
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure and magnetism of LaxSr2−xCo0.5Ir0.5O4−yHy (0 < x < 1) iridium-containing oxyhydride phases†
James I. Murrella,
Romain Wernert a,
Hank C. H. Wub,
Benjamin M. Huddartb,
Stephen J. Blundellb,
Ronald I. Smithc and
Michael A. Hayward*a
a,
Hank C. H. Wub,
Benjamin M. Huddartb,
Stephen J. Blundellb,
Ronald I. Smithc and
Michael A. Hayward*a
aDepartment of Chemistry, University of Oxford, Inorganic Chemistry Laboratory, South Parks Road, Oxford, OX1 3QR, UK. E-mail: michael.hayward@chem.ox.ac.uk
bDepartment of Physics, Clarendon Laboratory, University of Oxford, Parks Road, Oxford OX1 3PU, UK
cISIS Facility, Rutherford Appleton Laboratory, Chilton, Oxon OX11 0QX, UK
First published on 23rd July 2025
Abstract
Ruddlesden-Popper oxide phases in the LaxSr2−xCo0.5Ir0.5O4 (0 < x < 1) solid solution can be converted to the corresponding LaxSr2−xCo0.5Ir0.5O4−yHy oxyhydride phases, by topochemical reaction with LiH, in which the hydride ions are substituted exclusively onto the equatorial anion sites of the host framework. Analysis reveals that oxyhydride phases in the range 0.5 < x < 1 adopt LaxSr2−xCo0.5Ir0.5O2+xH2−x compositions which maintain a constant Co1+, Ir3+ oxidation-state combination (confirmed by Co K-edge XANES data), with the presence of low-spin d6 Ir3+ being consistent with the covalent stabilization of the metastable oxyhydride phases via strong Ir–H σ-bonds. Phases at the lanthanum-poor end of the solid solution (x < 0.5) adopt LaxSr2−xCo0.5Ir0.5O4−yHy compositions with lower hydride concentrations (y < 1.5). Magnetisation and μSR data indicate that all the LaxSr2−xCo0.5Ir0.5O4−yHy oxyhydride phases exhibit strong magnetic frustration, attributed to the large-scale cation and anion disorder, and resulting in glassy magnetic behaviour at low temperature.
Introduction
Complex transition-metal oxides with perovskite or related structures have been, and continue to be, extensively studied due to the wide range of physical and chemical properties they exhibit.1 The diverse and often complex physical behaviour of perovskite oxides can be attributed to the structural topology of these phases which consists of a network of apex-linked, transition-metal centred BO6 octahedra. This arrangement naturally co-aligns the d-orbitals of the transition metals on neighbouring BO6 units, both with each other and with the p-orbitals of the bridging oxide ions, facilitating the formation of relatively wide transition-metal d-bands and a variety of magnetic exchange couplings. Much work has gone into modifying the subtle structural distortions exhibited by materials with perovskitoid structures to tune the B–O–B bond angles, and thus modify the couplings between the transition-metal centres.Partial chemical substitution of the oxide ions in phases with perovskite-like structures also offers the opportunity to modify the behaviour of materials by both tuning the transition-metal oxidation states (in the case of aliovalent anion substitutions) and changing the inter-cation couplings which make use of the anion orbitals. The preparation of oxyhydride phases, via the partial replacement of O2− oxide ions with H− hydride ions, is particularly attractive given the strong contrast between the two anions. For example, the lower ionic charge and lower electronegativity of the hydride ion means oxyhydride phases tend to stabilize transition metals in lower oxidation states, and exhibit stronger magnetic super–exchange couplings, than all-oxide analogues.2,3 In addition, the lack of π-symmetry valence orbitals on the hydride ion, means that anion-ordered oxyhydride phases exhibit band structures which are qualitatively different to all-oxide analogues.3,4
Unfortunately, the vast majority of transition-metal oxyhydrides are unstable at ambient pressure with respect to decomposition via the reduction of the transition metal and formation of water (e.g. 2SrVO2H ⇒ 2SrO + VO + V + H2O)3 so must either be prepared via reductive topochemical anion exchange or via high pressure synthesis routes. Recently we have shown that late transition-metals can kinetically stabilize oxyhydride phases via strong M–H Sigma bonds, as observed initially in LaSrCoO3H0.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 and then subsequently in ruthenium and rhodium phases such as LaSr3NiRuO4H4,6 LaSrCoRuO3.2H1.87 and La0.5Sr1.5Mn0.5Rh0.5O3H.8 Building on this idea, we reported the first iridium-containing oxyhydride phases, Sr2Mn0.5Ir0.5O3.25H0.75 and Sr2Mn0.5Ir0.5O2.66H1.33, prepared via the reductive topochemical anion exchange of Sr2Mn0.5Ir0.5O4.9 Here we further investigate the formation and properties of iridium-containing oxyhydride phases by studying the anion exchange chemistry of the LaxSr2−xCo0.5Ir0.5O4 (0 < x < 1) series of phases.
