicate that the phosphorane (C2F5)3PF2 exhibits sufficient Lewis acidity to abstract fluoride from transition metal complexes, thereby serving as a viable precursor for the synthesis of cationic transition metal species incorporating the FAP− counteranion.1 Herein, we report the reactivity of IIa and IIb with selected different Lewis bases, focusing on the synthesis and characterization of N-heterocyclic carbene (NHC)- and cyclic (Alkyl)(amino)carbene (cAAC)-ligated copper cations featuring the weakly coordinating FAP− counteranion.
Results and discussion
The use of [(carbene)Cu(C6Me6)]+ as synthon for [(carbene)Cu]+
As our recent investigations already revealed that the hexamethylbenzene ligand of [(IDipp)Cu(C6Me6)]+FAP− (IIa) can be exchanged easily by other 2 VE (valence electron) donors, such as THF,1 we became interested in studying this behavior in more detail. Thus, we reacted IIa with selected nitrogen, oxygen, sulfur as well as phosphorus 2 VE donor ligands to probe their ability to replace C6Me6. Using this strategy, we isolated and fully characterized the copper FAP− salts [(IDipp)Cu(LB)]+FAP− (LB = NH3, 1; C6H12N2 = DABCO, 2; C7H10N2 = DMAP, 3; C4H4N2 = pyrazine, 4; C13H9N = acridine, 5; η1-OC13H9N = acridone, 6; C4H10S = SEt2, 7; C4H8S = THT, 8; PCy3, 9) as well as the dinuclear complex [{(IDipp)Cu}2(C2N3H3)2]2+2FAP− (10) (C2N3H3 = 1,2,4-triazole). All reactions occur at room temperature in dichloromethane or chloroform in yields of 40–87% (Scheme 1).
 | ||
| Scheme 1 Synthesis of [(IDipp)Cu(LB)]+FAP− (LB = NH3, 1; C6H12N2 = DABCO, 2; C7H10N2 = DMAP, 3; C4H4N2 = pyrazine, 4; C13H9N = acridine, 5; η1-OC13H9N = acridone, 6; C4H10S = SEt2, 7; C4H8S = THT, 8; PCy3, 9) (top) and [{(IDipp)Cu}2(C2N3H3)2]2+2FAP− (10, bottom, C2N3H3 = 1,2,4, triazole) via substitution of hexamethylbenzene of [(IDipp)Cu(C6Me6)]+FAP− (IIa). | ||
The most straight forward probe to confirm the formation of the complexes presented in Scheme 1 is a shift of the methyl resonances of the coordinated hexamethylbenzene in the 1H NMR spectra of the reaction mixtures. After work-up, the 1H NMR signal of hexamethylbenzene is absent. In the 1H NMR spectrum of [(IDipp)Cu(NH3)]+FAP− (1) the four iPr methyl groups of the Dipp ligand gave rise to two doublets at 1.24 and 1.25 ppm in CDCl3 and the corresponding methine protons were observed as a septet at 2.49 ppm. The signals of both phenyl groups were split into doublets (CHmeta) and triplets (CHpara) and were detected at 7.34 and 7.56 ppm, respectively. Additionally, the signals of the olefinic protons of the backbone were observed at 7.24 ppm. Besides the typical signals of the NHC ligand, the three protons of the ammine ligand in 1 gave rise to a broad resonance at 2.05 ppm in CDCl3, which is similar to δ(1H) of [(IDipp)Cu(NH3)][BF4] with 2.26 ppm.14 There are neither significant differences in the 19F and 31P NMR spectra of the mer-FAP− anion nor in the 1H and 13C{1H} NMR spectra of the IDipp ligand in any of these compounds. Likewise, there is no mentionable variation in the chemical shifts in dependence of whether nitrogen, sulfur or phosphorous binds towards the copper center. Table 1 summarizes selected chemical shifts of the 1H and 13C{1H} NMR spectra of the carbene ligand in the complex cations of [(IDipp)Cu(LB)]+FAP−.
| δ (13C{1H}) N–C–N | δ (1H) aryl-CHpara | δ (1H) aryl-CHmeta | δ (1H) N–CH–CH–N | δ (1H) iPr–CH | δ (1H) iPr–CH3 | |
|---|---|---|---|---|---|---|
| 1 | 177.5 | 7.56 | 7.34 | 7.24 | 2.49 | 1.24/1.25 |
| 2 | 176.6 | 7.56 | 7.34 | 7.28 | 2.44 | 1.21/1.26 |
| 3 | 178.3 | 7.57 | 7.36 | 7.29 | 2.55 | 1.24/1.27 |
| 4 | 177.2 | 7.60 | 7.35 | 7.29 | 2.51 | 1.07/1.25 |
| 5 | 177.5 | 7.57 | 7.54 | 7.50 | 2.67 | 1.17/1.33 |
| 6 | 177.9 | 7.59 | 7.38 | 7.31 | 2.62 | 1.26/1.28 |
| 7 | 176.2 | 7.56 | 7.35 | 7.32 | 2.50 | 1.22/1.27 |
| 8 | 176.3 | 7.58 | 7.36 | 7.32 | 2.49 | 1.21/1.27 |
| 9 | 178.0 | 7.53 | 7.34 | 7.32 | 2.54 | 1.24/1.26 |
| 10 | 181.8 | 7.65 | 7.35 | 7.23 | 2.52 | 0.94–1.06/1.22 |
In addition to multinuclear NMR spectroscopy, the NHC copper FAP− salts were characterized by using IR spectroscopy, HRMS, as well as elemental analysis (see ESI†). Furthermore, single crystals of 1, 3, 5, 6, 7, and 10 suitable for X-ray diffraction (XRD) were obtained (Fig. 1, 2 and Table 2). The central copper atom in the complexes 1, 3, 5, and 7 is linearly coordinated by the IDipp ligand and the nitrogen or sulfur atom of the neutral donor ligand with C1–Cu–N1 angles of 177.19(15)° (1), 175.45(9)° (3), and 179.62(7)° (5) and a C1–Cu–S angle of 176.49(8)° (7). Interestingly, the acridone ligand in 6 coordinates via the oxygen atom. The C1–Cu–O angle of 169.58(9)° is slightly distorted from a linear arrangement and the Cu–O distance amounts to 1.8313(16) Å. In 1, 3, 5, 6, and 7 the distances between the carbenic carbon atom and the copper atom are very close (<3σ) (1: 1.882(3) Å, 3: 1.875(2) Å, 5: 1.8789(18) Å, 6: 1.863(2) Å, 7: 1.887(3) Å) and similar to d(Cu–C) in other copper NHC complexes reported by Nolan et al. (1.884(2)–1.956(10) Å).15 The Cu–N1 bond distances in 1 (1.908(3) Å) and 5 (1.9038(15) Å) are slightly longer than d(Cu–N1) found in 3 (1.8785(19) Å), but within the standard range of precedent Cu–N bonds in NHC complexes.16,17
 | ||
| Fig. 1 Molecular structures of the complex cations of [(IDipp)Cu(NH3)]+FAP− (1, top left), [(IDipp)Cu(C7H10N2)]+FAP− (3, top right), [(IDipp)Cu(C13H9N)]+FAP− (5, middle left), [(IDipp)Cu(η1-OC13H9N)]+FAP− (6, middle right) and [(IDipp)Cu(C4H10S)]+FAP− (7, bottom) in the solid state (ellipsoids set at the 50% probability level; Dipp substituents are shown as wire-and-stick models). Hydrogen atoms (except the ones of NH3 in 1) and co-crystallized solvent molecules in the crystal structures of 1, 3, and 6 are omitted for clarity. Only one of two independent molecules in the asymmetric unit of 1 and 3 are shown. Selected bond length and angles are given in Table 2 and Fig. S1–S5 in the ESI.† | ||
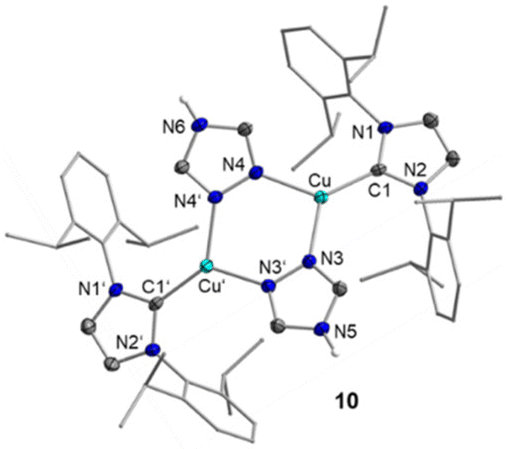 | ||
| Fig. 2 Molecular structure of the complex cation of [{(IDipp)Cu}2(C2N3H3)2]2+2FAP− (10) in the solid state (ellipsoids set at the 50% probability level; Dipp substituents are shown as wire-and-stick models). Hydrogen atoms except for the ones bound to N5 and N6 and a co-crystallized solvent molecule are omitted for clarity. Selected bond length and angles are collected in Table 2 and Fig. S6 in the ESI.† | ||
| Cu–C1 | Cu–N/O/S | C1–Cu–N/O/S | |
|---|---|---|---|
| 1 | 1.882(3) | 1.908(3) | 177.19(15) |
| 3 | 1.875(2) | 1.8785(19) | 175.45(9) |
| 5 | 1.8789(18) | 1.9038(15) | 179.62(7) |
| 6 | 1.863(2) | 1.8313(16) | 169.58(9) |
| 7 | 1.887(3) | 2.1705(8) | 176.49(8) |
| 10 | 1.937(6) | 2.038(5) | 128.50(19) |
| 2.035(4) | 132.12(19) | ||
| 11 | 1.900(4) | 1.927(3) | 176.68(15) |
| 12 | 1.890(3) | 1.906(2) | 170.45(12) |
The Cu–S bond distance of the central copper atom to sulfur in 7 of 2.1705(8) Å is similar to values observed for [(IMes)Cu(SSi(iPr)3)] (2.1336(4) Å) or [(IMes)Cu(SC(O)CH3)] (2.1483(9) Å).18 The dinuclear complex 10 crystallizes in the monoclinic space group P21/m with one dinuclear dicationic complex [{(IDipp)Cu}2(C2N3H3)2]2+, two mer-isomer FAP− counteranions, and three solvent molecules in the unit cell. Thus, a crystallographically imposed mirror plane is located perpendicular through the atoms N5 and N6. The Cu–C1 distance of 1.937(6) Å is slightly longer than the distances observed in the related mononuclear compounds discussed before (Table 2). Both [(IDipp)Cu]+ moieties are bridged by two 1,2,4-triazole ligands with angles of C1–Cu1–N3 128.50(19)° and C1–Cu1–N4 132.12(19)°, respectively, and bond distances of 2.038(5) and 2.035(4) Å between Cu and N3 or N4. These distances are longer compared to d(Cu–N) in 1, 3, and 5 (1.88–1.91 Å) due to the higher steric demand and the increased coordination number at copper in 10.
