
A comparative study on nido- and closo-carborane supported zinc-salen catalysts for the ROCOP of epoxides and anhydrides†
Jin-Bian
Xue‡
,
Jia-Ni
Wang‡
,
Ke-Cheng
Chen
,
Qian
Cheng
,
Jia-Ying
Zhang
and
Xu-Qiong
Xiao
 *
*
Key Laboratory of Organosilicon Chemistry and Material Technology, Ministry of Education, Zhejiang Key Laboratory of Organosilicon Material Technology, College of Material, Chemistry and Chemical Engineering, Hangzhou Normal University, No. 2318 Yuhangtang Rd. Hangzhou, 311121, Zhejiang, China. E-mail: xqxiao@hznu.edu.cn
First published on 16th May 2025
Abstract
Nido- and closo-carborane supported Zn–salen complexes (2–4) were prepared. The nido-C2B9 carborane anion supported Zn–salen complexes are superior to the closo-carborane supported ones in ROCOP of epoxides and anhydrides. These findings show the importance of electronic effects in the backbone of the salen ligands and offer guidance for future catalyst design.
Polyesters represent a class of polymers that are not only versatile but also hold potential for sustainability and biodegradability.1 These materials find extensive applications across various fields, ranging from packaging to biomedical devices. One particularly promising approach for synthesizing polyesters is the ring-opening copolymerization (ROCOP) of epoxides and cyclic anhydrides.2 This method offers significant advantages, including the ability to finely tune the properties of the resulting polyesters by modifying the structures of the epoxide and cyclic anhydride monomers. Furthermore, ROCOP proceeds under relatively mild reaction conditions.3 The continued advancement of this polymerization technique is largely driven by innovations in catalyst design, which play a crucial role in enhancing its efficiency and expanding its applicability.4 Although numerous catalytic systems have been developed, metallic catalysts based on salen ligands remain highly attractive due to their ease of accessibility, tunable electronic and steric properties, and ability to coordinate with a wide range of metals in various oxidation states. These features enable the design of versatile catalysts with alterable Lewis acidic metal centers and optimized coordination environments. Among these metal–salen catalysts, Zn-based complexes stand out for their low cost, low toxicity, controlled reactivity and selectivity and their abundance on Earth.5
On the other hand, 1,2-dicarba-closo-dodecaboranes (o-carboranes) and their derivatives have received considerable attention since their first synthesis in 1963, owing to their unique cage-like structures and applications in medicine, nonlinear optical materials, luminescent materials, and coordination/organometallic chemistry.6 Among these, closo- and nido-carboranes are the most extensively studied due to their distinct structural and electronic properties.7 For example, the o-carborane cluster as a whole can exhibit electron-withdrawing properties when attached to other systems like organometallic complexes or substituted derivatives, while nido-carboranes are stronger electron donors as they carry a negative charge.8 These remarkable differences are particularly important in coordination or organometallic complexes when they are employed as auxiliary ligands. In this contribution, we report the preparation of nido-C2B9 carborane anion- and o-carborane-supported zinc–salen complexes and a comparative study of their catalytic abilities in ROCOP of epoxides and anhydrides.
The o-carborane-supported salen ligands (1a–1d) were synthesized according to literature procedures.9 Complexation with zinc was initially carried out using the common zinc salt, Zn(OAc)2. Treatment of ligands 1a–1d with Zn(OAc)2 in ethanol at room temperature for 24 hours afforded the corresponding complexes 2a–2d. (Scheme 1) The absence of phenolic proton signals in the 1H NMR spectra confirmed successful complexation, while the 11B NMR spectra exhibited resonances between −10 ppm and −39 ppm, indicative of o-carborane cage opening.10 The formation of the zinc complexes was further corroborated by single-crystal X-ray diffraction (SCXRD) analysis (Fig. 1).
