
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Layered anion ordering in oxide chloride Ca2TlO3Cl†
Lars Botolv Ekeli Bråtalien a,
Ola Nilsena,
Dipankar Sahaa,
Chris Erik Mohna,
Athanasios Chatzitakisb and
Martin Valldor*a
a,
Ola Nilsena,
Dipankar Sahaa,
Chris Erik Mohna,
Athanasios Chatzitakisb and
Martin Valldor*a
aDepartment of Chemistry, Centre for Materials Science and Nanotechnology, University of Oslo, Sem Sælands vei 26, NO-0371 Oslo, Norway. E-mail: b.m.valldor@kjemi.uio.no
bDepartment of Chemistry, Centre for Materials Science and Nanotechnology, University of Oslo, Gaustadalléen 21, NO-0349 Oslo, Norway
First published on 7th July 2025
Abstract
Single crystals and polycrystals of the novel compound Ca2TlO3Cl can be obtained by gas-phase and solid-state reactions, respectively. The title compound's crystal structure is hexagonal (a = 3.8806(2) Å, c = 18.6168(7) Å, P63/mmc) as measured by single crystal X-ray diffraction. A Rietveld refinement on powder X-ray diffraction data confirms this crystal structure. The atomic arrangement can be described as a cation ordered anti-structure of ScAl3C3, with a homoleptically, five-fold oxygen coordinated Tl3+ that consitutes corner-sharing trigonal bipyramids. Between these Tl–O sheets, a Cl-layer is found, making the whole structure quasi-2D. Thermal expansion coefficients in the range 15–290 K, as estimated from X-ray powder diffraction data, reveal that the relative length of the crystallographic c-axis (α33) is more temperature dependent than that of the a-axis (α11) lengths. The brown color of the polycrystalline title compound was investigated by spectroscopic reflectometry measurements. These data suggest an optical band gap of either 1.4 eV (indirect) or about 2.4–2.7 eV (direct), depending on interpretation model. However, density funciton theory calculations deliver a band structure that indicates that indirect band gaps are most likely, but with a size of about 2.7 eV. This discrepancy is discussed as a result of further local excitation phenomena, like charge-transfers and crystal defects. Photo-electrochemical investigations suggest that the title compound has an n-type character and is electrochemically activated by solar light.
1. Introduction
A promising and relatively recent approach to designing inorganic crystalline materials involves exploring chemistries based on combining two simple anions.1–3 One aspect of the design is the crystal structure dimensionality, which might be connected to the stoichiometry ratio, R, between the two anions. For oxide chlorides, a 3D lattice is observed in anti-perovskites like Li3OCl (R = 1),4 where O2− and Cl− are arranged in a CsCl-type manner. However, 2D alternating layers of the two anions are found in Sr2CuO2Cl2 (R = 1)5 and in Sr2CuO2Cl (R = 2).6 In contrast, a 1D Cl-lattice (columns) is observed in La3WO6Cl3 (R = 2),7 and a 0D case with sole O2− in a Cl-matrix is displayed in Ca4OCl6 (R = 1/6).8 This suggests that the design of anion dimensionality depends on more parameters, which are still unclear. In Sr2Te4V2O13Cl2 (R = 6.5),9 the Cl− ions form a layer with four-fold rotational symmetry that seems to be rigid enough to enforce a pseudo-tetragonal symmetry despite the orthorhombic space group symmetry for the whole lattice. Evidently, more oxide chloride chemistry is needed to expand the experimental crystal structure basis before conclusions can be drawn of how to design anion lattice dimensionality; it has attracted interest because of the general assumption that the lattice dimensionality has great impact on electron conductivity and optical properties. Hence, making a route to control lattice dimensionality is desirable when trying to replace transparent semiconductors like indium-tin-oxides (ITO)10 in display applications.During explorative syntheses in the Ba–Ca–Tl–O–Cl system, hexagonally shaped single crystals of a novel compound were discovered. Here, we report the synthesis, the crystal structure, and fundamental properties of Ca2TlO3Cl.