5 and then subsequently in ruthenium and rhodium phases such as LaSr3NiRuO4H4,6 LaSrCoRuO3.2H1.87 and La0.5Sr1.5Mn0.5Rh0.5O3H.8 Building on this idea, we reported the first iridium-containing oxyhydride phases, Sr2Mn0.5Ir0.5O3.25H0.75 and Sr2Mn0.5Ir0.5O2.66H1.33, prepared via the reductive topochemical anion exchange of Sr2Mn0.5Ir0.5O4.9 Here we further investigate the formation and properties of iridium-containing oxyhydride phases by studying the anion exchange chemistry of the LaxSr2−xCo0.5Ir0.5O4 (0 < x < 1) series of phases.
Experimental
Synthesis of LaxSr2−xCo0.5Ir0.5O4 (0 ≤ x ≤ 1) phases
Samples of LaxSr2−xCo0.5Ir0.5O4 (x = 0, 0.25, 0.5, 0.75, 1) were prepared by a high-temperature ceramic synthesis method. Suitable stoichiometric ratios of La2O3 (99.999%, dried at 900 °C), SrCO3 (99.99%), Co3O4 (99.7%) and IrO2 (99.99%, dried for 2 hours at 700 °C) were thoroughly ground together in an agate pestle and mortar and pressed into pellets. Samples of Sr2Co0.5Ir0.5O4 were heated in air for 2 days at 950 °C, followed by 2 further 2-day periods at 1000 °C. Samples of La0.25Sr1.75Co0.5Ir0.5O4 were heated in air for 2 days at 1000 °C, followed by a further period of 2 days at 1100 °C and then 2 days at 1200 °C. Samples of La0.5Sr1.5Co0.5Ir0.5O4 and La0.75Sr1.25Co0.5Ir0.5O4 were heated in air for 2 days at 1000 °C, followed by 2 further periods of 2 days at 1300 °C. Samples of LaSrCo0.5Ir0.5O4 were heated for 2 days at 1000 °C in an approximately 0.5:99.5 O2:Ar atmosphere (to favour formation of Ir4+ whilst also minimising reduction to Ir metal), followed by 2 further cycles of 2 days at 1300 °C in the same atmosphere. All samples were reground and pressed into pellets between heating periods.
Topochemical reduction of LaxSr2−xCo0.5Ir0.5O4 (0 ≤ x ≤ 1) phases
Topochemical reduction of LaxSr2−xCo0.5Ir0.5O4 phases was performed using LiH. Initially, small-scale reactions were performed by grinding ∼200 mg of each LaxSr2−xCo0.5Ir0.5O4 phase with 4 mole-equivalents of LiH in an argon-filled glovebox. The resulting mixtures were then sealed in evacuated Pyrex ampoules and heated at 1 °C min−1 to temperatures in the range 150–400 °C for periods of 2 days, to assess reactivity, which was monitored by powder X-ray diffraction. CAUTION: Heating LiH mixtures in sealed apparatus can release H2 gas and thus represents an explosion hazard.Sr2Co0.5Ir0.5O4 was observed to begin reacting with LiH at 150 °C, with non-topochemical, decomposition reactions observed at temperatures above 250 °C. LaxSr2−xCo0.5Ir0.5O4 phases with 0.25 < x < 1 were observed to react with LiH at temperatures above 250 °C, with non-topochemical decomposition reactions occurring above 430 °C.
Large-scale, topochemically-reduced samples were prepared as follows: Sr2Co0.5Ir0.5O4−yHz was prepared by grinding ∼2 g of Sr2Co0.5Ir0.5O4 with 4.5 mole-equivalents of LiH in an argon-filled glove box, the resulting mixture was then sealed in an evacuated Pyrex ampoule and heated for 7 days at 195 °C. LaxSr2−xCo0.5Ir0.5O4−yHz (0.25 < x < 1) samples were prepared by grinding ∼2 g of the corresponding LaxSr2−xCo0.5Ir0.5O4 phase with 4.5 mole-equivalents of LiH in an argon-filled glovebox, sealing the resulting mixture in an evacuated Pyrex tube and heating for 2 days at 385 °C, followed by 2–4, 5-day periods at 410 °C. Samples were reground between heating periods and reaction progress was monitored by powder X-ray diffraction. Griding and heating cycles were continued until reactions were deemed complete. After reaction, samples were washed with MeOH under a nitrogen atmosphere, to remove LiOH by-products and any unreacted LiH.
