
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManganese germylene complexes: reactivity with dihydrogen, isonitriles, and dinitrogen†
Jeffrey S.
Price
and
David J. H.
Emslie
 *
*
Department of Chemistry, McMaster University, 1280 Main St West, Hamilton, Ontario L8S 4M1, Canada. E-mail: emslied@mcmaster.ca
First published on 11th June 2025
Abstract
The manganese germylene-hydride complexes [(dmpe)2MnH(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) GeR2)] (1a: R = Ph, 1b: R = Et) reacted with H2 (approx. 1.5 atm) to afford the ‘germyl dihdyride’ species [(dmpe)2MnH2(GeHR2)] (2a: R = Ph, 2b: R = Et) as an equilibrium mixture with the starting material (1a–b). In solution, 2a–b exist as a mixture of isomers where the major isomer (71–84%) is a trans hydrogermane hydride complex trans-[(dmpe)2MnH(HGeHR2)] (transHGe-2a–b). The minor isomer of 2a–b is tentatively assigned as the cis germyl dihydrogen complex cis-[(dmpe)2Mn(GeHR2)(H2)] (cis-2a–b), possibly in rapid equilibrium with a small amount of the germanate complex [(dmpe)2Mn(H2GeHR2)] (central-2a–b). DFT calculations were employed to gain insight into the nature of bonding in the isomers of 2a–b, and an X-ray crystal structure was obtained for trans-[(dmpe)2MnH(HGeHEt2)] (transHGe-2b) which co-crystallized with 1b. Reactions of 1a–b with D2 suggest a pathway that proceeds via conversion of 1a–b to a 5-coordinate germyl intermediate [(dmpe)2Mn(GeHR2)] (A) prior to reaction with H2/D2. Providing support for this pathway, intermediate A (R = Ph) was trapped via reactions of 1a with isonitriles, affording the manganese(I) germyl isonitrile complexes [(dmpe)2Mn(GeHPh2)(CNR)] (3a: R = tBu, 3b: R = o-xylyl, and 3c: R = nBu). These complexes formed as a mixture of cis and trans isomers, and X-ray quality crystals were obtained for cis-3a, cis-3b and trans-3c. Complexes 1a–b also reacted slowly with dinitrogen at room temperature to afford germyl dinitrogen complexes [(dmpe)2Mn(GeHR2)(N2)] (5a: R = Ph, 5b: R = Et). Compounds 5a–b were initially formed as cis isomers, but the trans isomer is the thermodynamic product in each case, and the cis and trans isomers were crystallographically characterized for both 5a and 5b. X-ray crystallography, IR spectroscopy, and DFT calculations were employed to compare metal–dinitrogen bonding in the cis and trans isomers of 5a–b. The silyl dinitrogen derivative [(dmpe)2Mn(SiHPh2)(N2)] (6) was also generated as a mixture of the cis and trans isomers, and the trans isomer was structurally characterized. The trans isomers of 5b and 6 show 55Mn–31P coupling in the 31P{1H} and 55Mn{1H} NMR spectra, affording 1
GeR2)] (1a: R = Ph, 1b: R = Et) reacted with H2 (approx. 1.5 atm) to afford the ‘germyl dihdyride’ species [(dmpe)2MnH2(GeHR2)] (2a: R = Ph, 2b: R = Et) as an equilibrium mixture with the starting material (1a–b). In solution, 2a–b exist as a mixture of isomers where the major isomer (71–84%) is a trans hydrogermane hydride complex trans-[(dmpe)2MnH(HGeHR2)] (transHGe-2a–b). The minor isomer of 2a–b is tentatively assigned as the cis germyl dihydrogen complex cis-[(dmpe)2Mn(GeHR2)(H2)] (cis-2a–b), possibly in rapid equilibrium with a small amount of the germanate complex [(dmpe)2Mn(H2GeHR2)] (central-2a–b). DFT calculations were employed to gain insight into the nature of bonding in the isomers of 2a–b, and an X-ray crystal structure was obtained for trans-[(dmpe)2MnH(HGeHEt2)] (transHGe-2b) which co-crystallized with 1b. Reactions of 1a–b with D2 suggest a pathway that proceeds via conversion of 1a–b to a 5-coordinate germyl intermediate [(dmpe)2Mn(GeHR2)] (A) prior to reaction with H2/D2. Providing support for this pathway, intermediate A (R = Ph) was trapped via reactions of 1a with isonitriles, affording the manganese(I) germyl isonitrile complexes [(dmpe)2Mn(GeHPh2)(CNR)] (3a: R = tBu, 3b: R = o-xylyl, and 3c: R = nBu). These complexes formed as a mixture of cis and trans isomers, and X-ray quality crystals were obtained for cis-3a, cis-3b and trans-3c. Complexes 1a–b also reacted slowly with dinitrogen at room temperature to afford germyl dinitrogen complexes [(dmpe)2Mn(GeHR2)(N2)] (5a: R = Ph, 5b: R = Et). Compounds 5a–b were initially formed as cis isomers, but the trans isomer is the thermodynamic product in each case, and the cis and trans isomers were crystallographically characterized for both 5a and 5b. X-ray crystallography, IR spectroscopy, and DFT calculations were employed to compare metal–dinitrogen bonding in the cis and trans isomers of 5a–b. The silyl dinitrogen derivative [(dmpe)2Mn(SiHPh2)(N2)] (6) was also generated as a mixture of the cis and trans isomers, and the trans isomer was structurally characterized. The trans isomers of 5b and 6 show 55Mn–31P coupling in the 31P{1H} and 55Mn{1H} NMR spectra, affording 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1:1:1 sextets and 1:4:6:4:1 quintets, respectively.
:1:1:1:1:1 sextets and 1:4:6:4:1 quintets, respectively.
Introduction
Germylene (:GeR2) ligands in transition metal complexes can serve as highly reactive sites, including for small molecule activation.1–4 Examples of reactivity involving germylene ligands include coordination of nucleophiles to germanium,5–8 Lewis base (RCN or pyridine) addition to a germylene hydride complex to afford a germyl complex,9 and migration of a hydrogen substituent in a GeHR ligand to the metal center to form a metallogermylene (LxHM-Ge-R).10,11 Germylene to germylyne transformations have also been demonstrated by α-chloride migration,12 loss of a hydride ligand and an alkyl substituent on germanium (as HR, where R = CH2PMe2),13 or dehydrogenation of an [LxMH(GeHR)] complex by reaction with an NHC, isocyanate or nitrile.7,8,14 Additionally, various reactions of [LxMH(GeHR)]x+ complexes with unsaturated substrates have been reported. For example, cationic complexes reacted with alkenes and alkynes to form hydrogermylation products,15 and neutral complexes reacted with aldehydes and ketones (R′2CO) to afford [LxMH(Ge(OCHR′2)R)] (the latter reactions were proposed to proceed by R′2CO coordination to Ge, nucleophilic attack at the carbonyl carbon by the metal hydride to form a germyl intermediate, and subsequent 1,1-deinsertion).5,9 Furthermore, [LxMH(GeHR)] complexes reacted with isocyanates or isothiocyanates to afford metallacyclic products containing a 5-membered MGeECN (E = O or S) or MGeNCS ring.8,9,16 Addition of H2O, MeOH, or NH3 across a germylene MGe bond has also been reported, affording germyl hydride complexes with an OH, OMe or NH2 substituent on germanium,17,18 and 2 + 2 cycloaddition has been observed or proposed in reactions of germylene complexes with CO2 and isothiocyanates.9,19 Such reactivity offers many avenues for Ge incorporation into larger molecules. However, compared to lighter silylene2,20–23 and (especially) carbene24–26 complexes, the reactivity of transition metal germylene species is far less explored.
Recently, our group isolated the first terminal base-free germylene complexes of manganese, [(dmpe)2MnH(GeR2)] (1a: R = Ph, 1b: R = Et), by hydrogermane addition to Girolami and Wilkinson's27,28 manganese hydride complex [(dmpe)2MnH(C2H4)] (top of Scheme 1),29 which serves as a source of [(dmpe)2MnEt].30 While 1a–b did not react with additional equivalents of H2GeR2, the monosubstituted derivative [(dmpe)2MnH(GeHnBu)] (1c) reacted with excess H3GenBu to produce a solution containing 1c and H3GenBu in equilibrium with the products of hydrogermane addition; trans-[(dmpe)2Mn(GeH2nBu)(HGeH2nBu)] and mer-[(dmpe)2MnH(GeH2nBu)2] (bottom of Scheme 1).29 This reactivity suggests that the germylene-hydride complex 1c exists in equilibrium with an undetected 5-coordinate manganese(I) germyl complex [(dmpe)2Mn(GeH2nBu)] which is responsible for the observed reactivity with hydrogermanes.
 | ||
| Scheme 1 Top: Synthesis of manganese germylene-hydride complexes [(dmpe)2MnH(GeR2)] (1a: R = Ph, 2b: Et). Bottom: Equilibria observed between germylene-hydride complex [(dmpe)2MnH(GeHnBu)] (1c) and the Mn(I) germyl complexes trans-[(dmpe)2Mn(GeH2nBu)(HGeH2nBu)] and mer-[(dmpe)2MnH(GeH2nBu)2] upon exposure to excess hydrogermane.29 | ||
We have previously reported the manganese silylene complexes [(dmpe)2MnH(SiR2)],31 which are lighter congeners of 1a–b, and their diverse (and in some cases highly unusual) reactivity with small molecules including H2, H2SiR2, C2H4, CO2, and C(NiPr)2.31–34 Herein, we report reactions of the germylene-hydride complexes [(dmpe)2MnH(GeR2)] (1a–b) with dihydrogen, isonitriles, and dinitrogen. Literature examples of reactivity of metal germylene complexes with these small molecules are scarce, though reactions with dihydrogen to form germyl hydride complexes have been reported for [(Et3P)2Pt(Ge{N(SiMe3)2}2)],19 the heterobimetallic complex [(OC)Rh(μ-H)(μ-dppm)2(μ-GePh2)Ir(CO)][CF3SO3],35 [(Cy3P)2RuH2(H2)(GePh2)] (in this case affording a mixture of germyl hydride and hydrogermane complexes),36 [(Dipp2ArMe2P)Pt{GeCl(Dipp2Ar)}],18 and [(IPr)Ni(κ2-P,Ge-Ph2PCH2SiiPr2N(Dipp)GeAr)].37 A reaction of the titanium germylene complex [(THF)TiCp2{GeSi(SiMetBu2)Si(SiMetBu2)2Si(SiMetBu2)}] with an isonitrile has also been reported, but simply resulted in substitution of the THF co-ligand.38
Results and discussion
Reactions with H2
Solutions of the manganese germylene-hydride complexes trans-[(dmpe)2MnH(GeR2)] (1a: R = Ph, 1b: R = Et)29 reacted with H2 (∼1.5 atm) at room temperature to afford the ‘germyl dihydride’ complexes [(dmpe)2MnH2(GeHR2)] (2a: R = Ph, 2b: R = Et); Scheme 2. These reactions only progressed to 87% (2a) or 73% (2b) conversion, reaching equilibrium with 1a–b and H2 within 3 days. This reactivity contrasts that of the silicon analogues which proceeded to completion (within a few minutes (R = Ph) or 24 hours (R = Et)) to form ‘silyl dihydride’ complexes that are stable towards H2 elimination.31,33
 | ||
| Scheme 2 Reactions of the germylene-hydride complexes [(dmpe)2MnH(GeR2)] (1a: R = Ph, 1b: R = Et) with dihydrogen to afford an equilibrium mixture containing isomers of [(dmpe)2MnH2(GeHR2)] (2a: R = Ph, 2b: R = Et). The major isomer of 2a–b was identified as the trans-hydrogermane hydride complex transHGe-2a,b. For consistency with previous literature, the hydrogermane ligand in this isomer is drawn as a σ-complex, despite existing on a continuum between a sigma complex and a germyl dihydride species resulting from Ge–H bond oxidative addition.39 The minor isomer of 2a–b is tentatively assigned as the cis germyl dihydrogen complex cis-[(dmpe)2Mn(GeHR2)(H2)] (cis-2a–b), possibly in rapid equilibrium with a small amount of the germanate complex [(dmpe)2Mn(H2GeHR2)] (central-2a–b; shown in gray); vide infra. The inset shows transH2 isomers which were not experimentally observed. | ||
At room temperature, compounds 2a–b gave rise to a single hydride signal in the 1H NMR spectrum and one broad 31P{1H} NMR signal. However, upon cooling to 176 K, three 1H NMR signals (2a: 5:5:2, 2b: 4:4:3 relative integration) were observed in the low-frequency (<0 ppm) region, and the 31P{1H} NMR spectrum contained one intense and three low intensity peaks (see Fig. 1 for 2a), indicative of a major and a minor isomer (vide infra).
 | ||
| Fig. 1 Variable temperature 1H NMR (500 MHz; left) and 31P{1H} NMR (202 MHz; right) spectra of the reaction mixture formed from the reaction of [(dmpe)2MnH(GePh2)] (1a; ‡) with H2 in d8-toluene (see Fig. S23† for 31P{1H} NMR spectra at all temperatures). This reaction affords the ‘germyl dihydride’ complex [(dmpe)2MnH2(GeHPh2)] (2a), as a mixture of rapidly exchanging isomers, in slow equilibrium with 1a and H2. Peaks attributed to transHGe-2a are indicated with the symbol †, whereas those for an isomer with a disphenoidal arrangement of the dmpe ligands (tentatively identified as cis-2a, possibly in rapid equilibrium with a small amount of central-2a–b; vide infra) are indicated with the symbol *. | ||
The dominant isomer of 2a–b in solution (84% for 2a, 71% for 2b) is the trans-hydrogermane hydride complex trans-[(dmpe)2MnH(HGeHR2)] (transHGe-2a: R = Ph, transHGe-2b: R = Et). At 176 K, this isomer gave rise to a single sharp peak in the 31P{1H} NMR spectrum (at 78 ppm (2a) or 76 ppm (2b)), indicative of an equatorial arrangement of the two dmpe ligands, and requiring rapid rotation of the hydrogermane ligand (which could alternatively be viewed as a hydride and a germyl ligand that are closely interacting) on the NMR timescale. 1H NMR signals for the major isomer include a single terminal GeH peak at 6.1 (2a) or 4.6 (2b) ppm, accompanied by two low frequency signals (−10.5 and −11.2 ppm for 2a, −11.0 and −11.3 ppm for 2b). In each case, the lower frequency signal is a well-defined quintet (2JH,P = 56–58 Hz) arising from the hydride ligand trans to the hydrogermane (positioned cis to four equivalent phosphines), whereas the higher frequency signal corresponds to the H atom of the manganese-coordinated Ge–H bond.
At 176 K, the lower symmetry isomer of 2a–b gave rise to single MnH (−13.3 (2a) or −13.9 (2b) ppm) and terminal GeH (6.05 (2a) or 4.55 (2b) ppm) 1H NMR environments integrating to 2H and 1H, respectively, and three 31P{1H} NMR signals with 1:1:2 integration, indicative of a disphenoidal arrangement of the phosphine donors (71–76 (2a) or 72–75 (2b) ppm). These data are consistent with two potential isomers: (a) cis-[(dmpe)2Mn(GeHR2)(H2)] (cis-2a–b) containing cis-disposed dihydrogen and germyl ligands, or (b) [(dmpe)2Mn(H2GeHR2)] (central-2a–b) featuring an anionic germanate (H2GeHR2−) ligand that coordinates to manganese via the two newly-formed Ge–H bonds (the germanate ligand could alternatively be viewed as a germyl ligand interacting closely with two flanking hydride ligands). X-ray crystal structures have been reported for Ru40 and Nb41 complexes existing as germyl dihydrogen or germyl dihydride complexes, similar to the cis and central isomers of 2a–b, respectively.
Maintaining a solution containing 1b and 2b in hexanes under an atmosphere of dihydrogen at −78 °C afforded crystals of trans-[(dmpe)2MnH(HGeHEt2)] (transHGe-2b) co-crystallized with the germylene-hydride starting material [(dmpe)2MnH(GeEt2)] (1b) in a 0.46:0.54 ratio (Fig. 2). To the best of our knowledge, this is the first X-ray crystal structure of a hydrogermane manganese complex, and a rare example of a monometallic transition metal complex featuring a terminal hydrogermane ligand.39,42–45 However, due to co-crystallization of 1b with transHGe-2b as well as disorder in the hydrogermane ligand in transHGe-2b (which together results in three overlapping Ge ellipsoids), the structure is only suitable to establish connectivity.
 | ||
| Fig. 2 Views of the (a) [(dmpe)2MnH(GeEt2)] (1b) and (b) transHGe-[(dmpe)2MnH(HGeHEt2)] (transHGe-2b) components in crystals containing both of these species. The germanium-containing ligand in the structure is disordered over three parts – the germylene (GeEt2) ligand in 1b (54.2(3)% occupancy; view a), the hydrogermane (HGeHEt2) ligand in transHGe-2b (40.2(3)% occupancy; view b), and another orientation of the hydrogermane ligand in transHGe-2b with 5.6(3)% occupancy (not shown; H atoms on this minor disorder component were not located from the difference map). Most hydrogen atoms have been omitted for clarity, with the exception of those on Mn and Ge which were located from the difference map and refined isotropically. Ellipsoids are shown at 50% probability. | ||
DFT calculations (ADF/AMS, gas phase, all-electron, TZ2P, PBE, ZORA, D3-BJ) were carried out on the aforementioned transHGe, cis, and central isomers of germyl dihydride complexes 2a–b, as well as an isomer with trans-disposed H2 and germyl ligands, trans-[(dmpe)2Mn(GeHR2)(H2)] (transH2-2a–b); Fig. 3and S175, Table S13.† These calculations identified transHGe-2a–b (the major species observed in solution by NMR spectroscopy) as the global minimum, and ΔG176K (176 K is the lowest temperature at which decoalesced NMR spectra were obtained) for conversion of transHGe-2a–b to the other isomers was 9–11 kJ mol−1 for cis-2a–b and central-2a–b, and 14–16 kJ mol−1 for transH2-2a–b (Fig. 4).
 | ||
| Fig. 3 Geometry optimized DFT calculated structures for the transHGe, central, cis, and transH2 isomers of [(dmpe)2MnH2(GeHEt2)] (2b), with P-methyl groups and most hydrogen atoms omitted for clarity. Spheres represent Mn (red), Ge (green), P (orange), and H (white), whereas carbon atoms are represented by grey vertices. Solid bonds are those with Mayer bond orders >0.40, while dashed bonds are those with Mayer bond orders between 0.13 and 0.23. | ||
 | ||
| Fig. 4 DFT calculated Gibbs free energy changes (at 176 K) for isomerization of trans-[(dmpe)2MnH(HGeHR2)] (transHGe-2a: R = Ph, transHGe-2b: R = Et) to the central, cis and transH2 isomers. Values for 2a are shown as red hollow squares, and values for 2b are shown as blue diamonds. | ||
The calculated structures of transHGe-2a–b feature a hydrogermane ligand which has undergone substantial but incomplete Ge–H bond oxidative addition. The terminal Mn–H, bridging Mn–H (bridging between Mn and Ge), and terminal Ge–H distances are 1.54–1.55, 1.56–1.57, and 1.58 Å, with Mayer bond orders of 0.82–0.83, 0.65, and 0.84 respectively, and the bridging Ge–H distances are 2.00–2.08 Å with Mayer bond orders of 0.22–0.23 (Fig. 5; left). These bridging Ge–H distances are similar to those in Mo (2.08(6) Å),39 Rh (2.13(3) Å),42 and Pd (2.04(8) Å)43 complexes that are described as σ-hydrogermane complexes with partial Ge–H bond oxidative addition. By contrast, they are elongated relative to those in a cationic Pt(II) σ-hydrogermane complex (XRD: 1.78(4) Å, DFT: 1.79 Å) which would be expected to feature very limited Ge–H bond oxidative addition.44 Conversely, they are shorter than the Ge–H distances (2.17(4)–2.23(6) Å) in a series of nickel complexes described as containing germyl and hydride ligands with a significant interligand interaction.37
 | ||
| Fig. 5 Mayer bond orders for Mn–E, Mn–H and E–H bonds (E = Ge or Si) in trans-[(dmpe)2MnH(HGeHR2)] (transHGe-2a–b) compared with those previously reported for the germyl hydrogermane complexes trans-[(dmpe)2Mn(GeH2R)(HGeH2R)] (R = Ph or nBu)29 and the hydrosilane hydride complexes trans-[(dmpe)2MnH(HSiHR2)] (R = Ph or Et).33 In all cases [Mn] is Mn(dmpe)2, and the same computational method (ADF/AMS, gas phase, all-electron, TZ2P, PBE, ZORA, D3-BJ) was employed. | ||
The Mn–Ge distances in transHGe-2a–b are 2.43 and 2.46 Å, with Mayer bond orders of 0.74 and 0.78 (Fig. 5). These bond distances are significantly longer than that in germylene complex 1a (2.2636(4) Å),29 but are comparable with those calculated for the three higher energy isomers (the cis, central and trans-H2 isomers) of 2a–b; 2.39–2.49 Å, with Mayer bond orders of 0.69–0.85 (Table S13†). They are also similar to the crystallographically determined Mn–Ge distances in the germyl complexes [(dmpe)2Mn(GeHR2)(L)] (R = Ph or Et, L = CNR or N2) discussed below (2.475(1)–2.538(9) Å). Similarities between the transition metal–germanium distances in germyl and hydrogermane complexes have previously been reported. For example, DFT calculations on [(κ2-H2PCH2CH2PH2)2Mo(CO)(L)] (L = hydrogermane or germyl hydride) complexes afforded Mo–Ge distances that are marginally longer in the germyl hydride isomers (by 0.01–0.04 Å) than in the lowest energy hydrogermane isomers.39
In Fig. 5, relevant Mayer bond orders in transHGe-2a–b are compared with those in the germyl hydrogermane complexes trans-[(dmpe)2Mn(GeH2R)(HGeH2R)] (R = Ph or nBu),29 and the hydrosilane hydride complexes trans-[(dmpe)2MnH(HSiHR2)] (R = Ph or Et).33 These comparisons reveal (a) very similar Mn–H, Mn–Ge and Ge–H bond orders in transHGe-2a–b and the germyl hydrogermane complexes, and (b) lower E–HMn (E = Ge or Si) bond orders in transHGe-2a–b compared to the hydrosilane hydride analogues (especially considering that the Mayer bond orders are marginally higher for the Ge–H versus Si–H bonds in free H2GeEt2 and H2SiEt2, respectively; 0.92 vs. 0.91), indicative of a greater degree of E–H bond oxidative addition in transHGe-2a–b. The trend in the relative degree of Si–H vs. Ge–H oxidative addition mirrors that previously noted for [(bisphosphine)2Mo(CO)(HER3)] (E = Si, Ge) complexes.39 The structures of the silicon analogues of transHGe-2a–b provide an important reference point given that the negative J29Si,1H coupling constants for these complexes (−41 and −54 Hz; determined using 29Si_edited 1H–1H COSY NMR spectroscopy) are indicative of dominant 1J coupling (rather than 2J coupling which would give rise to a positive coupling constant), supporting the non-classical nature of the complexes; J29Si,1H values ranging from 0 to −70 Hz are commonly considered to indicate activated Si–H bonds in nonclassical hydrosilane complexes.46–48
Reactions with D2
On a timescale of hours, reactions of [(dmpe)2MnH(GeR2)] (1a–b) with D2 in C6D6 or d8-toluene cleanly produced [(dmpe)2MnD2(GeHR2)] (d2-2a: R = Ph, d2-2b: R = Et), with hydrogen located exclusively in the terminal GeH bond in both (or all) isomers of the complexes. This reactivity with D2 is analogous to that of the silylene complex [(dmpe)2MnH(SiEt2)],31 and suggests that the formation of 2a–b proceeds via initial isomerization of 1a–b to a 5-coordinate germyl intermediate [(dmpe)2Mn(GeHR2)] (A), followed by H2/D2 oxidative addition. This contrasts the reactivity of D2 with the heterobimetallic μ-germylene hydride complex [(OC)Rh(μ-H)(μ-dppm)2(μ-GePh2)Ir(CO)]+, which occurred rapidly at room temperature to generate [(OC)Rh(μ-H)(μ-dppm)2(μ-GeDPh2)IrD(CO)]+ as the primary product (although an equal distribution of H/D across all three sites was reported after 2 days at room temperature).35
After 3 days in d8-toluene, the reaction of 1a with D2 (4–6 equiv.) to form d2-2a is at or near equilibrium, and low temperature 31P{1H} NMR spectroscopy revealed that the fast equilibrium between the major transHGe isomer§ and the minor species (the cis or central isomer, or a rapidly exchanging mixture of both) is shifted further towards the minor species compared with the reaction with H2 to form 2a, affording an equilibrium isotope effect (EIE = KH/KD) of 0.63–0.69 over the temperature range 176–198 K. Room temperature H/D scrambling (between the Ge–H and Mn–D positions in [(dmpe)2MnD2(GeHR2)]) was not observed for d2-2a, but heating overnight at 55–60 °C afforded baseline MnH134 signals (<2% relative to the GeH signals).