As the hexamethylbenzene ligand in the IDipp complex IIa is easily replaced by various 2 VE donor ligands, we expanded our study to the related cAAC-ligated complex [(cAACMe)Cu(C6Me6)]+FAP− (IIb). The reaction of IIb with DABCO or pyrazine resulted in the formation of dinuclear complexes [{(cAACMe)Cu}2(C6H12N2)]2+2FAP− (11) and [{(cAACMe)Cu}2 (C4H4N2)]2+2FAP− (12) in 62% (11) and 68% (12) yield, respectively. The reaction of IIb with DMAP or SEt2 afforded the mononuclear complexes [(cAACMe)Cu(C7H10N2)]+FAP− (13) and[(cAACMe)Cu(C4H10S)]+FAP− (14) in 57% (13) and 74% (14) yield (Scheme 2).
 | ||
| Scheme 2 Synthesis of [{(cAACMe)Cu}2(C6H12N2)]2+2FAP− (11), [{(cAACMe)Cu}2(C4H4N2)]2+2FAP− (12), [(cAACMe)Cu(C7H10N2)]+FAP− (13), and [(cAACMe)Cu(C4H10S)]+FAP− (14) via hexamethylbenzene substitution of [(cAACMe)Cu(C6Me6)]+FAP− (IIb). | ||
The complexes 11–14 were characterized by multinuclear NMR spectroscopy, elemental analysis, IR spectroscopy, and HRMS (13, 14). In analogy to the IDipp copper complexes 1–10 introduced above, the 19F and 31P NMR chemical shifts of mer-FAP− and the 1H and 13C{1H} chemical shifts of the cAACMe ligand of these compounds did not reveal significant differences. In case of 11 and 12, the signals for the carbene ligand were observed with a relative intensity of 2 with respect to the signals of DABCO or pyrazine. These findings match the results of the elemental analysis and X-ray diffraction, which are in accordance with dinuclear structures in solution and in the solid state. Single crystals of 11 and 12 suitable for XRD were obtained by diffusion of n-hexane into solutions of 11 or 12 in dichloromethane (Fig. 3 and Table 2). Both complexes 11 and 12 crystallize in the monoclinic space group P21/n and are located on an inversion center. Besides the coordination of the carbene ligand, the copper atom is coordinated to the nitrogen atom N1 of the DABCO or pyrazine ligand, respectively, with almost linear C1–Cu–N1 angles of 176.68(15)° (11) and 170.45(12)° (12). A comparison of both closely related structures shows that the Cu–C1 bond lengths (11: 1.900(4) Å; 12: 1.890(3) Å) and the Cu–N1 distances (11: 1.927(3) Å; 12: 1.906(2) Å) differ only marginally.
 | ||
| Fig. 3 Molecular structures of the complex cations of [{(cAACMe)Cu}2{C6H12N2}]2+2FAP− (11; left) and [{(cAACMe)Cu}2{C4H4N2}]2+2FAP− (12; right) in the solid state (ellipsoids set at the 50% probability level; Dipp ligands are shown as wire-and-stick models). Hydrogen atoms are omitted for clarity. Selected bond length and angles are collected in Table 2 and Fig. S7, S8 in the ESI.† | ||
Fluoride abstraction in the presence of neutral N- and O-donor ligands
Previous studies demonstrated that the percent buried volume model (%VBur), developed by Cavallo and colleagues, serves as a powerful descriptor for evaluating the steric properties of N-heterocyclic carbenes, phosphines, and related ligands in transition metal chemistry.19 Building on this approach, we introduced the LAB-Rep model, designed to assess steric repulsion between specific Lewis acid and base pairs.10 According to this model, the favored mer-trans isomer of (C2F5)3PF2–LB, with a percent buried volume of 67.7%Vbur, is considered to be very bulky, rendering the formation of LA/LB adducts with sterically hindered Lewis bases unlikely. Building on our previous studies,1 we employed the phosphorane (C2F5)3PF2 for the fluoride abstraction from fluoride complexes [(carbene)CuF], featuring IDipp, SIDipp, and cAACMe as carbene ligands, in the presence of a nucleophile. In the following we explore the question whether (C2F5)3PF2 can assist fluoride exchange reactions in complexes [(carbene)CuF] with 2 VE ligands, or if (i) a replacement of the neutral carbene ligand with the 2 VE ligand or (ii) the formation of Lewis acid/base pair of the 2 VE ligand and (C2F5)3PF2 prevails.The reaction of [(cAACMe)CuF] (Ic) with (C2F5)3PF2 in a solvent mixture of acetonitrile and dichloromethane afforded the acetonitrile adduct [(cAACMe)Cu(NCMe)]+FAP− (15), whereas the reaction of [(IDipp)CuF] (Ia) under similar conditions led to the three-coordinated complex [(IDipp)Cu(NCMe)2]+FAP− (16) (Scheme 3). This observation may seem contradictory to the assumption of the LAB-Rep model, as acetonitrile is considered a sterically non-demanding LB, and the formation of a phosphorane-acetonitrile adduct would be expected. However, the pronounced fluoride ion affinity of the phosphorane (405.5 kJ mol−1)1 seemingly favors fluoride abstraction over adduct formation, thus leading to the generation of [(IDipp)Cu(NCMe)2]+ and the weakly coordinating FAP− anion.
The sterically more demanding benzonitrile yielded the di-coordinated copper(I) complex [(IDipp)Cu(NCPh)]+FAP− (17). All reactions proceeded in good yields of 69% (15), 81% (16) and 82% (17), respectively (Scheme 3). Compounds 15–17 were fully characterized by 1H, 13C{1H}, 19F, and 31P NMR spectroscopy, IR spectroscopy, HRMS, and elemental analysis. The 19F and 31P NMR spectra confirm the formation of the mer-isomer of the FAP− anion, consistent with other previously reported FAP− complex salts.1,20
 | ||
| Scheme 3 Synthesis of [(carbene)Cu(LB)]+FAP− complexes 15–17via fluoride ion abstraction from [(carbene)CuF] using (C2F5)3PF2 in the presence of N-donor ligands. | ||
The IR spectrum of 17 displays a characteristic band at 2275 cm−1 for the NC stretching vibration, which is shifted 40 cm−1 to higher wavenumbers compared to non-coordinated benzonitrile (2234 cm−1).21 This shift can be rationalized by coordination to a Lewis-acidic center in conjunction with negligible π-back-bonding from copper to benzonitrile. Significant π-back-bonding would lead to a decrease in ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (CN). Higher wavenumbers for the CN stretch have been reported for other end-on coordinated Cu(I) complexes, previously.22 We discussed these phenomena for other cationic copper complexes previously. For example, the related complex [(IDipp)Cu(PhCCPh)]+FAP− shows a lack of π-back-bonding from the cationic [(IDipp)Cu]+ complex fragment to the alkyne, which was evident from very similar δ(13C) shifts of the CC unit of the free and coordinated alkyne (Δδalkyne ∼0.5 ppm), which indicates a rather weak copper–alkyne interaction.13
(CN). Higher wavenumbers for the CN stretch have been reported for other end-on coordinated Cu(I) complexes, previously.22 We discussed these phenomena for other cationic copper complexes previously. For example, the related complex [(IDipp)Cu(PhCCPh)]+FAP− shows a lack of π-back-bonding from the cationic [(IDipp)Cu]+ complex fragment to the alkyne, which was evident from very similar δ(13C) shifts of the CC unit of the free and coordinated alkyne (Δδalkyne ∼0.5 ppm), which indicates a rather weak copper–alkyne interaction.13
Single crystals suitable for XRD were obtained for compounds 16 and 17, and selected bonding parameters of the molecular structures (Fig. 4) are summarized in Table 3. The cationic complex [(IDipp)Cu(NCMe)2]+ in 16 exhibits a distorted trigonal-planar geometry at the metal atom with angels of 121.11(9)° (C1–Cu–N1), 131.08(10)° (C1–Cu–N2), and 107.71(9)° (N1–Cu–N2). In contrast, the copper atom in 17 adopts a linear environment with a C1–Cu–N1 angle of 178.03(7)°. Due to the reduced coordination number of copper in 17, the Cu–N1 distance of 1.8453(14) Å is significantly shorter than those observed in the tri-coordinated complex 16 (1.967(2) and 1.938(2) Å). The Cu–C1 bond length, however, is less sensitive to the coordination number, with 1.909(2) and 1.8848(15) Å in 16 and 17, respectively.
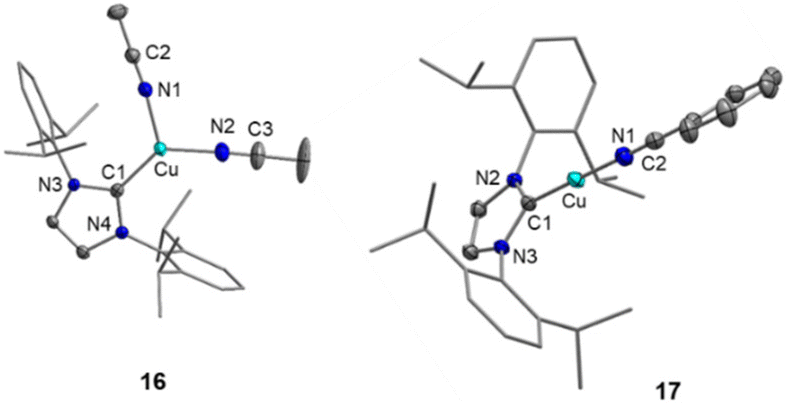 | ||
| Fig. 4 Molecular structures of the complex cations of [(IDipp)Cu(NCMe)2]+FAP− (16; left) and [(IDipp)Cu(NCPh)]+FAP− (17; right) in the solid state (ellipsoids set at the 50% probability level; Dipp substituents are shown as wire-and-stick models). Hydrogen atoms are omitted for clarity. Selected bond lengths and angles are collected in Table 3 and Fig. S9, S10 in the ESI.† | ||
| Cu–C1 | N/O | C1–Cu–N/O | |
|---|---|---|---|
| 16 | 1.909(2) | 1.967(2) | 121.11(9) |
| 1.938(2) | 131.08(10) | ||
| 17 | 1.8848(15) | 1.8453(14) | 178.03(7) |
| 19 | 1.893(3) | 1.935(3) | 171.88(13) |
| 20 | 1.882(3) | 1.942(3) | 175.47(12) |
| 21 | 1.8716(17) | 1.9492(15) | 175.97(7) |
| 23 | 1.8853(17) | 1.9038(15) | 174.25(6) |
| 24 | 1.878(2) | 1.916(2) | 172.2(1) |
| 26 | 1.873(3) | 2.054(2) | 142.16(11) |
| 1.876(3) | 2.027(2) | 145.85(11) | |
| 144.71(12) | |||
| 143.59(11) | |||
| 27 | 1.866(3) | 1.8624(19) | 167.61(9) |
Additionally, we explored the reaction of fluoride complexes Ia with (C2F5)3PF2 in the presence of aniline, and for Ic, with (C2F5)3PF2 in the presence of diphenylamine and the cationic copper complexes [(carbene)Cu(NH2Ph)]+FAP− (carbene = IDipp, 18; SIDipp, 19; cAACMe, 20) and [(IDipp)Cu(NHPh2)]+FAP− (21) were isolated in moderate to good yields (Scheme 4). The 1H NMR spectra of complexes 18–21 show characteristic broad singlets for the NH protons (18: 4.74 ppm, 19: 4.24 ppm, 20: 4.58 ppm, 21: 6.36 ppm), compared to free aniline (3.69 ppm)23 and diphenylamine (5.69 ppm).24 Further characterization was carried out using NMR and IR spectroscopy, HRMS and elemental analysis. SC-XRD confirmed the molecular structures of 19–21 (Fig. 5 and Table 3). The complex cations adopt linear structures in which copper is coordinated by both the carbene and the amino ligand, with C1–Cu–N1 angles of 171.88(13)° (19), 175.47(12)° (20), and 175.97(7)° (21). In 19 and 20, the Cu–N1 bond length (19: 1.935(3) Å; 20: 1.942(3) Å) is significantly shorter than d(Cu–N) reported for related aniline complexes [(dtbpe)Cu(NH2Ph)]+[BF4]− (2.010(2) Å; dtbpe = 1,2-bis(di-tertbutyl-phosphino)ethane)25 and [(JohnPhos)Cu (NH2Ph)][PF6] (1.964(2) Å; JohnPhos = 2-(di-tertbutyl-phosphino)-1,1′-biphenyle).26
 | ||
| Scheme 4 Synthesis of [(carbene)Cu(LB)]+FAP− complexes 18–20via fluoride ion abstraction from [(carbene)CuF] using (C2F5)3PF2 in the presence of amines. | ||
 | ||
| Fig. 5 Molecular structures of the complex cations of [(SIDipp)Cu(NH2Ph)]+FAP− (19, left), [(cAACMe)Cu(NH2Ph)]+FAP− (20, middle) and [(IDipp)Cu(NHPh2)]+FAP− (21, right) in the solid state (ellipsoids set at the 50% probability level; Dipp substituents are shown as wire-and-stick models). Hydrogen atoms except for those of the N atom of the aniline and diphenylamine ligands and a solvent molecule in the crystal structure of 19 are omitted for clarity. Only one of two independent cations in the asymmetric unit of 19 is shown. Selected bond lengths and angles are collected in Table 2 and Fig. S11–S13 in the ESI.† | ||
This difference may be attributed to the lower coordination number of copper in 19 and 20. The Cu–C1 distances in 19 (1.893(3) Å) and 20 (1.882(3) Å) are nearly identical within experimental error. Compound 21 displays slightly different bond lengths (Cu–N1: 1.9492(15) Å, Cu–C1: 1.8716(17) Å).