Characterisation
Sample purity and reaction progress were monitored by powder X-ray diffraction (PXRD), using a Bruker D8 Advance diffractometer with Cu Kα radiation and a Lynx-eye PSD detector. High-resolution synchrotron X-ray powder diffraction (SXRD) data were collected using instrument I11 at the Diamond Light Source. SXRD data were collected using Si-calibrated X-rays with an approximate wavelength of 0.825 Å from samples sealed in 0.3 mm diameter borosilicate glass capillaries. Neutron powder diffraction (NPD) data were collected using the POLARIS instrument10 at the ISIS neutron and muon source from samples sealed under argon in vanadium cans. Analysis of the diffraction data was performed using the Rietveld method utilizing the Topas Academic software suite (v7).11 DC and AC magnetisation data were collected using a Quantum Design MPMS-3 SQUID magnetometer. Thermogravimetric measurements were performed by heating powder samples at a rate of 10 °C min−1 under flowing oxygen using a PerkinElmer TGA 8000, with the exhaust gases monitored using a Hiden Analytical HPR-20 EGA mass spectrometer. Muon spin rotation (μSR) experiments were carried out at the Swiss Muon Source, PSI, Switzerland. Spin-polarised muons were implanted in the bulk of the sample and the time-dependence of their polarisation was monitored by recording the angular distribution of the subsequent positron decay.Co K-edge XANES spectra were collected using an EasyXAFS300+ laboratory spectrometer based on Rowland circle geometry and operating in transmission mode. The desired energy range was selected using an Si(511) spherically bent crystal analyser and the instrument's chamber was purged with He gas to minimize absorption. Samples were diluted in dried cellulose to reach appropriate mass loading, pelletized and packed in sealed plastic bags in order to avoid exposure to moist air. Spectra were energy calibrated using a Co foil and subsequently background subtracted and normalized using Athena from the demeter software package.12
Results
Structural characterisation of LaxSr2−xCo0.5Ir0.5O4 (0 ≤ x ≤ 1) phases
SXRD data collected from LaxSr2−xCo0.5Ir0.5O4 (x = 0, 0.25, 0.5, 0.75, 1) phases can be indexed using body-centred tetragonal cells, with lattice parameters consistent with n = 1 Ruddlesden-Popper phases. Data collected from Sr2Co0.5Ir0.5O4 and La0.5Sr1.5Co0.5Ir0.5O4 can be fit using previously reported, A- and B-site disordered structural models for these phases,13,14 to give good fits to the data, as detailed in the ESI.† Likewise, data from La0.25Sr1.75Co0.5Ir0.5O4, La0.75Sr1.25Co0.5Ir0.5O4 and LaSrCo0.5Ir0.5O4 can also be fitted well by analogous A- and B-site disordered, n = 1 Ruddlesden-Popper structural models as detailed in the ESI.† In the case of LaSrCo0.5Ir0.5O4 a small amount of iridium metal (1.3 weight percent) was observed in the sample, highlighting the challenge of stabilizing low oxidation states of iridium in oxides. The refined lattice parameters of the LaxSr2−xCo0.5Ir0.5O4 phases are listed in Table 1. The a-lattice parameters of the LaxSr2−xCo0.5Ir0.5O4 materials contract from x = 0 to x = 0.5 and then expand for x > 0.5, while the c-lattice parameters follow the opposite trend. The a-lattice parameter variation can be attributed to the competition between two competing factors. Firstly, the smaller ionic radius of La3+ compared to Sr2+ leads to a contraction of the a-lattice parameter with increasing x. However, substitution of Sr2+ with La3+ reduces the transition metals, expanding the Co–O and Ir–O bonds counter-acting the first trend. The combination of these two effects leads to the observed behaviour. The two-dimensional nature of the (Co/Ir)–O network means that the expansion of the (Co/Ir)–O bonds, driven by a reduction of the transition metals, doesn't have to lead to an expansion in the c-axis. Therefore, the lattice strain that would build up from competition between the opposing contraction of the A-site and expansion of the B-site, with increasing x, can be relieved by the c-axis following the opposite expansion/contraction trend to the a-axis.| x | LaxSr2−xCo0.5Ir0.5O4 | LaxSr2−xCo0.5Ir0.5O4−yHy | ||
|---|---|---|---|---|
| a (Å) | c (Å) | a (Å) | c (Å) | |
| 0 | 3.91778(6) | 12.4770(2) | 3.8058(3) | 13.037(1) |
| 0.25 | 3.88836(3) | 12.5844(1) | 3.6888(5) | 13.172(1) |
| 0.5 | 3.87999(4) | 12.5927(1) | 3.6596(5) | 13.266(2) |
| 0.75 | 3.90328(7) | 12.5893(2) | 3.6896(7) (3.7661(8)) | 13.233(2) (13.029(5)) |
| 1 | 3.89677(2) | 12.5777(1) | 3.7070(6) (3.7789(8)) | 13.342(4) (13.040(2)) |
Structural characterisation of LaxSr2−xCo0.5Ir0.5O4−yHy (0 ≤ x ≤ 1) phases
SXRD data collected from the products of reaction between LaxSr2−xCo0.5Ir0.5O4 (x = 0, 0.25, 0.5) phases and LiH could be indexed using body-centred tetragonal unit cells with reflection conditions consistent with I4/mmm space group symmetry. The lattice parameters of the reduced phases, listed in Table 1, exhibit a contraction in the xy-plane and an expansion parallel to the z-axis, compared to the corresponding ‘parent’ LaxSr2−xCo0.5Ir0.5O4 phases, consistent with the small size of the H− hydride ion, compared to the O2− oxide ion.SXRD data collected from the products of reaction between LaxSr2−xCo0.5Ir0.5O4 (x = 0.75, 1) phases and LiH could only be indexed using two closely related body-centred tetragonal unit cells, both of which have reflection conditions consistent with I4/mmm space group symmetry. Again, the lattice parameters of the reduced phases exhibit a contraction in the xy-plane and an expansion parallel to the z-axis, compared to the corresponding parent LaxSr2−xCo0.5Ir0.5O4 phase.