By contrast, allowing the reaction of 1b with D2 (4 equiv.) to proceed for 4 days in d8-toluene at room temperature afforded d2-2b as well as other isotopologues (d3-2b, and smaller amounts of d1-2b and 2b), giving rise to both GeH and MnH signals in the 1H NMR spectrum, as well as some HD and H2. These observations are consistent with slow hydrogermane (e.g. HDGeEt2) dissociation and re-coordination from dn-2b (perhaps from the transHGe isomer) as well as pathways involving D2/HD/H2 dissociation and re-coordination.
A low temperature (176 K) 1H{31P} NMR spectrum of the mixture of 2b, d1-2b, d2-2b, and d3-2b features a 1:1:1 triplet at −13.94 ppm overlapping with a broad singlet at slightly higher frequency (Fig. 6). These signals correspond to the minor isomer of dn-2b (n = 0–2), and the coupling constant for the 1:1:1 triplet is 28 Hz, indicative of an HD ligand.49,50 This signal is proposed to arise from the cis isomer of [(dmpe)2Mn(GeXEt2)(HD)] {X = D (d2-2b) and H (d1-2b); see Fig. 6}, and the 28 Hz coupling constant almost exactly matches that predicted49,51 based on the calculated H–H distance of 0.95 Å in cis-2b. The aforementioned broad singlet is proposed to arise from the cis isomer of [(dmpe)2Mn(GeXEt2)(H2)] {X = D (d1-2b) and H (2b)}, both of which contain an H2 ligand (see Fig. 6).
 | ||
| Fig. 6
1H and 1H{31P} NMR spectra (176 K; 500 MHz) of the mixtures formed from the room temperature reactions of [(dmpe)2MnH(GeEt2)] (1b) with (top) H2 or (bottom) D2 in d8-toluene after 4 days. The reaction with D2 afforded a mixture of d2-2b and d3-2b, as well as smaller amounts of d1-2b and 2b. Only the low frequency region of the spectra is shown (which does not contain any signals for d3-2b or isomers/isotopomers of d2-2b with a terminal Ge–H bond). The expansion on the right shows the low-frequency 1H{31P} signal for the minor isomer(s) of dn-2b (n = 0–2), which consists of a 1:1:1 triplet overlapping with a broad singlet. The 1:1:1 triplet is proposed to arise from isomers containing an HD ligand (d2-2b where X = D and d1-2b where X = H; X is the terminal H/D substituent on germanium), while the broad singlet is proposed to arise from isomers containing an H2 ligand (d1-2b where X = D and 2b). The H2 and HD complexes (cis isomers) may or may not be in rapid equilibrium with a small amount of a germanate species (central isomers). [Mn] = (dmpe)2Mn. | ||
While these data suggest that the minor isomer of 2b is the cis-dihydrogen complex (the cis isomer) rather than a germanate complex (the central isomer), the presence of a small amount of the central isomer (in rapid equilibrium with the cis isomer) cannot be excluded, given (a) the very similar energies of the cis and central isomers of 2a–b in DFT calculations, and (b) potential differences in the position of any cis–central equilibrium in reactions involving HD versus H2 or D2 (especially considering that low-temperature reactions with H2/D2 to form an H2/D2 complex typically exhibit a substantial inverse equilibrium isotope effect).52,53 This situation contrasts that for silicon analogues of 2a–b, where the spectroscopically identified isomers were trans-[(dmpe)2MnH(HSiHRR′)] (a transHSi isomer) and the silicate complex [(dmpe)2Mn(H2SiHRR′)] (a central isomer), both of which were crystallographically characterized.31
Reactions with isonitriles
The putative 5-coordinate germyl intermediate [(dmpe)2Mn(GeHR2)] (A) was trapped using isonitriles. For example, exposure of [(dmpe)2MnH(GePh2)] (1a) to an excess of tert-butyl isonitrile (CNtBu) afforded the germyl isonitrile complex [(dmpe)2Mn(GeHPh2)(CNR)] (3a: R = tBu) as a yellow solid in 42% yield (Scheme 3). Analogues of 3a featuring different isonitrile ligands (3b: R = o-xylyl, 3c: R = nBu) were also synthesized via the same route (Scheme 3) and characterized in situ by NMR spectroscopy.‡ The reactions to form 3a–c all proceeded to completion within 1 hour at room temperature.
 | ||
| Scheme 3 Syntheses of manganese(I) germyl isonitrile complexes [(dmpe)2Mn(GeHPh2)(CNR)] (3a–c; R = tBu, Xyl or nBu) and the previously reported54 hydride complex [(dmpe)2MnH(CNXyl)] (4). Xyl = o-xylyl. | ||
In solution, two sets of NMR signals were observed for 3a–c (Table S12†), consistent with a high symmetry trans isomer containing an equatorial belt of dmpe ligands (one 31P{1H} singlet was observed at 71–74 ppm), and a low symmetry cis isomer with multiple 31P NMR environments arising from disphenoidal dmpe coordination. The GeH signals in the 1H NMR spectra range from 5.06–5.27 ppm for the trans isomers and 5.45–5.48 for the cis isomers. Initially formed solutions of 3a–c contain a mixture of cis and trans isomers in ratios ranging from 83:17 to 77:23, which remained unchanged overnight at room temperature (though a different ratio was observed in recrystallized samples of 3a and 3c due to preferential crystallization of the cis isomers).
Heating the reaction mixture from the synthesis of [(dmpe)2Mn(GeHPh2)(CNXyl)] (3b; Xyl = o-xylyl) resulted in slow formation of the previously reported54 manganese(I) hydride complex [(dmpe)2MnH(o-xylyl)] (4) (70% conversion after heating for ∼12 h at 100 °C) accompanied by unidentified byproducts; Scheme 3. Recrystallization from hexanes afforded X-ray quality crystals of 4, with an Mn–H distance of 1.75(7) Å, an Mn–C distance of 1.789(6) Å, a C–N distance of 1.221(8) Å, and a C–N–C angle of 160.4(5)° (Fig. S168†). An X-ray crystal structure of this complex was previously published54 with a different unit cell (in a higher symmetry space group), featuring a perfectly straight Mn–C–N–C rod lying on a C2 axis.
X-ray crystal structures were obtained for 3a–c¶ as the cis (3a–b) or trans (3c) isomer; Fig. 7 and S167, Table S11.† In all cases, the hydrogen atoms on Ge were located from the difference map and refined isotropically. The Mn–Ge distances of 2.475(1)–2.481(1) Å (cis isomers of 3a–b) and 2.528(1) Å (trans-3c) are at the higher end of the range previously reported for neutral manganese(I) germyl complexes (2.37–2.53 Å),55 and in each case, appreciable bending of the isonitrile ligands (C–N–C = 144.9(4)–172.6(3)°) was observed.
 | ||
| Fig. 7 X-ray crystal structures of (a) cis-[(dmpe)2Mn(GeHPh2)(CNtBu)] (cis-3a) and (b) trans-[(dmpe)2Mn(GeHPh2)(CNnBu)] (trans-3c), with ellipsoids at 50% probability. Most hydrogen atoms have been omitted for clarity, except for those on Ge, which were located from the difference map and refined isotropically. | ||
Reactions with N2
While coordination of the highly donating isonitrile ligands to manganese(I) is not unexpected, similar reactivity was also observed with dinitrogen. The manganese germylene-hydride complexes trans-[(dmpe)2MnH(GeR2)] (1a: R = Ph, 1b: R = Et)29 reacted in solution with dinitrogen (∼1.5 atm) at room temperature to afford solutions of the germyl-dinitrogen complexes [(dmpe)2Mn(GeHR2)(N2)] (5a: R = Ph, 5b: R = Et); Scheme 4. These reactions are much slower than those involving isonitriles, reaching 95% (5a) or complete (5b) conversion to 5a–b after three days in the dark. Relative to the rich dinitrogen chemistry56 of other d6 transition metals such as Mo(0),57 Fe(II),58 and Re(I),59 far fewer Mn(I)–N2 complexes have been reported (see Scheme S1† and associated text).54,60–75
 | ||
| Scheme 4 Syntheses of manganese(I) germyl dinitrogen complexes [(dmpe)2Mn(GeHR2)(N2)] (5a: R = Ph, 5b: R = Et). | ||
In solution, a single set of NMR environments was observed for initially formed 5a and 5b (Table 1), consistent with a low symmetry cis isomer (featuring four distinct 31P NMR environments). However, the diphenyl derivative [(dmpe)2Mn(GeHPh2)(N2)] (5a) slowly isomerized to a higher symmetry trans isomer, affording a cis:trans ratio of ∼3:1 after 3 days at room temperature in the dark (i.e. over the duration of the reaction of 1a with N2), and further conversion to achieve a 3:7 ratio occurred over an additional 12 days. Significant room temperature isomerization was not observed for the diethyl derivative [(dmpe)2Mn(GeHEt2)(N2)] (5b) (upon complete consumption of 1b after 3 days, only ∼2% of the dinitrogen complex had isomerized to the trans isomer). However, the trans isomer became the dominant product after heating a solution of 5b under an atmosphere of N2 at 60 °C for 1 day. This indicates that for both 5a and 5b, the trans isomer is the thermodynamically favoured product. 1H NMR spectra of 5a–b feature GeH chemical shifts ranging from 3.02 to 5.04 ppm, where the values for each isomer of the diphenylgermyl derivative 5a are 1.72–1.75 ppm higher frequency than those of 5b, and the cis isomer GeH signal is shifted 0.63–0.66 ppm to higher frequency of the trans isomer in each case (Table 1).
| E/R | cis | trans | ||||
|---|---|---|---|---|---|---|
| δ(1H)EH/1JSi,H | δ(31P) | δ(55Mn) | δ(1H)EH/1JSi,H | δ(31P) | δ(55Mn)/1JP,Mn | |
|
a 55Mn{1H} data for 5a–b was obtained in d8-toluene.
|
||||||
| Ge/Ph (5a) | 5.40 | 72.0, 69.3, 68.7, 58.7 | −718a | 4.74 | 70.6 | −845a |
| Ge/Et (5b) | 3.65 | 75.5, 71.4, 65.4, 60.6 | −1011a | 3.02 | 72.3 | −1094/275 Hza |
| Si/Ph (6) | 5.53/141 Hz | 71.8, 68.5, 64.1, 58.7 | −973 | 4.82/140 Hz | 70.8 | −1098/270 Hz |
In contrast to the trans isomer of the germyl isonitrile complexes 3a–c and various other previously reported trans-[(dmpe)2MnRL] complexes where the equatorial array of 31P nuclei gave rise to a singlet,27,28,31,33,54 the 31P{1H} NMR signal arising from trans-5a–b was a very broad singlet (5a) or an approximately equal-intensity sextet (5b; left of Fig. 8) consistent with coupling to 55Mn (nat. abund. = 100%; I = 5/2). Furthermore, the trans isomer of [(dmpe)2Mn(GeHEt2)(N2)] (5b) afforded a quintet at −1094 ppm in the 55Mn{1H} NMR spectrum at ambient temperature (right of Fig. 8), indicative of a low electric field gradient at Mn.|| The manganese–phosphorous coupling constant (1J31P,55Mn) of 275 Hz in trans-5b is similar to those in previously reported alkyl tetracarbonyl manganese(I) complexes bearing a triphenylphosphine co-ligand (208–298 Hz).76,77 By contrast, trans-5a and cis-5a–b gave rise to broad singlets in the 55Mn{1H} NMR spectrum (Table 1), even upon heating to 97 °C.
 | ||
| Fig. 8
31P{1H} (left, 202 MHz) and 55Mn{1H} (right, 124 MHz) NMR spectra of [(dmpe)2Mn(GeHEt2)(N2)] (5b) at 370 and 298 K, respectively (d8-toluene, N2 atmosphere), showing the sextet or quintet arising from the trans isomer due to 31P–55Mn coupling, along with peaks arising from the cis isomer and from (left only) trans-[(dmpe)2MnH(GeEt2)] (1b; formed due to reversible N2 dissociation from 5b at elevated temperature). The 31P{1H} NMR spectrum was obtained at elevated temperature to reduce the extent of quadrupolar collapse for the trans-5b signal (note that signals for cis-5b, which are broadened at this temperature, are sharp singlets at low temperature as shown in Fig. S143† due to complete quadrupolar collapse of coupling to 55Mn and/or slowing of a fluxional process which renders the 31P atoms equivalent at high temperature). | ||
Chemical shifts in 55Mn NMR spectroscopy (δ55Mn) vary widely for Mn(I) complexes.78 For example, the trifluorophosphine complex [(F3P)5MnH]79 gave rise to a 55Mn NMR signal at −2953 ppm, compared with +1077 ppm for [CpMn(η6-cycloheptatriene)].80 In this work, the 55Mn chemical shifts for the cis and trans isomers of 5a–b (−718 to −1094 ppm) fall near the centre of this range. The 55Mn NMR signal for the diphenylgermyl derivative (5a) is 245–293 ppm more positive than the diethylgermyl derivative (5b), consistent with the trend previously noted for [(R3Sn)Mn(CO)5], where δ55Mn for the triphenylstannyl derivative is 50 ppm more positive than for the trimethylstannyl derivative.81 The significant difference (79–127 ppm) in 55Mn chemical shifts arising from cis and trans isomers in 5a–b highlights the sensitivity of the 55Mn nucleus to its electronic environment; similarly 55Mn chemical shifts of −920 and −637 ppm have been reported for trans and cis isomers, respectively, of [ClMn(TeMe)2(CO)3],82 and δ55Mn differences of up to 237 ppm have been reported for various diastereomers of manganese(I) carbonyl halide complexes bearing bidentate chalcogenoether ligands.82–85
Solutions of 5a–b in C6D6 are stable for days under an atmosphere of N2. However, in the absence of N2, these solutions suffered from slow dinitrogen loss to re-form the germylene-hydride starting materials 1a–b. This process occurred more rapidly for 5a than 5b; ∼8% conversion back to germylene-hydride was observed in a sealed J-young tube under argon after 6 hours or 2 days in the dark, respectively. Dissociation of N2 could also be promoted by exposure to light;86–89 after 6 hours under a medium pressure mercury vapour lamp, a sealed solution of 5b in a J-young tube converted to a 1.7:1 mixture of 1b and 5b, accompanied by a small amount of decomposition to unidentified species.
Complexes 5a–b were isolated in 54–58% yield as yellow solids by removal of solvent in vacuo in the dark (5a–b are stable in vacuo for short periods of time in the absence of light) followed by recrystallization at −30 °C under argon. Analytical purity was confirmed by combustion elemental analysis (EA) and 2D powder X-ray diffraction (PXRD), and no germylene-hydride starting material was detected by IR spectroscopy (Nujol mull). Diffractograms of the bulk solids indicated that 5a was isolated as an approximate 1:3 ratio of the cis and trans isomers, whereas 5b was isolated almost exclusively as the cis isomer. In the solid state, 5a–b proved to be reasonably stable to dinitrogen loss when kept under an argon atmosphere. For example, no decomposition was observed for 5b after 2 years in a sealed vial at −30 °C as measured by PXRD and EA.
X-ray crystal structures were obtained for both the cis and trans isomers of 5a and 5b (Fig. 9 and S169;†Table 2). These complexes are octahedral with end-on terminal dinitrogen ligands, and (in all cases except trans-5a) the hydrogen atom on Ge was located from the difference map and refined isotropically. The manganese–germanium distances of 2.4795(6)–2.538(9) Å are at the higher end of the range previously reported for neutral manganese(I) germyl complexes (2.37–2.53 Å),55 and are similar to those in the isonitrile complexes 3a–b (2.475(1)–2.528(1) Å). The Mn–N distances are somewhat longer in the cis isomers (1.841(2) and 1.822(2) Å) than the trans isomers (1.806(2) and 1.79(2) Å), but the N–N distances (1.120(4)–1.139(3) Å) in all four complexes are equal within 3 standard deviations. To the best of our knowledge, the only other crystallographically characterized examples of neutral Mn(I) complexes containing a terminal dinitrogen ligand are the cymantrene derivative [CpMn(CO)2(N2)]90 (Mn–N = 1.8418(4) Å; N–N = 1.1144(1) Å)91 and the octahedral tetraphosphine hydride complex trans-[(dmpe)2MnH(N2)] (Mn–N = 1.817(5) Å; N–N = 1.127(7) Å),54 which feature similar Mn–N and N–N distances to those in 5a–b.