2. Experimental
2.1. Synthesis
All synthesis preparations were performed inside an argon filled glovebox (O2 and H2O < 0.1 ppm) because several of the constituents are air sensitive.Sample 1: The prime single crystals of the title compound were found by reacting two powders that were spatially separated inside a silica ampoule: one powder, consisting of a homogenized powder mixture of BaO2 (84% min. Thermo Sci. Chem.) and CaCl2 (4N Thermo Fisher Sci.) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry was placed in a corundum crucible, while the other powder (2 relative moles of TlCl, 5N ABCR) was placed outside the crucible at the bottom of a silica ampoule. The sample filled ampoule was plugged by a septum before lowering the inner Ar pressure to ∼0.25 bar using a syringe. Subsequently, the ampoule was sealed with a hydrogen/oxygen torch and placed in a muffle furnace. The sample was heated up during 10 h to 750 °C, where it was soaking for 10 hours before cooling to 550 °C over 40 hours and finally set to room temperature, which was reached in about an hour. Note, that BaO2 decomposes and forms O2 gas under the synthesis conditions, which limits the amount of sample per ampoule volume to avoid any risks for ampoule breakage.
:1 stoichiometry was placed in a corundum crucible, while the other powder (2 relative moles of TlCl, 5N ABCR) was placed outside the crucible at the bottom of a silica ampoule. The sample filled ampoule was plugged by a septum before lowering the inner Ar pressure to ∼0.25 bar using a syringe. Subsequently, the ampoule was sealed with a hydrogen/oxygen torch and placed in a muffle furnace. The sample was heated up during 10 h to 750 °C, where it was soaking for 10 hours before cooling to 550 °C over 40 hours and finally set to room temperature, which was reached in about an hour. Note, that BaO2 decomposes and forms O2 gas under the synthesis conditions, which limits the amount of sample per ampoule volume to avoid any risks for ampoule breakage.
Sample 2: To synthesise the title compound in relatively pure form, a stoichiometric (1:3:1) powder consisting of Tl2O3 (96% Thermo Fisher Sci.), CaO (in-house decomposition at 900 °C overnight of CaCO3 99% Merck), and CaCl2 was homogenized using an agate mortar and placed directly in the silica ampoule, before sealing it as mentioned above. However, the temperature programs were altered: the sample was heated up to 500 °C over 10 hours and soaked for 10 hours before cooling it down to room temperature over approximately 40 hours. There was no visible reaction between CaO and the silica tube at the chosen reaction temperature.
Note, Tl compounds, especially Tl+ containing ones, are hazardous for humans, because this ion resembles Na+ and K+ in metabolisms. To avoid any contamination of the lab, these high temperature reactions should be performed in closed ampoules and in fume-hoods.
2.2. X-ray single crystal diffraction
Single crystals were selected from Sample 1 under an optical microscope and mounted onto cryo-loop (MiTiGen) holder using highly viscous oil (NVHO-1). The specimen was investigated at room-temperature with a Bruker D8 VENTURE single-crystal diffractometer with a MoKα Incoatec microfocus X-ray source using a PHOTON 100 detector. The JANA2020 software11 was used to find an ab initio structure model (superflip), and, subsequently, refine it against observed data intensities.2.3. X-ray powder diffraction
For the Rietveld refinement investigation, the finely crushed powder from Sample 2 was dispersed in isopropanol and further ground using an agate mortar and pestle to form a thick slurry. This slurry was drop-cast onto a low-background silicon crystal sample holder and allowed to air dry. Care was taken to ensure the sample surface was flat to minimize errors due to sample height. Data were collected using a Bruker D8 Advance diffractometer in Bragg–Brentano geometry, utilizing a rotating anode Cu radiation source and an XE-T detector with energy filtering technique for fluorescence suppression. The X-ray tube was operated at 45 kV 120 mA without a monochromator. Data acquisition covered a 2θ range of 5° to 120°, with a step size of 0.01° and a dwell time of 0.3 seconds per step. The sample holder was continuously rotated during PXRD data acquisition to improve particle statistics and minimize preferred orientation effects. Phases were identified using Diffract.EVA V7.3 equipped with ICDD data base PDF4+:2025 for PXRD. Rietveld refinement, including a quantification of the identified phases, were carried out with software package TOPAS 7.12 Fundamental parameters were used to describe instrumental broadening. A starting model derived from the single-crystal structure solution was used as the initial input for the Rietveld refinement. A Chebyshev polynomial of 5th order was used to fit the background. Symmetry-allowed atomic coordinates, thermal parameters, isotropic strain, and crystallite sizes were refined during the process. The occupancies of Tl, Ca, and Cl were refined independently to verify the validity of the model. The resulting values were close to 1 and were therefore fixed to 1 during the refinement. Effects from preferred orientations were tried during the refinement, but they did not improve the results. Hence, preferred orientation was assumed to be insignificant in this case.Low temperature X-ray powder diffraction data were collected using a Huber G670 Guinier camera equipped with a Germanium monochromator and an imaging plate detector, operating in transmission geometry with Cu Kα1 radiation (λ = 1.54059 Å). A thin layer of the Sample 2 was placed between two Mylar films and clamped onto a sample holder designed for the cryogenic setup on the diffractometer. The sample was cooled to 15 K using a closed-loop helium cryojet, and the temperature was stabilized at 15 K for one hour before measurements. Variable-temperature PXRD data were collected at 5 K intervals during the heating cycle up to 290 K, with an exposure time of 5 minutes and a dwell time of 5 minutes at each temperature. The same measurement parameters were applied during the cooling cycle. Instrumental calibration was performed using an NIST LaB6 standard, and the corresponding parameter file was employed for Rietveld refinement. For structural analysis, the model derived from the single-crystal structure solution was used as the initial input for Rietveld refinement against the 15 K dataset using software package of TOPAS. This refined model was subsequently applied to surface Rietveld refinement of all collected datasets.