In an attempt to determine the oxygen content of the reduced phases, thermogravimetric data were collected as a function of temperature while heating samples under oxygen, as described in detail in the ESI (Fig. S6–S10†). All samples exhibit a mass gain under these conditions and exhaust gas monitoring reveals that the x = 0 and x = 0.25 samples release water on oxidation, while the x = 0.5, x = 0.75 and x = 1 samples release both water and hydrogen on oxidation, indicating that all 5 reduced phases are oxyhydrides rather than oxygen-deficient oxides. Analysis of samples after these measurements revealed that in common with previously reported Sr2Mn0.5Ir0.5OxHy phases,9 the TGA conditions did not lead to complete oxidation of samples. Thus, the oxygen contents listed in Table 2, determined from the relative mass gain of samples (which assume complete oxidation back to the corresponding LaxSr2−xCo0.5Ir0.5O4 phase) should be considered as maximum limits for the value of the oxygen content, with real values being significantly lower.
| x | Composition from TGA data | Composition from NPD data |
|---|---|---|
| 0 | Sr2Co0.5Ir0.5O3.47Hy | Sr2Co0.5Ir0.5O3.25H0.75 |
| 0.25 | La0.25Sr1.75Co0.5Ir0.5O3.16Hy | La0.25Sr1.75Co0.5Ir0.5O2.75H1.25 |
| 0.5 | La0.5Sr1.5Co0.5Ir0.5O2.97Hy | La0.5Sr1.5Co0.5Ir0.5O2.5H1.5 |
| 0.75 | La0.75Sr1.25Co0.5Ir0.5O2.92Hy | La0.75Sr1.25Co0.5Ir0.5O2.75H1.25 (La0.75Sr1.25Co0.5Ir0.5O3.4H0.6) |
| 1 | LaSrCo0.5Ir0.5O2.96Hy | LaSrCo0.5Ir0.5O3H (LaSrCo0.5Ir0.5O3.5H0.5) |
NPD data collected from the reduced LaxSr2−xCo0.5Ir0.5O4−yHz phases could be indexed using the tetragonal unit cells used to index the SXRD data. Thus, models based on the structures of the corresponding LaxSr2−xCo0.5Ir0.5O4 phases were refined against the data, with the occupancies of the anion sites allowed to vary. These models achieved good fits to the data and yielded structures with full oxygen occupancy on the apical (4e) anion sites, but reduced occupancy on the equatorial (4c) anion sites. Thus, hydride ions were introduced onto the equatorial anion sites along with the constraint that the oxide and hydride occupancies summed to 1, i.e. compositions of LaxSr2−xCo0.5Ir0.5O4−yHy. These models refined smoothly to give good fits to the data and yielded compositions listed in Table 2, which have oxygen contents that are lower than the maximal values determined from the TGA data. In the case of the x = 0.75 and x = 1 samples, it was possible to refine 2-phase models in which the structures and compositions of the two phases could be refined independently. In both cases the data indicate samples contain a more reduced majority phase (weight fraction >80%) and a less reduced minority phase, as shown in Table 2, suggesting that the reduction reactions did not reach completion during the preparation of the samples. Full details of the refined structures of the LaxSr2−xCo0.5Ir0.5O4−yHy phases are given in the ESI,† along with plots of fits to the diffraction data. Fits to the data collected from Sr2Co0.5Ir0.5O3.25H0.75 are shown in Fig. 1 as a representative data set.
 | ||
| Fig. 1 Observed, calculated and difference plots from the structural refinement of Sr2Co0.5Ir0.5O3.25H0.75 against NPD data collected at room temperature from 2 of the detector banks of the POLARIS instrument. Inset show representation of the structure of Sr2Co0.5Ir0.5O3.27H0.73. | ||
Fig. 2 shows XANES data collected from the cobalt K-edges of Sr2Co0.5Ir0.5O3.25H0.75, La0.5Sr1.5Co0.5Ir0.5O2.5H1.5 and LaSrCo0.5Ir0.5O3H along with data from Sr2CoIrO6 and YBaCo2O5 to act as Co3+ and Co2.5+ standards respectively. The edge position of Sr2Co0.5Ir0.5O3.25H0.75 is at lower energy than either of the two standards, consistent with a Co2+ oxidation state, while the edges of La0.5Sr1.5Co0.5Ir0.5O2.5H1.5 and LaSrCo0.5Ir0.5O3H are at even lower energy, consistent with an oxidation state of Co1+ in these materials.