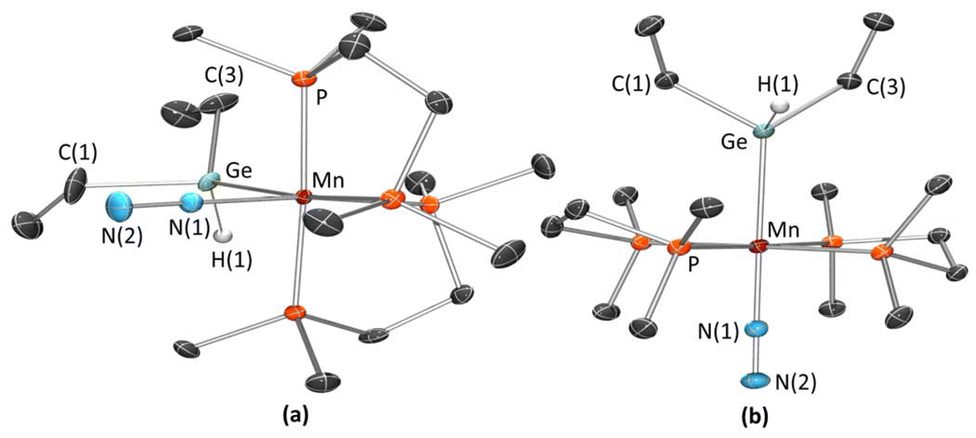 | ||
| Fig. 9 X-ray crystal structures of (a) cis-[(dmpe)2Mn(GeHEt2)(N2)] (cis-5b) and (b) trans-[(dmpe)2Mn(GeHEt2)(N2)] (trans-5b), with ellipsoids at 50% probability. Most hydrogen atoms have been omitted for clarity, with the exception of those on Ge which were located from the difference map and refined isotropically. For cis-5b, the dmpe ligands are disordered over two positions, and only the dominant position (81.0(2)%) is shown. | ||
| R | cis | trans | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mn–Ge | Mn–N | N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N N |
Ge–Mn–N | Mn–N–N | Mn–Ge | Mn–N | NN |
Ge–Mn–N | Mn–N–N | |
| a For trans-5a, two sets of values are provided for some bond metrics due to a 2-part disorder of the germyl ligand. | ||||||||||
| Ph (5a)a | 2.4867(8) (2.51) {0.81} | 1.841(2) (1.80) {1.01} | 1.120(4) (1.14) {2.47} | 85.95(7) (86.3) | 176.2(2) (176.4) | 2.538(9), 2.51(1) (2.51) {0.87} | 1.79(2) (1.79) {1.11} | 1.14(3) (1.14) {2.45} | 165.0(8), 169.7(8) (175.9) | 177(2) (179.6) |
| Et (5b) | 2.4795(6) (2.50) {0.80} | 1.822(2) (1.80) {1.04} | 1.124(3) (1.14) {2.48} | 86.65(5) (86.6) | 178.8(2) (178.2) | 2.5089(9) (2.51) {0.86} | 1.806(2) (1.79) {1.12} | 1.139(3) (1.14) {2.43} | 177.58(6) (177.4) | 179.4(2) (178.9) |
IR spectroscopy (Nujol mull) was employed as a more sensitive tool to investigate differences in Mn–N2 bonding, and peaks were assigned to cis or trans isomers based on relative peak intensity (compared to expected ratios derived from PXRD). IR spectra of 5a–b included ν(NN) stretches at 2010, 1973, 1989, and 1968 cm−1 (calcd 2045, 2026, 2040, and 2018 cm−1) for cis-5a, trans-5a, cis-5b, and trans-5b, respectively.** These data point to increased π-backdonation to N2 in the trans complexes. The N–N stretches for both isomers of 5a–b are lower than in previously reported cyclopentadienyl-containing (2078–2169 cm−1)64,66,90,92 and cationic (2146–2167 cm−1)73,74 manganese(I) complexes with terminal end-on N2 ligands. However, they are slightly higher than in [(dmpe)2MnH(κ1-N2)] (νNN = 1947 cm−1).54 For broader comparison, the range of ν(NN) values for end-on transition metal dinitrogen complexes typically spans from 2250 to 1800 cm−1 (cf. 2330 cm−1 for free N2).56
DFT calculations (ADF/AMS, gas phase, all-electron, TZ2P, PBE, ZORA, D3-BJ) were carried out to further probe the nature of Mn–N2 bonding in 5a–b. Geometry optimization of both the cis and trans isomers of 5a–b afforded structures with Mn–Ge, Mn–N, and N–N distances within 0.04 Å of the crystallographically determined values, along with Ge–Mn–N and Mn–N–N angles within three standard deviations of the XRD metrics (with the exception of Ge–Mn–N in trans-5a, which involved a disordered germyl ligand in the X-ray crystal structure).
Mayer bond orders for the Mn–N and NN bonds in the trans isomers are 0.08–0.10 higher and 0.02–0.05 lower, respectively, than in the cis isomers (Table 2), indicative of increased π-backdonation in the former (trans isomers). This is consistent with the trend indicated by N–N stretching frequencies (vide supra), which are mirrored by the DFT calculated NN stretching frequencies which are 19–22 cm−1 lower for the trans isomers of 5a–b.
Bonding between the N2 ligands and (dmpe)2Mn(GeHR2) fragments was further investigated via fragment interaction calculations using the energy decomposition analysis (EDA)93 method of Ziegler and Rauk (Table 3). This approach affords an overall interaction energy, ΔEint, which is divided into five components, as shown in eqn (1).94,95 In this analysis, ΔEelec represents the electrostatic interaction energy (calculated using frozen charge distributions for both fragments), ΔEPauli corresponds to Pauli repulsion, ΔEorb is the orbital interaction energy (this term includes all contributions resulting from intrafragment polarization), ΔEdisp is the dispersion interaction energy, and ΔEprep is the energy needed to bring the fragments from their optimum geometries to their geometries in the unfragmented complex.
| ΔEint = ΔEelec + ΔEPauli + ΔEorb + ΔEdisp + ΔEprep | (1) |
| GeHR2 | cis-5a | trans-5a | cis-5b | trans-5b | |
|---|---|---|---|---|---|
| GeHPh2 | GeHEt2 | ||||
| EDA | ΔEint | −117 | −163 | −149 | −173 |
| ΔEelec | −321 | −338 | −320 | −339 | |
| ΔEorb | −391 | −422 | −395 | −427 | |
| ΔEPauli | 545 | 575 | 540 | 572 | |
| ΔEdisp | −15 | −14 | −14 | −14 | |
| ΔEprep | 57 | 28 | 32 | 26 | |
| BSSE | 9 | 9 | 9 | 9 | |
| Hirsh | [M] | 0.29 | 0.34 | 0.31 | 0.35 |
| N2 | −0.29 | −0.34 | −0.31 | −0.35 | |
| ETS-NOCV | ΔEσ | −118 (30%) | −119 (28%) | −119 (30%) | −114 (27%) |
| ΔEπ(1) | −129 (33%) | −138 (33%) | −131 (33%) | −142 (33%) | |
| ΔEπ(2) | −116 (30%) | −133 (32%) | −119 (30%) | −139 (32%) | |
| Other | −28 (7%) | −32 (7%) | −27 (7%) | −32 (8%) | |
Overall interaction energies for N2 coordination are −117 (5a) and −149 (5b) kJ mol−1 for the cis isomers and −163 (5a) and −173 (5b) kJ mol−1 for the trans isomers. Stronger N2 bonding to manganese in the trans isomers is driven by stronger electrostatic and orbital contributions (ΔEelec and ΔEorb are more negative by 17–19 and 31–32 kJ mol−1, respectively), partially offset by increased Pauli repulsion (ΔEPauli is more positive by 30–32 kJ mol−1). The less negative ΔEint for the Mn–N2 bond in cis-5a relative to cis-5b is driven primarily by a higher preparation energy for the (dmpe)2Mn(GeHPh2) fragment. This is due to stabilization of the geometry optimized fragment through a γ-hydride interaction involving an ortho C–H bond of a phenyl substituent on germanium (illustrated by a Mn⋯H bond distance of 1.91 Å and a Mayer bond order of 0.24; Fig. S176†).
The deformation density (Δρ) associated with the orbital interaction component (ΔEorb) from fragment interaction calculations was further divided using the Extended Transition State and Natural Orbitals for Chemical Valence (ETS-NOCV) method (Table 3), affording a textbook example of end-on N2 bonding to a transition metal. Deformation density isosurfaces and the main fragment orbital contributors for the two isomers of 5a are shown in Fig. 10 (similar figures for 5b, and the NOCVs associated with each ETS-NOCV contribution, are shown in Fig. S179–182†). In each case, three contributions of similar energy (27–33% of ΔEorb in all cases) were elucidated. One of these (Δρσ) involves σ-donation from the HOMO of dinitrogen to the LUMO of the (dmpe)2Mn(GeHR2) fragment, whereas the other two contributions (Δρπ(1) and Δρπ(2)) involve orthogonal π-backdonation from occupied manganese d orbitals to the vacant π* orbitals of dinitrogen. The relative energies of these σ-donation and π-backdonation contributions are similar to those reported for other end-on N2 complexes.96–100
 | ||
| Fig. 10 Deformation density contributions (shown in green/yellow) and the main fragment orbital contributions (shown in red/blue or orange/turquoise) to bonding between a (dmpe)2Mn(GeHPh2) fragment and an N2 fragment in the cis- (left) and trans- (right) isomers of [(dmpe)2Mn(GeHPh2)(N2)] (5a). Three major interactions were observed (Δρσ, Δρπ(1) and Δρπ(2)). Deformation density isosurfaces (set to 0.003) correspond to increased (green) and decreased (yellow) electron density relative to the non-interacting fragments. Orbital isosurfaces are set to 0.03. | ||
The aforementioned differences in the magnitude of ΔEorb between the cis and trans isomers of 5a–b is driven exclusively by the two π interactions, which are stronger (by 7–17 kJ mol−1) in the trans isomers. More negative Hirshfeld charges on the N2 fragment (from fragment interaction calculations) in the trans isomers (−0.34 to −0.35, versus −0.29 to −0.31 for the cis isomers; Table 3) also indicate an increase in charge transfer from the (dmpe)2Mn(GeHR2) fragment to N2, consistent with increased π-backdonation in the trans isomers.
Reaction of N2 with [(dmpe)2MnH(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) SiPh2)]
SiPh2)]
Given that the germylene-hydride complexes (1a–b) reacted with N2, we were interested to probe whether a silylene-hydride analogue would engage in analogous reactivity. Indeed, [(dmpe)2MnH(SiPh2)] (which exists in solution as an equilibrium mixture of the cis and trans isomers)31 also reacts with dinitrogen to afford complexes tentatively identified by NMR spectroscopy as cis and trans isomers of the silyl dinitrogen complex [(dmpe)2Mn(SiHPh2)(N2)] (6); Scheme 5. This reactivity reflects that previously reported for another d6 silylene-hydride complex, [(Cp*)(iPr2MeP)FeH(SiHTrip)], which reversibly coordinates N2.101
 | ||
| Scheme 5 Synthesis (in situ) of the manganese(I) silyl dinitrogen complex [(dmpe)2Mn(SiHPh2)(N2)] (6) as a mixture of the cis and trans isomers. The starting material [(dmpe)2MnH(SiPh2)] exists as a mixture of the trans (minor) and cis (major) isomers; only one isomer is shown. | ||
Unfortunately, the synthesis of the silylene-hydride starting material affords an inseparable mixture of this complex with the silyl dihydride species [(dmpe)2MnH2(SiHPh2)]31 (which does not react with N2 under mild conditions), so complex 6 was generated as a mixture with [(dmpe)2MnH2(SiHPh2)]. X-ray quality crystals of trans-6 (Fig. S170†) were obtained from this mixture, showing that trans-6 is isostructural to the germyl derivative trans-5a, including statistically equivalent Mn–N and N–N distances. The Mn–Si distances of 2.457(3)–2.469(6) Å in trans-6 are significantly elongated relative to those in the previously reported manganese(I) SiHnR3−n (n = 1–2) complexes; the primary silyl complexes [(dmpe)2Mn(SiH2R)(CNR′)] (R = Ph, nBu, R′ = tBu, o-xylyl; 2.3552(5)–2.3618(5) Å),32 and the more electron-poor tetracarbonyl phosphine complex [(OC)4Mn(SiHPh2)(PPh3)] (2.410(1) Å).102
Selected NMR spectroscopic data for 6 (Table 1) includes 1H NMR SiH environments at 5.53 (cis-6, 1JH,Si = 141 Hz) and 4.82 (trans-6, 1JH,Si = 140 Hz and 3JH,P = 8.6 Hz) ppm, and 29Si NMR signals at 36.6 (cis-6) and 33.9 (trans-6) ppm. Similar to the NMR spectra of diethylgermyl derivative 5b, the cis isomer of 6 gave rise to multiple 31P{1H} NMR resonances and a broad singlet in the 55Mn{1H} NMR spectrum at −971 ppm, while trans-6 afforded a single 31P{1H} NMR signal at 70.8 ppm which shows coupling to 55Mn in the process of quadrupolar collapse and a quintet in the 55Mn{1H} NMR spectrum at −1098 ppm (1JMn,P = 270 Hz; Fig. S164 and S157†). The shift in 55Mn NMR signals to more negative frequency (by 253–255 ppm) relative to those of germyl derivative 5a mirrors the previously reported trend in the tetryl-ligated manganese(I) carbonyl complexes [(R3E)Mn(CO)5] (R = Cl, C6F5, or Ph, E = Si or Ge), where δ55Mn is more negative (by 40–330 ppm) in the silyl derivatives.103 However, this trend differs from those for manganese(I) complexes which vary in the identity of a halide82–84,104,105 or chalcogenoether82–84,106 ligand, where more negative 55Mn chemical shifts were observed proceeding down the group.
Summary and conclusions
Reactions of the germylene-hydride complexes [(dmpe)2MnH(GeR2)] (R = Ph or Et; 1a–b) with H2 afforded [(dmpe)2MnH2(GeHR2)] (2a–b) in slow equilibrium with the starting materials. This reactivity contrasts that of the silicon analogues which reacted completely to form ‘silyl dihydride’ complexes that are stable towards H2 elimination. In solution, complexes 2a–b exist as multiple isomers in rapid equilibrium, which were characterized by low temperature NMR spectroscopy. The major isomer of 2a–b is trans-[(dmpe)2MnH(HGeHR2)] (transHGe-2a–b) featuring trans-disposed hydride and hydrogermane ligands, and DFT calculations indicate a higher degree of Ge–H bond oxidative addition in transHGe-2a–b compared to Si–H bond oxidative addition in the previously reported hydrosilane hydride analogues trans-[(dmpe)2MnH(HSiHR2)]. The minor isomer of 2a–b features a disphenoidal arrangement of the phosphine donors, and is tentatively assigned as the cis germyl dihydrogen complex cis-[(dmpe)2Mn(GeHR2)(H2)] (cis-2a–b); the presence of this isomer is supported by the observation of a 1:1:1 triplet with a large (28 Hz) JH,D coupling constant in the low-temperature 1H{31P} NMR spectrum of a mixture containing partially deuterated isotopologues of 2b (where the triplet is attributed to [(dmpe)2Mn(GeXEt2)(HD)] {X = D (d2-2b) and H (d1-2b)}, both of which contain an HD ligand). However, the presence of a small amount of the germanate complex [(dmpe)2Mn(H2GeHR2)] (central-2a–b), in rapid equilibrium with the cis isomer, cannot be excluded, given (a) the very similar energies of the cis and central isomers of 2a–b in DFT calculations, and (b) potential differences in the position of any cis–central equilibrium in reactions involving HD versus H2 or D2.
An X-ray crystal structure was obtained for transHGe-2b co-crystallized with the starting germylene complex 1b. To the best of our knowledge, this is the first crystallographically characterized example of a manganese hydrogermane complex, and more generally, transHGe-2a–b are rare examples of monometallic transition metal complexes featuring a terminal hydrogermane ligand. Reactions of 1a–b with D2 initially afforded only [(dmpe)2MnD2(GeHR2)] (d2-2a–b), suggesting that the formation of 2a–b proceeds via initial isomerization of the germylene-hydride complexes to a 5-coordinate manganese(I) germyl intermediate [(dmpe)2Mn(GeHR2)] (A). This intermediate was trapped by reacting [(dmpe)2MnH(GePh2)] (1a) with isonitriles to afford the germyl isonitrile complexes [(dmpe)2Mn(GeHPh2)(CNR)] (3a–c). These complexes were formed as mixtures of cis and trans isomers, and X-ray crystal structures were obtained for either isomer depending on the isonitrile used.
Germylene-hydride complexes 1a–b also reacted slowly with dinitrogen to afford the manganese(I) germyl dinitrogen complexes [(dmpe)2Mn(GeHR2)(N2)] (5a–b). Complexes 5a–b were initially formed as cis isomers, but the trans isomer became dominant over time in solution (at room temperature or with mild heating). The N2 ligands in 5a–b are labile in solution, but the complexes are reasonably stable in the solid state. X-ray crystal structures were obtained for both isomers of 5a and 5b, providing rare examples of crystallographically characterized manganese(I) terminal N2 complexes. The silylene complex [(dmpe)2MnH(SiPh2)] was also shown to react with N2 to form the silyl dinitrogen derivative [(dmpe)2Mn(SiHPh2)(N2)] (6) as a mixture of cis and trans isomers.
Unusually, NMR spectra of trans-5b and trans-6 showed 1-bond coupling between 31P (100% abundance, I = 1/2) and 55Mn (100% abundance; I = 5/2), resulting in an approximate 1:1:1:1:1:1 sextet in the 31P{1H} NMR spectra (at elevated temperature) and a 1:4:6:4:1 quintet in the 55Mn{1H} NMR spectra. Such coupling has not been observed in other [(dmpe)2MnXL] (X = an anionic ligand; L = a neutral ligand) complexes. Bonding between Mn and N2 was studied through XRD, IR spectroscopy, and DFT calculations (including ETS-NOCV analysis), revealing stronger N2 coordination in the trans isomers. This results from larger electrostatic and orbital contributions (ΔEelec and ΔEorb) to bonding, partly offset by increased Pauli repulsion (ΔEPauli), where the larger ΔEorb stems from enhanced π-backdonation.
Experimental
General methods
An argon-filled MBraun UNIlab glove box equipped with a −30 °C freezer was employed for the manipulation and storage of all oxygen- and moisture-sensitive compounds. Air-sensitive preparative reactions were performed on a double-manifold high-vacuum line equipped with a two stage Welch 1402 belt-drive vacuum pump (ultimate pressure 1 × 10−4 Torr) using standard techniques.107 The vacuum was measured periodically using a Kurt J. Lesker 275i convection enhanced Pirani gauge. Residual oxygen and moisture was removed from the argon stream by passage through an Oxisorb-W scrubber from Matheson Gas Products.Benzene was purchased from Sigma-Aldrich, hexanes and toluene were purchased from Caledon, and deuterated solvents were purchased from ACP Chemicals. Benzene, hexanes, and toluene were initially dried and distilled at atmospheric pressure from sodium/benzophenone (first two) or sodium (toluene). All solvents were stored over an appropriate drying agent (benzene, toluene, d8-toluene, C6D6 = Na/Ph2CO; hexanes = Na/Ph2CO/tetraglyme) and introduced to reactions or solvent storage flasks via vacuum transfer with condensation at −78 °C.
Cl2GePh2, Cl2GeEt2, H2SiPh2, dmpe, 1,4-dioxane, ethylmagnesium chloride solution (2.0 M in diethyl ether), D2, tert-butyl isonitrile, o-xylyl isonitrile, and n-butyl isonitrile were purchased from Sigma-Aldrich. Manganese dichloride was purchased from Strem Chemicals. [(dmpe)2MnH(GePh2)] (1a),29 [(dmpe)2MnH(GeEt2)] (1b),29 and [(dmpe)2MnH(SiPh2)]31 were prepared according to literature procedures, as were reagents used in their preparation; [(dmpe)2MnH(C2H4)],27,28 H2GePh2,108 and H2GeEt2.29 Argon, H2, and N2 were purchased from PraxAir.
NMR spectroscopy was performed on Bruker AV-500 and AV-600 spectrometers. Spectra were obtained at 298 K unless otherwise indicated. 1H NMR spectra were referenced relative to SiMe4 through a resonance of the protio impurity of the solvent: C6D6 (δ 7.16 ppm) and d8-toluene (δ 2.08, 6.97, 7.01, and 7.09 ppm; at lower or higher temperatures the peak at 2.08 ppm was used). 13C NMR spectra were referenced relative to SiMe4 through a resonance of the solvent: C6D6 (δ 128.06 ppm) and d8-toluene (δ 20.43, 125.13, 127.96, 128.87, and 137.48 ppm; at lower temperatures the peak at 20.43 ppm was used). 2H NMR spectra were referenced through a resonance of the solvent C6D6 (δ 7.16 ppm) and the CD3 peak in d8-toluene (δ 2.08 ppm). The 29Si, 31P, and 55Mn NMR spectra were referenced to SiMe4 (1 vol% in CDCl3; Ξ = 19.867187%), 85% aqueous H3PO4 (Ξ = 40.480742%), and conc. KMnO4(aq) (Ξ = 24.789218%), respectively, by indirect referencing from a 1H NMR spectrum.109 NMR chemical shift abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, quin. = quintet, m = multiplet, app. = apparent, br. = broad.
Combustion elemental analyses were performed at the University of Calgary.
IR spectroscopy was performed in a nujol mull sandwiched between CaF2 plates using a ThermoScientific Nicolet iS5 spectrometer on transmission mode. Spectra collection and viewing was done using OMNIC.
Single-crystal X-ray crystallographic analyses were performed on crystals coated in Paratone oil and mounted on a STOE IPDS II diffractometer with an image plate detector or a Bruker Dual Source D8 Venture diffractometer using the IμS 3.0 Mo source at 70 W with a HELIOS Mo focusing optic (ELM33) in the McMaster Analytical X-Ray (MAX) Diffraction Facility. A semi-empirical absorption correction was applied using redundant and symmetry related data. Raw data was processed using XPREP (as part of the APEX4 v2022.10-0 software), and solved by intrinsic (SHELXT)110 methods. Structures were completed by difference Fourier synthesis and refined with full-matrix least-squares procedures based on F2. In all cases, non-hydrogen atoms were refined anisotropically and hydrogen atoms were generated in ideal positions and then updated with each cycle of refinement (with the exception of hydrogen atoms on Ge and Mn, which were located from the difference map and refined isotropically). Refinement was performed with SHELXL111 in Olex2.112
2D powder X-ray diffraction was performed on a Bruker D8 Discover diffractometer equipped with a Vantec 500 area detector and a focused Cu source with Kα radiation (λ = 1.54056 Å) operated at 40 kV and 40 mA, or a Bruker D8 Venture diffractometer equipped with a PHOTON CMOS (complementary metal oxide semiconductor) area detector and a Incoatec IμS Cu source with Kα radiation (λ = 1.54184 Å) operated at 50 kV and 1.10 mA. The samples were packed in a 0.5 mm o.d. special glass (SG; wall thickness 0.01 mm) capillary tube for X-ray diffraction (purchased from Charles Supper Co.) and sealed by inverting to submerge the open end in a pool of Apiezon H-grease within the glovebox. The powder diffractograms were generated using Gadds and/or Diffrac.eva. Rietveld refinement (PV_TCHZ peak types between 5.5° and 35°, LP factor of 23.7, 6th order PO spherical harmonics for preferred orientation, and a Chebychev order 9 background) was performed using Topas.