Moreover, the reactivity of the fluoride complexes [(IDipp)CuF] (Ia) and [(cAACMe)CuF] (Ic) towards (C2F5)3PF2 in the presence of one equivalent of pyridine or fluorinated pyridine derivates was investigated. Fluoride abstraction followed by pyridine coordination led to isolation and full characterization of the cationic complexes [(IDipp)Cu(NC5H5)]+FAP− (22), [(cAACMe)Cu(NC5H5)]+FAP− (23), [(IDipp)Cu(NC5H3F2)]+FAP− (24), and [(IDipp)Cu(NC5H2F3)]+FAP− (25) in yields of 68–80% (Scheme 5). The reaction of the highly fluorinated 2,3,5,6-tetrafluoropyridine with Ia in the presence of (C2F5)3PF2 did not result in [(IDipp)Cu(NC5HF4)]+FAP− but [{(IDipp)Cu}2]2+2FAP− was obtained. The low basicity and thus poor coordination ability of tetrafluoropyridine favors the coordination of the IDipp substituent of the carbene ligand, resulting in the dimeric dicationic complex [{(IDipp)Cu}2]2+.1 The formation of 22–25 was confirmed by 1H, 13C{1H}, 19F, and 31P NMR spectroscopy. A decreasing resonance frequency of the hydrogen and carbon nuclei in meta-position of pyridine with an increasing number of fluorine substituents was observed (δ(1H): 22: 7.77/7.50 ppm, 24: 7.03 ppm; 25: 6.75 ppm; δ(13C): 22: 147.5/126.8 ppm; 24: 108.3 ppm; 25: 98.1 ppm). In addition, single crystals of 23 and 24 suitable for XRD studies were obtained (Fig. 6).
 | ||
| Scheme 5 Synthesis of [(carbene)Cu(LB)]+FAP− complexes 22–25via fluoride ion abstraction from [(carbene)CuF] using (C2F5)3PF2 in the presence of (partially fluorinated) pyridine. | ||
 | ||
| Fig. 6 Molecular structures of the complex cations of [(cAACMe)Cu(NC5H5)]+FAP− (23; left) and [(IDipp)Cu(NC5H3F2)]+FAP− (24; right) in the solid state (ellipsoids set at the 50% probability level; Dipp substituents are shown as wire-and-stick models). Hydrogen atoms are omitted for clarity. Selected bond lengths and angles are collected in Table 3 and Fig. S14, S15 in the ESI.† | ||
The solid-state structures of 23 and 24 confirm the coordination of pyridine and the linear geometry at copper, with C1–Cu–N1 angles of 174.25(6)° (23) and 172.2(1)° (24). The Cu–C1 distances in 23 (1.8853(17) Å) and 24 (1.878(2) Å) are within the typical range compared to related complexes.13,27 In contrast, the Cu–N1 bond in 23 (1.9038(15) Å) is slightly shorter than in 24 (1.916(2) Å), which mirrors the reduced basicity of 2,6-difluoropyridine relative to pyridine. The Cu–C1 and Cu–N1 bond lengths in 23 are slightly longer than those reported by Steffen et al. for the related pyridine complex [(IDipp)Cu(NC5H5)][BF4] (Cu–C1: 1.872(2) Å; Cu–N1: 1.8900(18) Å).27
The stabilization of the carbene copper cations [(IDipp)Cu]+ by oxygen donor ligands is demonstrated by the formation of [{(IDipp)Cu(μ-ONC5H5)}2]2+2FAP− (26), [(IDipp)Cu(η1OCPh2)]+FAP− (27), and [(cAACMe)Cu(THF)]+FAP− (28) depicted in Scheme 6. Fluoride abstraction from [(IDipp)CuF] (Ia) with (C2F5)3PF2 in the presence of pyridine-N-oxide yielded the dinuclear pyridine-N-oxide-bridged complex 26 in 81% yield. The reaction of Ia with (C2F5)3PF2 in the presence of benzophenone afforded the mononuclear complex 27 in 79% yield and the reaction of [(cAACMe)CuF] (Ic) with the phosphorane in Et2O/THF afforded 28 in 68% yield. The 1H NMR analysis of 26 revealed broadening of the pyridine-N-oxide resonances at room temperature. However, at −36.5 °C, three well resolved resonances were observed at 7.71 (pyridine-aryl-CparaH), 7.47 (pyridine-aryl-CorthoH), and 7.38 ppm (pyridine-aryl-CmetaH) (see Fig. S118 in the ESI†). SC-XRD confirmed the dinuclear structure of 26 in the solid state, revealing a central Cu2O2 core, in which two [(IDipp)Cu]+ cations are bridged by two pyridine-N-oxide ligands (Fig. 7). These extended bond distances indicate the absence of significant Cu⋯Cu bonding interactions. The Cu–O–Cu angles in 26 are 108.47(11)° and 107.84(10)°, respectively. The benzophenone ligand in [(IDipp)Cu(η1-OCPh2)]+FAP− (27) adopts an end-on η1-coordination mode in the solid state and in solution. Such a shift in δ(13C) is a hallmark of benzophenone ligands coordinated via the oxygen atom in an end-on η1-fashion.28
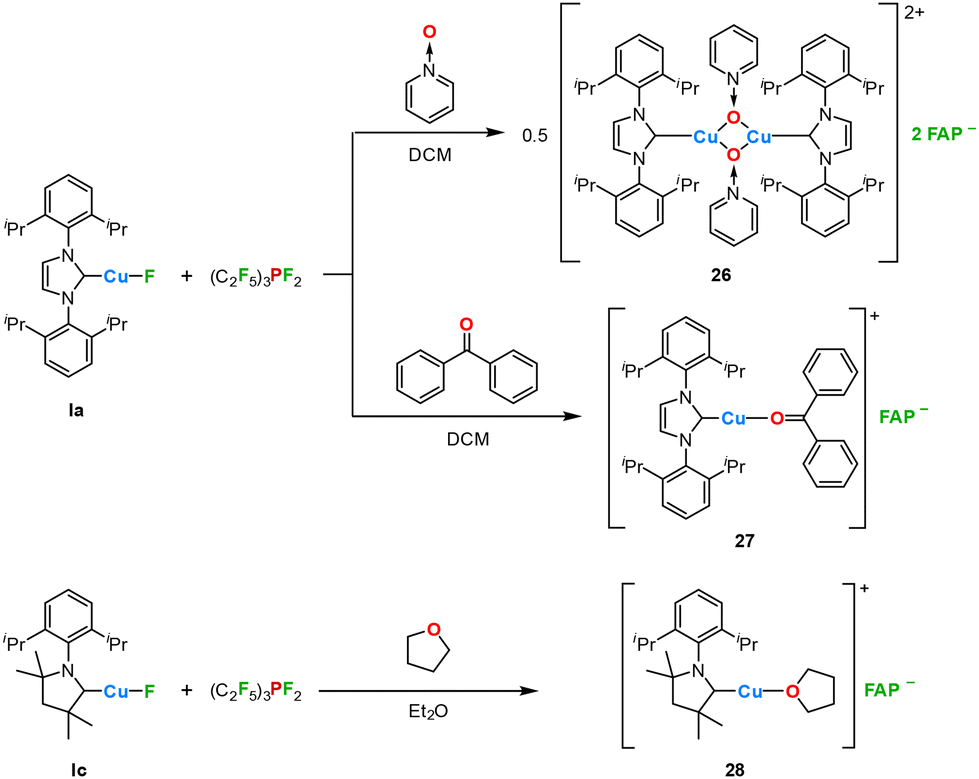 | ||
| Scheme 6 Synthesis of complexes 26–28via fluoride ion abstraction from [(carbene)CuF] using (C2F5)3PF2 in the presence of O-donor ligands. | ||
 | ||
| Fig. 7 Molecular structures of the complex cations of [{(IDipp)Cu(μ-ONC5H5)}2]2+2FAP− (26; left) and [(IDipp)Cu(η1-OCPh2)]+FAP− (27; right) in the solid state (ellipsoids set at the 50% probability level; Dipp substituents are shown as wire-and-stick models). Hydrogen atoms and two co-crystallized solvent molecules in the crystal structure of 26 are omitted for clarity. Selected bond lengths and angles are collected in Table 3 and Fig. S16, S17 in the ESI.† | ||
In contrast, a side-on η2-coordination of the OC moiety would induce a shift to lower resonance frequency,29,30 as previously observed for the nickel complexes [(NHC)2Ni(η2-OCPh2)] (NHC = IiPr, IMes) which we have reported earlier.30 In these nickel complexes, the [(NHC)2Ni] unit exhibits strong π-back-donation, favoring side-on coordination. SC-XRD experiments of 27 further confirm the end-on η1-coordination mode in the solid state (Fig. 7). The η1-hapticity of the oxygen atom suggests minimal or no π-back-donation from the [(IDipp)Cu]+ cation, which is consistent with the contracted and energetically low-lying d-orbitals characteristic for the closed-shell copper(I) center. The O–C2 bond length in 27 (1.246(3) Å) remains nearly unchanged compared to free benzophenone (cf. distances of benzophenone: 1.23(1) Å),31 indicating negligible electron density donation from copper to the carbonyl moiety. The Cu–O bond distance of 1.8624(19) Å is slightly longer than those observed in the alkoxide complexes [(IDipp)Cu(OX)] (X = Et: 1.799(3) Å;16tBu: 1.8104(13) Å;32 CH(Me)Ph: 1.794(3) Å (ref. 33)) or carboxylato complexes [(IDipp)Cu(OX)] (X = C(O)H: 1.848(2) Å;34 C(O)Me:1.850(3) Å (ref. 35)), as expected for a ketone ligand. The Cu–O–C2 bond angle of 133.48(18)° is consistent with oxygen lone-pair coordination to the [(IDipp)Cu]+ center.