 | ||
| Fig. 2 XANES data collected from the Co K-edges of Sr2Co0.5Ir0.5O3.25H0.75, La0.5Sr1.5CoIrO2.5H1.5 and LaSrCo0.5Ir0.5O3H with data from YBaCo2O5 and Sr2CoIrO6 acting as Co2.5+ and Co3+ standard respectively. | ||
Magnetic characterisation of LaxSr2−xCo0.5Ir0.5O4−yHy phases
Magnetisation-field data collected from LaxSr2−xCo0.5Ir0.5O4−yHy phases at 300 K are sigmoidal, consistent with the presence of 3–6 mol% ferromagnetic elemental cobalt, as shown in the ESI.† Thus, magnetisation data were collected using a ‘ferrosubtraction’ method described in detail in the ESI.† The paramagnetic susceptibility of LaxSr2−xCo0.5Ir0.5O4−yHy phases measured by this procedure are plotted in Fig. 3a and b. | ||
| Fig. 3 Paramagnetic susceptibility of LaxSr2−xCo0.5Ir0.5O4−yHy phases obtained via the ‘ferrosubtraction’ method. | ||
The paramagnetic susceptibility of Sr2Co0.5Ir0.5O3.25H0.75 (Fig. 3a) can be fit by the Curie–Weiss law with the addition of a temperature independent component, in the range 35 < T/K < 200, to yield values of C = 1.17(1) cm3 K mol−1, θ = −30.5(6) K, K = 0.0077(5) cm3 mol−1, broadly consistent with the value expected for a combination of Co2+ and Ir4+/5+ with the temperature independent contribution being consistent with the strong spin–orbit coupling expected for both Co2+ and Ir4+/5+ centres. Below this temperature range the susceptibility increases sharply and magnetisation-field data collected at 5 K, after cooling in an applied field of 5 T, are off-set from the origin (Fig. S16†), suggesting a transition to a glassy magnetic state at low temperature. AC susceptibility data collected from Sr2Co0.5Ir0.5O3.25H0.75 (Fig. 4a) show a strong frequency dependence consistent with the freezing of a spin glass at T ∼ 20 K and μSR data (Fig. 4b) show a change in relaxation between 27.5 and 35 K, consistent with such a transition.
 | ||
| Fig. 4 (a) The real component of AC susceptibility data collected as a function of temperature from Sr2Co0.5Ir0.5O3.25H0.75 (b) μSR data collected from Sr2Co0.5Ir0.5O3.25H0.75, La0.5Sr1.5Co0.5Ir0.5O2.5H1.5 and LaSrCo0.5Ir0.5O3H. | ||
Plots of the paramagnetic susceptibility of the remaining LaxSr2−xCo0.5Ir0.5O4−yHy phases (Fig. 3) become increasingly temperature independent with increasing lanthanum content, so cannot be fit by the Curie–Weiss law. In addition, with the exception of the data from the x = 1 sample which shows a sharp increase in magnitude for T < 60 K, none of the data show obvious features indicative of phase transitions, such as a precession signal resulting from static ordered moments (as found in ordered materials).2,3 μSR data collected from the x = 0.5 and x = 1 samples (Fig. 4b) exhibit a slowing down of spin fluctuations on cooling, but that the dynamics saturate below around 40 to 50 K. It is hard to definitively describe the magnetic behaviour of the oxyhydride phases from these data. However, on balance, given the large-scale chemical disorder in the materials, we think glassy behaviour at low temperature is the most likely scenario which is consistent with the data. This is consistent with the observation that magnetisation-field data collected from all reduced phases at 5 K, after cooling in an applied field of 5 T, are off-set from the origin (Fig. S17–S20†).
Discussion
Reaction between LaxSr2−xCo0.5Ir0.5O4 phases and LiH leads to topochemical, hydride-for-oxide, reductive anion-exchange and the formation of the corresponding LaxSr2−xCo0.5Ir0.5O4−yHy phases, with the hydride ions residing in the equatorial anion sites of the phases formed. The selective substitution of equatorial, not axial, oxide ions by hydride ions is also observed for other transition-metal oxyhydride phases with n = 1 Ruddlesden-Popper structures, such as the 3d transition-metal systems LaSrCoO3H0.7,2 Sr3CoO4.33H0.84,15 Sr2VO3H3,16 and LaSrMnO3.3H0.717 or the 3d/4d transition-metal phases LaSr3NiRuO4H4,6 LaSr3CoRuO4H418 and La0.5Sr1.5Mn0.5Rh0.5O3H.8
This strong preference for locating hydride ions within the equatorial anion sites of A2BO4 n = 1 Ruddlesden-Popper phases can be rationalized by observing that the equatorial anion sites have an A4B2 coordination compared to the A5B coordination of the axial sites. Thus, replacing the equatorial O2− oxide ions with H− hydride ions lowers the bond valence sum of the B-cations more (changes more B-anion interactions) than the equivalent change to the axial anion sites. Given that the B-cations are reduced on hydride-for-oxide exchange and the A-cations are not, locating the hydride ions on the equatorial sites leads to oxyhydride phases with lower lattice strain. The same argument has been made previously to rationalise the analogous preference for locating anion vacancies on the equatorial sites of A2BO4 phases.19
Recently we reported that Sr2Mn0.5Ir0.5O2.66H1.33 adopts an n = 1 Ruddlesden-Popper structure in which some of the hydride ions reside in axial anion sites, violating the ‘equatorial hydride’ selectivity observed for other A2BO4−xHx phases.9 In this instance it is argued that a strong trans-influence arising from strong Ir–H Sigma bonds disfavours the local mer configuration (which forces two hydride ligands to be trans to each other) which the IrO3H3 units must adopt if the hydride ions are only to be located on equatorial sites of A2BO4−xHx phases with x > 1. In addition, it was also noted that strong spin–orbit coupling favours the pseudo degeneracy of the 5dxy, 5dxz and 5dyz orbitals which occurs when IrO3H3 units adopt a fac configuration, which actively favours locating anions on axial coordination sites.