2.4. Scanning electron microscopy
The EDX data and SEM images were collected in a Hitachi SU8230 SEM. An aluminium SEM pin sample holder was taped with a carbon tape, sample was dropped onto the tape and excess powder was knocked off before mounting the pin and inserting it into the SEM. For images the working distance was 8 mm. To capture images a method of fast scan picture integration was used to avoid possible charge build up that could blur or put streaking the picture. EDX was performed at a working distance of 15 mm and acceleration voltage of 15 kV.2.5. UV-vis spectroscopy
UV-Vis absorbance data was acquired from diffuse reflectance measurements using the Kubelka–Munk method,13 which was measured using a Flame-S spectrometer from Ocean optics, equipped with a HD-2000 combined halogen lamp and deuterium lamp from Mikropack and optical fibers. The samples consisted of lightly pressed powder and acquisition was performed with the incident light normal to the sample surface and the detector at a 45° angle. Lightly pressed BaSO4 was used as white reference.2.6. Computational details
Ab initio calculations of the band structure and optical properties of Ca2TlO3Cl were carried out at the Perdew–Burke–Enterzhof (PBE), the Modified Becke–Johnson potential (MBJ),14 the Heud–Scurzia–Erntzerhof (HSE06)15 and quasiparticle (GW)16,17 levels of theory using projected augmented waves as implemented in the VASP program.18,19 The 5d106s26p1, 2s22p63s2, 3s24p5 and 2s24p5 electrons were incorporated in as valence shell for Tl, Ca, Cl and O respectively. Structural optimisations were carried out using HSE0615 in which 25% of the PBE exchange is replaced by the exact Hartree–Fock exchange energy. The PBE exchange term is divided into long- and short-range contributions defined by a screening parameter, ω = 0.2 Å−1. The band structures were also calculated at the quasiparticle GW level of theory in a single shot using the self-consistent G0W0 approximation.17,18 Motivated by previous benchmark studies showing that the meta-GGA MBJ predicts band gaps with similar – or often better – accuracy than HSE06,20 we also report band gaps calculated using MBJ. The structural optimisations were carried out using a 9 × 9 × 1 gamma centred k-mesh to sample the 1st Brillouin zone and an energy cut-off of 500 eV. The structure (i.e. volume, cell-shape and basic atom positions) were fully relaxed structure and then reoptimized to ensure that all forces were converged to less than 0.005 eV Å−1. Additional test calculations carried out by increasing the k-mesh to 9 × 9 × 3 and the energy cutoff to 700 eV had negligible effect on the calculated structural properties. Structural parameters, reported in ESI (S1),† are in excellent agreement with those found experimentally. Band structure calculations using MBJ, HSE06, and G0W0 were also carried out using an energy cut of 500 eV, a 9 × 9 × 1 mesh with a density of 0.04 Å−1 along the high-symmetry points in the 1st Brillouin zone. The band gaps are also calculated at the quasiparticle GW level of theory in a single shot using the self-consistent G0W0 approximation.16,17 Optical properties such as absorption coefficients are calculated from the frequency dependent dielectric matrix as implemented in VASP. The calculated band gaps together with the location of the critical points are tabulated in the ESI (S2),† along with the calculated absorption coefficients (S3).†2.7. Photo-electrochemical measurements
The working electrode (WE) consisting of the oxychloride Ca2TlO3Cl was prepared by firstly making an ink, which was drop-casted on FTO glass (Ossila, TEC 10 FTO Coated Glass, unpatterned, 20 × 15 × 1.0 mm3) covering an area of approx. 1 cm2. To prepare the ink, 50 mg of powder were added to 1.25 mL water, 0.65 mL isopropanol, and 15 μL of 5 wt% Nafion. The dispersion was sonicated for at least 30 minutes to obtain a homogeneous ink. Then, 50 μL of ink were applied on the FTO glass, yielding a visually homogenous film of Ca2TlO3Cl. The electrode was left to dry in ambient air and used as received in a three-electrode photo-electrochemical (PEC) cell. A saturated calomel electrode (SCE) and a Pt wire were used as reference and counter electrodes, respectively. The supporting electrolyte in all PEC tests was 0.5 M Na2SO4. All PEC measurements were performed in an Ivium Vertex potentiostat/galvanostat, under 1 sun simulated solar light (Newport Oriel® LCS-100 solar simulator equipped with a 100 W ozone-free xenon lamp and an AM 1.5G filter). A Si PV reference cell (Newport 91150V-KG5) was used to calibrate the light intensity at 100 mW cm−2 (1 sun). All potentials were corrected against the reversible hydrogen electrode (RHE) considering that water electrolysis takes place thermodynamically at 1.23 V vs. RHE. The potentials vs. the SCE were corrected vs. RHE according to the Nernst equation (1):| ERHE = ESCE + 0.059 × pH + 0.242 V, | (1) |