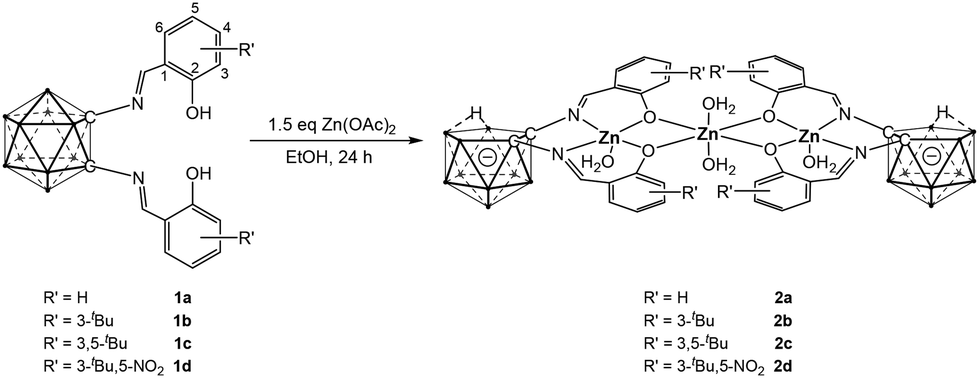 | ||
| Scheme 1 Synthesis of trinuclear zinc complexes 2a–2d. | ||
 | ||
| Fig. 1 Molecular structure of complex 2a (some of the hydrogen atoms are omitted for clarity; ellipsoids are set at the 30% probability level). Selected bond lengths (Å) and angles (°): C(1)–C(2) 1.594(6), Zn(1)–Zn(2) 3.1277(7), Zn(1)–O(1) 1.993(3), Zn(1)–O(2) 1.981(3), Zn(1)–N(1) 2.042(3), Zn(1)–N(2) 2.044(3), Zn(2)–O(1) 2.103(3), and Zn(2)–O(2) 2.123(3); O(1)–Zn(1)–O(3) 97.09(12), O(1)–Zn(1)–N(1) 89.98(12), O(1)–Zn(1)–N(2) 152.95(13), O(2)–Zn(1)–O(1) 81.70(11), O(2)–Zn(1)–O(3) 100.86(12), O(2)–Zn(1)–N(1) 150.72(13), O(2)–Zn(1)–N(2) 91.72(12), N(1)–Zn(1)–O(3) 108.03(13), N(1)–Zn(1)–N(2) 83.02(13) and N(2)–Zn(1)–O(3) 109.93(13). | ||
The molecular structure of complex 2a is shown in Fig. 1 (see also Fig. S59,† with selected bond lengths and angles in the caption). The solid-state structure clearly reveals that the salen-Zn units are supported by nido-C2B9 carborane anions, consistent with the 11B NMR data. Remarkably, two of these nido-carborane-supported salen–Zn moieties are bridged by a third Zn(II) ion, forming a trinuclear complex. The two nido-carborane units adopt a face-to-face arrangement, with their [ONNO] coordination planes nearly parallel. Together with the third Zn(II) ion, these two C2B3 and two [ONNO] planes form a channel-like structure. Unlike previously reported multinuclear Zn(II) complexes, which often feature a dangling methoxy group as an additional donor ligand,11 no such group is present here. The Zn(1) center adopts a pentacoordinate environment with a τ5 value of 0.04,12 suggesting a near-ideal square pyramidal geometry, whereas Zn(2) displays a hexacoordinate geometry. The Zn(1)–Zn(2) distance is 312.77(7) pm, shorter than the sum of their van der Waals radii (410 pm), suggesting a weak metal–metal interaction.
To obtain both nido-C2B9 carborane anion- and o-carborane-supported salen complexes, complexation reactions were subsequently carried out using ZnCl2. Treatment of ligands 1a–1d with anhydrous ZnCl2 in the presence of Et3N under reflux in THF yielded yellow complexes. (Scheme 2) The 11B NMR spectra displayed resonances in the range of −9.56 to −35.08 ppm, and the 1H NMR spectrum exhibited a broad signal at −2.3 ppm, both indicative of cage opening of the closo-carborane. The molecular structure of the anionic part of complex 3b is shown in Fig. 2 (see also Fig. S60†). It is clearly shown that the Zn(1) center was supported by the nido-C2B9 carborane anion and adopted a pentacoordinate geometry, with a chloride ligand occupying the apical position in addition to the four donor atoms of the salen ligand. The calculated τ5 value is 0.33, indicating a distorted square pyramidal geometry. The asymmetric unit of 3b clearly contains two [HNEt3] moieties.