All prepared complexes are air sensitive, and their products upon reaction with air are malodorous. Therefore, unless otherwise indicated, all syntheses were conducted under an atmosphere of argon.
DFT calculations
All calculated structures were fully optimized with the ADF/AMS DFT package (SCM, version 2020.102 for 2a–b or 2024.102 for 5a–b).113,114 For 2a–b, energy minima were located for four (transHGe isomer) or three (cis and central isomers) rotamers, and only data from the minima leading to the lowest Gibbs free energy of formation at 176 K is discussed. Calculations were conducted in the gas phase within the generalized gradient approximation (GGA) using the 1996 Perdew–Burke–Ernzerhof exchange and correlation functional (PBE),115 the scalar zeroth-order regular approximation (ZORA)116–120 for relativistic effects, and Grimme's DFT-D3-BJ dispersion correction.121,122 Geometry optimizations were conducted using all-electron triple-ζ basis sets with two polarization functions (TZ2P), fine integration grids (Becke123,124 verygood), and stricter-than-default convergence criteria (gradients = 0.0001, step = 0.002). Calculations were restricted.Visualization of the computational results was performed using the ADF/AMS-GUI (SCM) or Biovia Discovery Studio Visualizer. Orbitals and deformation densities were generated with a fine grid using the densf auxiliary program.
Analytical frequency calculations125–127 were conducted on all geometry optimized structures (including geometry optimized fragments) to ensure that the geometry optimization led to an energy minimum.
Bonding was analyzed in more detail using a fragment approach (energy decomposition analysis128,129 with ETS-NOCV analysis130–133) that considered the interaction of neutral (dmpe)2Mn(GeHR2) fragments with neutral N2 ligands. Fragments were generated from the TZ2P geometry optimized structures of each complex, geometries were frozen, and single-point calculations (as well as the EDA/ETS-NOCV calculations) were conducted using the same parameters as for the geometry optimizations. Preparation energies (ΔEprep) were obtained for all fragments by allowing the fragments to adopt equilibrium geometries (using the same method previously described for geometry optimization). Basis set superposition errors (BSSEs) were calculated through the use of ghost atoms with no nuclear charge and no electrons to contribute to the molecule (using the molecular fragments method).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) GePh2)] (1a) with H2 to afford mixtures containing 1a, H2, and [(dmpe)2MnH2(GeHPh2)] (2a).
Approx. 10 mg of [(dmpe)2MnH(GePh2)] (1a) was dissolved in approx. 0.6 mL of C6D6 or d8-toluene, and the solution placed in a J-young NMR tube. The mixture was freeze/pump/thawed three times, and placed under 1 atm of H2 at −95 °C, sealed, and warmed to room temperature. This reaction was monitored over time in situ by NMR spectroscopy and had reached equilibrium within 3 days with 87% conversion to 2a, at which time the solution had turned light orange. 1H NMR (C6D6, 600 MHz, 298 K): δ 7.97 (d of d, 4H, 3JH,H 7.9 Hz, 4JH,H 1.4 Hz, o-Ph), 7.22 (t, 4H, 3JH,H 7.4 Hz, m-Ph), 7.11 (t of t, 2H, 3JH,H 7.3 Hz, 4JH,H 1.4 Hz, p-Ph), 5.90 (m, 1H, Ge
GePh2)] (1a) with H2 to afford mixtures containing 1a, H2, and [(dmpe)2MnH2(GeHPh2)] (2a).
Approx. 10 mg of [(dmpe)2MnH(GePh2)] (1a) was dissolved in approx. 0.6 mL of C6D6 or d8-toluene, and the solution placed in a J-young NMR tube. The mixture was freeze/pump/thawed three times, and placed under 1 atm of H2 at −95 °C, sealed, and warmed to room temperature. This reaction was monitored over time in situ by NMR spectroscopy and had reached equilibrium within 3 days with 87% conversion to 2a, at which time the solution had turned light orange. 1H NMR (C6D6, 600 MHz, 298 K): δ 7.97 (d of d, 4H, 3JH,H 7.9 Hz, 4JH,H 1.4 Hz, o-Ph), 7.22 (t, 4H, 3JH,H 7.4 Hz, m-Ph), 7.11 (t of t, 2H, 3JH,H 7.3 Hz, 4JH,H 1.4 Hz, p-Ph), 5.90 (m, 1H, Ge![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) ), 1.32 (m, 8H, PC2), 1.18 (br. s, 24H, PC3), −11.09 (br. m, 2H, Mn). 1H NMR (d8-toluene, 500 MHz, 298 K): δ 7.85 (d of d, 4H, 3JH,H 6.5 Hz, 4JH,H 1.3 Hz, o-Ph), 7.14 (t, 4H, 3JH,H 7.3 Hz, m-Ph), 7.05 (t of t, 2H, 3JH,H 7.3 Hz, 4JH,H 1.9 Hz, p-Ph), 5.78 (m, 1H, Ge), 1.30 (m, 8H, PC2), 1.15 (br. s, 24H, PC3), −11.13 (br. m, 2H, Mn). 13C{1H} NMR (d8-toluene, 126 MHz, 298 K): δ 155.21 (s, i-Ph), 135.81 (s, o-Ph), 127.40 (s, m-Ph), 126.07 (s, p-Ph), 32.70 (m, P
), 1.32 (m, 8H, PC2), 1.18 (br. s, 24H, PC3), −11.09 (br. m, 2H, Mn). 1H NMR (d8-toluene, 500 MHz, 298 K): δ 7.85 (d of d, 4H, 3JH,H 6.5 Hz, 4JH,H 1.3 Hz, o-Ph), 7.14 (t, 4H, 3JH,H 7.3 Hz, m-Ph), 7.05 (t of t, 2H, 3JH,H 7.3 Hz, 4JH,H 1.9 Hz, p-Ph), 5.78 (m, 1H, Ge), 1.30 (m, 8H, PC2), 1.15 (br. s, 24H, PC3), −11.13 (br. m, 2H, Mn). 13C{1H} NMR (d8-toluene, 126 MHz, 298 K): δ 155.21 (s, i-Ph), 135.81 (s, o-Ph), 127.40 (s, m-Ph), 126.07 (s, p-Ph), 32.70 (m, P![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) H2), 25.60 (m, PH3). 31P{1H} NMR (d8-toluene, 202 MHz, 298 K):δ 76.18 (s). transHGe-2a: 1H NMR (d8-toluene, 500 MHz, 176 K): δ 8.05 (d, 4H, 3JH,H 7.1 Hz, o-Ph), 7.27 (t, 4H, 3JH,H 7.2 Hz, m-Ph), 7.14 (t, 2H, 3JH,H 7.2 Hz, p-Ph), 6.05 (d, 1H, 2JH,H 15.8 Hz, Ge), 1.46, 1.21 (2 × br. s, 4H, PC2), 1.24, 0.96 (2 × s, 12H, PC3), −10.53 (m, 1H, MnGe), −11.19 (quin, 1H, 2JH,P 57.9 Hz, Mn). 13C{1H} NMR (d8-toluene, 126 MHz, 176 K): δ 154.76 (s, i-Ph), 135.24 (s, o-Ph), 127.51 (s, m-Ph), 126.20 (s, p-Ph), 31.43 (m, PH2), 28.66, 21.51 (2 × s, PH3). 31P{1H} NMR (d8-toluene, 202 MHz, 176 K): δ 78.14 (s). Isomers(s) of2awith a disphenoidal arrangement of the phosphine donors (selected data): 1H NMR (d8-toluene, 500 MHz, 176 K): δ 8.32 (d, 2H, 3JH,H 6.9 Hz, o-Ph), 7.99 (d, 2H, 3JH,H 7.0 Hz, o-Ph), 7.42 (t, 2H, 3JH,H 7.1 Hz, m-Ph), 7.24 (t, 2H, 3JH,H 7.3 Hz, m-Ph), 5.49 (m, 1H, Ge), 1.52, 1.26, 1.00, 0.81, 0.57 (5 × s, 3H, PH3), 1.39 (d, 3H, 2JH,P 5.6 Hz, PC3), 0.74 (d, 3H, 2JH,P 4.6 Hz, PC3), 0.72 (d, 3H, 2JH,P 4.0 Hz, PC3), −13.29 (br. s, 2H, Mn). 13C{1H} NMR (d8-toluene, 126 MHz, 176 K): δ 156.67 (s, i-Ph), 136.67 (s, o-Ph), 127.09 (s, m-Ph), 29.94 (d, JC,P 20.2 Hz, PH3), 28.17, 25.10, 23.04, 21.18 (4 × s, PH3), 27.57 (d, JC,P 18.5 Hz, PH3), 21.90 (d, JC,P 10.0 Hz, PH3), 19.51 (d, JC,P 15.5 Hz, PH3). 31P{1H} NMR (d8-toluene, 202 MHz, 176 K):δ 76.11, 73.42 (2 × s, 1P), 71.22 (s, 2P).
GeEt2)] (1b) with H2 to afford mixtures containing 1b, H2, and [(dmpe)2MnH2(GeHEt2)] (2b).
This was done in an analogous fashion to the reaction between 1a and H2, using [(dmpe)2MnH(GeEt2)] (1b) in place of 1a, and resulting in 73% conversion to 2b after reaching equilibrium (after 2 days), at which time the solution had turned yellow. 1H NMR (C6D6, 600 MHz, 298 K): δ 4.28 (s, 1H, Ge), 1.61 (t, 6H, 3JH,H 7.8 Hz, CH2C3), 1.35 (m, 8H, PC2), 1.23 (s, 24H, PC3), 1.09, 1.00 (2 × m, 2H, C2CH3), −11.93 (m, 2H, Mn). 1H NMR (d8-toluene, 500 MHz, 298 K): δ 4.15 (s, 1H, Ge), 1.41 (t, 6H, 3JH,H 7.8 Hz, CH2C3), 1.33 (m, 8H, PC2), 1.21 (s, 24H, PC3), 0.99, 0.91 (2 × m, 2H, C2CH3), −12.00 (m, 2H, Mn). 13C{1H} NMR (d8-toluene, 126 MHz, 298 K): δ 33.54 (m, PH2), 25.48 (s, PH3), 16.10 (s, H2CH3), 14.59 (s, CH2H3). 31P{1H} NMR (d8-toluene, 202 MHz, 298 K): δ 75.53 (s). transHGe-2b: 1H NMR (d8-toluene, 500 MHz, 176 K): δ 4.55 (s, Ge, 1H), 1.73 (m, 6H, CH2C3), 1.52, 1.24 (2 × br. s, 4H, PC2), 1.32, 1.10 (2 × s, PC3, 12H), 1.22, 1.01 (2 × C2CH3),†† −11.01 (m, 1H, MnGe), −11.28 (quin, 1H, 2JH,P 56.0 Hz, Mn). 13C{1H} NMR (d8-toluene, 126 MHz, 176 K): δ 32.73 (m, PH2), 27.52 (m, PH3), 22.37 (s, PH3), 15.23 (s, H2CH3), 14.28 (s, CH2H3). 31P{1H} NMR (d8-toluene, 202 MHz, 176 K): δ 76.49 (s). Isomers(s) of2bwith a disphenoidal arrangement of the phosphine donors (selected data): 1H NMR (d8-toluene, 500 MHz, 176 K): δ 3.86 (s, 1H, Ge), 2.08 (m, 3H, CH2C3), 1.91, 1.28, 1.12 (3 × PC3),†† 1.37, 0.99, 0.95, 0.94 0.76 (5 × s, 3H, PC3), 1.36 (C2CH3),†† −13.85 (br. s, 2H, Mn). 13C{1H} NMR (d8-toluene, 126 MHz, 176 K): δ 25.03 (d, JC,P 12.8 Hz, PH3), 22.69 (d, JC,P 11.8 Hz, PH3), 22.66 (m, PH3), 16.89 (s, CH2H3), 16.06 (s, PH3). 31P{1H} NMR (d8-toluene, 202 MHz, 176 K): δ 74.65 (s, 2P), 72.68, 71.97 (2 × s, 1P).
GeR2)] (R = Ph: 1a, R = Et: 1b) with D2 to afford mixtures containing 1a or 1b, D2, and [(dmpe)2MnD2(GeHR2)] (R = Ph: d2-2a, R = Et: d2-2b).
These reactions were conducted in an analogous fashion to those described above for reactions of 1a–b with H2 (in C6D6 or d8-toluene), but using D2 in place of H2. To monitor deuterium scrambling in 2a, the reaction (in C6D6) was heated in the J-young tube at 60 °C and periodically analysed by NMR spectroscopy at room temperature. 1H and 31P{1H} NMR spectra at 298 K in C6D6 of 2a–b mirror those of 1a–b, with the exception that the Mn1H NMR environment is not present and the Ge1H NMR signal shifted slightly to 5.87 (d2-2a) or 4.24 (d2-2b) ppm. [(dmpe)2MnD2(HGePh2)] (d2-2a): 2H NMR (C6D6, 77 MHz, 298 K): δ −11.28 (app. t, 2JD,P 5.0 Hz, Mn
H2), 25.60 (m, PH3). 31P{1H} NMR (d8-toluene, 202 MHz, 298 K):δ 76.18 (s). transHGe-2a: 1H NMR (d8-toluene, 500 MHz, 176 K): δ 8.05 (d, 4H, 3JH,H 7.1 Hz, o-Ph), 7.27 (t, 4H, 3JH,H 7.2 Hz, m-Ph), 7.14 (t, 2H, 3JH,H 7.2 Hz, p-Ph), 6.05 (d, 1H, 2JH,H 15.8 Hz, Ge), 1.46, 1.21 (2 × br. s, 4H, PC2), 1.24, 0.96 (2 × s, 12H, PC3), −10.53 (m, 1H, MnGe), −11.19 (quin, 1H, 2JH,P 57.9 Hz, Mn). 13C{1H} NMR (d8-toluene, 126 MHz, 176 K): δ 154.76 (s, i-Ph), 135.24 (s, o-Ph), 127.51 (s, m-Ph), 126.20 (s, p-Ph), 31.43 (m, PH2), 28.66, 21.51 (2 × s, PH3). 31P{1H} NMR (d8-toluene, 202 MHz, 176 K): δ 78.14 (s). Isomers(s) of2awith a disphenoidal arrangement of the phosphine donors (selected data): 1H NMR (d8-toluene, 500 MHz, 176 K): δ 8.32 (d, 2H, 3JH,H 6.9 Hz, o-Ph), 7.99 (d, 2H, 3JH,H 7.0 Hz, o-Ph), 7.42 (t, 2H, 3JH,H 7.1 Hz, m-Ph), 7.24 (t, 2H, 3JH,H 7.3 Hz, m-Ph), 5.49 (m, 1H, Ge), 1.52, 1.26, 1.00, 0.81, 0.57 (5 × s, 3H, PH3), 1.39 (d, 3H, 2JH,P 5.6 Hz, PC3), 0.74 (d, 3H, 2JH,P 4.6 Hz, PC3), 0.72 (d, 3H, 2JH,P 4.0 Hz, PC3), −13.29 (br. s, 2H, Mn). 13C{1H} NMR (d8-toluene, 126 MHz, 176 K): δ 156.67 (s, i-Ph), 136.67 (s, o-Ph), 127.09 (s, m-Ph), 29.94 (d, JC,P 20.2 Hz, PH3), 28.17, 25.10, 23.04, 21.18 (4 × s, PH3), 27.57 (d, JC,P 18.5 Hz, PH3), 21.90 (d, JC,P 10.0 Hz, PH3), 19.51 (d, JC,P 15.5 Hz, PH3). 31P{1H} NMR (d8-toluene, 202 MHz, 176 K):δ 76.11, 73.42 (2 × s, 1P), 71.22 (s, 2P).
GeEt2)] (1b) with H2 to afford mixtures containing 1b, H2, and [(dmpe)2MnH2(GeHEt2)] (2b).
This was done in an analogous fashion to the reaction between 1a and H2, using [(dmpe)2MnH(GeEt2)] (1b) in place of 1a, and resulting in 73% conversion to 2b after reaching equilibrium (after 2 days), at which time the solution had turned yellow. 1H NMR (C6D6, 600 MHz, 298 K): δ 4.28 (s, 1H, Ge), 1.61 (t, 6H, 3JH,H 7.8 Hz, CH2C3), 1.35 (m, 8H, PC2), 1.23 (s, 24H, PC3), 1.09, 1.00 (2 × m, 2H, C2CH3), −11.93 (m, 2H, Mn). 1H NMR (d8-toluene, 500 MHz, 298 K): δ 4.15 (s, 1H, Ge), 1.41 (t, 6H, 3JH,H 7.8 Hz, CH2C3), 1.33 (m, 8H, PC2), 1.21 (s, 24H, PC3), 0.99, 0.91 (2 × m, 2H, C2CH3), −12.00 (m, 2H, Mn). 13C{1H} NMR (d8-toluene, 126 MHz, 298 K): δ 33.54 (m, PH2), 25.48 (s, PH3), 16.10 (s, H2CH3), 14.59 (s, CH2H3). 31P{1H} NMR (d8-toluene, 202 MHz, 298 K): δ 75.53 (s). transHGe-2b: 1H NMR (d8-toluene, 500 MHz, 176 K): δ 4.55 (s, Ge, 1H), 1.73 (m, 6H, CH2C3), 1.52, 1.24 (2 × br. s, 4H, PC2), 1.32, 1.10 (2 × s, PC3, 12H), 1.22, 1.01 (2 × C2CH3),†† −11.01 (m, 1H, MnGe), −11.28 (quin, 1H, 2JH,P 56.0 Hz, Mn). 13C{1H} NMR (d8-toluene, 126 MHz, 176 K): δ 32.73 (m, PH2), 27.52 (m, PH3), 22.37 (s, PH3), 15.23 (s, H2CH3), 14.28 (s, CH2H3). 31P{1H} NMR (d8-toluene, 202 MHz, 176 K): δ 76.49 (s). Isomers(s) of2bwith a disphenoidal arrangement of the phosphine donors (selected data): 1H NMR (d8-toluene, 500 MHz, 176 K): δ 3.86 (s, 1H, Ge), 2.08 (m, 3H, CH2C3), 1.91, 1.28, 1.12 (3 × PC3),†† 1.37, 0.99, 0.95, 0.94 0.76 (5 × s, 3H, PC3), 1.36 (C2CH3),†† −13.85 (br. s, 2H, Mn). 13C{1H} NMR (d8-toluene, 126 MHz, 176 K): δ 25.03 (d, JC,P 12.8 Hz, PH3), 22.69 (d, JC,P 11.8 Hz, PH3), 22.66 (m, PH3), 16.89 (s, CH2H3), 16.06 (s, PH3). 31P{1H} NMR (d8-toluene, 202 MHz, 176 K): δ 74.65 (s, 2P), 72.68, 71.97 (2 × s, 1P).
GeR2)] (R = Ph: 1a, R = Et: 1b) with D2 to afford mixtures containing 1a or 1b, D2, and [(dmpe)2MnD2(GeHR2)] (R = Ph: d2-2a, R = Et: d2-2b).
These reactions were conducted in an analogous fashion to those described above for reactions of 1a–b with H2 (in C6D6 or d8-toluene), but using D2 in place of H2. To monitor deuterium scrambling in 2a, the reaction (in C6D6) was heated in the J-young tube at 60 °C and periodically analysed by NMR spectroscopy at room temperature. 1H and 31P{1H} NMR spectra at 298 K in C6D6 of 2a–b mirror those of 1a–b, with the exception that the Mn1H NMR environment is not present and the Ge1H NMR signal shifted slightly to 5.87 (d2-2a) or 4.24 (d2-2b) ppm. [(dmpe)2MnD2(HGePh2)] (d2-2a): 2H NMR (C6D6, 77 MHz, 298 K): δ −11.28 (app. t, 2JD,P 5.0 Hz, Mn![[D with combining low line]](https://www.rsc.org/images/entities/i_char_0044_0332.gif) ). [(dmpe)2MnD2(HGeEt2)] (d2-2b): 2H NMR (C6D6, 77 MHz, 298 K): δ −12.16 (quin, 2JD,P 4.7 Hz, Mn).
GeEt2)] (1b) and transHGe-[(dmpe)2MnH(HGeHEt2)] (transHGe-2b).
11.7 mg (0.02 mmol) of [(dmpe)2MnH(GeEt2)] (1b) was dissolved in 0.35 mL of toluene, and the solution was placed in a 25 mL Schlenk flask, freeze/pump/thawed three times, placed under 1 atm of H2 at −78 °C, sealed, and warmed to room temperature. The reaction mixture was then stirred for 2 days, after which the solvent was removed in vacuo, ∼0.5 mL of hexanes was added, and the solution was placed under 1 atm of H2 at −78 °C and sealed. Maintaining the solution at −78 °C for a few hours afforded large yellow X-ray quality crystals containing a mixture of 1b and transHGe-2b (due to disorder between two germyl groups and a germylene group in a 0.402(3):0.056(3):0.542(3) ratio).