3. Results and discussion
3.1. Material
From the initial single crystal growth experiment (Sample 1), two different crystals were observed: agglomerates of brown needles of Ca2Tl2O521 alongside with hexagonal, thick plates of light brown transparent appearance that proved to be the novel title compound, Ca2TlO3Cl. Note that larger crystals of the title compound were formed when reacting two separate powders, probably meaning that the crystal growth involves gaseous species. All crystals were formed where BaO2 and CaCl2 initially were placed – here, within the crucible – so, TlCl was transported into the crucible during heating. It is also interesting to observed that both major products contain Tl3+ although TlCl was used as starting material. This implies that BaO2 is sufficiently strong as oxidizing agent (in a closed ampoule) to raise the oxidation state of thallium from +1 to +3, which could be useful for future reference.22
The resulting powder of Sample 2 had dominantly a cinnamon like appearance, but with minor impurity stains of white. When trying different reaction conditions, higher temperatures up to 750 °C resulted in increasing amounts of white, secondary phase (TlCl) covering the ampoule walls. This decomposition at higher temperatures is believed to follow reaction (2), producing CaO alongside TlCl and oxygen gas in the ampoule.
| Ca2TlO3Cl(s) → 2CaO(s) + TlCl(s) + ½O2 (g) | (2) |
Both Sc3+ and In3+ were tried instead of Tl3+ under the same reaction condition as for Sample 2, but these chemical substitutions were not successful.
3.2. Crystal structure of Ca2TlO3Cl
The crystal structure of the title compound can be described on basis of anion close packing (Fig. 1): three oxygen layers are arranged in ABA fashion followed by a C layers of chloride ions, while the next trilayer of oxide ions is BAB with a subsequent C layer of chloride ions, again. This means that this structure can be described as a mixture of cubic and hexagonal close packed layers, despite having two differently sized anions. Note that the chloride ions are found in layers separating the oxide sheets, making the global structure into a 2D material by anion ordering. Ca2+ is found on a singular Wyckoff site (4f) with a heteroleptic, 7 coordination to 4 oxide ions (3 × 2.29 Å + 1 × 2.64 Å) on one side and 3 chloride ions (all 3.02 Å) on the opposite, as compared to the Ca–O distance of 2.40 Å in CaO23 and the Ca–Cl distances in CaCl2 in the range 2.70–2.77 Å.24 Obviously, the heteroleptic coordination allows for shorter Ca–O and longer Ca–Cl distances, as compared to the homoleptic coordination. This is probably due to the different charges of O2− and Cl−, where the former results in stronger electrostatic attraction. For future research, the coordination of the Ca site in this compound, which can be described as a distorted, monocapped fac-octahedron, might be interesting for specific doping with smaller rare-earth elements. | ||
| Fig. 1 The crystal structures of the title compound (left), as compared to ScAl3C3 (right) to emphasize their similarities. The unit cells are indicated by thin, black lines, and the Ca coordination is indicated by interatomic connections. | ||
In contrast, Tl3+ is situated in the middle of the oxide sheets, half-ways between two tetrahedral voids, making the coordination homoleptic to oxide ions (3 × 2.24 Å + 2 × 2.14 Å). In Tl2O3, Tl3+ is six-fold coordinated and the corresponding distances range from 2.14 to 2.47 Å,25 making it obvious that the Tl-coordination in the title compound is close to expectancy, but the distances are a bit shorter on average because of the lower coordination number of 5.