Given that both the strong trans influence and strong spin–orbit coupling arise from the presence of iridium, it may be expected that other iridium-containing, A2BO4−xHx oxyhydride phases with x > 1 would also violate the equatorial hydride rule. However, as detailed above La0.25Sr1.75Co0.5Ir0.5O2.75H1.25, La0.5Sr1.5Co0.5Ir0.5O2.5H1.5 and La0.75Sr1.25Co0.5Ir0.5O2.75H1.25 adopt structures in which the hydride ions are located exclusively on equatorial sites, despite containing iridium. We suggest this restoration of the equatorial hydride preference can be attributed to the increased A-site charge in the La1−xSr2−xCo0.5Ir0.5O4−yHy phases, compared to Sr2Mn0.5Ir0.5O2.66H1.33, which enhances the bond-valence-sum/strain effects disfavouring the location of hydride ions on axial anion sites. However, further study is required to fully understand the anion configuration preferences of iridium-containing oxyhydride phases.
Table 2 shows that the degree of hydride exchange (y) in LaxSr2−xCo0.5Ir0.5O4−yHy phases depends strongly on the La:Sr ratio (x), taking a maximum value of y = 1.5 at x = 0.5. Fig. 5 plots the observed LaxSr2−xCo0.5Ir0.5O4−yHy phases on a composition-space diagram, with the La:Sr A-site ratio varying along the x-axis, and the O:H ratio varying along the y-axis. Thus, moving to the right, or moving down on the diagram requires a reduction in transition-metal oxidation states, and moving left, or moving up on the diagram requires an oxidation of the transition-metals. It can be seen on Fig. 5 that La0.5Sr1.5Co0.5Ir0.5O2.5H1.5, La0.75Sr1.25Co0.5Ir0.5O2.75H1.25 and LaSrCo0.5Ir0.5O3H all lie on the line for which the sum of the cobalt and iridium oxidation states is equal to 4. XANES data (Fig. 2) indicate a cobalt oxidation state of Co1+ for La0.5Sr1.5Co0.5Ir0.5O2.5H1.5 and La0.75Sr1.25Co0.5Ir0.5O2.75H1.25, consistent with the observation that all the LaxSr2−xCo0.5Ir0.5O4−yHy phases in the range 0.5 < x < 1 have a Co1+, Ir3+ transition-metal oxidation-state combination.
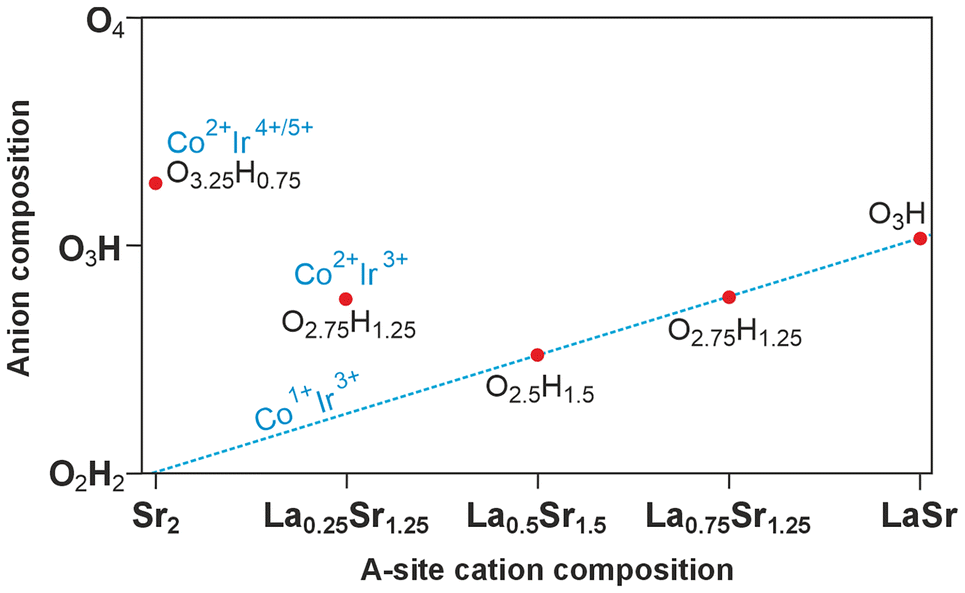 | ||
| Fig. 5 Composition diagram of LaxSr2−xCo0.5Ir0.5O4−yHy phases. | ||
As noted above, the low-spin d6 configuration of Ir3+ is consistent with the ‘covalent stabilization’ of transition-metal oxyhydrides observed previously for ruthenium and rhodium phases.6,8 It is interesting to note that in the case of the LaxSr2−xCo0.5Ir0.5O4−yHy (0.5 < x < 1) phases shown here, the transition-metal oxidation states appear to define the final hydride concentration, with compounds maintaining the Co1+, Ir3+ combination as x rises by lowering the extent of hydride-for-oxide anion exchange. This contrasts with the behaviour of the LaxSr4−xNiRuO4H4 series in which the ‘O4H4’ anion composition of the x = 1 phase, LaSr3Ni1+Ru2+O4H4, remains constant across the 0.5 < x < 1.5 range, with increases in Sr2+ content (x < 1) accommodated by the partial oxidation of Ni1+ to Ni2+ and increases in La3+ content (x > 1) by the partial reduction of Ru2+ to Ru1+.20
This contrasting behaviour of the Ir and Ru systems can be rationalized by observing that the enhanced M–X σ-bonding of Ir compared to Ru makes the ligand-field splitting of 5d Ir much greater than 4d Ru and thus makes the (t2g)6(eg)1 configuration of Ir2+ ions energetically unfeasible. As a result, it is not possible to reduce the Co1+, Ir3+ oxidation-state combination in LaxSr2−xCo0.5Ir0.5O4−yHy phases as x increases, so the hydride concentration must decline.