 | ||
| Scheme 2 Synthesis of zinc complexes 3a–3d. | ||
 | ||
| Fig. 2 Molecular structure of complex 3b (some of the hydrogen atoms are omitted for clarity; ellipsoids are set at the 30% probability level). Selected bond lengths (Å) and angles(°): C(1)–C(2) 1.584(4), Zn(1)–Cl(1) 2.3454(13), Zn(1)–O(1) 2.027(2), Zn(1)–O(2) 1.955(2), Zn(1)–N(1) 2.133(3), and Zn(1)–N(2) 2.107(2); O(1)–Zn(1)–Cl(1) 107.51(7), O(1)–Zn(1)–N(1) 82.54(9), O(1)–Zn(1)–N(2) 137.85(9), O(2)–Zn(1)–Cl(1) 103.43(8), O(2)–Zn(1)–O(1) 95.10(8), O(2)–Zn(1)–N(1) 157.74(10), O(2)–Zn(1)–N(2) 88.52(9), N(1)–Zn(1)–Cl(1) 98.35(8), N(2)–Zn(1)–Cl(1) 112.41(7), and N(2)–Zn(1)–N(1) 78.91(10). | ||
A structural comparison between complexes 2a and 3b suggests that Zn(2) in 2a plays a similar role to the triethylammonium cation in 3b, serving to balance the negative charge of the nido-C2B9 carborane moieties. The conversion of 2 to 3 was achieved by treatment with excess [HNEt3]Cl in CH2Cl2. It is documented that elevated temperatures induced cage opening. Consequently, reactions of 1 with ZnCl2 in the presence of 2 equivalents of Et3N were performed at room temperature (Scheme 3). The 11B NMR spectrum showed peaks between −7.22 ppm and −15.68 ppm, indicating that deboronation did not occur, while 1H NMR showed persistent phenolic proton signals with reduced integration, suggesting incomplete deprotonation. To address this, the reaction was repeated using excess Et3N (ca. 6 equivalents), successfully affording complexes 4 (Scheme 3). Alternatively, deprotonation of 1 with n-BuLi followed by ZnCl2 addition also afforded complexes 4. However, the method outlined in Scheme 3 avoids the use of n-BuLi, providing a milder synthetic route.
 | ||
| Scheme 3 Synthesis of zinc complexes 4a–4d. | ||
Single crystals suitable for XRD analysis for 4a were obtained by slow diffusion of hexane into a dichloromethane solution. The molecular structure of 4a is depicted in Fig. 3 (see also Fig. S61†). It clearly demonstrates that the salen ligand is supported by a closo-carborane framework. The Zn centers adopt an ideal square pyramidal geometry, as indicated by a τ5 value of 0.00. The C1–C1A bond length is 166.7(5) pm, whereas the corresponding distances in complexes 2a and 3b are 159.4(6) and 158.4(4) pm, respectively. For comparison, the typical C–C bond distance in o-carborane is approximately 162 pm.13 These values clearly indicate that the nido- and closo-carborane ligands exert different electronic effects on the salen framework, which may, in turn, influence the catalytic activity of the corresponding complexes.
 | ||
| Fig. 3 Molecular structure of complex 4a (some of the hydrogen atoms are omitted for clarity; ellipsoids are set at the 30% probability level). Selected bond lengths (Å) and angles(°): C1–C(1A) 1.667(5), Zn1–O1 1.973(2), Zn1–O2 2.012(3), Zn1–N1 2.124(2), N1–C1 1.421(3), and N1–C2 1.291(3); O(1A)–Zn1–O1 92.87(13), O(1A)–Zn1–O2 102.20(9), O1–Zn1–O2 102.20(9), O1–Zn1–N1 87.59(9), O1–Zn1–N(1A) 153.88(10), O(1A)–Zn1–N1 153.88(10), O(1A)–Zn1–N(1A) 87.59(9), O2–Zn1–N(1A) 103.21(9), O2–Zn1–N1 103.21(9), and N(1A)–Zn1–N1 80.87(12). | ||
With complexes 2–4 in hand, their catalytic activities in the ROCOP of phthalic anhydride (PA) and cyclohexene oxide (CHO) were evaluated and compared (Table 1). Initial reactions were carried out using a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:100:100 for the Zn catalyst, co-catalyst (bis(triphenylphosphine)iminium chloride, PPNCl), PA, and CHO, respectively, at an elevated temperature of 80 °C. As shown in entries 1–3 (Table 1), all complexes 2a–4a exhibited activity for the ROCOP, affording polyesters with conversions ranging from 46% to 71%. A possible mechanism is proposed in Scheme 3. It involves activation of the epoxide by the metal center, followed by nucleophilic attack by a halide or carboxylate. This generated an alkoxide intermediate, which subsequently reacts with an anhydride to form a new ester linkage, incorporating one complete repeat unit and regenerating both a carboxylate and an open coordination site on the metal centre (Scheme 4).