The structure of Ca2TlO3Cl can be described as a cation ordered anti-structure of ScAl3C3,26 where Ca2+ and Tl3+ in the former occupy the C4− sites in the latter (Fig. 1). Note, that the carbide ions in ScAl3C3 occupy two Wyckoff positions, thus allowing for a perfect ordering of the cations in the title compound, despite having the same space group (P63/mmc). The data from the single crystal experiment and refinement can be found in Table 1.
| Chemical formula | Ca2TlO3Cl |
| Form. weight (g mol−1) | 367.99 |
| T (K) | 293 |
| λ (Å) | 0.71073 |
| Crystal system | Hexagonal |
| Space group | P63/mmc (no. 194) |
| a (Å) | 3.8806(2) |
| c (Å) | 18.6168(7) |
| V (Å3) | 242.79(2) |
| Z | 2 |
| ρ (g cm−3) | 5.0336 |
| R(obs), R(all) | 0.0163, 0.0211 |
| Rw(obs), Rw(all) | 0.0450, 0.0460 |
| GOF(obs), GOF(all) | 1.7900, 1.8069 |
| hole, peak (e Å−3) | 3.80, −3.31 |
| index ranges | −5 ≤ h ≤ 7, −6 ≤ k ≤ 6, −32 ≤ l ≤ 31 |
| Site, Wyckoff, x, y, z, U11, U22, U33, U12, U13 = U23 = 0 | Tl1, 2c, 2/3, 1/3, 3/4, 0.00704(9), 0.00704(9), 0.0057(1), 0.00352(4) |
| Ca1, 4f, 1/3, 2/3, 0.6084(2), 0.0071(2), 0.0071(2), 0.0116(4), 0.0036(1) | |
| Cl1, 2a, 0, 0, 1/2, 0.0149(4), 0.0149(4), 0.0104(6), 0.0075(2) | |
| O1, 4f, 2/3, 1/3, 0.6353(0), 0.011(1), 0.011(1), 0.007(1), 0.0055(5) | |
| O2, 2d, 1/3, 2/3, 3/4, 0.005(1), 0.005(1), 0.016(3), 0.0027(6) | |
| CSD | 2446417 |
3.3. Rietveld refinement on Ca2TlO3Cl, Sample 2
Powder X-ray diffraction data of Sample 2 were subject to Rietveld refinement, and the result is shown in Fig. 2. The title compound dominates the Bragg scattering intensities. However, minor secondary phases are present, identified as 1.4(2) vol% CaO and 1.0(3) vol% TlCl, which can be explained by a minor decomposition according to reaction (2) above. One unidentified intensity, marked with * in Fig. 2, is also observed, but its origin remains unclear. The refined parameters from the Rietveld simulation can be found in the ESI (S4).† As the sample purity is above 97% further investigations were carried out, as presented below. From the Rietveld refinement we extract the unit cell parameters a = 3.88036(5) Å and c = 18.6466(4) Å, which are minimally different than the single crystal measurement. Minor differences are expected when using two different diffraction techniques and reflecting on that the powder data come from an average of many crystals as compared to the single crystal specimen.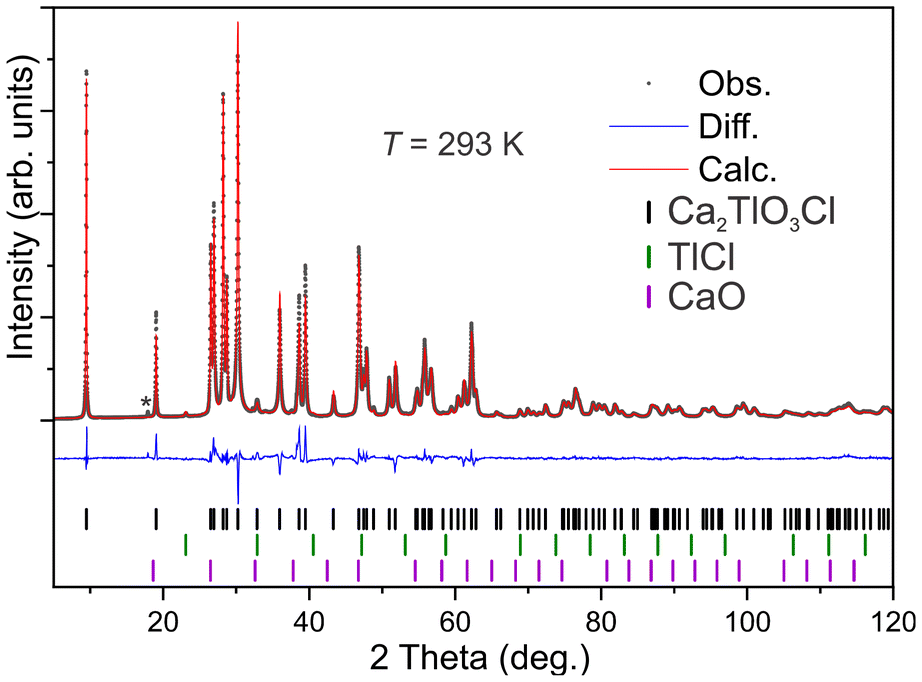 | ||
| Fig. 2 Rietveld refinement of Ca2TlO3Cl, indicating two secondary phases and an unknown intensity (*). The wavelengths were Cu Kalpha1,2 (λ = 1.54056, 1.54439 Å). | ||
3.4. Thermal expansion of Ca2TlO3Cl, Sample 2
The powder diffraction data of Sample 2 were obtained from about 15 K up to room-temperature to extract the thermal expansion coefficients. A flood-schematics with all data is shown in the ESI (S5).† From the obtained data, it is obvious that there is no structural change, and the hexagonal unit cell persists down to lowest measured temperature. However, there seems to be two different ranges of thermal expansion, at either side of 115 K.The observed thermal expansions are within expectancy for insulating ceramics. However, there is a clear anisotropy in the expansion coefficients: below 115 K, α33 is almost a magnitude larger than α11 (Table 2). A possible explanation is that the bonds within the ab-plane are relatively more rigid as compared to those between the same planes. When considering the crystal structure, it is tempting to assume that the Tl–O layers represent the stiffest structural unit, while the Ca–Cl sublattice is less strongly bound together, agreeing with intuition when comparing the electrostatic forces: Tl is +3 and O is −2, giving a stronger attraction, relative to interactions between Ca (+2) and Cl (−1).