GePh2)] (1a) was dissolved in 10 mL of benzene and placed in a 50 mL storage flask. 21.4 mg (0.26 mmol) of tert-butyl isonitrile was added and the reaction mixture was stirred for 1.5 hours at room temperature in the dark. The solvent was removed in vacuo, and the resulting yellow solid was dried in vacuo for 1 hour and washed with 4 mL of hexanes. Recrystallization of the residue (which did not dissolve in hexanes) at −30 °C from a solution in toluene layered with the hexanes that had been used to wash the crude material afforded 36.0 mg (0.05 mmol, 42%) of cis-3a as X-ray quality yellow crystals. The reaction of 1a with CNtBu affords 3a in near quantitative spectroscopic yield (as a mixture of cis and trans isomers), so the low yield is due to losses during crystallization. (b) 15.3 mg (0.03 mmol) of [(dmpe)2MnH(GePh2)] (1a) and 4.4 mg (0.05 mmol) of tert-butyl isonitrile were dissolved in approx. 0.6 mL of C6D6, placed in a J-young tube, and the reaction mixture was monitored over time at room temperature by NMR spectroscopy. After 30 minutes, the reaction mixture contained cis-3a and trans-3a in a 3:1 ratio, which remained constant over 4 days. cis-3a: 1H NMR (C6D6, 600 MHz, 298 K): δ 8.20, 8.15 (2 × d of d, 2H, 3JH,H 7.6 Hz, 4JH,H 1.0 Hz, o-Ph), 7.25, 7.22 (2 × t, 2H, 3JH,H 7.9 Hz, m-Ph), 7.12 (t, 1H, 3JH,H 7.6 Hz, p-Ph), 7.08 (t, 1H, 3JH,H 7.4 Hz, p-Ph), 5.45 (t of d, 1H, 3JH,P 8.6 and 5.2 Hz, Ge), 1.80, 1.62 (2 × m, 1H, PC2), 1.71 (d, 3H, 2JH,P 6.7 Hz, PC3), 1.69 (d, 3H, 2JH,P 6.9 Hz, PC3), 0.94–1.49 (m, 6H, PC2), 1.34, 1.16 (2 × d, 3H, 2JH,P 6.6 Hz, PC3), 1.20 (d, 3H, 2JH,P 5.9 Hz, PC3), 1.10 (d, 3H, 2JH,P 5.8 Hz, PC3), 1.03 (s, 9H, C(C3)3), 0.97 (d, 3H, 2JH,P 4.6 Hz, PC3), 0.82 (d, 3H, 2JH,P 4.8 Hz, PC3). 13C{1H} NMR (C6D6, 151 MHz, 298 K): δ 158.29 (d, 3JC,P 2.6 Hz, i-Ph), 157.74 (d, 3JC,P 3.4 Hz, i-Ph), 137.65, 137.47 (2 × s, o-Ph), 127.23, 127.10 (2 × s, m-Ph), 125.23, 125.20 (2 × s, p-Ph), 54.94 (s, Me3), 34.62 (app. t of d, JC,P 20.7 and 5.5 Hz, PH2), 33.64 (app. t of d, JC,P 23.3 and 7.5 Hz, PH2), 33.13 (app. t, JC,P 20.4 Hz, PH2), 31.46 (s, C(H3)3), 30.62 (app. t, JC,P 19 Hz, PH2), 24.31 (d of d, JC,P 14.1 and 5.2 Hz, PH3), 23.92 (d, JC,P 12.4 Hz, PH3), 23.43 (d of d, JC,P 16.0 and 5.8 Hz, PH3), 23.04 (d, JC,P 8.0 Hz, PH3), 22.40 (d of d, JC,P 17.9 and 6.0 Hz, PH3), 21.73 (m, PH3), 21.01 (d of m, JC,P 20.5 Hz, PH3). 31P{1H} NMR (C6D6, 243 MHz, 298 K): δ 75.78, 56.99 (2 × s, 1P), 70.53 (s, 2P). trans-3a: 1H NMR (C6D6, 500 MHz, 298 K): δ 8.00 (d, 4H, 3JH,H 7.1 Hz, o-Ph), 7.19 (m, m-Ph), 5.12 (quin., 1H, 3JH,P 7.7 Hz, Ge), 1.85, 1.44 (2 × m, 4H, PC2), 1.40, 1.18 (2 × s, 12H, PC3), 0.98 (s, 9H, C(C3)3). 13C{1H} NMR (C6D6, 126 MHz, 298 K): δ 158.83 (s, i-Ph), 137.10 (s, o-Ph), 127.38 (s, m-Ph), 125.36 (s, p-Ph), 54.55 (s, Me3), 32.52 (m, PH2), 31.36 (s, C(H3)3), 21.72 (m, PH3). 31P{1H} NMR (C6D6, 202 MHz, 298 K): δ 72.71 (s). Anal. found (calcd): C, 52.31 (52.28); H, 8.00 (7.87); N, 2.10 (2.08).
GePh2)] (1a; for 3b 11.6 mg/0.02 mmol, and for 3c 13.3 mg/0.02 mmol) and free isonitrile (for 3b 5.2 mg/0.04 mmol of o-xylyl isontrile, and for 3c 7.0 mg/0.08 mmol of n-butyl isonitrile) were dissolved in approx. 0.6 mL of C6D6, placed in a J-young tube, and the reaction mixtures were monitored over time at room temperature by NMR spectroscopy. Both reactions were complete after 1 hour. cis-3b: 1H NMR (C6D6, 600 MHz, 298 K): δ 8.07 (d of m, 2H, 3JH,H 7.2 Hz, o-Ph), 8.03 (d of d, 2H, 3JH,H 7.8 Hz, 4JH,H 1.2 Hz, o-Ph), 7.13 (t, 2H, 3JH,H 7.4 Hz, m-Ph), 7.07 (m, 2H, p-Ph), 7.06 (m, 2H, m-Ph), 6.90 (d, 2H, 3JH,H 7.5 Hz, xylyl-m), 6.80 (t, 1H, 3JH,H 7.5 Hz, xylyl-p), 5.48 (q, 1H, 3JH,P 6.6 Hz, Ge), 2.10 (s, 6H, xylyl-C3), 1.72 (d, 3H, 2JH,P 7.0 Hz, PC3), 1.63 (d, 3H, 2JH,P 6.8 Hz, PC3), 1.47–1.61 (m, 2H, PC2), 1.51, 1.13 (2 × d, 3H, 2JH,P 6.2 Hz, PC3), 1.04–1.40 (m, 5H, PC2), 1.19 (d, 3H, 2JH,P 5.9 Hz, PC3), 0.98 (d, 3H, 2JH,P 6.1 Hz, PC3), 0.90, 0.80 (2 × d, 3H, 2JH,P 4.8 Hz, PC3), 0.88 (m, 1H, PC2). 13C{1H} NMR (C6D6, 151 MHz, 298 K):δ 156.22 (s, i-Ph), 155.39 (d, 3JC,P 4.0 Hz, i-Ph), 137.78, 137.19 (2 × s, o-Ph), 133.69 (s, xylyl-i), 133.11 (s, xylyl-o), 128.16 (xylyl-m),†† 127.22, 127.10 (2 × s, m-Ph), 125.41, 125.36 (2 × s, p-Ph), 122.62 (s, xylyl-p), 33.81 (app. t, JC,P 19.9 Hz, PH2), 33.17 (m, PH2), 31.86 (app. t, JC,P 20.1 Hz, PH2), 23.68 (d of m, JC,P 16.0 Hz, PH3), 22.50, 21.74, 21.05 (3 × m, PH3), 20.72 (s, xylyl-H3). 31P{1H} NMR (C6D6, 243 MHz, 298 K):δ 70.57 (s, 2P), 67.94, 53.44 (2 × s, 1P). trans-3b: 1H NMR (C6D6, 600 MHz, 298 K): δ 7.95 (d of d, 4H, 3JH,H 7.7 Hz, 4JH,H 1.2 Hz, o-Ph), 7.18 (t, 4H, 3JH,H 7.3 Hz, m-Ph), 7.07 (m, 2H, p-Ph), 6.83 (d, 2H, 3JH,H 7.5 Hz, xylyl-m), 6.69 (t, 1H, 3JH,H 7.5 Hz, xylyl-p), 5.27 (quin., 1H, 3JH,P 7.3 Hz, Ge), 2.24 (s, 6H, xylyl-C3), 1.71, 1.52 (2 × m, 4H, PC2), 1.41, 1.21 (2 × s, 12H, PC3). 13C{1H} NMR (C6D6, 151 MHz, 298 K):δ 157.67 (s, i-Ph), 137.32 (s, o-Ph), 133.81 (s, xylyl-i), 132.11 (s, xylyl-o), 128.68 (s, xylyl-m), 127.50 (s, m-Ph), 125.60 (s, p-Ph), 122.25 (s, xylyl-p), 32.86 (m, PH2), 22.49, 21.91 (2 × m, PH3), 20.63 (s, xylyl-H3). 31P{1H} NMR (C6D6, 243 MHz, 298 K):δ 70.57 (s). cis-3c: 1H NMR (C6D6, 600 MHz, 298 K): δ 8.21, 8.13 (2 × d, 2H, 3JH,H 7.8 Hz, o-Ph), 7.27, 7.22 (2 × t, 2H, 3JH,H 7.5 Hz, m-Ph), 7.15 (t, 1H, 3JH,H 7.5 Hz, p-Ph), 7.11 (t, 1H, 3JH,H 7.3 Hz, p-Ph), 5.46 (app. t of d, 1H, 3JH,P 8.6 and 4.7 Hz, Ge), 3.16 (m, 2H, C2CH2CH2CH3), 1.75, 1.61, 140 (3 × m, 1H, PC2), 1.64, 1.18 (2 × d, 3H, 2JH,P 6.2 Hz, PC3), 1.62 (d, 3H, 2JH,P 6.5 Hz, PC3), 1.39, 1.14, 1.06 (3 × d, 3H, 2JH,P 6.0 Hz, PC3), 1.21 (m, 2H, CH2C2CH2CH3), 1.19 (m, 2H, CH2CH2C2CH3), 1.07–1.12 (m, 3H, PC2), 0.92–1.05 (m, 2H, PC2), 0.95 (d, 3H, 2JH,P 4.7 Hz, PC3), 0.81 (d, 3H, 2JH,P 4.9 Hz, PC3), 0.79 (t, 3H, 3JH,H 7.1 Hz, CH2CH2CH2C3). 13C{1H} NMR (C6D6, 151 MHz, 298 K):δ 157.61, 157.41 (2 × s, i-Ph), 137.63, 137.28 (2 × s, o-Ph), 127.20, 127.13 (2 × s, m-Ph), 125.30, 125.27 (2 × s, p-Ph), 45.12 (s, H2CH2CH2CH3), 34.22, 33.41 (2 × m, PH2), 33.08 (s, CH2H2CH2CH3), 30.48 (app. t, JC,P 19.3 Hz, PH2), 24.47, 23.01, 22.85, 21.34, 21.11, 20.95 (6 × m, PH3), 23.53 (d, JC,P 13.2 Hz, PH3), 22.38 (d of d, JC,P 20.3 and 5.6 Hz, PH3), 20.40 (s, CH2CH2H2CH3), 13.76 (s, CH2CH2CH2H3). 31P{1H} NMR (C6D6, 243 MHz, 298 K):δ 76.11, 53.33 (2 × s, 1P), 71.59 (s, 2P). trans-3c(selected):1H NMR (C6D6, 600 MHz, 298 K): δ 7.96 (d, 4H, 3JH,H 7.1 Hz, o-Ph), 5.06 (quin., 1H, 3JH,P 15.5 Hz, Ge), 1.81, 1.43 (2 × m, 4H, PC2), 1.36, 1.16 (2 × s, 12H, PC3). 13C{1H} NMR (C6D6, 151 MHz, 298 K):δ 137.01 (s, o), 127.42, 125.45 (2 × s, m and p), 32.26 (m, PH2), 22.97, 21.30 (2 × m, PH3). 31P{1H} NMR (C6D6, 243 MHz, 298 K):δ 73.77 (s).
:3:73 ratio. The solvent was then removed in vacuo, the resulting solid was extracted with hexanes, and the mother liquors were stored at −30 °C to afford X-ray quality yellow crystals of 4. The remaining solid (which did not dissolve in hexanes) was dissolved in minimal toluene, and that solution was stored at −30 °C to afford yellow crystals from which an X-ray structure of cis-3b was obtained.
GePh2)] (1a) was dissolved in 5 mL of benzene, and the solution was transferred to a 50 mL storage flask. 25.1 mg (0.30 mmol) of n-butyl isonitrile was added, and the reaction was stirred for 1 hour at room temperature. Removal of the solvent in vacuo afforded an orange oil which was dried in vacuo for 1 hour at room temperature. The oil was extracted with 1.5 mL of hexanes to afford an orange solution which was stored at −30 °C to afford yellow crystals from which an X-ray structure of trans-3c was obtained. Recrystallization of the residue (which did not dissolve in hexanes) at −30 °C from toluene layered with hexanes afforded 4.7 mg (0.01 mmol) of cis-3c, in >95% purity (measured by NMR spectroscopy).
GePh2)] (1a) was dissolved in 5 mL of benzene and placed in a 100 mL storage flask. The mixture was freeze/pump/thawed three times, placed under 1 atm of N2 at −95 °C, sealed, and warmed to room temperature. After stirring for 5 days at room temperature in the dark, the solvent was removed in vacuo. The resulting solid was recrystallized from a concentrated solution of toluene layered with a concentrated solution of hexanes at −30 °C to afford 28.4 mg (0.05 mmol, 54%) of 5a as yellow crystals. X-ray quality crystals of cis-5a were obtained by recrystallization from a concentrated solution in toluene layered with hexanes at −30 °C. X-ray quality crystals of trans-5a were obtained by serendipitous N2 addition to a reaction mixture formed from the combination of approx. 10 mg of 1a with H2 in d8-toluene, followed by removal of the solvent in vacuo and recrystallization from toluene at −30 °C. NMR spectra were obtained under an atmosphere of N2. cis-5a: ν(NN): 2010 cm−1, ν(Ge–H): 1860 cm−1 (very broad). 1H NMR (C6D6, 600 MHz, 298 K): δ 8.22, 8.11 (2 × d, 2H, 3JH,H 7.0 Hz, o-Ph), 7.34, 7.24 (2 × t, 2H, 3JH,H 7.4 Hz, m-Ph), 7.17 (t, 3JH,H 7.7 Hz, p-Ph),‡‡ 7.12 (t, 1H, 3JH,H 7.3 Hz, p-Ph), 5.40 (q, 1H, 3JH,P 5.4 Hz, Ge), 0.70–1.74 (m, 8H, PC2), 1.48 (d, 6H, 2JH,P 6.8 Hz, PC3), 1.29 (PC3),†† 1.09 (d, 3H, 2JH,P 6.1 Hz, PC3), 0.99 (d, 3H, 2JH,P 6.5 Hz, PC3), 0.93 (d, 3H, 2JH,P 5.9 Hz, PC3), 0.85 (d, 3H, 2JH,P 5.1 Hz, PC3), 0.53 (d, 3H, 2JH,P 5.3 Hz, PC3). 13C{1H} NMR (C6D6, 151 MHz, 298 K): δ 154.87, 153.61 (2 × s, i-Ph), 137.80, 137.22 (2 × s, o-Ph), 127.49, 127.34 (2 × s, m-Ph), 125.84 (s, p-Ph), 125.73 or 125.68 (s, p-Ph),§§ 33.18, 29.30 (2 × m, PH2), 32.53 (app. t, JC,P 20.8 Hz, PH2), 23.09, 21.90, 21.80, 16.52 (4 × m, PH3), 22.64 (d of d, JC,P 18.5 and 5.8 Hz, PH3), 19.87 (d, JC,P 16.8 Hz, PH3), 17.45 (d, JC,P 18.7 Hz, PH3), 15.31 (d of d, JC,P 17.3 and 4.8 Hz, PH3). 31P{1H} NMR (C6D6, 243 MHz, 298 K):δ 71.98, 69.33, 68.72, 58.71 (4 × s, 1P). 31P{1H} NMR (d8-toluene, 202 MHz, 223 K): δ 72.47, 70.27, 68.91, 58.75 (4 × s, 1P). 55Mn{1H} NMR (d8-toluene, 124 MHz, 298 K): δ −718 (br. s). 55Mn{1H} NMR (d8-toluene, 124 MHz, 223 K): δ −812 (br. s). 55Mn{1H} NMR (d8-toluene, 124 MHz, 370 K): δ −601 (br. s). trans-5a: ν(NN): 1973 cm−1, ν(Ge–H): 1860 cm−1 (very broad). 1H NMR (C6D6, 600 MHz, 298 K): δ 7.81 (d, 4H, 3JH,H 7.0 Hz, o-Ph), 7.12 (t, 4H, 3JH,H 7.3 Hz, m-Ph), 7.04 (t, 2H, 3JH,H 7.2 Hz, p-Ph), 4.74 (quin., 1H, 3JH,P 7.7 Hz, Ge), 1.80, 1.38 (2 × m, 4H, PC2), 1.29, 1.16 (2 × s, 12H, PC3). 13C{1H} NMR (C6D6, 151 MHz, 298 K): δ 156.81 (s, i-Ph), 136.69 (s, o-Ph), 127.49 (s, m-Ph), 125.73 or 125.68 (s, p-Ph),§§ 31.35 (m, PH2), 20.28, 16.48 (2 × m, PH3). 31P{1H} NMR (C6D6, 243 MHz, 298 K): δ 70.63 (br. s). 31P{1H} NMR (d8-toluene, 202 MHz, 223 K): δ 71.37 (s). 31P{1H} NMR (d8-toluene, 202 MHz, 370 K): δ 70.2 (broad multiplet with nearly indiscernible coupling due to quadrupolar collapse). 55Mn{1H} NMR (d8-toluene, 124 MHz, 298 K): δ −845 (br. s). 55Mn{1H} NMR (d8-toluene, 124 MHz, 223 K): δ −991 (br. s). 55Mn{1H} NMR (d8-toluene, 124 MHz, 370 K): δ −730 (br. s). Anal. found (calcd): C, 47.25 (47.17); H, 7.14 (7.09); N, 4.42 (4.58). Repetition of EA after 2.7 years sealed under argon at −30 °C: Anal. found (calcd): C, 46.96 (47.17); H, 7.20 (7.09); N, 3.84 (4.58).
GeEt2)] (1b) was dissolved in 10 mL of benzene and transferred to a 100 mL storage flask. The mixture was freeze/pump/thawed three times, placed under 1 atm of N2 at −95 °C, sealed, and warmed to room temperature. After stirring for 3 days at room temperature in the dark, the solvent was removed in vacuo. The resulting solid was recrystallized using 5 mL of hexanes at −30 °C to afford 65.3 mg (0.13 mmol, 58%) of cis-5b as yellow crystals. X-ray quality crystals of trans-5b were obtained by heating a mixture of approx. 10 mg of 1b under N2 for 4 days at 60 °C in C6D6 (which resulted in cis–trans isomerization to a 22:78 ratio), followed by removal of the solvent in vacuo and recrystallization from a concentrated solution of hexanes at −30 °C. NMR spectra were obtained under an atmosphere of N2. cis-5b: ν(NN): 1989 cm−1, ν(Ge–H): 1823 cm−1. 1H NMR (C6D6, 600 MHz, 298 K): δ 3.65 (m, 1H, Ge), 1.83, 1.82 (2 × t, 3H, 3JH,H 7.8 Hz, CH2C3), 1.63 (m, 1H, PC2), 1.54 (d, 3H, 2JH,P 7.1 Hz, PC3), 1.23–1.56 (m, 4H, PC2), 1.50, 1.33, 1.32, 1.27 (4 × m, 1H, C2CH3) 1.47 (d, 3H, 2JH,P 6.7 Hz, PC3), 1.14 (d, 3H, 2JH,P 6.3 Hz, PC3), 1.12 (d, 3H, 2JH,P 5.8 Hz, PC3), 1.06 (d, 3H, 2JH,P 6.5 Hz, PC3), 1.03 (d, 3H, 2JH,P 5.9 Hz, PC3), 0.97 (d, 3H, 2JH,P 5.0 Hz, PC3), 0.70–0.94 (m, 3H, PC2), 0.58 (d, 3H, 2JH,P 5.2 Hz, PC3). 13C{1H} NMR (C6D6, 151 MHz, 298 K):δ 33.97 (m, PH2), 31.36 (app. t, JC,P 19.7 Hz, PH2), 28.90 (d of d, JC,P 23.5 and 16.4 Hz, PH2), 22.69 (app. d of t, JC,P 13.1 and 2.7 Hz, PH3), 21.99 (d of d, JC,P 13.1 and 5.0 Hz, PH3), 21.38 (d of m, JC,P 12.2 Hz, PH3), 20.98, 16.45 (2 × m, PH3), 20.64 (d, JC,P 13.9 Hz, PH3), 16.53, 16.02 (2 × s, CH2H3), 15.78 (app. d of t, JC,P 20.7 and 3.3 Hz, PH3), 15.32 (d of d, JC,P 15.3 and 5.2 Hz, PH3), 12.47, 10.98 (2 × s, H2CH3). 31P{1H} NMR (C6D6, 243 MHz, 298 K): δ 75.51, 71.37, 65.43, 60.62 (4 × s, 1P). 31P{1H} NMR (d8-toluene, 202 MHz, 223 K): δ 75.98, 71.85, 65.68, 60.76 (4 × s, 1P). 55Mn{1H} NMR (d8-toluene, 124 MHz, 298 K): δ −1011 (br. s). 55Mn{1H} NMR (d8-toluene, 124 MHz, 223 K): δ −1070 (br. s). 55Mn{1H} NMR (d8-toluene, 124 MHz, 370 K): δ −906 (br. s). trans-5b: ν(NN): 1968 cm−1ν(Ge–H): not detected. 1H NMR (C6D6, 600 MHz, 298 K):δ 3.02 (m, 1H, Ge), 1.56, 1.34 (2 × m, 4H, PC2), 1.54 (t, 6H, 3JH,H 7.7 Hz, CH2C3), 1.35, 1.21 (2 × s, 12H, PC3), 0.66 (m, 4H, C2CH3). 13C{1H} NMR (C6D6, 151 MHz, 298 K):δ 31.55 (quin, JC,P 11.7 Hz, PH2), 20.40, 16.53 (2 × m, PH3), 16.74 (s, CH2H3), 15.38 (s, H2CH3). 31P{1H} NMR (C6D6, 243 MHz, 298 K): δ 72.51 (m). 31P{1H} NMR (d8-toluene, 202 MHz, 370 K): δ 71.79 (approx. 1:1:1:1:1:1 sextet, 1JP,Mn 274 Hz). 55Mn{1H} NMR (d8-toluene, 124 MHz, 298 K): δ −1094 (quin, 1JP,Mn 275 Hz). 55Mn{1H} NMR (d8-toluene, 124 MHz, 223 K): δ −1168 (s). 55Mn{1H} NMR (d8-toluene, 124 MHz, 370 K): δ −982 (quin, 1JP,Mn 274 Hz). Anal. found (calcd): C, 37.33 (37.31); H, 8.49 (8.42); N, 5.48 (5.44). Repetition of EA after 1000 days sealed in argon at −30 °C Anal. found (calcd): C, 37.16 (37.31); H, 8.56 (8.42); N, 5.10 (5.44).