Conclusions
Ligand exchange reactions of [(IDipp)Cu(C6Me6)]+FAP− (IIa) were studied in some detail leading to the copper FAP− salts [(IDipp)Cu(LB)]+FAP− (LB = NH3, 1; C6H12N2 = DABCO, 2; C7H10N2 = DMAP, 3; C4H4N2 = pyrazine, 4; C13H9N = acridine, 5; η1-OC13H9N = acridone, 6; C4H10S = SEt2, 7; C4H8S = THT, 8; PCy3, 9) as well as the dinuclear 1,2,4-triazole complex [{(IDipp)Cu}2(C2N3H3)2]2+2FAP− (10). Similarly, [(cAACMe)Cu(C6Me6)]+FAP− (IIb) was used for the synthesis of [(cAACMe)Cu(LB)]+FAP− (LB = C7H10N2, 13; C4H10S, 14) via replacement of the C6Me6 ligand. The reactions of DABCO or pyrazine with IIa afforded [{(cAACMe)Cu}2(C6H12N2)]2+2FAP− (11) and [{(cAACMe)Cu}2(C4H4N2)]2+2FAP− (12). In all cases the cationic copper(I) complexes [(carbene)Cu(LB)]+ were stabilized by the weakly coordinating tris(pentafluoroethyl)trifluorophosphate anion (FAP− anion, [(C2F5)3PF3]−). Furthermore, we extended the use of the readily available Lewis acid tris(pentafluoroethyl)difluorophosphorane (C2F5)3PF2 as fluoride abstraction reagent for the generation of cationic copper(I) complexes. The reactions of (C2F5)3PF2 with [(IDipp)CuF] (Ia), [(SIDipp)CuF] (Ib), or [(cAACMe)CuF] (Ic) in the presence of different Lewis bases (LB), i.e. nitriles, amines, fluorinated and non-fluorinated pyridines, and oxygen donor ligands, were studied. The complex salts [(IDipp)Cu(LB)]+FAP− (LB = (NCMe)2, 16; NCPh, 17; NH2Ph, 18; NHPh2, 21; NC5H5, 22; NC5H3F2, 24; NC5H2F3, 25; η1-OCPh2, 27), [(SIDipp) Cu(NH2Ph)]+FAP− (19), and [(cAACMe)Cu(LB)]+FAP− (LB = NCMe, 15; NH2Ph, 20; NC5H5, 23; THF, 28) were isolated and characterized. In addition, the dinuclear complex [{(IDipp)Cu(μ-ONC5H5)}2]2+2FAP− (26) was isolated. This approach was investigated for different 2 VE nitrogen and oxygen donor ligands. In none of these cases the carbene ligand was replaced with the 2 VE ligand employed, the formation of Lewis acid/base pair of the 2 VE ligand and (C2F5)3PF2 was never observed in the presence of the copper fluoride, and the FAP− anion coordinated in none of these cases to the copper cations. Hence, mixtures of [(carbene)CuF] and (C2F5)3PF2 serve as synthon for [(carbene)Cu]+.
Experimental
General considerations
All reactions and subsequent manipulations involving organometallic reagents were carried out under an argon atmosphere by using standard Schlenk techniques or in a Glovebox (Innovative Technology Inc., and MBraun Uni Lab).36 All reactions were performed in oven-dried glassware. Elemental analyses were performed in the microanalytical laboratory of the Institute of Inorganic Chemistry of the University Würzburg with an Elementar vario micro cube. High-resolution mass spectroscopy (HRMS) was performed on a Thermo Scientific Exactive Plus mass spectrometer, equipped with an Orbitrap Mass Analyzer. NMR spectra were recorded on a Bruker Avance 400 (1H, 400.1 MHz; 13C, 100.6 MHz; 19F, 376.8 MHz; 31P, 162.0 MHz), a Bruker Avance 500 (1H, 500.1 MHz; 13C, 125.8 MHz; 19F, 470.5 MHz; 31P, 202.4 MHz) and a Bruker Avance 600 (1H, 600.2 MHz; 13C, 150.9 MHz; 19F, 564.7 MHz; 31P, 242.9 MHz) spectrometer using CD2Cl2 or CDCl3 as solvent. Assignment of the 1H NMR and 13C{1H} NMR data was supported by 1H, 1H and 13C{1H},1H correlation experiments. 13C NMR spectra were recorded broad-band proton-decoupled (13C{1H}) at 298 K, if not otherwise noted. Chemical shifts are listed in parts per million (ppm), reported relative to TMS and were calibrated against residual solvent signals (δ(1H): CDHCl2 5.32, CHCl3 7.26; δ(13C): CD2Cl2 53.84, CDCl3 77.16)37 or external CFCl3 (δ(19F): 0) and 85% H3PO4 (δ(31P): 0). If not otherwise noted 19F and 31P NMR spectra were not proton decoupled. Coupling constants are quoted in Hertz. Infrared spectra were recorded under an argon atmosphere on solid samples on a Bruker Alpha FT-IR spectrometer by using an ATR unit at room temperature. Values are given in cm−1. All solvents for synthetic reactions were HPLC grade, further treated to remove traces of water using an Innovative Technology Inc. Pure-Solv Solvent Purification System. CD2Cl2 and CDCl3 were purchased from Sigma-Aldrich and stored over molecular sieve. The compounds [(IDipp)CuF] (Ia),38 [(SIDipp)CuF] (Ib),13 [(cAACMe)CuF] (Ic),13 [(IDipp)Cu(C6Me6)]+FAP− (IIa)1 and [(cAACMe)Cu(C6Me6)]+FAP− (IIb)13 were prepared according to literature procedures. Commercially available (C2F5)3PF2 was used or the phosphorane was synthesized via electrochemical fluorination (ECF) starting from triethyl phosphine as reported in the literature.39 All other starting materials were purchased from commercial sources and used without further purification.Preparation of compounds
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) C13H9N)]+FAP− (6).
A solution of IIa (120 mg, 113 μmol) and acridone (22.1 mg, 113 μmol) in dichloro methane (5 mL) was stirred for 4 h at room temperature. All volatiles were removed under reduced pressure and the remaining solid was suspended in n-hexane (10 mL) and the product was filtered off. The product was washed with n-hexane (2 × 5 mL) and dried in vacuo to yield 6 (108 mg, 98.9 μmol, 87%) as a green solid. Single crystals of 6 suitable for X-ray diffraction were obtained by diffusion of n-hexane into a solution of 6 in chloroform. 1H NMR (400.1 MHz, CDCl3, 298 K): δ [ppm] = 1.26 (d, 12 H, 3JHH = 7.7 Hz, iPr–CH3), 1.28 (d, 12 H, 3JHH = 7.7 Hz, iPr–CH3), 2.62 (sept, 4 H, 3JHH = 6.2 Hz, iPr–CH), 7.13 (t, 2 H, 3JHH = 6.5 Hz, acridone–CH), 7.32 (s, 2 H, N–CH–CH–N), 7.38 (d, 4 H, 3JHH = 7.8 Hz, IDipp-aryl-CmetaH), 7.59 (t, 2 H, 3JHH = 7.6 Hz, IDipp-aryl-CparaH), 7.67 (d, 2 H, acridone–CH), 7.76 (t, 3 H, acridone–CH), 8.00 (d, 2 H, 3JHH = 6.2 Hz, acridone–CH), 9.71 (s, 1 H, acridone–NH); 13C{1H} NMR (125.8 MHz, CDCl3, 298 K): δ [ppm] = 24.0 (iPr–CH3), 25.0 (iPr–CH3), 29.0 (iPr–CH), 118.9 (acridone–CH), 123.5 (acridone–CH), 124.3 (N–CH–CH–N), 124.7 (IDipp-aryl-Cmeta), 131.3 (IDipp-aryl-Cpara), 134.2 (IDipp-aryl-Cipso), 135.5 (acridone–CH), 140.9 (acridone–C), 145.8 (IDipp-aryl-Cortho), 177.9 (N–C–N); one out of four signals for the acridone–CH-groups, one out of two signals for the quaternary carbon atoms as well as the signal for the carbonyl carbon atom were not detected; 19F NMR (376.8 MHz, CDCl3, 298 K): δ [ppm] = −43.5 (dm, 1 F, 1JPF = 889 Hz, PF), −80.1 (m, 3 F, CF3), −81.8 (m, 6 F, CF3), −87.9 (dm, 2 F, 1JPF = 902 Hz, PF2), −115.6 (dm, 2 F, 2JPF = 78 Hz, CF2), −115.8 (dm, 4 F, 2JPF = 98 Hz, CF2); 31P NMR (202.4 MHz, CDCl3, 298 K): δ [ppm] = −145.8 (tdm, 1JPF = 902 Hz, 1JPF = 889 Hz); IR ([cm−1]): 3390 (w), 3179 (vw), 3076 (vw), 2965 (w), 2929 (w), 2872 (vw), 1629 (m), 1593 (w), 1531 (m), 1468 (m), 1414 (w), 1386 (vw), 1365 (vw), 1351 (w), 1311 (m), 1297 (m), 1261 (w), 1216 (vs), 1184 (s), 1162 (s), 1137 (s), 1098 (s), 1087 (s), 1062 (m), 1027 (vw), 968 (m), 937 (w), 862 (vw), 812 (s), 801 (s), 757 (m), 744 (m), 719 (m), 670 (w), 660 (w), 636 (m), 617 (s), 580 (m), 549 (m), 535 (m), 505 (w), 493 (w), 440 (w), 429 (w), 421 (vw); HRMS (ESI) m/z [M]+ calcd for C40H45CuN3O: 646.2859, found: 646.2843; m/zFAP− calcd for C6F18P: 444.9450, found: 444.9451; elemental analysis calcd (%) for C46H45CuF18N3OP: C 50.58, H 4.15, N 3.85; found: C 50.13, H 4.38, N 3.29.
C13H9N)]+FAP− (6).