| T range [K] | α11 [K−1] | α33 [K−1] |
|---|---|---|
| 15–115 | 7.47 × 10−7 | 7.86 × 10−6 |
| 115–290 | 7.99 × 10−6 | 1.71 × 10−5 |
3.5. Scanning electron microscopy on Ca2TlO3Cl, Sample 2
The crystal morphology was dominantly hexagonal plates, as shown in Fig. 4. The plates in Sample 2 had sizes of up to 1–5 μm, but plate thickness varied considerably. As an average of about 40 elemental analyses on different crystallites, the Ca-normalized composition was estimated to Ca2.0(2)Tl1.1(1)Cl0.8(1)Ox, where oxygen was disregarded due to its strong absorption dependence as most of the examined crystallites had non-horizonal surface alignments. Hence, considering the expected, poor conductivity of the title compound, the elemental analysis is within measurement errors of theoretical starting composition. | ||
| Fig. 3 Lattice parameters of Ca2TlO3Cl from temperature dependent powder diffraction data. Standard deviations are smaller than the markers. The red lines indicate linear fits, given in Table 1 text. | ||
 | ||
| Fig. 4 Scanning electron microscopy photo of a hexagonal crystal in a polycrystalline sample of Ca2TlO3Cl. The scale bar is added for size comparison. | ||
3.6. UV-vis spectroscopy on Ca2TlO3Cl, Sample 2
The diffuse reflectance of the sample shows a steadily increasing absorption from long to short wavelengths all over the visible spectrum, until about 300 nm where it becomes transparent. The raw data are shown in the ESI (S6).† The wide range in absorption over the visible spectrum indicates a more complex optical absorption process than what can be explained by band gaps alone. Hence, we expect the optical properties to be influenced by defects, colour centres, local charge transfers (O to Tl) or others in addition to its inherent band gap. To obtain more insight into possible optical transitions the reflectance spectra were converted to absorption using the Kubelka–Munk method. Subsequently, this was used as basis for plotting the optical data for direct and indirect transitions, Fig. 5. The plots are not directly conclusive, but this method results in possible direct band gaps of 2.4 or 2.7 eV, or indirect band gap of 1.4 eV. Only judging from these plots, the visibly most convincing one is the indirect band gap. However, these estimated band gaps are all notably different than what obtained from DFT calculations, supporting that other effects than band gaps are influencing the optical properties of this material. | ||
| Fig. 5 Plots of reflection spectroscopy data of Ca2TlO3Cl, calculated as indirect band gap. The inset shows the same data calculated as direct band gap(s). | ||
3.7. Density functional theory calculations