). [(dmpe)2MnD2(HGeEt2)] (d2-2b): 2H NMR (C6D6, 77 MHz, 298 K): δ −12.16 (quin, 2JD,P 4.7 Hz, Mn).
GeEt2)] (1b) and transHGe-[(dmpe)2MnH(HGeHEt2)] (transHGe-2b).
11.7 mg (0.02 mmol) of [(dmpe)2MnH(GeEt2)] (1b) was dissolved in 0.35 mL of toluene, and the solution was placed in a 25 mL Schlenk flask, freeze/pump/thawed three times, placed under 1 atm of H2 at −78 °C, sealed, and warmed to room temperature. The reaction mixture was then stirred for 2 days, after which the solvent was removed in vacuo, ∼0.5 mL of hexanes was added, and the solution was placed under 1 atm of H2 at −78 °C and sealed. Maintaining the solution at −78 °C for a few hours afforded large yellow X-ray quality crystals containing a mixture of 1b and transHGe-2b (due to disorder between two germyl groups and a germylene group in a 0.402(3):0.056(3):0.542(3) ratio).
GePh2)] (1a) was dissolved in 10 mL of benzene and placed in a 50 mL storage flask. 21.4 mg (0.26 mmol) of tert-butyl isonitrile was added and the reaction mixture was stirred for 1.5 hours at room temperature in the dark. The solvent was removed in vacuo, and the resulting yellow solid was dried in vacuo for 1 hour and washed with 4 mL of hexanes. Recrystallization of the residue (which did not dissolve in hexanes) at −30 °C from a solution in toluene layered with the hexanes that had been used to wash the crude material afforded 36.0 mg (0.05 mmol, 42%) of cis-3a as X-ray quality yellow crystals. The reaction of 1a with CNtBu affords 3a in near quantitative spectroscopic yield (as a mixture of cis and trans isomers), so the low yield is due to losses during crystallization. (b) 15.3 mg (0.03 mmol) of [(dmpe)2MnH(GePh2)] (1a) and 4.4 mg (0.05 mmol) of tert-butyl isonitrile were dissolved in approx. 0.6 mL of C6D6, placed in a J-young tube, and the reaction mixture was monitored over time at room temperature by NMR spectroscopy. After 30 minutes, the reaction mixture contained cis-3a and trans-3a in a 3:1 ratio, which remained constant over 4 days. cis-3a: 1H NMR (C6D6, 600 MHz, 298 K): δ 8.20, 8.15 (2 × d of d, 2H, 3JH,H 7.6 Hz, 4JH,H 1.0 Hz, o-Ph), 7.25, 7.22 (2 × t, 2H, 3JH,H 7.9 Hz, m-Ph), 7.12 (t, 1H, 3JH,H 7.6 Hz, p-Ph), 7.08 (t, 1H, 3JH,H 7.4 Hz, p-Ph), 5.45 (t of d, 1H, 3JH,P 8.6 and 5.2 Hz, Ge), 1.80, 1.62 (2 × m, 1H, PC2), 1.71 (d, 3H, 2JH,P 6.7 Hz, PC3), 1.69 (d, 3H, 2JH,P 6.9 Hz, PC3), 0.94–1.49 (m, 6H, PC2), 1.34, 1.16 (2 × d, 3H, 2JH,P 6.6 Hz, PC3), 1.20 (d, 3H, 2JH,P 5.9 Hz, PC3), 1.10 (d, 3H, 2JH,P 5.8 Hz, PC3), 1.03 (s, 9H, C(C3)3), 0.97 (d, 3H, 2JH,P 4.6 Hz, PC3), 0.82 (d, 3H, 2JH,P 4.8 Hz, PC3). 13C{1H} NMR (C6D6, 151 MHz, 298 K): δ 158.29 (d, 3JC,P 2.6 Hz, i-Ph), 157.74 (d, 3JC,P 3.4 Hz, i-Ph), 137.65, 137.47 (2 × s, o-Ph), 127.23, 127.10 (2 × s, m-Ph), 125.23, 125.20 (2 × s, p-Ph), 54.94 (s, Me3), 34.62 (app. t of d, JC,P 20.7 and 5.5 Hz, PH2), 33.64 (app. t of d, JC,P 23.3 and 7.5 Hz, PH2), 33.13 (app. t, JC,P 20.4 Hz, PH2), 31.46 (s, C(H3)3), 30.62 (app. t, JC,P 19 Hz, PH2), 24.31 (d of d, JC,P 14.1 and 5.2 Hz, PH3), 23.92 (d, JC,P 12.4 Hz, PH3), 23.43 (d of d, JC,P 16.0 and 5.8 Hz, PH3), 23.04 (d, JC,P 8.0 Hz, PH3), 22.40 (d of d, JC,P 17.9 and 6.0 Hz, PH3), 21.73 (m, PH3), 21.01 (d of m, JC,P 20.5 Hz, PH3). 31P{1H} NMR (C6D6, 243 MHz, 298 K): δ 75.78, 56.99 (2 × s, 1P), 70.53 (s, 2P). trans-3a: 1H NMR (C6D6, 500 MHz, 298 K): δ 8.00 (d, 4H, 3JH,H 7.1 Hz, o-Ph), 7.19 (m, m-Ph), 5.12 (quin., 1H, 3JH,P 7.7 Hz, Ge), 1.85, 1.44 (2 × m, 4H, PC2), 1.40, 1.18 (2 × s, 12H, PC3), 0.98 (s, 9H, C(C3)3). 13C{1H} NMR (C6D6, 126 MHz, 298 K): δ 158.83 (s, i-Ph), 137.10 (s, o-Ph), 127.38 (s, m-Ph), 125.36 (s, p-Ph), 54.55 (s, Me3), 32.52 (m, PH2), 31.36 (s, C(H3)3), 21.72 (m, PH3). 31P{1H} NMR (C6D6, 202 MHz, 298 K): δ 72.71 (s). Anal. found (calcd): C, 52.31 (52.28); H, 8.00 (7.87); N, 2.10 (2.08).
GePh2)] (1a; for 3b 11.6 mg/0.02 mmol, and for 3c 13.3 mg/0.02 mmol) and free isonitrile (for 3b 5.2 mg/0.04 mmol of o-xylyl isontrile, and for 3c 7.0 mg/0.08 mmol of n-butyl isonitrile) were dissolved in approx. 0.6 mL of C6D6, placed in a J-young tube, and the reaction mixtures were monitored over time at room temperature by NMR spectroscopy. Both reactions were complete after 1 hour. cis-3b: 1H NMR (C6D6, 600 MHz, 298 K): δ 8.07 (d of m, 2H, 3JH,H 7.2 Hz, o-Ph), 8.03 (d of d, 2H, 3JH,H 7.8 Hz, 4JH,H 1.2 Hz, o-Ph), 7.13 (t, 2H, 3JH,H 7.4 Hz, m-Ph), 7.07 (m, 2H, p-Ph), 7.06 (m, 2H, m-Ph), 6.90 (d, 2H, 3JH,H 7.5 Hz, xylyl-m), 6.80 (t, 1H, 3JH,H 7.5 Hz, xylyl-p), 5.48 (q, 1H, 3JH,P 6.6 Hz, Ge), 2.10 (s, 6H, xylyl-C3), 1.72 (d, 3H, 2JH,P 7.0 Hz, PC3), 1.63 (d, 3H, 2JH,P 6.8 Hz, PC3), 1.47–1.61 (m, 2H, PC2), 1.51, 1.13 (2 × d, 3H, 2JH,P 6.2 Hz, PC3), 1.04–1.40 (m, 5H, PC2), 1.19 (d, 3H, 2JH,P 5.9 Hz, PC3), 0.98 (d, 3H, 2JH,P 6.1 Hz, PC3), 0.90, 0.80 (2 × d, 3H, 2JH,P 4.8 Hz, PC3), 0.88 (m, 1H, PC2). 13C{1H} NMR (C6D6, 151 MHz, 298 K):δ 156.22 (s, i-Ph), 155.39 (d, 3JC,P 4.0 Hz, i-Ph), 137.78, 137.19 (2 × s, o-Ph), 133.69 (s, xylyl-i), 133.11 (s, xylyl-o), 128.16 (xylyl-m),†† 127.22, 127.10 (2 × s, m-Ph), 125.41, 125.36 (2 × s, p-Ph), 122.62 (s, xylyl-p), 33.81 (app. t, JC,P 19.9 Hz, PH2), 33.17 (m, PH2), 31.86 (app. t, JC,P 20.1 Hz, PH2), 23.68 (d of m, JC,P 16.0 Hz, PH3), 22.50, 21.74, 21.05 (3 × m, PH3), 20.72 (s, xylyl-H3). 31P{1H} NMR (C6D6, 243 MHz, 298 K):δ 70.57 (s, 2P), 67.94, 53.44 (2 × s, 1P). trans-3b: 1H NMR (C6D6, 600 MHz, 298 K): δ 7.95 (d of d, 4H, 3JH,H 7.7 Hz, 4JH,H 1.2 Hz, o-Ph), 7.18 (t, 4H, 3JH,H 7.3 Hz, m-Ph), 7.07 (m, 2H, p-Ph), 6.83 (d, 2H, 3JH,H 7.5 Hz, xylyl-m), 6.69 (t, 1H, 3JH,H 7.5 Hz, xylyl-p), 5.27 (quin., 1H, 3JH,P 7.3 Hz, Ge), 2.24 (s, 6H, xylyl-C3), 1.71, 1.52 (2 × m, 4H, PC2), 1.41, 1.21 (2 × s, 12H, PC3). 13C{1H} NMR (C6D6, 151 MHz, 298 K):δ 157.67 (s, i-Ph), 137.32 (s, o-Ph), 133.81 (s, xylyl-i), 132.11 (s, xylyl-o), 128.68 (s, xylyl-m), 127.50 (s, m-Ph), 125.60 (s, p-Ph), 122.25 (s, xylyl-p), 32.86 (m, PH2), 22.49, 21.91 (2 × m, PH3), 20.63 (s, xylyl-H3). 31P{1H} NMR (C6D6, 243 MHz, 298 K):δ 70.57 (s). cis-3c: 1H NMR (C6D6, 600 MHz, 298 K): δ 8.21, 8.13 (2 × d, 2H, 3JH,H 7.8 Hz, o-Ph), 7.27, 7.22 (2 × t, 2H, 3JH,H 7.5 Hz, m-Ph), 7.15 (t, 1H, 3JH,H 7.5 Hz, p-Ph), 7.11 (t, 1H, 3JH,H 7.3 Hz, p-Ph), 5.46 (app. t of d, 1H, 3JH,P 8.6 and 4.7 Hz, Ge), 3.16 (m, 2H, C2CH2CH2CH3), 1.75, 1.61, 140 (3 × m, 1H, PC2), 1.64, 1.18 (2 × d, 3H, 2JH,P 6.2 Hz, PC3), 1.62 (d, 3H, 2JH,P 6.5 Hz, PC3), 1.39, 1.14, 1.06 (3 × d, 3H, 2JH,P 6.0 Hz, PC3), 1.21 (m, 2H, CH2C2CH2CH3), 1.19 (m, 2H, CH2CH2C2CH3), 1.07–1.12 (m, 3H, PC2), 0.92–1.05 (m, 2H, PC2), 0.95 (d, 3H, 2JH,P 4.7 Hz, PC3), 0.81 (d, 3H, 2JH,P 4.9 Hz, PC3), 0.79 (t, 3H, 3JH,H 7.1 Hz, CH2CH2CH2C3). 13C{1H} NMR (C6D6, 151 MHz, 298 K):δ 157.61, 157.41 (2 × s, i-Ph), 137.63, 137.28 (2 × s, o-Ph), 127.20, 127.13 (2 × s, m-Ph), 125.30, 125.27 (2 × s, p-Ph), 45.12 (s, H2CH2CH2CH3), 34.22, 33.41 (2 × m, PH2), 33.08 (s, CH2H2CH2CH3), 30.48 (app. t, JC,P 19.3 Hz, PH2), 24.47, 23.01, 22.85, 21.34, 21.11, 20.95 (6 × m, PH3), 23.53 (d, JC,P 13.2 Hz, PH3), 22.38 (d of d, JC,P 20.3 and 5.6 Hz, PH3), 20.40 (s, CH2CH2H2CH3), 13.76 (s, CH2CH2CH2H3). 31P{1H} NMR (C6D6, 243 MHz, 298 K):δ 76.11, 53.33 (2 × s, 1P), 71.59 (s, 2P). trans-3c(selected):1H NMR (C6D6, 600 MHz, 298 K): δ 7.96 (d, 4H, 3JH,H 7.1 Hz, o-Ph), 5.06 (quin., 1H, 3JH,P 15.5 Hz, Ge), 1.81, 1.43 (2 × m, 4H, PC2), 1.36, 1.16 (2 × s, 12H, PC3). 13C{1H} NMR (C6D6, 151 MHz, 298 K):δ 137.01 (s, o), 127.42, 125.45 (2 × s, m and p), 32.26 (m, PH2), 22.97, 21.30 (2 × m, PH3). 31P{1H} NMR (C6D6, 243 MHz, 298 K):δ 73.77 (s).
:3:73 ratio. The solvent was then removed in vacuo, the resulting solid was extracted with hexanes, and the mother liquors were stored at −30 °C to afford X-ray quality yellow crystals of 4. The remaining solid (which did not dissolve in hexanes) was dissolved in minimal toluene, and that solution was stored at −30 °C to afford yellow crystals from which an X-ray structure of cis-3b was obtained.
GePh2)] (1a) was dissolved in 5 mL of benzene, and the solution was transferred to a 50 mL storage flask. 25.1 mg (0.30 mmol) of n-butyl isonitrile was added, and the reaction was stirred for 1 hour at room temperature. Removal of the solvent in vacuo afforded an orange oil which was dried in vacuo for 1 hour at room temperature. The oil was extracted with 1.5 mL of hexanes to afford an orange solution which was stored at −30 °C to afford yellow crystals from which an X-ray structure of trans-3c was obtained. Recrystallization of the residue (which did not dissolve in hexanes) at −30 °C from toluene layered with hexanes afforded 4.7 mg (0.01 mmol) of cis-3c, in >95% purity (measured by NMR spectroscopy).
GePh2)] (1a) was dissolved in 5 mL of benzene and placed in a 100 mL storage flask. The mixture was freeze/pump/thawed three times, placed under 1 atm of N2 at −95 °C, sealed, and warmed to room temperature. After stirring for 5 days at room temperature in the dark, the solvent was removed in vacuo. The resulting solid was recrystallized from a concentrated solution of toluene layered with a concentrated solution of hexanes at −30 °C to afford 28.4 mg (0.05 mmol, 54%) of 5a as yellow crystals. X-ray quality crystals of cis-5a were obtained by recrystallization from a concentrated solution in toluene layered with hexanes at −30 °C. X-ray quality crystals of trans-5a were obtained by serendipitous N2 addition to a reaction mixture formed from the combination of approx. 10 mg of 1a with H2 in d8-toluene, followed by removal of the solvent in vacuo and recrystallization from toluene at −30 °C. NMR spectra were obtained under an atmosphere of N2. cis-5a: ν(NN): 2010 cm−1, ν(Ge–H): 1860 cm−1 (very broad). 1H NMR (C6D6, 600 MHz, 298 K): δ 8.22, 8.11 (2 × d, 2H, 3JH,H 7.0 Hz, o-Ph), 7.34, 7.24 (2 × t, 2H, 3JH,H 7.4 Hz, m-Ph), 7.17 (t, 3JH,H 7.7 Hz, p-Ph),‡‡ 7.12 (t, 1H, 3JH,H 7.3 Hz, p-Ph), 5.40 (q, 1H, 3JH,P 5.4 Hz, Ge), 0.70–1.74 (m, 8H, PC2), 1.48 (d, 6H, 2JH,P 6.8 Hz, PC3), 1.29 (PC3),†† 1.09 (d, 3H, 2JH,P 6.1 Hz, PC3), 0.99 (d, 3H, 2JH,P 6.5 Hz, PC3), 0.93 (d, 3H, 2JH,P 5.9 Hz, PC3), 0.85 (d, 3H, 2JH,P 5.1 Hz, PC3), 0.53 (d, 3H, 2JH,P 5.3 Hz, PC3). 13C{1H} NMR (C6D6, 151 MHz, 298 K): δ 154.87, 153.61 (2 × s, i-Ph), 137.80, 137.22 (2 × s, o-Ph), 127.49, 127.34 (2 × s, m-Ph), 125.84 (s, p-Ph), 125.73 or 125.68 (s, p-Ph),§§ 33.18, 29.30 (2 × m, PH2), 32.53 (app. t, JC,P 20.8 Hz, PH2), 23.09, 21.90, 21.80, 16.52 (4 × m, PH3), 22.64 (d of d, JC,P 18.5 and 5.8 Hz, PH3), 19.87 (d, JC,P 16.8 Hz, PH3), 17.45 (d, JC,P 18.7 Hz, PH3), 15.31 (d of d, JC,P 17.3 and 4.8 Hz, PH3). 31P{1H} NMR (C6D6, 243 MHz, 298 K):δ 71.98, 69.33, 68.72, 58.71 (4 × s, 1P). 31P{1H} NMR (d8-toluene, 202 MHz, 223 K): δ 72.47, 70.27, 68.91, 58.75 (4 × s, 1P). 55Mn{1H} NMR (d8-toluene, 124 MHz, 298 K): δ −718 (br. s). 55Mn{1H} NMR (d8-toluene, 124 MHz, 223 K): δ −812 (br. s). 55Mn{1H} NMR (d8-toluene, 124 MHz, 370 K): δ −601 (br. s). trans-5a: ν(NN): 1973 cm−1, ν(Ge–H): 1860 cm−1 (very broad). 1H NMR (C6D6, 600 MHz, 298 K): δ 7.81 (d, 4H, 3JH,H 7.0 Hz, o-Ph), 7.12 (t, 4H, 3JH,H 7.3 Hz, m-Ph), 7.04 (t, 2H, 3JH,H 7.2 Hz, p-Ph), 4.74 (quin., 1H, 3JH,P 7.7 Hz, Ge), 1.80, 1.38 (2 × m, 4H, PC2), 1.29, 1.16 (2 × s, 12H, PC3). 13C{1H} NMR (C6D6, 151 MHz, 298 K): δ 156.81 (s, i-Ph), 136.69 (s, o-Ph), 127.49 (s, m-Ph), 125.73 or 125.68 (s, p-Ph),§§ 31.35 (m, PH2), 20.28, 16.48 (2 × m, PH3). 31P{1H} NMR (C6D6, 243 MHz, 298 K): δ 70.63 (br. s). 31P{1H} NMR (d8-toluene, 202 MHz, 223 K): δ 71.37 (s). 31P{1H} NMR (d8-toluene, 202 MHz, 370 K): δ 70.2 (broad multiplet with nearly indiscernible coupling due to quadrupolar collapse). 55Mn{1H} NMR (d8-toluene, 124 MHz, 298 K): δ −845 (br. s). 55Mn{1H} NMR (d8-toluene, 124 MHz, 223 K): δ −991 (br. s). 55Mn{1H} NMR (d8-toluene, 124 MHz, 370 K): δ −730 (br. s). Anal. found (calcd): C, 47.25 (47.17); H, 7.14 (7.09); N, 4.42 (4.58). Repetition of EA after 2.7 years sealed under argon at −30 °C: Anal. found (calcd): C, 46.96 (47.17); H, 7.20 (7.09); N, 3.84 (4.58).