The hydride content of Sr2Co0.5Ir0.5O3.25H0.75 and La0.25Sr1.75Co0.5Ir0.5O2.75H1.25 do not appear to be defined by the transition metal oxidation states, but are instead limited by the robustness of the (La/Sr)–Co–Ir–O framework, with the reaction conditions required to achieve higher levels of hydride-for-oxide anion exchange leading to non-topochemical reactions and the destruction of the n = 1 Ruddlesden-Popper structure.
Magnetisation and μSR data collected from LaxSr2−xCo0.5Ir0.5O4−yHy phases (Fig. 3 and 4) are indicative of strong interactions between local spins which appear to be strongly frustrated – a combination of behaviours which is particularly evident in phases with x > 0.25. The ‘parent’ phases Sr2Co0.5Ir0.5O4 and La0.5Sr1.5Co0.5Ir0.5O4 exhibit spin glass behaviour at low temperature13,14 consistent with disorder of the Co and Ir centres within the n = 1 Ruddlesden-Popper framework, with magnetic frustration arising from competition between Co–O–Co, Co–O–Ir and Ir–O–Ir super-exchange interactions.
It may be expected that the reduction of the iridium centres on oxyhydride formation, to adopt diamagnetic, low-spin, Ir3+ configurations, may reduce the frustration in the system. However, diamagnetic Ir3+ can participate in super–super–exchange coupling in which the empty σ-symmetry iridium d-orbitals provide a pathway to couple neighbouring Co centres in an antiferromagnetic manner, as shown in Fig. 6a, by a mechanism analogous to the super–super–exchange couplings mediated by Ir5+ in double perovskite phases such as LaSrNiIrO6.21 This super–super–exchange interaction, utilizing O 2p or H 1s anion orbitals, acts to couple next–next–nearest neighbouring cobalt centres in an antiferromagnetic manner, a coupling which would lead to a ‘Type II’ antiferromagnetic structure in a rocksalt ordered array of Co and Ir centres.22,23 However, in a cation disordered system, this super–super–exchange coupling competes with simple nearest-neighbour super–exchange couplings between cobalt centres (Fig. 6b) and is likely to be one source of the strong magnetic frustration observed in the LaxSr2−xCo0.5Ir0.5O4−yHy phases.
 | ||
| Fig. 6 (a) Super–super–exchange coupling between next–next–nearest neighbour cobalt centres mediated by empty Ir σ-symmetry d-orbitals (b) nearest-neighbour super–exchange couplings between cobalt centres. | ||
The substitution of oxide ions by hydride ions is not expected to change the sign of any of the dominant σ-symmetry exchange interactions, but is likely to strengthen them due to the enhanced covalency of M–H compared to M–O interactions.
Conclusions
Reaction between LaxSr2−xCo0.5Ir0.5O4 (0 < x < 1) phases and LiH yields the corresponding LaxSr2−xCo0.5Ir0.5O4−yHy oxyhydride phases via topochemical anion exchange at the equatorial anion site. Phases in the range 0.5 < x < 1 have anion compositions defined by a Co1+, Ir3+ transition-metal oxidation-state combination, consistent with covalent stabilization of the oxyhydride phase via strong Ir–H σ-bonds. Magnetisation data indicate strong magnetic interactions between local cobalt spins, which are strongly frustrated, consistent with the disorder on both the cation and anion lattices.Author contributions
JIM prepared the samples and performed the structural and chemical analysis, RW assisted with the XANES data, SJB, HCHW and BMH collected the μSR data and managed the analysis of this data, RIS assisted with the collection of the NPD data, MAH conceived the study, wrote the manuscript and supervised the study.Conflicts of interest
There are no conflicts to declare.Data availability
Neutron Powder diffraction data can be found at: (https://doi.org/10.5286/ISIS.E.RB2310613). Additional data are available in the ESI.†Acknowledgements
Diffraction experiments at the Diamond Light Source were performed as part of the Block Allocation Group award “Oxford Solid State Chemistry BAG to probe composition–structure–property relationships in solids” (CY25166). Experiments at the ISIS pulsed neutron facility were supported by a beam time allocation from the STFC (https://doi.org/10.5286/ISIS.E.RB2310613). JIM acknowledges funding from the EPSRC Centre for Doctoral Training in Inorganic Chemistry for Future Manufacturing (OxICFM), EP/S023828/1. HCHW, BMH and SJB acknowledge funding from UK Research and Innovation (UKRI) under the UK government's Horizon Europe guarantee funding (Grant No. EP/X025861/1).References
- R. J. D. Tilley, Perovskites: Structure-Property Relationships, John Wiley and Sons, Chichester, 2016 Search PubMed.