In the ESI (S2),† the calculated band gaps using PBE, MBJ, HSE06 and G0W0 are presented. The band gap calculated using either MBJ or HSE06 is 2.7 eV in agreement with that calculated at the quasiparticle G0W0 method (2.8 eV) suggesting that the PBE bandgap of about 1.7 eV is severely underestimated consistent with previous extensive benchmark tests.27 The band structure shown in Fig. 6 shows that the fundamental band gap is indirect with a conduction band minimum at Γ and nearly degenerate valence-band maxima at K and H. The density of states shows that the top of the valence bands is mainly a mix of O 2p and Tl 5d states whereas the antibonding hybridisation of the lowest conduction bands at 2.7 to 6 eV are dominated by O 2p and Tl 6s states. | ||
| Fig. 6 (a) Band-structure of Ca2TlO3Cl, as estimated by HSE06, and (b) the density of states resolved in elemental contributions. | ||
The fundamental gaps calculated using hybrid HSE06 and quasiparticle G0W0 suggests that Ca2TlO3Cl can absorb visible light but the predicted fundamental gaps are apparently inconsistent with the sample colour and optical gaps predicted from the Tauc plots (Fig. 5). Formation of defect states in the mid-gap range (such as colour centres frequently observed in many halides) and/or possible local charge-transfer between Tl and O can possibly explain a broader absorption of visible light. Larger dispersion near the conduction band minimum at Γ in the band-structure of Ca2TlO3Cl points to an intrinsic n-type conductor with lower effective electron masses than effective hole masses.
3.8. Photo-electrochemical properties on Ca2TlO3Cl, Sample 2
To evaluate the semiconducting nature of Ca2TlO3Cl, a series of PEC measurements were conducted. A straightforward, as well as indicative measurement is the open circuit potential (OCP), which is shown in Fig. 7. We observe that upon light exposure the OCP shifts towards more negative potentials, indicating an n-type character of the material. The range of the potential shift is around 10–20 mV, implying a limited band bending under the studied conditions (0.5 M Na2SO4(aq)). Therefore, we expect limited photocurrent production under potentiodynamic conditions, as it is seen in Fig. 8 and the linear sweep voltammograms (LSVs) therein. The inset of Fig. 8 shows the photocurrent response, which is in the range of 0.5 μA cm−2, of Ca2TlO3Cl under chopped illumination (1 sun) in the potential window from 0.80 V vs. RHE to 0.95 V vs. RHE. Striking is the large cathodic currents observed at potentials lower that 0.80 V vs. RHE. In this region the material is in the accumulation region (further correlating with an n-type character) and these significant dark and reductive currents may partly originate from the reduction of Tl3+ to Tl+. | ||
| Fig. 7 Open-circuit-voltage measurement of Ca2TlO3Cl under dark and solar simulated conditions. | ||
 | ||
| Fig. 8 LSV curves in the dark (black line) and under illumination (orange line) in the potential region from approx. 0.70 V vs. RHE to 0.95 V vs. RHE. Scan rate 10 mV s−1 in 0.5 M Na2SO4. Inset: Close up of the selected potential region. | ||
Steady state photocurrent densities are given in the chronoamperometric curves of Fig. 9. Again, the photocurrent range of the order of 0.5 μA cm−2 that is increasing with increasing potential, followed by an increase in the dark/background current too. The latter correlates well with the electroactive behavior observed at more cathodic potentials and it could be possibly attributed to the redox behavior of Tl. This is again important for future studies, when redox-active cations are desired.