A solution of IIa (120 mg, 113 μmol) and acridone (22.1 mg, 113 μmol) in dichloro methane (5 mL) was stirred for 4 h at room temperature. All volatiles were removed under reduced pressure and the remaining solid was suspended in n-hexane (10 mL) and the product was filtered off. The product was washed with n-hexane (2 × 5 mL) and dried in vacuo to yield 6 (108 mg, 98.9 μmol, 87%) as a green solid. Single crystals of 6 suitable for X-ray diffraction were obtained by diffusion of n-hexane into a solution of 6 in chloroform. 1H NMR (400.1 MHz, CDCl3, 298 K): δ [ppm] = 1.26 (d, 12 H, 3JHH = 7.7 Hz, iPr–CH3), 1.28 (d, 12 H, 3JHH = 7.7 Hz, iPr–CH3), 2.62 (sept, 4 H, 3JHH = 6.2 Hz, iPr–CH), 7.13 (t, 2 H, 3JHH = 6.5 Hz, acridone–CH), 7.32 (s, 2 H, N–CH–CH–N), 7.38 (d, 4 H, 3JHH = 7.8 Hz, IDipp-aryl-CmetaH), 7.59 (t, 2 H, 3JHH = 7.6 Hz, IDipp-aryl-CparaH), 7.67 (d, 2 H, acridone–CH), 7.76 (t, 3 H, acridone–CH), 8.00 (d, 2 H, 3JHH = 6.2 Hz, acridone–CH), 9.71 (s, 1 H, acridone–NH); 13C{1H} NMR (125.8 MHz, CDCl3, 298 K): δ [ppm] = 24.0 (iPr–CH3), 25.0 (iPr–CH3), 29.0 (iPr–CH), 118.9 (acridone–CH), 123.5 (acridone–CH), 124.3 (N–CH–CH–N), 124.7 (IDipp-aryl-Cmeta), 131.3 (IDipp-aryl-Cpara), 134.2 (IDipp-aryl-Cipso), 135.5 (acridone–CH), 140.9 (acridone–C), 145.8 (IDipp-aryl-Cortho), 177.9 (N–C–N); one out of four signals for the acridone–CH-groups, one out of two signals for the quaternary carbon atoms as well as the signal for the carbonyl carbon atom were not detected; 19F NMR (376.8 MHz, CDCl3, 298 K): δ [ppm] = −43.5 (dm, 1 F, 1JPF = 889 Hz, PF), −80.1 (m, 3 F, CF3), −81.8 (m, 6 F, CF3), −87.9 (dm, 2 F, 1JPF = 902 Hz, PF2), −115.6 (dm, 2 F, 2JPF = 78 Hz, CF2), −115.8 (dm, 4 F, 2JPF = 98 Hz, CF2); 31P NMR (202.4 MHz, CDCl3, 298 K): δ [ppm] = −145.8 (tdm, 1JPF = 902 Hz, 1JPF = 889 Hz); IR ([cm−1]): 3390 (w), 3179 (vw), 3076 (vw), 2965 (w), 2929 (w), 2872 (vw), 1629 (m), 1593 (w), 1531 (m), 1468 (m), 1414 (w), 1386 (vw), 1365 (vw), 1351 (w), 1311 (m), 1297 (m), 1261 (w), 1216 (vs), 1184 (s), 1162 (s), 1137 (s), 1098 (s), 1087 (s), 1062 (m), 1027 (vw), 968 (m), 937 (w), 862 (vw), 812 (s), 801 (s), 757 (m), 744 (m), 719 (m), 670 (w), 660 (w), 636 (m), 617 (s), 580 (m), 549 (m), 535 (m), 505 (w), 493 (w), 440 (w), 429 (w), 421 (vw); HRMS (ESI) m/z [M]+ calcd for C40H45CuN3O: 646.2859, found: 646.2843; m/zFAP− calcd for C6F18P: 444.9450, found: 444.9451; elemental analysis calcd (%) for C46H45CuF18N3OP: C 50.58, H 4.15, N 3.85; found: C 50.13, H 4.38, N 3.29.
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) CMe)]+FAP− (15).
The phosphorane (C2F5)3PF2 (77.0 μL, 327 μmol) was added at room temperature to a solution of Ic (120 mg, 326 μmol) in acetonitrile (5 mL). The reaction mixture was stirred for 2 h at room temperature. All volatiles were removed under reduced pressure and the remaining solid was suspended in n-hexane (5 mL) and the product was filtered off. The product was washed with n-hexane (5 mL) and dried in vacuo to yield 15 (188 mg, 225 μmol, 69%) as a colorless solid. 1H NMR (500.1 MHz, CDCl3, 298 K): δ [ppm] = 1.19 (d, 6 H, 3JHH = 6.8 Hz, iPr–CH3), 1.34 (d, 6 H, 3JHH = 6.8 Hz, iPr–CH3), 1.38 (s, 6 H, N–C(CH3)2), 1.42 (s, 6 H, Cu–C–C(CH3)2), 2.10 (s, 2 H, CH2), 2.22 (s, 3 H, NC–CH3), 2.75 (sept, 2 H, 3JHH = 6.8 Hz, iPr–CH), 7.31 (d, 2 H, 3JHH = 7.8 Hz, cAACMe-aryl-CmetaH), 7.48 (t, 1 H, 3JHH = 7.8 Hz, cAACMe-aryl-CparaH); 13C{1H}-NMR (125.8 MHz, CDCl3, 298 K): δ [ppm] = 2.1 (NC–CH3), 22.3 (iPr–CH3), 27.3 (iPr–CH3), 28.0 (Cu–C–C(CH3)2), 29.18 (iPr–CH/N–C(CH3)2), 29.20 (iPr–CH/N–C(CH3)2), 49.2 (CH2), 54.2 (Cu–C–C(CH3)2), 83.1 (N–C(CH3)2), 118.2 (NC), 125.2 (cAACMe-aryl-Cmeta), 130.7 (cAACMe-aryl-Cpara), 133.7 (cAACMe-aryl-Cipso), 145.0 (cAACMe-aryl-Cortho), 245.5 (N–C–Cu); 19F-NMR (470.5 MHz, CDCl3, 298 K): δ [ppm] = −45.0 (dm, 1 F, 1JPF = 892 Hz, PF), −80.1 (m, 3 F, CF3), −81.8 (m, 6 F, CF3), −88.4 (dm, 2 F, 1JPF = 903 Hz, PF2), −115.8 (dm, 2 F, 2JPF = 83 Hz, CF2), −116.3 (dm, 4 F, 2JPF = 98 Hz, CF2); 31P-NMR (202.4 MHz, CDCl3, 298 K): δ [ppm] = −147.3 (tdm, 1JPF = 903 Hz, 1JPF = 892 Hz); IR (ATR [cm−1]): 2975 (w), 2948 (w), 2873 (vw), 2323 (vw), 2297 (vw), 1588 (vw), 1524 (vw), 1460 (w), 1388 (vw), 1372 (vw), 1364 (vw), 1309 (w), 1296 (w), 1210 (s), 1180 (s), 1135 (s), 1124 (s), 1097 (m), 1068 (m), 962 (m), 932 (vw), 808 (s), 762 (m), 722 (s), 618 (vs), 580 (m), 533 (m), 494 (w), 467 (vw), 438 (vw), 428 (w); HRMS (ESI) m/z [M]+ calcd for C22H34CuN2: 389.2018, found: 389.2002; m/zFAP− calcd for C6F18P: 444.9450, found: 444.9444; elemental analysis calcd (%) for C28H34CuF18N2P: (gefunden): C 40.27, H 4.10, N 3.35; found: C 40.81, H 4.26, N 3.51.
CMe)2]+FAP− (16).
The phosphorane (C2F5)3PF2 (46.5 μL, 197 μmol) was added at room temperature to a solution of Ia (93.0 mg, 197 μmol) in acetonitrile (3 mL). The reaction mixture was stirred for 1.5 h at room temperature. All volatiles were removed under reduced pressure and the product was dried in vacuo to yield 16 (157 mg, 160 μmol, 81%) as a colorless solid. Single crystals of 16 suitable for X-ray diffraction were obtained by vapor diffusion of n-pentane into a solution of 16 in toluene. 1H NMR (500.1 MHz, CDCl3, 298 K): δ [ppm] = 1.23 (d, 12 H, 3JHH = 6.9 Hz, iPr–CH3) overlap with 1.25 (d, 12 H, 3JHH = 6.9 Hz, iPr–CH3), 1.99 (s, 6 H, NC-CH3), 2.51 (sept, 4 H, 3JHH = 6.9 Hz, iPr–CH), 7.27 (s, 2 H, N–CH–CH–N), 7.36 (d, 4 H, 3JHH = 7.8 Hz, IDipp-aryl-CmetaH), 7.55 (t, 2 H, 3JHH = 7.8 Hz, IDipp-aryl-CparaH); 13C{1H} NMR (125.8 MHz, CD2Cl2, 298 K): δ [ppm] = 2.3 (NC–CH3), 24.0 (iPr–CH3), 24.9 (iPr–CH3), 29.1 (iPr–CH), 117.3 (NC), 124.4 (N–CH–CH–N), 124.6 (IDipp-aryl-Cmeta), 131.1 (IDipp-aryl-Cpara), 134.6 (IDipp-aryl-Cipso), 146.2 (IDipp-aryl-Cortho), 178.3 (N–C–N); 19F NMR (470.5 MHz, CD2Cl2, 298 K): δ [ppm] = −45.1 (dm, 1 F, 1JPF = 889 Hz, PF), −80.6 (m, 3 F, CF3), −82.3 (m, 6 F, CF3), −88.6 (dm, 2 F, 1JPF = 902 Hz, PF2), −116.1 (dm, 2 F, 2JPF = 83 Hz, CF2), −116.7 (dm, 4 F, 2JPF = 98 Hz, CF2); 31P NMR (202.4 MHz, CD2Cl2, 298 K): δ [ppm] = −147.7 (tdm, 1JPF = 902 Hz, 1JPF = 889 Hz); IR ([cm−1]): 3186 (vw), 3151 (vw), 2967 (m), 2931 (w), 2871 (w), 2314 (vw), 1681 (vw), 1580 (vw), 1552 (vw), 1471 (m), 1408 (w), 1385 (w), 1365 (w), 1329 (w), 1310 (m), 1258 (vw), 1213 (vs), 1189 (vs), 1138 (s), 1127 (s), 1088 (s), 1061 (m), 967 (m), 949 (w), 937 (w), 806 (s), 763 (s), 742 (m), 724 (vs), 695 (w), 636 (m), 617 (vs), 602 (m), 580 (m), 560 (vw), 534 (w), 506 (w), 495 (w), 443 (w), 429 (w), 422 (w); HRMS (ESI) m/z [M − CH3CN]+ calcd for. C29H39CuN3: 492.2440, found: 492.2427; m/zFAP− calcd for C6F18P: 444.9450, found: 444.9458; elemental analysis calcd (%) for C37H42CuF18N4P: C 45.38, H 4.32, N 5.72; found: C 45.75, H 4.38, N 5.12.
CPh)]+FAP− (17).