:2:100:100 for the Zn catalyst, co-catalyst (bis(triphenylphosphine)iminium chloride, PPNCl), PA, and CHO, respectively, at an elevated temperature of 80 °C. As shown in entries 1–3 (Table 1), all complexes 2a–4a exhibited activity for the ROCOP, affording polyesters with conversions ranging from 46% to 71%. A possible mechanism is proposed in Scheme 3. It involves activation of the epoxide by the metal center, followed by nucleophilic attack by a halide or carboxylate. This generated an alkoxide intermediate, which subsequently reacts with an anhydride to form a new ester linkage, incorporating one complete repeat unit and regenerating both a carboxylate and an open coordination site on the metal centre (Scheme 4).
 | ||
| Scheme 4 Proposed mechanism for ROCOP. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat. | Co-cat.b | T (°C) | t (h) | Yieldc | M n (kDa) | PDId |
| a Polymerization was performed in 5 mL of toluene with PPNCl as the cocatalyst. b Molar ratio to the catalyst. c Yield = weight of polymer obtained/weight of monomer used. d M n and PDI were determined by GPC analysis calibrated with standard polystyrene samples. | |||||||
| 1 | 2a | 2 | 80 | 16 | 46% | 1.4 | 1.13 |
| 2 | 3a | 2 | 80 | 16 | 71% | 3.4 | 1.76 |
| 3 | 4a | 2 | 80 | 16 | 40% | 1.4 | 1.13 |
| 4 | 3b | 2 | 80 | 16 | 85% | 1.7 | 1.24 |
| 5 | 3c | 2 | 80 | 16 | 74% | 1.4 | 1.17 |
| 6 | 3d | 2 | 80 | 16 | 67% | 1.1 | 1.16 |
| 7 | 3b | 1 | 80 | 16 | 40% | 1.8 | 1.47 |
| 8 | 3b | 4 | 80 | 16 | 90% | 2.4 | 1.05 |
| 9 | 3b | 4 | 80 | 1 | 18% | 1.3 | 1.01 |
| 10 | 3b | 4 | 80 | 8 | 63% | 1.3 | 1.29 |
| 11 | 3b | 4 | 80 | 24 | 87% | 1.3 | 1.38 |
| 12 | 3b | 4 | 50 | 16 | 14% | 1.7 | 1.16 |
| 13 | 3b | 4 | 110 | 16 | 98% | 1.9 | 1.37 |

The catalytic activity decreased in the order 3a > 2a > 4a. Notably, the nido-carborane-supported mononuclear complex 3a exhibited the highest activity under these conditions. These results suggest that nido-carborane-supported salen ligands outperform their closo-carborane counterparts due to differing electronic effects. The nido-carborane acts as an electron-donating group due to its negative charge, whereas closo-carborane is electron-withdrawing, as electron density can delocalize over its 3D cage structure. Comparing the two nido-carborane-based complexes 2a and 3a, it appears that the molecular structure plays a critical role. As discussed above, the structure of 2a forms a “channel” through the coordination of two salen–Zn moieties with a third bridging Zn center. This channel likely limits substrate accessibility, thereby reducing catalytic efficiency.