 | ||
| Fig. 9 Chronoamperometry at three different applied potentials (0.85, 0.09 and 0.95 V vs. RHE) under chopped illumination in 0.5 M Na2SO4. Inset: Overall response indicating the large cathodic currents at more negative potentials (0.85 V vs. RHE) and significant anodic spikes upon shifting of the applied potentials at more positive values. | ||
4. Conclusions
Single crystals of the novel compound Ca2TlO3Cl can be grown via a gas phase transport reaction between BaO2, CaO and TlCl. A relatively pure sample of the same compound can be synthesized through a solid-state reaction in closed ampoules using CaO, Tl2O3 and CaCl2 as starting materials. The crystal structure of Ca2TlO3Cl can be described as an anti-structure of ScAl3C3 and its lattice is layered (2D) by the ordering of anions. The light absorption in the visible range is probably due to an indirect band gap of about 1.4 eV but could also be affected by local charge-transfer phenomena and crystal defects. Exposure to simulated sun light activates the material and reveals that it is (dominantly) an n-type semiconductor.Conflicts of interest
The authors declare no conflict of interests.Data availability
Crystallographic data for the subject compound has been deposited at the CCDC repository under ID 2446417† and can be obtained from https://www.ccdc.cam.ac.uk/structures/.Additional data supporting this article have been included as part of the ESI.†
Raw data that were used in this report can be obtained by requesting them through an email to the corresponding author.
Acknowledgements
The atomistic simulations were performed on resources provided by Sigma2 – the National Infrastructure for High-Performance Computing and Data Storage in Norway (grant number NN2916K). Single crystal and PXRD Data collection at RECX, the Norwegian Center for X-ray diffraction and scattering (Research Council of Norway, project 208896) is acknowledged.References
- M. Valldor, Inorganics, 2016, 4, 23 CrossRef.
- J. K. Harada, N. Charles, K. R. Poeppelmeier and J. M. Rondinelli, Adv. Mater., 2019, 31, 1805295 CrossRef PubMed.
- H. Kageyama, K. Hayashi, K. Maeda, J. P. Attfield, Z. Hiroi, J. M. Rondinelli and K. R. Poeppelmeier, Nat. Commun., 2018, 9, 772 CrossRef PubMed.
- J. Zhang, J. Han, J. Zhu, Z. Lin, M. H. Braga, L. L. Daemen, L. Wang and Y. Zhao, Inorg. Chem. Commun., 2014, 48, 140–143 CrossRef CAS.
- L. L. Miller, X. L. Wang, S. X. Wang, C. Stassis, D. C. Johnston, J. Faber and C. K. Loong, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 1921–1925 CrossRef CAS PubMed.
- H. Müller-Buschbaum and W. Leib, Monatsh. Chem., 1985, 116, 1–5 CrossRef.
- L. H. Brixner, H. Y. Chen and C. M. Foris, J. Solid State Chem., 1982, 44, 99–107 CrossRef CAS.
- H.-J. Meyer, G. Meyer and M. Simon, Z. Anorg. Allg. Chem., 1991, 596, 89–92 CrossRef CAS.
- J. A. Sannes and M. Valldor, Chem. Mater., 2024, 36, 2355–2362 CrossRef CAS.
- Z. Chen, W. Li, R. Li, Y. Zhang, G. Xu and H. Cheng, Langmuir, 2013, 29, 13836–13842 CrossRef CAS PubMed.
- V. Petříček, L. Palatinus, J. Plášil and M. Dušek, Z. Kristallogr. - Cryst. Mater., 2023, 238, 271–282 CrossRef.
- A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- R. Alcaraz de la Osa, I. Iparragirre, D. Ortiz and J. M. Saiz, ChemTexts, 2019, 6, 2 CrossRef.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2006, 124, 221101 CrossRef PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- L. Hedin, Phys. Rev., 1965, 139, A796–A823 CrossRef.
- M. Shishkin and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 035101 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. Borlido, J. Schmidt, A. W. Huran, F. Tran, M. A. L. Marques and S. Botti, npj Comput. Mater., 2020, 6, 96 CrossRef CAS.
- F. Goutenoire, V. Caignaert, M. Hervieu and B. Raveau, J. Solid State Chem., 1995, 119, 134–141 CrossRef CAS.
- Y. Pelovsky, V. Raynova, I. Gruncharov and I. Dombalov, J. Therm. Anal., 1991, 37, 841–847 CrossRef.
- I. Oftedal, Z. Phys. Chem., 1927, 128U, 154–158 CrossRef.
- A. K. V. Bever and W. Nieuwenkamp, Z. Kristallogr. - Cryst. Mater., 1935, 90, 374–376 CrossRef.
- P. Papamantellos, Z. Kristallogr. - Cryst. Mater., 1968, 126, 143–146 CrossRef.
- A. O. Tsokol’, O. I. Bodak, E. P. Marusin and M. G. Baivel'man, Kristallografiya, 1986, 791–792 Search PubMed.
- M. Shishkin and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 235102 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2446417. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt01111f |
| This journal is © The Royal Society of Chemistry 2025 |