- M. A. Hayward, E. J. Cussen, J. B. Claridge, M. Bieringer, M. J. Rosseinsky, C. J. Kiely, S. J. Blundell, I. M. Marshall and F. L. Pratt, Science, 2002, 295, 1882–1884 CrossRef CAS PubMed.
- F. Denis Romero, A. Leach, J. S. Moller, F. Foronda, S. Blundell and M. A. Hayward, Angew. Chem., Int. Ed., 2014, 53, 7556–7559 CrossRef CAS PubMed.
- T. Yamamoto, D. Zeng, T. Kawakami, V. Arcisauskaite, K. Yata, M. A. Patino, N. Izumo, J. E. McGrady, H. Kageyama and M. A. Hayward, Nat. Commun., 2017, 8, 1217 CrossRef PubMed.
- C. A. Bridges, G. R. Darling, M. A. Hayward and M. J. Rosseinsky, J. Am. Chem. Soc., 2005, 127, 5996–6011 CrossRef CAS PubMed.
- L. Jin, M. Lane, D. Zeng, F. K. K. Kirschner, F. Lang, P. Manuel, S. J. Blundell, J. E. McGrady and M. A. Hayward, Angew. Chem., Int. Ed., 2018, 57, 5025–5028 CrossRef CAS PubMed.
- Z. L. Liang, M. Batuk, F. Orlandi, P. Manuel, J. Hadermann and M. A. Hayward, Inorg. Chem., 2024, 63, 12910–12919 CrossRef CAS PubMed.
- L. Jin and M. A. Hayward, Chem. Commun., 2019, 55, 4861–4864 RSC.
- J. I. Murrell and M. A. Hayward, Inorg. Chem., 2024, 63, 22308–22314 CrossRef CAS PubMed.
- R. I. Smith, S. Hull, M. G. Tucker, H. Y. Playford, D. J. McPhail, S. P. Waller and S. T. Norberg, Rev. Sci. Instrum., 2019, 90, 115101 CrossRef CAS PubMed.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. E. Page and M. A. Hayward, Inorg. Chem., 2049, 58, 6336–6343 CrossRef PubMed.
- D. Mikhailova, Z. W. Hu, C. Y. Kuo, S. Oswald, K. M. Mogare, S. Agrestini, J. F. Lee, C. W. Pao, S. A. Chen, J. M. Lee, S. C. Haw, J. M. Chen, Y. F. Liao, H. Ishii, K. D. Tsuei, A. Senyshyn and H. Ehrenberg, Eur. J. Inorg. Chem., 2017, 587–595, DOI:10.1002/ejic.201600970.
- R. M. Helps, N. H. Rees and M. A. Hayward, Inorg. Chem., 2010, 49, 11062–11068 CrossRef CAS PubMed.
- J. Bang, S. Matsuishi, H. Hiraka, F. Fujisaki, T. Otomo, S. Maki, J. Yamaura, R. Kumai, Y. Murakami and H. Hosono, J. Am. Chem. Soc., 2014, 136, 7221–7224 CrossRef CAS PubMed.
- C. Tassel, Y. Goto, D. Watabe, Y. Tang, H. C. Lu, Y. Kuno, F. Takeiri, T. Yamamoto, C. M. Brown, J. Hester, Y. Kobayashi and H. Kageyama, Angew. Chem., Int. Ed., 2016, 55, 9666–9669 CrossRef PubMed.
- L. Jin, M. Batuk, F. K. K. Kirschner, F. Lang, S. J. Blundell, J. Hadermann and M. A. Hayward, Inorg. Chem., 2019, 58, 14863–14870 CrossRef CAS PubMed.
- M. A. Hayward, Semicond. Sci. Technol., 2014, 29, 064010 CrossRef CAS.
- L. Jin and M. A. Hayward, Angew. Chem., Int. Ed., 2020, 59, 2076–2079 CrossRef CAS PubMed.
- X. M. D. Irujo-Labalde, J. E. Page, H. W. T. Morgan, J. E. McGrady and M. A. Hayward, J. Solid State Chem., 2022, 315, 123477–123482 CrossRef.
- N. Narayanan, D. Mikhailova, A. Senyshyn, D. M. Trots, R. Laskowski, P. Blaha, K. Schwarz, H. Fuess and H. Ehrenberg, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 024403 CrossRef.
- D. Mikhailova, N. Narayanan, W. Gruner, A. Voss, A. Senyshyn, D. M. Trots, H. Fuess and H. Ehrenberg, Inorg. Chem., 2010, 49, 10348–10356 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5dt00979k |
| This journal is © The Royal Society of Chemistry 2025 |