The phosphorane (C2F5)3PF2 (75.0 μL, 319 μmol) was added at room temperature to a solution of Ia (150 mg, 318 μmol) and benzonitrile (32.6 μL, 319 μmol) in dichloro methane (5 mL). The reaction mixture was stirred for 3 h at room temperature. All volatiles were removed under reduced pressure and the remaining solid was suspended in n-hexane (5 mL) and the product was filtered off. The product was washed with n-hexane (2 × 5 mL) and dried in vacuo to yield 17 (235 mg, 262 μmol, 82%) as a colorless solid. Single crystals of 17 suitable for X-ray diffraction were obtained by vapor diffusion of n-pentane into a saturated solution of 17 in toluene. 1H NMR (500.1 MHz, CDCl3, 298 K): δ [ppm] = 1.26 (d, 12 H, 3JHH = 6.9 Hz, iPr–CH3), 1.27 (d, 12 H, 3JHH = 6.9 Hz, iPr–CH3), 2.51 (sept, 4 H, 3JHH = 6.9 Hz, iPr–CH), 7.30 (s, N–CH–CH–N), 7.37 (d, 4 H, 3JHH = 7.8 Hz, IDipp-aryl-CmetaH), 7.54 (tbr, 2 H, 3JHH = 7.6 Hz, benzonitrile-aryl-CmetaH), 7.58 (t, 2 H, 3JHH = 7.8 Hz, IDipp-aryl-CparaH), 7.65 (dbr, 2 H, 3JHH = 7.6 Hz, benzonitrile-aryl-CorthoH), 7.77 (tbr, 1 H, 3JHH = 7.6 Hz, benzonitrile-aryl-CparaH); 13C{1H} NMR (125.8 MHz, CDCl3, 298 K): δ [ppm] = 23.8 (iPr–CH3), 25.2 (iPr–CH3), 28.9 (iPr–CH), 106.9 (benzonitrile-aryl-Cipso), 118.3 (NC), 124.71 (N–CH–CH–N), 124.74 (IDipp-aryl-Cmeta), 130.0 (benzonitrile-aryl-Cmeta), 131.4 (IDipp-aryl-Cpara), 133.6 (benzonitrile-aryl-Cortho), 133.7 (IDipp-aryl-Cipso), 136.3 (benzonitrile-aryl-Cpara), 145.7 (IDipp-aryl-Cortho), 175.5 (N–C–N); 19F NMR (470.5 MHz, CDCl3, 298 K): δ [ppm] = −45.3 (dm, 1 F, 1JPF = 892 Hz, PF), −80.2 (m, 3 F, CF3), −81.8 (m, 6 F, CF3), −88.6 (dm, 2 F, 1JPF = 903 Hz, PF2), −115.9 (dm, 2 F, 2JPF = 82 Hz, CF2), −116.4 (dm, 4 F, 2JPF = 98 Hz, CF2); 31P NMR (202.4 MHz, CDCl3, 298 K): δ [ppm] = −147.3 (tdm, 1JPF = 903 Hz, 1JPF = 892 Hz); IR ([cm−1]): 3186 (vw), 3134 (vw), 2967 (w), 2927 (w), 2874 (w), 2275 (w), 1597 (w), 1547 (vw), 1468 (w), 1414 (w), 1389 (vw), 1367 (vw), 1312 (m), 1215 (vs), 1184 (s), 1138 (s), 1125 (m), 1111 (w), 1089 (m), 1061 (w), 963 (m), 934 (w), 805 (s), 758 (s), 743 (m), 716 (vs), 702 (w), 681 (w), 636 (w), 618 (vs), 601 (w), 581 (w), 551 (w), 534 (w), 505 (w), 494 (vw), 441 (w), 429 (w), 420 (w); HRMS (ESI) m/z [M]+ calcd for C34H41CuN3: 554.2597, found: 554.2583; m/zFAP− calcd for C6F18P: 444.9450, found: 444.9426; elemental analysis calcd (%) for C40H41CuF18N3P: C 48.03, H 4.13, N 4.20; found: C 48.23, H 4.07, N 4.42.
CPh2)]+FAP− (27).
The phosphorane (C2F5)3PF2 (75.0 μL, 319 μmol) was added at room temperature to a solution of Ia (150 mg, 318 μmol) and benzophenone (58.0 mg, 318 μmol) in dichloro methane (5 mL). The reaction mixture was stirred for 2 h at room temperature. All volatiles were removed under reduced pressure and the remaining solid was suspended in n-hexane (5 mL) and the product was filtered off. The product was washed with n-hexane (2 × 5 mL) and dried in vacuo to yield 27 (272 mg, 252 μmol, 79%) as an off-white solid. Single crystals of 27 suitable for X-ray diffraction were obtained by diffusion of n-hexane into a solution of 27 in 1,2-difluorobenzene. 1H NMR (500.1 MHz, CDCl3, 298 K): δ [ppm] = 1.13 (d, 12 H, 3JHH = 6.8 Hz, iPr–CH3), 1.24 (d, 12 H, 3JHH = 6.8 Hz, iPr–CH3), 2.50 (sept, 4 H, 3JHH = 6.8 Hz, iPr–CH), 7.28 (s, 2 H, N–CH–CH–N), 7.34 (d, 4 H, 3JHH = 7.8 Hz, IDipp-aryl-CmetaH), 7.39 (m, 4 H, benzophenone-aryl-CmetaH), 7.52 (dd, 4 H, 3JHH = 8.4 Hz, 4JHH = 1.2 Hz, benzophenone-aryl-CorthoH), 7.59 (t, 2 H, 3JHH = 7.8 Hz, IDipp-aryl-CparaH), 7.70 (tt, 2 H, 3JHH = 7.5 Hz, 4JHH = 1.2 Hz, benzophenone-aryl-CparaH); 13C{1H} NMR (125.8 MHz, CDCl3, 298 K): δ [ppm] = 23.8 (iPr–CH3), 25.0 (iPr–CH3), 28.9 (iPr–CH), 124.7 (N–CH–CH–N/IDipp-aryl-Cmeta), 124.8 (N–CH–CH–N/IDipp-aryl-Cmeta), 129.4 (benzophenone-aryl-Cmeta), 131.1 (benzophenone-aryl-Cortho), 131.3 (IDipp-aryl-Cpara), 134.0 (IDipp-aryl-Cipso), 135.6 (benzophenone-aryl-Cipso), 136.0 (benzophenone-aryl-Cpara), 145.7 (IDipp-aryl-Cortho), 175.7 (N–C–N), 206.3 (OC); 19F NMR (470.5 MHz, CDCl3, 298 K): δ [ppm] = −45.3 (dm, 1 F, 1JPF = 891 Hz, PF), −80.2 (m, 3 F, CF3), −81.8 (m, 6 F, CF3), −88.7 (dm, 2 F, 1JPF = 903 Hz, PF2), −115.9 (dm, 2 F, 2JPF = 83 Hz, CF2), −116.4 (dm, 4 F, 2JPF = 98 Hz, CF2); 31P NMR (202.4 MHz, CDCl3, 298 K): δ [ppm] = −147.2 (tdm, 1JPF = 903 Hz, 1JPF = 891 Hz); IR ([cm−1]): 3137 (vw), 2964 (w), 2927 (w), 2872 (w), 1591 (w), 1558 (m), 1493 (vw), 1461 (w), 1451 (w), 1416 (w), 1388 (vw), 1366 (vw), 1333 (m), 1293 (m), 1212 (vs), 1177 (vs), 1142 (s), 1126 (s), 1098 (s), 1061 (m), 1026 (vw), 999 (vw), 972 (m), 958 (m), 926 (w), 850 (vw), 817 (s), 810 (s), 761 (s), 746 (m), 721 (s), 706 (s), 681 (w), 651 (w), 637 (w), 617 (vs), 580 (m), 532 (w), 496 (w), 438 (w), 429 (w), 411 (vw); HRMS (ESI) m/z [M]+ calcd for C40H46CuN2O: 633.2906, found: 633.2888; m/zFAP− calcd for C6F18P: 444.9450, found: 444.9430; elemental analysis calcd (%) for C46H46CuF18N2OP: C 51.19, H 4.30, N 2.60; found: C 51.79, H 4.30, N 2.88.
CMe)]+FAP− (15).
The phosphorane (C2F5)3PF2 (77.0 μL, 327 μmol) was added at room temperature to a solution of Ic (120 mg, 326 μmol) in acetonitrile (5 mL). The reaction mixture was stirred for 2 h at room temperature. All volatiles were removed under reduced pressure and the remaining solid was suspended in n-hexane (5 mL) and the product was filtered off. The product was washed with n-hexane (5 mL) and dried in vacuo to yield 15 (188 mg, 225 μmol, 69%) as a colorless solid. 1H NMR (500.1 MHz, CDCl3, 298 K): δ [ppm] = 1.19 (d, 6 H, 3JHH = 6.8 Hz, iPr–CH3), 1.34 (d, 6 H, 3JHH = 6.8 Hz, iPr–CH3), 1.38 (s, 6 H, N–C(CH3)2), 1.42 (s, 6 H, Cu–C–C(CH3)2), 2.10 (s, 2 H, CH2), 2.22 (s, 3 H, NC–CH3), 2.75 (sept, 2 H, 3JHH = 6.8 Hz, iPr–CH), 7.31 (d, 2 H, 3JHH = 7.8 Hz, cAACMe-aryl-CmetaH), 7.48 (t, 1 H, 3JHH = 7.8 Hz, cAACMe-aryl-CparaH); 13C{1H}-NMR (125.8 MHz, CDCl3, 298 K): δ [ppm] = 2.1 (NC–CH3), 22.3 (iPr–CH3), 27.3 (iPr–CH3), 28.0 (Cu–C–C(CH3)2), 29.18 (iPr–CH/N–C(CH3)2), 29.20 (iPr–CH/N–C(CH3)2), 49.2 (CH2), 54.2 (Cu–C–C(CH3)2), 83.1 (N–C(CH3)2), 118.2 (NC), 125.2 (cAACMe-aryl-Cmeta), 130.7 (cAACMe-aryl-Cpara), 133.7 (cAACMe-aryl-Cipso), 145.0 (cAACMe-aryl-Cortho), 245.5 (N–C–Cu); 19F-NMR (470.5 MHz, CDCl3, 298 K): δ [ppm] = −45.0 (dm, 1 F, 1JPF = 892 Hz, PF), −80.1 (m, 3 F, CF3), −81.8 (m, 6 F, CF3), −88.4 (dm, 2 F, 1JPF = 903 Hz, PF2), −115.8 (dm, 2 F, 2JPF = 83 Hz, CF2), −116.3 (dm, 4 F, 2JPF = 98 Hz, CF2); 31P-NMR (202.4 MHz, CDCl3, 298 K): δ [ppm] = −147.3 (tdm, 1JPF = 903 Hz, 1JPF = 892 Hz); IR (ATR [cm−1]): 2975 (w), 2948 (w), 2873 (vw), 2323 (vw), 2297 (vw), 1588 (vw), 1524 (vw), 1460 (w), 1388 (vw), 1372 (vw), 1364 (vw), 1309 (w), 1296 (w), 1210 (s), 1180 (s), 1135 (s), 1124 (s), 1097 (m), 1068 (m), 962 (m), 932 (vw), 808 (s), 762 (m), 722 (s), 618 (vs), 580 (m), 533 (m), 494 (w), 467 (vw), 438 (vw), 428 (w); HRMS (ESI) m/z [M]+ calcd for C22H34CuN2: 389.2018, found: 389.2002; m/zFAP− calcd for C6F18P: 444.9450, found: 444.9444; elemental analysis calcd (%) for C28H34CuF18N2P: (gefunden): C 40.27, H 4.10, N 3.35; found: C 40.81, H 4.26, N 3.51.
CMe)2]+FAP− (16).