GeEt2)] (1b) was dissolved in 10 mL of benzene and transferred to a 100 mL storage flask. The mixture was freeze/pump/thawed three times, placed under 1 atm of N2 at −95 °C, sealed, and warmed to room temperature. After stirring for 3 days at room temperature in the dark, the solvent was removed in vacuo. The resulting solid was recrystallized using 5 mL of hexanes at −30 °C to afford 65.3 mg (0.13 mmol, 58%) of cis-5b as yellow crystals. X-ray quality crystals of trans-5b were obtained by heating a mixture of approx. 10 mg of 1b under N2 for 4 days at 60 °C in C6D6 (which resulted in cis–trans isomerization to a 22:78 ratio), followed by removal of the solvent in vacuo and recrystallization from a concentrated solution of hexanes at −30 °C. NMR spectra were obtained under an atmosphere of N2. cis-5b: ν(NN): 1989 cm−1, ν(Ge–H): 1823 cm−1. 1H NMR (C6D6, 600 MHz, 298 K): δ 3.65 (m, 1H, Ge), 1.83, 1.82 (2 × t, 3H, 3JH,H 7.8 Hz, CH2C3), 1.63 (m, 1H, PC2), 1.54 (d, 3H, 2JH,P 7.1 Hz, PC3), 1.23–1.56 (m, 4H, PC2), 1.50, 1.33, 1.32, 1.27 (4 × m, 1H, C2CH3) 1.47 (d, 3H, 2JH,P 6.7 Hz, PC3), 1.14 (d, 3H, 2JH,P 6.3 Hz, PC3), 1.12 (d, 3H, 2JH,P 5.8 Hz, PC3), 1.06 (d, 3H, 2JH,P 6.5 Hz, PC3), 1.03 (d, 3H, 2JH,P 5.9 Hz, PC3), 0.97 (d, 3H, 2JH,P 5.0 Hz, PC3), 0.70–0.94 (m, 3H, PC2), 0.58 (d, 3H, 2JH,P 5.2 Hz, PC3). 13C{1H} NMR (C6D6, 151 MHz, 298 K):δ 33.97 (m, PH2), 31.36 (app. t, JC,P 19.7 Hz, PH2), 28.90 (d of d, JC,P 23.5 and 16.4 Hz, PH2), 22.69 (app. d of t, JC,P 13.1 and 2.7 Hz, PH3), 21.99 (d of d, JC,P 13.1 and 5.0 Hz, PH3), 21.38 (d of m, JC,P 12.2 Hz, PH3), 20.98, 16.45 (2 × m, PH3), 20.64 (d, JC,P 13.9 Hz, PH3), 16.53, 16.02 (2 × s, CH2H3), 15.78 (app. d of t, JC,P 20.7 and 3.3 Hz, PH3), 15.32 (d of d, JC,P 15.3 and 5.2 Hz, PH3), 12.47, 10.98 (2 × s, H2CH3). 31P{1H} NMR (C6D6, 243 MHz, 298 K): δ 75.51, 71.37, 65.43, 60.62 (4 × s, 1P). 31P{1H} NMR (d8-toluene, 202 MHz, 223 K): δ 75.98, 71.85, 65.68, 60.76 (4 × s, 1P). 55Mn{1H} NMR (d8-toluene, 124 MHz, 298 K): δ −1011 (br. s). 55Mn{1H} NMR (d8-toluene, 124 MHz, 223 K): δ −1070 (br. s). 55Mn{1H} NMR (d8-toluene, 124 MHz, 370 K): δ −906 (br. s). trans-5b: ν(NN): 1968 cm−1ν(Ge–H): not detected. 1H NMR (C6D6, 600 MHz, 298 K):δ 3.02 (m, 1H, Ge), 1.56, 1.34 (2 × m, 4H, PC2), 1.54 (t, 6H, 3JH,H 7.7 Hz, CH2C3), 1.35, 1.21 (2 × s, 12H, PC3), 0.66 (m, 4H, C2CH3). 13C{1H} NMR (C6D6, 151 MHz, 298 K):δ 31.55 (quin, JC,P 11.7 Hz, PH2), 20.40, 16.53 (2 × m, PH3), 16.74 (s, CH2H3), 15.38 (s, H2CH3). 31P{1H} NMR (C6D6, 243 MHz, 298 K): δ 72.51 (m). 31P{1H} NMR (d8-toluene, 202 MHz, 370 K): δ 71.79 (approx. 1:1:1:1:1:1 sextet, 1JP,Mn 274 Hz). 55Mn{1H} NMR (d8-toluene, 124 MHz, 298 K): δ −1094 (quin, 1JP,Mn 275 Hz). 55Mn{1H} NMR (d8-toluene, 124 MHz, 223 K): δ −1168 (s). 55Mn{1H} NMR (d8-toluene, 124 MHz, 370 K): δ −982 (quin, 1JP,Mn 274 Hz). Anal. found (calcd): C, 37.33 (37.31); H, 8.49 (8.42); N, 5.48 (5.44). Repetition of EA after 1000 days sealed in argon at −30 °C Anal. found (calcd): C, 37.16 (37.31); H, 8.56 (8.42); N, 5.10 (5.44).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :trans ratios for reactions of 1a–b with N2 to form [(dmpe)2Mn(GeHR2)(N2)] (5a: R = Ph, 5b: R = Et).
Approx. 10 mg of [(dmpe)2MnH(GeR2)] (1a: R = Ph, 1b: R = Et) was dissolved in approx. 0.6 mL of C6D6 and the mixture was transferred to a J-young tube. The solutions were freeze/pump/thawed three times, placed under 1 atm of N2 at −95 °C, sealed, and warmed to room temperature. Conversion of 1a–b to 5a–b, along with cis:trans ratios of 5a–b, were monitored by 1H NMR spectroscopy at various intervals. In the case of the reaction involving 1b, the reaction mixture was also heated at 60 °C for various times and monitored by 1H NMR spectroscopy.
SiPh2)] and [(dmpe)2MnH2(SiHPh2)] in a 6:1 ratio was dissolved in approx. 0.6 mL of C6D6 and the mixture was transferred to a J-young tube. The mixture was freeze/pump/thawed three times, placed under 1 atm of N2 at −95 °C, sealed, and warmed to room temperature. The resulting solution was analyzed by NMR spectroscopy in situ after sitting at room temperature for 2 hours, and contained [(dmpe)2MnH2(SiHPh2)], cis-[(dmpe)2Mn(SiHPh2)(N2)] (cis-6), and trans-[(dmpe)2Mn(SiHPh2)(N2)] (trans-6) in a 1:5.7:2.3 ratio. X-ray quality crystals of trans-6 were obtained by removing the solvent in vacuo, washing with 1 mL of hexanes, and recrystallization of the residue from 1 mL of toluene at −30 °C. cis-6(selected): 1H NMR (C6D6, 600 MHz, 298 K): δ 8.24 (d of d, 2H, 3JH,H 7.7 Hz, 4JH,H 1.3 Hz, o-Ph), 8.15 (d of d, 2H, 3JH,H 8.0 Hz, 4JH,H 1.3 Hz, o-Ph), 7.32 (t, 2H, 3JH,H 7.7 Hz, m-Ph), 7.24 (t, 2H, 3JH,H 7.6 Hz, m-Ph), 7.16 (m, 2H, p-Ph), 5.53 (m w. 29Si sat., 1H, 1JH,Si 141 Hz, Si), 1.35, 1.30 (2 × d, 3H, JH,P 6.6 Hz, PC3), 1.33 (d, 3H, JH,P 7.2 Hz, PC3), 1.17 (d, 3H, JH,P 5.6 Hz, PC3), 0.91 (d, 3H, JH,P 5.2 Hz, PC3), 0.90 (d, 3H, JH,P 4.7 Hz, PC3), 0.53 (d, 3H, JH,P 5.4 Hz, PC3). 13C{1H} NMR (C6D6, 151 MHz, 298 K):δ 152.75, 151.83 (2 × s, i-Ph), 137.25, 136.92 (2 × s, o-Ph), 127.18 (s, m-Ph), 126.14 (s, p-Ph), 34.25 (t, JC,P 21.8 Hz, PH2), 32.90, 32.06 (2 × m, PH2), 29.77 (d of d, JC,P 24.5 and 16.0 Hz, PH2), 24.71 (d of d, JC,P 13.2 and 4.8 Hz, PH3), 23.05 (d of d, JC,P 13.2 and 6.3 Hz, PH3), 22.57 (m, PH3), 19.75 (d, JC,P 19.7 Hz, PH3), 16.86 (d, JC,P 18.4 Hz, PH3), 15.95 (d of d, JC,P 18.5 and 5.4 Hz, PH3), 15.26 (d of d, JC,P 17.6 and 4.7 Hz, PH3). 29Si NMR (data from29Si–1H HMBC in C6D6, 119 MHz, 298 K):δ 36.0. 31P{1H} NMR (C6D6, 243 MHz, 298 K):δ 71.78, 68.50, 64.12, 58.67 (4 × s, 1P). 55Mn{1H} NMR (C6D6, 124 MHz, 298 K): δ −973 (br. s). 55Mn{1H} NMR (C6D6, 124 MHz, 360 K): δ −881 (br. s). trans-6: 1H NMR (C6D6, 600 MHz, 298 K): δ 7.78 (d of d, 4H, 3JH,H 7.8 Hz, 4JH,H 1.4 Hz, o-Ph), 7.11 (t, 4H, 3JH,H 7.3 Hz, m-Ph), 7.04 (t of t, 2H, 3JH,H 7.3 Hz, 4JH,H 1.4 Hz, p-Ph), 4.82 (quin. w. 29Si sat., 1H, 3JH,P 8.6 Hz, 1JH,Si 140 Hz, Si), 1.79, 1.35 (2 × m, 4H, PC2), 1.27, 1.17 (2 × s, 12H, PC3). 13C{1H} NMR (C6D6, 151 MHz, 298 K):δ 154.10 (s, i-Ph), 136.39 (s, o-Ph), 127.31 (s, m-Ph), 126.01 (s, p-Ph), 31.31 (m, PH2), 20.55, 17.08 (2 × m, PH3). 29Si NMR (data from29Si–1H HMBC in C6D6, 119 MHz, 298 K):δ 33.9. 31P{1H} NMR (C6D6, 243 MHz, 298 K):δ 70.8 (m). 31P{1H} NMR (C6D6, 202 MHz, 360 K):δ 70.39 (approx. 1:1:1:1:1:1 sextet, 1JP,Mn 269 Hz). 55Mn{1H} NMR (C6D6, 124 MHz, 298 K): δ −1098 (quin, 1JP,Mn 270 Hz). 55Mn{1H} NMR (C6D6, 124 MHz, 360 K): δ −1006 (quin, 1JP,Mn 270 Hz).
GeR2)] (1a: R = Ph, 1b: R = Et) and [(dmpe)2MnH(SiPh2)].
Approximately 15 mg of [(dmpe)2MnH(GePh2)] (1a), [(dmpe)2MnH(GeEt2)] (1b), or an 8:1 mixture of [(dmpe)2MnH(SiPh2)] and [(dmpe)2MnH2(SiHPh2)] were dissolved in approx. 0.6 mL of (for 1a–b) d8-toluene or (for the silicon-containing species) C6D6, and analyzed by NMR spectroscopy. 1a: 55Mn{1H} NMR (d8-toluene, 124 MHz, 298 K): δ −1176 (br. s). 55Mn{1H} NMR (d8-toluene, 124 MHz, 370 K): δ −1107 (br. s). 1b: 55Mn{1H} NMR (d8-toluene, 124 MHz, 298 K): δ −1303 (br. s). 55Mn{1H} NMR (d8-toluene, 124 MHz, 370 K): δ −1226 (br. s). [(dmpe)2MnH(SiPh2)]: 55Mn{1H} NMR (C6D6, 124 MHz, 298 K): δ −548 (br. s).
:trans ratios for reactions of 1a–b with N2 to form [(dmpe)2Mn(GeHR2)(N2)] (5a: R = Ph, 5b: R = Et).
Approx. 10 mg of [(dmpe)2MnH(GeR2)] (1a: R = Ph, 1b: R = Et) was dissolved in approx. 0.6 mL of C6D6 and the mixture was transferred to a J-young tube. The solutions were freeze/pump/thawed three times, placed under 1 atm of N2 at −95 °C, sealed, and warmed to room temperature. Conversion of 1a–b to 5a–b, along with cis:trans ratios of 5a–b, were monitored by 1H NMR spectroscopy at various intervals. In the case of the reaction involving 1b, the reaction mixture was also heated at 60 °C for various times and monitored by 1H NMR spectroscopy.
SiPh2)] and [(dmpe)2MnH2(SiHPh2)] in a 6:1 ratio was dissolved in approx. 0.6 mL of C6D6 and the mixture was transferred to a J-young tube. The mixture was freeze/pump/thawed three times, placed under 1 atm of N2 at −95 °C, sealed, and warmed to room temperature. The resulting solution was analyzed by NMR spectroscopy in situ after sitting at room temperature for 2 hours, and contained [(dmpe)2MnH2(SiHPh2)], cis-[(dmpe)2Mn(SiHPh2)(N2)] (cis-6), and trans-[(dmpe)2Mn(SiHPh2)(N2)] (trans-6) in a 1:5.7:2.3 ratio. X-ray quality crystals of trans-6 were obtained by removing the solvent in vacuo, washing with 1 mL of hexanes, and recrystallization of the residue from 1 mL of toluene at −30 °C. cis-6(selected): 1H NMR (C6D6, 600 MHz, 298 K): δ 8.24 (d of d, 2H, 3JH,H 7.7 Hz, 4JH,H 1.3 Hz, o-Ph), 8.15 (d of d, 2H, 3JH,H 8.0 Hz, 4JH,H 1.3 Hz, o-Ph), 7.32 (t, 2H, 3JH,H 7.7 Hz, m-Ph), 7.24 (t, 2H, 3JH,H 7.6 Hz, m-Ph), 7.16 (m, 2H, p-Ph), 5.53 (m w. 29Si sat., 1H, 1JH,Si 141 Hz, Si), 1.35, 1.30 (2 × d, 3H, JH,P 6.6 Hz, PC3), 1.33 (d, 3H, JH,P 7.2 Hz, PC3), 1.17 (d, 3H, JH,P 5.6 Hz, PC3), 0.91 (d, 3H, JH,P 5.2 Hz, PC3), 0.90 (d, 3H, JH,P 4.7 Hz, PC3), 0.53 (d, 3H, JH,P 5.4 Hz, PC3). 13C{1H} NMR (C6D6, 151 MHz, 298 K):δ 152.75, 151.83 (2 × s, i-Ph), 137.25, 136.92 (2 × s, o-Ph), 127.18 (s, m-Ph), 126.14 (s, p-Ph), 34.25 (t, JC,P 21.8 Hz, PH2), 32.90, 32.06 (2 × m, PH2), 29.77 (d of d, JC,P 24.5 and 16.0 Hz, PH2), 24.71 (d of d, JC,P 13.2 and 4.8 Hz, PH3), 23.05 (d of d, JC,P 13.2 and 6.3 Hz, PH3), 22.57 (m, PH3), 19.75 (d, JC,P 19.7 Hz, PH3), 16.86 (d, JC,P 18.4 Hz, PH3), 15.95 (d of d, JC,P 18.5 and 5.4 Hz, PH3), 15.26 (d of d, JC,P 17.6 and 4.7 Hz, PH3). 29Si NMR (data from29Si–1H HMBC in C6D6, 119 MHz, 298 K):δ 36.0. 31P{1H} NMR (C6D6, 243 MHz, 298 K):δ 71.78, 68.50, 64.12, 58.67 (4 × s, 1P). 55Mn{1H} NMR (C6D6, 124 MHz, 298 K): δ −973 (br. s). 55Mn{1H} NMR (C6D6, 124 MHz, 360 K): δ −881 (br. s). trans-6: 1H NMR (C6D6, 600 MHz, 298 K): δ 7.78 (d of d, 4H, 3JH,H 7.8 Hz, 4JH,H 1.4 Hz, o-Ph), 7.11 (t, 4H, 3JH,H 7.3 Hz, m-Ph), 7.04 (t of t, 2H, 3JH,H 7.3 Hz, 4JH,H 1.4 Hz, p-Ph), 4.82 (quin. w. 29Si sat., 1H, 3JH,P 8.6 Hz, 1JH,Si 140 Hz, Si), 1.79, 1.35 (2 × m, 4H, PC2), 1.27, 1.17 (2 × s, 12H, PC3). 13C{1H} NMR (C6D6, 151 MHz, 298 K):δ 154.10 (s, i-Ph), 136.39 (s, o-Ph), 127.31 (s, m-Ph), 126.01 (s, p-Ph), 31.31 (m, PH2), 20.55, 17.08 (2 × m, PH3). 29Si NMR (data from29Si–1H HMBC in C6D6, 119 MHz, 298 K):δ 33.9. 31P{1H} NMR (C6D6, 243 MHz, 298 K):δ 70.8 (m). 31P{1H} NMR (C6D6, 202 MHz, 360 K):δ 70.39 (approx. 1:1:1:1:1:1 sextet, 1JP,Mn 269 Hz). 55Mn{1H} NMR (C6D6, 124 MHz, 298 K): δ −1098 (quin, 1JP,Mn 270 Hz). 55Mn{1H} NMR (C6D6, 124 MHz, 360 K): δ −1006 (quin, 1JP,Mn 270 Hz).
GeR2)] (1a: R = Ph, 1b: R = Et) and [(dmpe)2MnH(SiPh2)].
Approximately 15 mg of [(dmpe)2MnH(GePh2)] (1a), [(dmpe)2MnH(GeEt2)] (1b), or an 8:1 mixture of [(dmpe)2MnH(SiPh2)] and [(dmpe)2MnH2(SiHPh2)] were dissolved in approx. 0.6 mL of (for 1a–b) d8-toluene or (for the silicon-containing species) C6D6, and analyzed by NMR spectroscopy. 1a: 55Mn{1H} NMR (d8-toluene, 124 MHz, 298 K): δ −1176 (br. s). 55Mn{1H} NMR (d8-toluene, 124 MHz, 370 K): δ −1107 (br. s). 1b: 55Mn{1H} NMR (d8-toluene, 124 MHz, 298 K): δ −1303 (br. s). 55Mn{1H} NMR (d8-toluene, 124 MHz, 370 K): δ −1226 (br. s). [(dmpe)2MnH(SiPh2)]: 55Mn{1H} NMR (C6D6, 124 MHz, 298 K): δ −548 (br. s).
Data availability
Data supporting this article is included in the ESI.† Crystallographic data for trans-2a/1a, cis-3a, cis-3b, trans-3c, 4, cis-5a, trans-5a, cis-5b, trans-5b, and 6 has been deposited at the CCDC with deposition numbers 2447494–2447503,† respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. J. H. E. thanks NSERC of Canada for a Discovery Grant and Dr Yurij Mozharivskyj for access to his X-ray diffractometer. We are also grateful to Dr James Britten and Dr Gary Schrobilgen for helpful advice regarding X-ray crystal structures and analysis of NMR spectra involving coupling to quadrupolar nuclei, respectively, and Dr Novan Gray for assistance with high temperature NMR spectroscopy.References
- J. Baumgartner and C. Marschner, Rev. Inorg. Chem., 2014, 34, 119–152 CAS.
- V. Y. Lee, Eur. J. Inorg. Chem., 2022, e202200175 CrossRef CAS.
- M. Ghosh, N. Sen and S. Khan, ACS Omega, 2022, 7, 6449–6454 CrossRef CAS PubMed.
- T. J. Hadlington, Chem. Soc. Rev., 2024, 53, 9738–9831 RSC.
- H. Hashimoto, T. Tsubota, T. Fukuda and H. Tobita, Chem. Lett., 2009, 38, 1196–1197 CrossRef CAS.
- H. Sakaba, Y. Arai, K. Suganuma and E. Kwon, Organometallics, 2013, 32, 5038–5046 CrossRef CAS.
- T. Fukuda, H. Hashimoto and H. Tobita, J. Organomet. Chem., 2017, 848, 89–94 CrossRef CAS.
- T. P. Dhungana, H. Hashimoto, M. Ray and H. Tobita, Organometallics, 2020, 39, 4350–4361 CrossRef CAS.
- T. P. Dhungana, H. Hashimoto and H. Tobita, Dalton Trans., 2017, 46, 8167–8179 RSC.
- M. Widemann, S. Jeggle, M. Auer, K. Eichele, H. Schubert, C. P. Sindlinger and L. Wesemann, Chem. Sci., 2022, 13, 3999–4009 RSC.
- K. K. Pandey, M. Lein and G. Frenking, J. Am. Chem. Soc., 2003, 125, 1660–1668 CrossRef CAS PubMed.
- A. C. Filippou, U. Chakraborty and G. Schnakenburg, Chem. – Eur. J., 2013, 19, 5676–5686 CrossRef CAS PubMed.
- A. C. Filippou, N. Weidemann, A. I. Philippopoulos and G. Schnakenburg, Angew. Chem., Int. Ed., 2006, 45, 5987–5991 CrossRef CAS PubMed.
- H. Hashimoto, T. Fukuda, H. Tobita, M. Ray and S. Sakaki, Angew. Chem., Int. Ed., 2012, 51, 2930 CrossRef CAS PubMed.
- M. E. Fasulo and T. D. Tilley, Chem. Commun., 2012, 48, 7690–7692 RSC.
- H. Hashimoto, T. Fukuda and H. Tobita, New J. Chem., 2010, 34, 1723–1730 RSC.
- P. G. Hayes, R. Waterman, P. B. Glaser and T. D. Tilley, Organometallics, 2009, 28, 5082–5089 CrossRef CAS.
- S. Bajo, E. Soto, M. Fernández-Buenestado, J. López-Serrano and J. Campos, Nat. Commun., 2024, 15, 9656 CrossRef CAS PubMed.
- K. E. Litz, K. Henderson, R. W. Gourley and M. M. B. Holl, Organometallics, 1995, 14, 5008–5010 CrossRef CAS.
- S. Grumbine and T. D. Tilley, in Progress in Organosilicon Chemistry, ed. B. Marciniec and J. Chojnowski, Gordan and Breach, Amsterdam, 1995, pp. 133–146 Search PubMed.
- P. Gaspar and R. West, in The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, John Wiley & Sons, Chichester, UK, 1998, vol. 2, ch. 43, pp. 2463–2568 Search PubMed.
- M. Okazaki, H. Tobita and H. Ogino, Dalton Trans., 2003, 493–506 RSC.
- R. Waterman, P. G. Hayes and T. D. Tilley, Acc. Chem. Res., 2007, 40, 712–719 CrossRef CAS PubMed.