The catalytic performance of complexes 3a–3d was further investigated. Substituents on the phenyl ring also influenced the ROCOP activity, with complex 3b outperforming 3a, 3c, and 3d (entries 2 and 4–6). In our previous work, Al-based complexes with unsubstituted phenyl rings showed superior activity in the cycloaddition of epoxides and CO2 at atmospheric pressure. We propose that the enhanced performance of 3b may be attributed to an optimal balance of steric and electronic effects.
The effect of the catalyst-to-cocatalyst ratio was also examined. As shown in entries 4, 7, and 8, increasing the ratio from 1:1 to 1:4 led to a progressive improvement in yield. Thus, a 1:4 catalyst/cocatalyst ratio was adopted as optimal. It is important to note that the cocatalyst alone was inactive under similar conditions. Extending the reaction time improved yields (entries 4 and 9–11); however, beyond 16 hours, the increase was marginal (85% vs. 87%), establishing 16 hours as the optimal reaction time. Lowering the temperature to 50 °C significantly reduced the conversion to 14% (entry 12), while increasing the temperature to 110 °C improved the conversion to 98% (entry 13), making 110 °C the preferred temperature. This enhancement is likely due to increased solubility and mobility of both the polymer and phthalic anhydride at higher temperatures. Based on these findings, the optimal reaction conditions were established as a 1:4 catalyst/cocatalyst ratio, a reaction time of 16 hours, and a temperature of 110 °C. Notably, all resulting polyesters exhibited narrow molecular weight distributions, with polydispersity indices (PDIs) ranging from 1.01 to 1.76. Following optimization of the model PA/CHO ROCOP, various co-monomers were explored to assess the scope for producing structurally diverse polyesters. As summarized in Table 2, catalyst 3b demonstrated good to high activity in the ROCOP of epichlorohydrin, 4-vinylcyclohexene oxide, and 1,2-epoxyhexane with PA.
|
|
||||
|---|---|---|---|---|
| Entry | Substrates | Yieldb | M n (kDa) | PDIc |
|
a Polymerization was performed in 5 mL of toluene. Molar ratio of catalyst to co-catalyst: 1:4. Temperature: 110 °C; reaction time: 16 hours.
b Yield = weight of polymer obtained/weight of monomer used.
c
M
n and PDI were determined by GPC analysis calibrated with standard polystyrene samples.
|
||||
| 1 | I | 50% | 2.6 | 1.87 |
| 2 | II | 87% | 2.3 | 1.29 |
| 3 | III | 72% | 2.9 | 1.29 |

In summary, a series of zinc complexes supported by nido- and closo-carborane-functionalized salen ligands were successfully synthesized. Structural analyses revealed that the closo-carborane-supported complexes are mononuclear, while the nido-carborane-supported complexes can adopt either mononuclear or trinuclear architectures. In all cases, the [ONNO]-coordinated zinc centers are pentacoordinate, with an oxygen-containing solvent molecule occupying the axial position. These complexes exhibited high catalytic activity in the ROCOP of epoxides with PA at elevated temperatures, affording polyesters with narrow polydispersity indices. Under comparable conditions, the nido-carborane-supported zinc complexes demonstrated superior catalytic performance compared to their closo-carborane counterparts, which is attributed to the electron-donating character of the negatively charged nido-C2B9 carborane ligand. These findings provide valuable insights into the structure–activity relationship in carborane-based catalysts for ROCOP and may offer a foundation for the rational design of advanced catalysts. Comparative studies on nido- and closo-carborane-supported ligands in other systems and their catalytic abilities are currently ongoing in our laboratory.
Data availability
The experimental details, NMR spectra, and X-ray crystallographic details that support the findings of this work have been uploaded as part of the ESI.†CCDC 2444573–2444575 for 2a, 3b and 4a contain the supplementary crystallographic data for this paper.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (Grant No. 22471050 and 22171063) and the Natural Science Foundation of Zhejiang Province (Grant No. LY22B010002).References
- (a) C. Shi, E. C. Quinn, W. T. Diment and E. Y. X. Chen, Chem. Rev., 2024, 124, 4393–4478 CrossRef CAS PubMed; (b) C. V. Aarsen, A. Liguori, R. Mattsson, M. H. Sipponen and M. Hakkarainen, Chem. Rev., 2024, 124, 8473–8515 CrossRef CAS PubMed.