The phosphorane (C2F5)3PF2 (46.5 μL, 197 μmol) was added at room temperature to a solution of Ia (93.0 mg, 197 μmol) in acetonitrile (3 mL). The reaction mixture was stirred for 1.5 h at room temperature. All volatiles were removed under reduced pressure and the product was dried in vacuo to yield 16 (157 mg, 160 μmol, 81%) as a colorless solid. Single crystals of 16 suitable for X-ray diffraction were obtained by vapor diffusion of n-pentane into a solution of 16 in toluene. 1H NMR (500.1 MHz, CDCl3, 298 K): δ [ppm] = 1.23 (d, 12 H, 3JHH = 6.9 Hz, iPr–CH3) overlap with 1.25 (d, 12 H, 3JHH = 6.9 Hz, iPr–CH3), 1.99 (s, 6 H, NC-CH3), 2.51 (sept, 4 H, 3JHH = 6.9 Hz, iPr–CH), 7.27 (s, 2 H, N–CH–CH–N), 7.36 (d, 4 H, 3JHH = 7.8 Hz, IDipp-aryl-CmetaH), 7.55 (t, 2 H, 3JHH = 7.8 Hz, IDipp-aryl-CparaH); 13C{1H} NMR (125.8 MHz, CD2Cl2, 298 K): δ [ppm] = 2.3 (NC–CH3), 24.0 (iPr–CH3), 24.9 (iPr–CH3), 29.1 (iPr–CH), 117.3 (NC), 124.4 (N–CH–CH–N), 124.6 (IDipp-aryl-Cmeta), 131.1 (IDipp-aryl-Cpara), 134.6 (IDipp-aryl-Cipso), 146.2 (IDipp-aryl-Cortho), 178.3 (N–C–N); 19F NMR (470.5 MHz, CD2Cl2, 298 K): δ [ppm] = −45.1 (dm, 1 F, 1JPF = 889 Hz, PF), −80.6 (m, 3 F, CF3), −82.3 (m, 6 F, CF3), −88.6 (dm, 2 F, 1JPF = 902 Hz, PF2), −116.1 (dm, 2 F, 2JPF = 83 Hz, CF2), −116.7 (dm, 4 F, 2JPF = 98 Hz, CF2); 31P NMR (202.4 MHz, CD2Cl2, 298 K): δ [ppm] = −147.7 (tdm, 1JPF = 902 Hz, 1JPF = 889 Hz); IR ([cm−1]): 3186 (vw), 3151 (vw), 2967 (m), 2931 (w), 2871 (w), 2314 (vw), 1681 (vw), 1580 (vw), 1552 (vw), 1471 (m), 1408 (w), 1385 (w), 1365 (w), 1329 (w), 1310 (m), 1258 (vw), 1213 (vs), 1189 (vs), 1138 (s), 1127 (s), 1088 (s), 1061 (m), 967 (m), 949 (w), 937 (w), 806 (s), 763 (s), 742 (m), 724 (vs), 695 (w), 636 (m), 617 (vs), 602 (m), 580 (m), 560 (vw), 534 (w), 506 (w), 495 (w), 443 (w), 429 (w), 422 (w); HRMS (ESI) m/z [M − CH3CN]+ calcd for. C29H39CuN3: 492.2440, found: 492.2427; m/zFAP− calcd for C6F18P: 444.9450, found: 444.9458; elemental analysis calcd (%) for C37H42CuF18N4P: C 45.38, H 4.32, N 5.72; found: C 45.75, H 4.38, N 5.12.
CPh)]+FAP− (17).
The phosphorane (C2F5)3PF2 (75.0 μL, 319 μmol) was added at room temperature to a solution of Ia (150 mg, 318 μmol) and benzonitrile (32.6 μL, 319 μmol) in dichloro methane (5 mL). The reaction mixture was stirred for 3 h at room temperature. All volatiles were removed under reduced pressure and the remaining solid was suspended in n-hexane (5 mL) and the product was filtered off. The product was washed with n-hexane (2 × 5 mL) and dried in vacuo to yield 17 (235 mg, 262 μmol, 82%) as a colorless solid. Single crystals of 17 suitable for X-ray diffraction were obtained by vapor diffusion of n-pentane into a saturated solution of 17 in toluene. 1H NMR (500.1 MHz, CDCl3, 298 K): δ [ppm] = 1.26 (d, 12 H, 3JHH = 6.9 Hz, iPr–CH3), 1.27 (d, 12 H, 3JHH = 6.9 Hz, iPr–CH3), 2.51 (sept, 4 H, 3JHH = 6.9 Hz, iPr–CH), 7.30 (s, N–CH–CH–N), 7.37 (d, 4 H, 3JHH = 7.8 Hz, IDipp-aryl-CmetaH), 7.54 (tbr, 2 H, 3JHH = 7.6 Hz, benzonitrile-aryl-CmetaH), 7.58 (t, 2 H, 3JHH = 7.8 Hz, IDipp-aryl-CparaH), 7.65 (dbr, 2 H, 3JHH = 7.6 Hz, benzonitrile-aryl-CorthoH), 7.77 (tbr, 1 H, 3JHH = 7.6 Hz, benzonitrile-aryl-CparaH); 13C{1H} NMR (125.8 MHz, CDCl3, 298 K): δ [ppm] = 23.8 (iPr–CH3), 25.2 (iPr–CH3), 28.9 (iPr–CH), 106.9 (benzonitrile-aryl-Cipso), 118.3 (NC), 124.71 (N–CH–CH–N), 124.74 (IDipp-aryl-Cmeta), 130.0 (benzonitrile-aryl-Cmeta), 131.4 (IDipp-aryl-Cpara), 133.6 (benzonitrile-aryl-Cortho), 133.7 (IDipp-aryl-Cipso), 136.3 (benzonitrile-aryl-Cpara), 145.7 (IDipp-aryl-Cortho), 175.5 (N–C–N); 19F NMR (470.5 MHz, CDCl3, 298 K): δ [ppm] = −45.3 (dm, 1 F, 1JPF = 892 Hz, PF), −80.2 (m, 3 F, CF3), −81.8 (m, 6 F, CF3), −88.6 (dm, 2 F, 1JPF = 903 Hz, PF2), −115.9 (dm, 2 F, 2JPF = 82 Hz, CF2), −116.4 (dm, 4 F, 2JPF = 98 Hz, CF2); 31P NMR (202.4 MHz, CDCl3, 298 K): δ [ppm] = −147.3 (tdm, 1JPF = 903 Hz, 1JPF = 892 Hz); IR ([cm−1]): 3186 (vw), 3134 (vw), 2967 (w), 2927 (w), 2874 (w), 2275 (w), 1597 (w), 1547 (vw), 1468 (w), 1414 (w), 1389 (vw), 1367 (vw), 1312 (m), 1215 (vs), 1184 (s), 1138 (s), 1125 (m), 1111 (w), 1089 (m), 1061 (w), 963 (m), 934 (w), 805 (s), 758 (s), 743 (m), 716 (vs), 702 (w), 681 (w), 636 (w), 618 (vs), 601 (w), 581 (w), 551 (w), 534 (w), 505 (w), 494 (vw), 441 (w), 429 (w), 420 (w); HRMS (ESI) m/z [M]+ calcd for C34H41CuN3: 554.2597, found: 554.2583; m/zFAP− calcd for C6F18P: 444.9450, found: 444.9426; elemental analysis calcd (%) for C40H41CuF18N3P: C 48.03, H 4.13, N 4.20; found: C 48.23, H 4.07, N 4.42.
CPh2)]+FAP− (27).
The phosphorane (C2F5)3PF2 (75.0 μL, 319 μmol) was added at room temperature to a solution of Ia (150 mg, 318 μmol) and benzophenone (58.0 mg, 318 μmol) in dichloro methane (5 mL). The reaction mixture was stirred for 2 h at room temperature. All volatiles were removed under reduced pressure and the remaining solid was suspended in n-hexane (5 mL) and the product was filtered off. The product was washed with n-hexane (2 × 5 mL) and dried in vacuo to yield 27 (272 mg, 252 μmol, 79%) as an off-white solid. Single crystals of 27 suitable for X-ray diffraction were obtained by diffusion of n-hexane into a solution of 27 in 1,2-difluorobenzene. 1H NMR (500.1 MHz, CDCl3, 298 K): δ [ppm] = 1.13 (d, 12 H, 3JHH = 6.8 Hz, iPr–CH3), 1.24 (d, 12 H, 3JHH = 6.8 Hz, iPr–CH3), 2.50 (sept, 4 H, 3JHH = 6.8 Hz, iPr–CH), 7.28 (s, 2 H, N–CH–CH–N), 7.34 (d, 4 H, 3JHH = 7.8 Hz, IDipp-aryl-CmetaH), 7.39 (m, 4 H, benzophenone-aryl-CmetaH), 7.52 (dd, 4 H, 3JHH = 8.4 Hz, 4JHH = 1.2 Hz, benzophenone-aryl-CorthoH), 7.59 (t, 2 H, 3JHH = 7.8 Hz, IDipp-aryl-CparaH), 7.70 (tt, 2 H, 3JHH = 7.5 Hz, 4JHH = 1.2 Hz, benzophenone-aryl-CparaH); 13C{1H} NMR (125.8 MHz, CDCl3, 298 K): δ [ppm] = 23.8 (iPr–CH3), 25.0 (iPr–CH3), 28.9 (iPr–CH), 124.7 (N–CH–CH–N/IDipp-aryl-Cmeta), 124.8 (N–CH–CH–N/IDipp-aryl-Cmeta), 129.4 (benzophenone-aryl-Cmeta), 131.1 (benzophenone-aryl-Cortho), 131.3 (IDipp-aryl-Cpara), 134.0 (IDipp-aryl-Cipso), 135.6 (benzophenone-aryl-Cipso), 136.0 (benzophenone-aryl-Cpara), 145.7 (IDipp-aryl-Cortho), 175.7 (N–C–N), 206.3 (OC); 19F NMR (470.5 MHz, CDCl3, 298 K): δ [ppm] = −45.3 (dm, 1 F, 1JPF = 891 Hz, PF), −80.2 (m, 3 F, CF3), −81.8 (m, 6 F, CF3), −88.7 (dm, 2 F, 1JPF = 903 Hz, PF2), −115.9 (dm, 2 F, 2JPF = 83 Hz, CF2), −116.4 (dm, 4 F, 2JPF = 98 Hz, CF2); 31P NMR (202.4 MHz, CDCl3, 298 K): δ [ppm] = −147.2 (tdm, 1JPF = 903 Hz, 1JPF = 891 Hz); IR ([cm−1]): 3137 (vw), 2964 (w), 2927 (w), 2872 (w), 1591 (w), 1558 (m), 1493 (vw), 1461 (w), 1451 (w), 1416 (w), 1388 (vw), 1366 (vw), 1333 (m), 1293 (m), 1212 (vs), 1177 (vs), 1142 (s), 1126 (s), 1098 (s), 1061 (m), 1026 (vw), 999 (vw), 972 (m), 958 (m), 926 (w), 850 (vw), 817 (s), 810 (s), 761 (s), 746 (m), 721 (s), 706 (s), 681 (w), 651 (w), 637 (w), 617 (vs), 580 (m), 532 (w), 496 (w), 438 (w), 429 (w), 411 (vw); HRMS (ESI) m/z [M]+ calcd for C40H46CuN2O: 633.2906, found: 633.2888; m/zFAP− calcd for C6F18P: 444.9450, found: 444.9430; elemental analysis calcd (%) for C46H46CuF18N2OP: C 51.19, H 4.30, N 2.60; found: C 51.79, H 4.30, N 2.88.
Crystallographic details
Crystal data were collected on a Bruker X8 Apex-2 diffractometer with a CCD area detector and graphite monochromated Mo-Kα radiation or a Rigaku XtaLAB Synergy-DW diffractometer with an Hy-Pix-6000HE detector and monochromated Cu-Kα radiation equipped with an Oxford Cryo 800 cooling unit. Crystals were immersed in a film of perfluoropolyether oil on a glass fiber MicroMount™ (MiTeGen) and data were collected at 100 K. Images were processed with Bruker or CrySalis software packages and equivalent reflections were merged. Corrections for Lorentz-polarization effects and absorption were performed if necessary and the structures were solved by direct methods. Subsequent difference Fourier syntheses revealed the positions of all other non-hydrogen atoms. Structures were solved by using the ShelXTL software package.40 All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were assigned to idealized geometric positions and were included in structure factors calculations.Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 2444352 (1), 2444336 (3), 2444351 (5), 2444348 (6), 2444349 (7), 2444343 (10), 2444350 (11), 2444347 (12), 2444346 (16), 2444340 (17), 2444345 (19), 2444342 (20), 2444338 (21), 2444341 (23), 2444344 (24), 2444339 (26), 2444337 (27).