- D. J. Cardin, B. Cetinkaya and M. F. Lappert, Chem. Rev., 1972, 72, 545–574 Search PubMed.
- D. J. Cardin, B. Çetinkaya, M. J. Doyle and M. F. Lappert, Chem. Soc. Rev., 1973, 2, 99–144 Search PubMed.
- T. Strassner, Top. Organomet. Chem., 2004, 1–20 CAS.
- G. S. Girolami, G. Wilkinson, M. Thornton-Pett and M. B. Hursthouse, J. Am. Chem. Soc., 1983, 105, 6752–6753 CrossRef CAS.
- G. S. Girolami, C. G. Howard, G. Wilkinson, H. M. Dawes, M. Thornton-Pett, M. Motevalli and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1985, 921–929 RSC.
- J. S. Price, I. Vargas-Baca, D. J. H. Emslie and J. F. Britten, Dalton Trans., 2023, 52, 14880–14895 RSC.
- J. S. Price, D. J. H. Emslie, I. Vargas-Baca and J. F. Britten, Organometallics, 2018, 37, 3010–3023 CrossRef CAS.
- J. S. Price, D. J. H. Emslie and J. F. Britten, Angew. Chem., Int. Ed., 2017, 56, 6223–6227 CrossRef CAS.
- J. S. Price and D. J. H. Emslie, Chem. Sci., 2019, 10, 10853–10869 RSC.
- J. S. Price, D. J. H. Emslie and B. Berno, Organometallics, 2019, 38, 2347–2362 CrossRef CAS.
- J. S. Price and D. J. H. Emslie, Organometallics, 2020, 39, 4618–4628 CrossRef CAS.
- H. M. Mobarok, R. McDonald, M. J. Ferguson and M. Cowie, Inorg. Chem., 2012, 51, 4020–4034 Search PubMed.
- K. A. Smart, E. Mothes-Martin, L. Vendier, R. N. Perutz, M. Grellier and S. Sabo-Etienne, Organometallics, 2015, 34, 4158–4163 Search PubMed.
- P. M. Keil, S. Ezendu, A. Schulz, M. Kubisz, T. Szilvási and T. J. Hadlington, J. Am. Chem. Soc., 2024, 146, 23606–23615 CrossRef CAS.
- V. Y. Lee, R. Sakai, K. Takanashi, O. A. Gapurenko, R. M. Minyaev, H. Gornitzka and A. Sekiguchi, Angew. Chem., Int. Ed., 2021, 60, 3951–3955 CrossRef CAS PubMed.
- J. L. Vincent, S. Luo, B. L. Scott, R. Butcher, C. J. Unkefer, C. J. Burns, G. J. Kubas, A. Lledós, F. Maseras and J. Tomàs, Organometallics, 2003, 22, 5307–5323 CrossRef CAS.
- R. Herrmann, T. Braun and S. Mebs, Eur. J. Inorg. Chem., 2014, 2014, 4826–4835 CrossRef CAS.
- A. Antiñolo, F. Carrillo-Hermosilla, A. Castel, M. Fajardo, J. Fernández-Baeza, M. Lanfranchi, A. Otero, M. A. Pellinghelli, G. Rima, J. Satgé and E. Villaseñor, Organometallics, 1998, 17, 1523–1529 CrossRef.
- N. Zhang, R. S. Sherbo, G. S. Bindra, D. Zhu and P. H. M. Budzelaar, Organometallics, 2017, 36, 4123–4135 CrossRef CAS.
- J. Takaya and N. Iwasawa, Eur. J. Inorg. Chem., 2018, 2018, 5012–5018 CrossRef CAS.
- C. J. Laglera-Gándara, P. Ríos, F. J. Fernández-de-Córdova, M. Barturen, I. Fernández and S. Conejero, Inorg. Chem., 2022, 61, 20848–20859 CrossRef PubMed.
- M. A. Esteruelas, A. M. López, E. Oñate and E. Raga, Angew. Chem., Int. Ed., 2022, 61, e202204081 CrossRef CAS PubMed.
- U. Schubert, Adv. Organomet. Chem., 1990, 30, 151–187 CrossRef CAS.
- S. K. Ignatov, N. H. Rees, B. R. Tyrrell, S. R. Dubberley, A. G. Razuvaev, P. Mountford and G. I. Nikonov, Chem. – Eur. J., 2004, 10, 4991–4999 CrossRef CAS PubMed.
- P. Meixner, K. Batke, A. Fischer, D. Schmitz, G. Eickerling, M. Kalter, K. Ruhland, K. Eichele, J. E. Barquera-Lozada, N. P. M. Casati, F. Montisci, P. Macchi and W. Scherer, J. Phys. Chem. A, 2017, 121, 7219–7235 CrossRef CAS PubMed.
- S. Gründemann, H.-H. Limbach, G. Buntkowsky, S. Sabo-Etienne and B. Chaudret, J. Phys. Chem. A, 1999, 103, 4752–4754 Search PubMed.
- R. H. Crabtree, Chem. Rev., 2016, 116, 8750–8769 CrossRef CAS PubMed.
- D. M. Heinekey, J. Labelled Compd. Radiopharm., 2007, 50, 1063–1071 Search PubMed.
- B. R. Bender, G. J. Kubas, L. H. Jones, B. I. Swanson, J. Eckert, K. B. Capps and C. D. Hoff, J. Am. Chem. Soc., 1997, 119, 9179–9190 Search PubMed.
- G. Parkin, Acc. Chem. Res., 2009, 42, 315–325 Search PubMed.
- C. Perthuisot, M. Fan and W. D. Jones, Organometallics, 1992, 11, 3622–3629 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B, 2016, 72, 171–179 Search PubMed.
- Y. Tanabe and Y. Nishibayashi, in Transition Metal–Dinitrogen Complexes, ed. Y. Nishibayashi, Wiley-VCH, Weinheim, Germany, 2019, ch. 1, pp. 1–77 Search PubMed.
- A. D. Piascik and A. E. Ashley, in Transition Metal–Dinitrogen Complexes, ed. Y. Nishibayashi, Wiley-VCH, Weinheim, Germany, 2019, ch. 6, pp. 285–335 Search PubMed.
- N. Mézailles, in Transition Metal–Dinitrogen Complexes, ed. Y. Nishibayashi, Wiley-VCH, Weinheim, Germany, 2019, ch. 4, pp. 221–269 Search PubMed.
- E. A. Ison, in Transition Metal–Dinitrogen Complexes, ed. Y. Nishibayashi, Wiley-VCH, Weinheim, Germany, 2019, ch. 5, pp. 271–284 Search PubMed.
- Q. Le Dé, D. A. Valyaev and A. Simonneau, Chem. – Eur. J., 2024, 30, e202400784 Search PubMed.
- D. Sellmann, Angew. Chem., Int. Ed. Engl., 1972, 11, 534–534 CrossRef CAS.
- S. M. Howdle and M. Poliakoff, J. Chem. Soc., Chem. Commun., 1989, 1099–1101 RSC.
- J. A. Banister, P. D. Lee and M. Poliakoff, Organometallics, 1995, 14, 3876–3885 Search PubMed.
- P. D. Lee, J. L. King, S. Seebald and M. Poliakoff, Organometallics, 1998, 17, 524–533 CrossRef CAS.
- R. K. Merwin, A. C. Ontko, J. F. Houlis and D. M. Roddick, Polyhedron, 2004, 23, 2873–2878 CrossRef CAS.
- J. A. Banister, M. W. George, S. Grubert, S. M. Howdle, M. Jobling, F. P. A. Johnson, S. L. Morrison, M. Poliakoff, U. Schubert and J. R. Westwell, J. Organomet. Chem., 1994, 484, 129–135 CrossRef CAS.
- S. M. Howdle, M. A. Healy and M. Poliakoff, J. Am. Chem. Soc., 1990, 112, 4804–4813 CrossRef CAS.
- Y. Zheng, W. Wang, J. Lin, Y. She and K. Fu, J. Phys. Chem., 1992, 96, 9821–9827 Search PubMed.
- W. T. Boese and P. C. Ford, Organometallics, 1994, 13, 3525–3531 Search PubMed.
- J. B. Eastwood, L. A. Hammarback, M. T. McRobie, I. P. Clark, M. Towrie, I. J. S. Fairlamb and J. M. Lynam, Dalton Trans., 2020, 49, 5463–5470 Search PubMed.
- B. H. G. Swennenhuis, R. Poland, N. J. DeYonker, C. E. Webster, D. J. Darensbourg and A. A. Bengali, Organometallics, 2011, 30, 3054–3063 CrossRef CAS.
- Y.-Q. Zou, S. Chakraborty, A. Nerush, D. Oren, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2018, 8, 8014–8019 CrossRef CAS PubMed.
- W. A. King, X.-L. Luo, B. L. Scott, G. J. Kubas and K. W. Zilm, J. Am. Chem. Soc., 1996, 118, 6782–6783 CrossRef CAS.
- W. A. King, B. L. Scott, J. Eckert and G. J. Kubas, Inorg. Chem., 1999, 38, 1069–1084 CrossRef CAS PubMed.
- K. D. Welch, W. G. Dougherty, W. S. Kassel, D. L. DuBois and R. M. Bullock, Organometallics, 2010, 29, 4532–4540 CrossRef CAS.
- P. DeShong, G. A. Slough, D. R. Sidler, P. J. Rybczynski, W. Von Philipsborn, R. W. Kunz, B. E. Bursten and T. W. Clayton Jr., Organometallics, 1989, 8, 1381–1388 CrossRef CAS.
- J. D. Cotton and R. D. Markwell, Inorg. Chim. Acta, 1990, 175, 187–191 CrossRef CAS.
- Z.-L. Xue and T. M. Cook, in Comprehensive Inorganic Chemistry III, ed. J. Reedijk and K. R. Poeppelmeier, Elsevier, Oxford, 3rd edn, 2023, pp. 660–744 Search PubMed.
- W. J. Miles Jr., B. B. Garrett and R. J. Clark, Inorg. Chem., 1969, 8, 2817 CrossRef.
- B. Wrackmeyer, T. Hofmann and M. Herberhold, J. Organomet. Chem., 1995, 486, 255 CrossRef CAS.
- S. Onaka, T. Miyamoto and Y. Sasaki, Bull. Chem. Soc. Jpn., 1971, 44, 1851 Search PubMed.
- W. Levason, S. D. Orchard and G. Reid, Organometallics, 1999, 18, 1275–1280 CrossRef CAS.
- J. Connolly, M. K. Davies and G. Reid, J. Chem. Soc., Dalton Trans., 1998, 3833–3838 Search PubMed.
- J. Connolly, G. W. Goodban, G. Reid and A. M. Z. Slawin, J. Chem. Soc., Dalton Trans., 1998, 2225–2232 Search PubMed.
- W. Levason, L. P. Ollivere, G. Reid, N. Tsoureas and M. Webster, J. Organomet. Chem., 2009, 694, 2299–2308 CrossRef CAS.
- X.-X. Zhao, J. Wei and Z. Xi, Chin. J. Chem., 2023, 41, 2400–2407 Search PubMed.
- A. H. Klahn-Oliva, R. D. Singer and D. Sutton, J. Am. Chem. Soc., 1986, 108, 3107 Search PubMed.
- J. J. Carbó, O. Eisenstein, C. L. Higgitt, A. H. Klahn, F. Maseras, B. Oelckers and R. N. Perutz, J. Chem. Soc., Dalton Trans., 2001, 1452–1461 Search PubMed.
- J. A. Calladine, O. Torres, M. Anstey, G. E. Ball, R. G. Bergman, J. Curley, S. B. Duckett, M. W. George, A. I. Gilson, D. J. Lawes, R. N. Perutz, X.-Z. Sun and K. P. C. Vollhardt, Chem. Sci., 2010, 1, 622–630 RSC.
- D. Sellmann, Angew. Chem., Int. Ed. Engl., 1971, 10, 919–919 CrossRef.
- Q. Le Dé, A. Bouammali, C. Bijani, L. Vendier, I. del Rosal, D. A. Valyaev, C. Dinoi and A. Simonneau, Angew. Chem., Int. Ed., 2023, 62, e202305235 CrossRef.
- C. Barrientos-Penna and D. Sutton, J. Chem. Soc., Chem. Commun., 1980, 111–112 RSC.
- F. M. Bickelhaupt and E. J. Baerends, in Rev. Comput. Chem, ed. D. B. Boyd and K. B. Lipkowitz, Wiley-VCH, New York, 2000, pp. 1–86 Search PubMed.
- T. Ziegler and A. Rauk, Inorg. Chem., 1979, 18, 1755–1759 CrossRef CAS.
- T. Ziegler and A. Rauk, Inorg. Chem., 1979, 18, 1558–1565 CrossRef CAS.
- S. M. N. V. T. Gorantla and K. C. Mondal, ACS Omega, 2021, 6, 33932–33942 CrossRef CAS PubMed.
- S. M. N. V. T. Gorantla and K. C. Mondal, ACS Omega, 2021, 6, 33389–33397 CrossRef CAS PubMed.
- K. Devi, S. M. N. V. T. Gorantla and K. C. Mondal, Eur. J. Inorg. Chem., 2022, 2022, e202100931 CrossRef CAS.
- S. M. N. V. T. Gorantla, H. S. Karnamkkott, S. Arumugam, S. Mondal and K. C. Mondal, J. Comput. Chem., 2023, 44, 43–60 CrossRef CAS PubMed.
- H. S. Karnamkkott, S. M. N. V. T. Gorantla and K. C. Mondal, J. Chem. Sci., 2024, 136, 55 CrossRef CAS.
- P. W. Smith and T. D. Tilley, J. Am. Chem. Soc., 2018, 140, 3880–3883 CrossRef CAS PubMed.
- X. Yang and C. Wang, Angew. Chem., Int. Ed., 2018, 57, 923–928 CrossRef CAS PubMed.
- G. M. Bancroft, H. C. Clark, R. G. Kidd, A. T. Rake and H. G. Spinney, Inorg. Chem., 1973, 12, 728 CrossRef CAS.
- F. Calderazzo, E. A. C. Lucken and D. F. Williams, J. Chem. Soc. A, 1967, 154 RSC.
- M. K. Davies, M. C. Durrant, W. Levason, G. Reid and R. L. Richards, J. Chem. Soc., Dalton Trans., 1999, 1077–1084 RSC.
- J. Connolly, A. R. J. Genge, W. Levason, S. D. Orchard, S. J. A. Pope and G. Reid, J. Chem. Soc., Dalton Trans., 1999, 2343–2352 RSC.
- B. J. Burger and J. E. Bercaw, in Experimental Organometallic Chemistry - A Practicum in Synthesis and Characterization, American Chemical Society, Washington, D.C., 1987, vol. 357, pp. 79–98 Search PubMed.
- K. V. Zaitsev, I. P. Gloriozov, Y. F. Oprunenko, E. K. Lermontova and A. V. Churakov, J. Organomet. Chem., 2019, 897, 217–227 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 Search PubMed.
- ADF 2020.102, SCM, Theoretical Chemistry,Vrije Universiteit, Amsterdam, The Netherlands, https://www.scm.com Search PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. Van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 Search PubMed.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 Search PubMed.
- E. van Lenthe, A. Ehlers and E.-J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 Search PubMed.
- E. van Lenthe, J. G. Snijders and E. J. Baerends, J. Chem. Phys., 1996, 105, 6505–6516 Search PubMed.
- E. van Lenthe, R. van Leeuwen, E. J. Baerends and J. G. Snijders, Int. J. Quantum Chem., 1996, 57, 281–293 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1988, 88, 2547–2553 Search PubMed.
- M. Franchini, P. H. T. Philipsen and L. Visscher, J. Comput. Chem., 2013, 34, 1819–1827 Search PubMed.
- A. Bérces, R. M. Dickson, L. Y. Fan, H. Jacobsen, D. Swerhone and T. Ziegler, Comput. Phys. Commun., 1997, 100, 247–262 Search PubMed.
- H. Jacobsen, A. Bérces, D. P. Swerhone and T. Ziegler, Comput. Phys. Commun., 1997, 100, 263–276 Search PubMed.
- S. K. Wolff, Int. J. Quantum Chem., 2005, 104, 645–659 Search PubMed.
- T. Ziegler and A. Rauk, Inorg. Chem., 1979, 18, 1755–1759 CrossRef CAS.
- T. Ziegler and A. Rauk, Inorg. Chem., 1979, 18, 1558–1565 CrossRef CAS.
- M. Mitoraj and A. Michalak, Organometallics, 2007, 26, 6576–6580 Search PubMed.
- A. Michalak, M. Mitoraj and T. Ziegler, J. Phys. Chem. A, 2008, 112, 1933–1939 CrossRef CAS PubMed.
- M. P. Mitoraj, A. Michalak and T. Ziegler, Organometallics, 2009, 28, 3727–3733 CrossRef CAS.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS PubMed.
- C. J. Jameson, in eMagRes, ed. R. K. Harris and R. L. Wasylishen, John Wiley & Sons, Ltd., 2007 Search PubMed.
- A. J. Jordan, C. M. Wyss, J. Bacsa and J. P. Sadighi, Organometallics, 2016, 35, 613–616 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Overview of literature Mn(I) dinitrogen chemistry, selected NMR and IR spectra, SCD and PXRD data, and DFT results. CCDC 2447494–2447503. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt01025j |
| ‡ The reaction mixture formed from n-butyl isonitrile and 1a contained a significant unidentified byproduct comprising ∼30% of the mixture, which featured four 31P{1H} NMR environments and a very high frequency 1H NMR signal (consistent with a CH(NR)135 or GeHR29 ligand). Removal of the solvent in vacuo followed by washing with hexanes and recrystallization from toluene layered with hexanes at −30 °C resulted in isolation (though not with analytical purity) of cis-3c. Selected NMR data for the unidentified impurity are as follows: 1H NMR (C6D6, 600 MHz, 298 K, integrals normalized to 1H for the peak at 11.60 ppm): δ 11.60 (d of d, 1H, JH,P 9.3 and JH,P 3.6 Hz), 7.89, 7.79 (2 × t, 2H, JH,H 7.1 Hz), 3.78, 3.59 (2 × m, 1H), 1.46 (d, 3H, JH,P 4.8 Hz), 1.33 (d, 3H, JH,P 5.1 Hz), 1.29 (d, 3H, JH,P 6.4 Hz), 1.10 (d, 3H, JH,P 5.9 Hz), 0.82 (d, 3H, JH,P 4.6 Hz). 13C{1H} NMR (C6D6, 151 MHz, 298 K): δ 159.42, 158.79, 134.23, 133.69, 127.78, 127.69, 126.32, 125.95, 58.58 (9 × s), 35.79 (app. t, JC,P 22.1 Hz), 34.85 (d, JC,P 13.4 Hz), 33.88, 20.29 (2 × m), 26.14 (d, JC,P 9.1 Hz), 25.74 (d of d, JC,P 12.8 and 6.9 Hz), 19.16 (d, JC,P 7.4 Hz). 31P{1H} NMR (C6D6, 243 MHz, 298 K): δ 80.84, 72.74, 72.37, 62.64 (4 × s, 1P). |
| § The 31P NMR signal for transHGe-[(dmpe)2MnD(DGeHPh2)] (d2-transHGe-2a) is shifted 0.4 ppm to higher frequency than in the protio isotopomer transHGe-2a. This is relatively unusual, since secondary NMR isotopic shifts usually proceed to lower frequency upon substitution of a nearby atom with a heavier isotope.134 The average 31P NMR environment observed at 298 K for all isomers of 2a, as well as 31P NMR signals for the minor species in solution, are too broad for a similarly small secondary NMR isotopic shift to be measured. |
| ¶ The X-ray crystal structure of 3b contains two independent and essentially isostructural molecules in the unit cell. One of these has disorder in the dmpe ligands which could not be completely modelled from the difference map. Therefore, only bond metrics for 3b from the molecule without disorder are included in the discussion and in Table S11.† |
| || The 55Mn{1H} NMR chemical shifts for cis-5a, trans-5a, cis-5b and trans-5b increase significantly as the temperature is increased, from −812, −991, −1070, and −1168 ppm, respectively, at 223 K, to −601, −730, −906, and −982 ppm, respectively, at 370 K. Below 272 K, the quintet arising from trans-5b begins to broaden significantly due to quadrupolar collapse. At elevated temperature, the 55Mn{1H} NMR spectra of 5a–b also contain a peak arising from [(dmpe)2MnH(GeR2)] (1a–b), which is in equilibrium with 5a–b due to reversible N2 dissociation. At 370 K, the 55Mn{1H} NMR signals arising from 1a–b are broad singlets at −1107 ppm for 1a and −1226 ppm for 1b (at 298 K, the signals are shifted to −1176 and −1303 ppm, respectively). |
| ** IR spectroscopy νNN peaks for the two isomers of 5b overlap, and values reported here were obtained by peak fitting using Gaussian peak shapes and a quadratic baseline correction, which resulted in the lowest standard error and F statistic relative to alternative peak fitting methods using any combination of Gaussian, Voigt, or Gaussian/Lorentzian peak shapes, and zero, constant, linear, or quadratic background corrections (in all cases, νNN ranged from 1988–1989 cm−1 for the cis isomer and 1959–1968 cm−1 for the trans isomer). |
| †† This NMR environment was located using 2D NMR spectroscopy, and thus no integration and/or environment splitting information is provided. |
| ‡‡ This NMR signal could not be integrated because of overlap with a residual solvent signal. |
| §§ The p-Ph 13C NMR signals for the cis and trans isomers are too close in chemical shift to determine which signals correspond to which isomer. |
| This journal is © The Royal Society of Chemistry 2025 |