- Z. B. Xie, Z. J. Yang, C. Y. Hu, F. Q. Bai, N. N. Li, Z. W. Wang, S. T. Ku, X. Pang, X. S. Chen and X. H. Wang, J. Am. Chem. Soc., 2025, 147, 12115–12126 CrossRef PubMed.
- (a) F. Vidal, S. Smith and C. K. Williams, J. Am. Chem. Soc., 2023, 145, 13888–13900 CrossRef CAS PubMed; (b) G. L. Gregory and C. K. Williams, Macromolecules, 2022, 55, 2290–2299 CrossRef CAS PubMed.
- (a) J. R. Lamb, A. K. Hubbell, S. N. MacMillan and G. W. Coates, J. Am. Chem. Soc., 2020, 142, 8029–8035 CrossRef CAS PubMed; (b) J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed; (c) H.-Y. Ji, B. Wang, L. Pan and Y.-S. Li, Green Chem., 2018, 20, 641–648 RSC; (d) R. Xie, Y. Wang, S. Li, B. Li, J. Xu, J. Liu, Y. He, G. W. Yang and G. P. Wu, Angew. Chem., Int. Ed., 2024, e202404207 CAS; (e) Y. Y. Zhang, G. W. Yang, R. Xie, L. Yang, B. Li and G. P. Wu, Angew. Chem., Int. Ed., 2020, 59, 23291–23298 CrossRef CAS PubMed; (f) S. Li, S. Y. Liu, R. Yan, R. Xie, Q. Q. Xue, B. Li and G. P. Wu, Chem. – Eur. J., 2025, 31, e202500571 CrossRef CAS PubMed.
- (a) M. Winkler, C. Romain, M. A. R. Meier and C. K. Williams, Green Chem., 2015, 17, 300–306 RSC; (b) P. K. Saini, C. Romain, Y. Zhu and C. K. Williams, Polym. Chem., 2014, 5, 6068–6075 RSC.
- (a) T. L. Heying, J. W. Ager, S. L. Clark, D. J. Mangold, H. L. Goldstein, M. Hillman, R. J. Polak and J. W. Szymanski, Inorg. Chem., 1963, 2, 1089–1092 CrossRef CAS; (b) R. N. Grimes, Carboranes, Elsevier, Amsterdam, 3rd edn, 2016 Search PubMed; (c) R. N. Grimes, Chem. Rev., 1992, 92, 251–268 CrossRef CAS; (d) S. Liu, Y. F. Han and G. X. Jin, Chem. Soc. Rev., 2007, 36, 1543–1560 RSC; (e) Z. Qiu and Z. Xie, Chem. Soc. Rev., 2022, 51, 3164–3180 RSC; (f) Z. Z. Qiu and Z. W. Xie, Acc. Chem. Res., 2021, 54, 4065–4079 CrossRef CAS PubMed; (g) R. Cheng, J. Zhang, H. Zhang, Z. Qiu and Z. Xie, Nat. Commun., 2021, 12, 7146 CrossRef CAS PubMed; (h) I. Guerrero, Z. Kelemen, C. Viñas, I. Romero and F. Teixidor, Chem. – Eur. J., 2020, 26, 5027–5036 CrossRef CAS PubMed.