Data availability
The data supporting this article have been included as part of the manuscript and the ESI.† Crystallographic data are also deposited in the form of CIF files at the Cambridge Crystallographic Data Centre with reference number CCDC 2444352 (1), 2444336 (3), 2444351 (5), 2444348 (6), 2444349 (7), 2444343 (10), 2444350 (11), 2444347 (12), 2444346 (16), 2444340 (17), 2444345 (19), 2444342 (20), 2444338 (21), 2444341 (23), 2444344 (24), 2444339 (26), 2444337 (27).Conflicts of interest
The authors declare no conflict to declare.Acknowledgements
This work was supported by the Julius-Maximilians-Universität Würzburg.References
- S. A. Föhrenbacher, M. J. Krahfuss, L. Zapf, A. Friedrich, N. V. Ignat'ev, M. Finze and U. Radius, Chem. – Eur. J., 2021, 27, 3504–3516 CrossRef PubMed.
- G. N. Lewis, Valence and the Structure of Atoms and Molecules, Chemical Catalog Company, New York, 1923 CrossRef; G. N. Lewis, J. Franklin Inst., 1938, 226, 293–313 CrossRef.
- (a) A. G. Massey and A. J. Park, J. Organomet. Chem., 1964, 2, 245–250 CrossRef CAS; (b) H. Jacobsen, H. Berke, S. Döring, G. Kehr, G. Erker, R. Fröhlich and O. Meyer, Organometallics, 1999, 18, 1724–1735 CrossRef CAS; (c) G. C. Welch, T. Holtrichter-Roessmann and D. W. Stephan, Inorg. Chem., 2008, 47, 1904–1906 CrossRef CAS PubMed.
- (a) G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed; (b) P. A. Chase and D. W. Stephan, Angew. Chem., Int. Ed., 2008, 47, 7433–7437 CrossRef CAS PubMed; (c) P. A. Chase, A. L. Gille, T. M. Gilbert and D. W. Stephan, Dalton Trans., 2009, 38, 7179–7188 RSC; (d) S. Kronig, E. Theuergarten, D. Holschumacher, T. Bannenberg, C. G. Daniliuc, P. G. Jones and M. Tamm, Inorg. Chem., 2011, 50, 7344–7359 CrossRef CAS PubMed.
- Q. Sun, C. G. Daniliuc, C. Mück-Lichtenfeld, K. Bergander, G. Kehr and G. Erker, J. Am. Chem. Soc., 2020, 142, 17260–17264 CrossRef CAS PubMed.
- (a) A. E. Ashley, A. L. Thompson and D. O'Hare, Angew. Chem., Int. Ed., 2009, 48, 9839–9843 CrossRef CAS PubMed; (b) C. M. Mömming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643–6646 CrossRef PubMed; (c) A. L. Travis, S. C. Binding, H. Zaher, T. A. Q. Arnold, J.-C. Buffet and D. O'Hare, Dalton Trans., 2013, 42, 2431–2437 RSC.
- E. Theuergarten, T. Bannenberg, M. D. Walter, D. Holschumacher, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2014, 43, 1651–1662 RSC.
- E. Otten, R. C. Neu and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 9918–9919 CrossRef CAS PubMed.
- M. Sajid, A. Klose, B. Birkmann, L. Liang, B. Schirmer, T. Wiegand, H. Eckert, A. J. Lough, R. Fröhlich, C. G. Daniliuc, S. Grimme, D. W. Stephan, G. Kehr and G. Erker, Chem. Sci., 2013, 4, 213–219 RSC.
- L. Zapf, M. Riethmann, S. A. Föhrenbacher, M. Finze and U. Radius, Chem. Sci., 2023, 14, 2275–2288 RSC.
- (a) K. Schwedtmann, G. Zanoni and J. J. Weigand, Chem. – Asian J., 2018, 13, 1388–1405 CrossRef CAS PubMed; (b) T. Böttcher and G.-V. Röschenthaler, J. Fluor. Chem., 2015, 171, 4–11 CrossRef; (c) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed; (d) A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed; (e) W. Levason, F. M. Monzittu and G. Reid, Coord. Chem. Rev., 2019, 391, 90–130 CrossRef CAS.
- S. A. Föhrenbacher, V. Zeh, M. J. Krahfuss, N. V. Ignat'ev, M. Finze and U. Radius, Eur. J. Inorg. Chem., 2021, 1941–1960 CrossRef.
- M. Riethmann, S. A. Föhrenbacher, H. Keiling, N. V. Ignat'ev, M. Finze and U. Radius, Inorg. Chem., 2024, 63, 8351–8365 CrossRef CAS PubMed.
- H. Ibrahim, R. Guillot, F. Cisnetti and A. Gautier, Chem. Commun., 2014, 50, 7154–7156 RSC.
- S. Díez-González, E. C. Escudero-Adán, J. Benet-Buchholz, E. D. Stevens, A. M. Z. Slawin and S. P. Nolan, Dalton Trans., 2010, 39, 7595–7606 RSC.
- L. A. Goj, E. D. Blue, C. Munro-Leighton, T. B. Gunnoe and J. L. Petersen, Inorg. Chem., 2005, 44, 8647–8649 CrossRef CAS PubMed.
- (a) J. P. Coyle, E. R. Sirianni, I. Korobkov, G. P. A. Yap, G. Dey and S. T. Barry, Organometallics, 2017, 36, 2800–2810 CrossRef CAS; (b) G. C. Fortman, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2010, 29, 3966–3972 CrossRef CAS.
- S. J. Ferrara, B. Wang, E. Haas, K. W. LeBlanc, J. T. Mague and J. P. Donahue, Inorg. Chem., 2016, 55, 9173–9177 CrossRef CAS PubMed.
- (a) H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC; (b) A. Gómez-Suárez, D. J. Nelson and S. P. Nolan, Chem. Commun., 2017, 53, 3385–3385 RSC; (c) S. Díez-González and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874–883 CrossRef; (d) L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed; (e) A. C. Hillier, W. J. Sommer, B. S. Yong, J. L. Petersen, L. Cavallo and S. P. Nolan, Organometallics, 2003, 22, 4322–4326 CrossRef CAS; (f) A. Poater, F. Ragone, S. Giudice, C. Costabile, R. Dorta, S. P. Nolan and L. Cavallo, Organometallics, 2008, 27, 2679–2681 CrossRef CAS; (g) SambVca 2.1 web application, https://www.molnac.unisa.it/OMtools/sambvca2.1/index.html).
- S. Solyntjes, B. Neumann, H.-G. Stammler, N. Ignat'ev and B. Hoge, Eur. J. Inorg. Chem., 2016, 3999–4010 CrossRef CAS.
- J.-F. Guttenberger and W. Strohmeier, Chem. Ber., 1967, 100, 2807–2811 CrossRef CAS.
- (a) E. J. Fernández, A. Laguna, J. M. López-de-Luzuriaga, M. Monge, M. Montiel, M. E. Olmos and M. Rodríguez-Castillo, Dalton Trans., 2009, 38, 7509–7518 RSC; (b) J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos, D. Pascual and M. Rodríguez-Castillo, Inorg. Chem., 2011, 50, 6910–6921 CrossRef PubMed; (c) L. Li, P. S. Lopes, V. Rosa, C. A. Figueira, M. A. N. D. A. Lemos, M. T. Duarte, T. Avilés and P. T. Gomes, Dalton Trans., 2012, 41, 5144–5154 RSC.
- S. Ahammed, A. Saha and B. C. Ranu, J. Org. Chem., 2011, 76, 7235–7239 CrossRef CAS PubMed.
- D. S. Surry and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 10354–10355 CrossRef CAS PubMed.
- E. D. Blue, A. Davis, D. Conner, T. B. Gunnoe, P. D. Boyle and P. S. White, J. Am. Chem. Soc., 2003, 125, 9435–9441 CrossRef CAS PubMed.
- A. Grirrane, E. Álvarez, H. García and A. Corma, Angew. Chem., Int. Ed., 2014, 53, 7253–7258 CrossRef CAS PubMed.
- A. Liske, L. Wallbaum, T. Hölzel, J. Föller, M. Gernert, B. Hupp, C. Ganter, C. M. Marian and A. Steffen, Inorg. Chem., 2019, 58, 5433–5445 CrossRef CAS PubMed.
- (a) W. J. Evans, C. H. Fujimoto, M. A. Johnston and J. W. Ziller, Organometallics, 2002, 21, 1825–1831 CrossRef CAS; (b) W.-S. Sheen, C.-N. Kuo, S.-H. Hsieh and H.-M. Gau, J. Chin. Chem. Soc., 2004, 51, 719–722 CrossRef CAS; (c) D. L. Coombs, S. Aldridge, A. Rossin, C. Jones and D. J. Willock, Organometallics, 2004, 23, 2911–2926 CrossRef CAS.
- (a) A. Flores-Gaspar, P. Pinedo-González, M. G. Crestani, M. Muñoz-Hernández, D. Morales-Morales, B. A. Warsop, W. D. Jones and J. J. García, J. Mol. Catal. A: Chem., 2009, 309, 1–11 CrossRef CAS; (b) S. Sabater, M. J. Page, M. F. Mahon and M. K. Whittlesey, Organometallics, 2017, 36, 1776–1783 CrossRef CAS; (c) T. J. Hadlington, T. Szilvási and M. Driess, Chem. Commun., 2018, 54, 9352–9355 RSC.
- L. Tendera, T. Schaub, M. J. Krahfuss, M. W. Kuntze-Fechner and U. Radius, Eur. J. Inorg. Chem., 2020, 3194–3207 CrossRef CAS.
- E. B. Fleischer, N. Sung and S. Hawkinson, J. Phys. Chem., 1968, 72, 4311–4312 CrossRef CAS.
- N. P. Mankad, D. S. Laitar and J. P. Sadighi, Organometallics, 2004, 23, 3369–3371 CrossRef CAS.
- M. Trose, F. Nahra, A. Poater, D. B. Cordes, A. M. Z. Slawin, L. Cavallo and C. S. J. Cazin, ACS Catal., 2017, 7, 8176–8183 CrossRef CAS.
- L. Zhang, J. Cheng and Z. Hou, Chem. Commun., 2013, 49, 4782–4784 RSC.
- N. P. Mankad, T. G. Gray, D. S. Laitar and J. P. Sadighi, Organometallics, 2004, 23, 1191–1193 CrossRef CAS.
- J. Attner and U. Radius, Chem. – Eur. J., 2001, 7, 783–790 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- J. R. Herron and Z. T. Ball, J. Am. Chem. Soc., 2008, 130, 16486–16487 CrossRef CAS PubMed.
- N. Ignat'ev and P. Sartori, J. Fluor. Chem., 2000, 103, 57–61 CrossRef.
- G. Sheldrick, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data and NMR spectra. CCDC 2444352 (1), 2444336 (3), 2444351 (5), 2444348 (6), 2444349 (7), 2444343 (10), 2444350 (11), 2444347 (12), 2444346 (16), 2444340 (17), 2444345 (19), 2444342 (20), 2444338 (21), 2444341 (23), 2444344 (24), 2444339 (26) and 2444337 (27). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00904a |
| This journal is © The Royal Society of Chemistry 2025 |