- (a) H. Y. Ren, N. N. Zhou, W. L. Ma, P. Zhang, D. S. Tu, C. S. Lu and H. Yan, J. Am. Chem. Soc., 2024, 146, 26543–26555 CrossRef CAS PubMed; (b) H. T. Zhang, Y. Gao, Y. N. Ma and X. N. Chen, Org. Chem. Front., 2024, 11, 6706–6711 RSC; (c) F. Chen, W. Guo, Y.-N. Ma and X. Chen, Chem. Commun., 2024, 60, 614–617 RSC; (d) Y. N. Ma, H. Z. Ren, Y. X. Wu, N. Li, F. J. Chen and X. N. Chen, J. Am. Chem. Soc., 2023, 145, 7331–7342 CrossRef CAS PubMed; (e) P. Farras, E. J. Juarez-Perez, M. Lepsik, R. Luque, R. Nuñez and F. Teixidor, Chem. Soc. Rev., 2012, 41, 3445–3463 RSC; (f) H. B. Yang, Y. Guo, K. Cao, Q. J. Jiang, C. C. Teng, D. Y. Zhu and S. H. Wang, Chem. Commun., 2024, 60, 1124–1127 RSC; (g) K. Cao and C. Y. Zhang, Eur. J. Org. Chem., 2022, e202200737 CrossRef CAS; (h) J. Jiao, P. He, T. L. Yang, T. W. Zhang, L. H. Wang, T. Han, Y. Nie, Z. Y. Lin and P. F. Li, Org. Chem. Front., 2023, 10, 5965–5970 RSC; (i) J. X. Li, L. J. Zhou, Y. P. Zhang, Y. X. Luo, M. Wu, P. F. Cui and G. X. Jin, Eur. J. Inorg. Chem., 2025, 28, e202400752 CrossRef CAS; (j) D. K. Semyonov, G. K. Slushko, M. Y. Stogniy, S. A. Anufriev, I. A. Godovikov, K. Y. Suponitsky, V. I. Bregadze and I. B. Sivaev, Organometallics, 2023, 42, 2522–2530 CrossRef CAS.
- (a) A. M. Spokoyny, Pure Appl. Chem., 2013, 85, 903–919 CrossRef CAS PubMed; (b) Z. Z. Cui, B. N. Wang, J. X. Li, R. L. Pang, Y. R. Kang and X. Q. Xiao, Chin. J. Chem., 2021, 39, 2410–2416 CrossRef CAS.
- (a) Y. R. Kang, B. N. Wang, R. X. Nan, Y. W. Li, Z. L. Zhu and X. Q. Xiao, Inorg. Chem., 2022, 61, 8806–8814 CrossRef CAS PubMed; (b) B. N. Wang, Z. L. Zhu, M. J. Liang, Y. K. Ren, J. B. Xue, J. Y. Zhang, F. Qi and X. Q. Xiao, Inorg. Chem., 2024, 63, 5481–5486 CrossRef CAS PubMed; (c) R. X. Nan, Y. W. Li, Z. L. Zhu, F. Qi and X. Q. Xiao, J. Am. Chem. Soc., 2023, 145, 15538–15546 CrossRef CAS PubMed; (d) Y. K. Ren, Y. Li, M. J. Liang, J. W. Ma, Z. X. Niu and X. Q. Xiao, Inorg. Chem., 2025, 64 DOI:10.1021/acs.inorgchem.5c00935.
- (a) T. Y. He and R. A. Musah, Polyhedron, 2019, 163, 171–177 CrossRef CAS; (b) L. J. Todd and A. R. Siedle, Prog. Nucl. Magn. Reson. Spectrosc., 1979, 13, 87–176 CrossRef.
- (a) A. Thevenon, J. A. Garden, A. J. P. White and C. K. Williams, Inorg. Chem., 2015, 54, 11906–11915 CrossRef CAS PubMed; (b) M. Ghosh, S. Biswas, M. Roy, S. Biswas, P. Ghosh, S. Koner, S. Mandal and S. Saha, New J. Chem., 2021, 45, 12296–12304 RSC.
- (a) N. Ikpo, S. M. Barbon, M. W. Drover, L. N. Dawe and F. M. Kerton, Organometallics, 2012, 31, 8145–8158 CrossRef CAS; (b) A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- (a) R. Pang, J. Li, Z. Cui, C. Zheng, Z. Li, W. Chen, F. Qi, L. Su and X. Q. Xiao, Dalton Trans., 2019, 48, 7242–7248 RSC; (b) J. Li, R. Pang, Z. Li, G. Lai, X. Q. Xiao and T. Müller, Angew. Chem., Int. Ed., 2019, 58, 1397–1401 CrossRef CAS PubMed; (c) D. Buzsáki, D. Gál, B. Szathmári, T. Holczbauer, A. Udvardy, J. K. Szilágyiné, D. Kargin, C. Bruhn, R. Pietschnig and Z. Kelemen, Inorg. Chem. Front., 2025, 12, 1822–1830 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, X-ray crystallographic data. CCDC 2444573–2444575. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00946d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |