
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWhen catalysis meets spintronic: the emergence of spintro-catalysis and the potential of mixed-anion spinels
Ulrich Aschauer*a and
Houria Kabbour *b
*b
aDepartment of Chemistry and Physics of Materials, University of Salzburg, 5020 Salzburg, Austria. E-mail: ulrichjohannes.aschauer@plus.ac.at
bNantes Université, CNRS, Institut des Matériaux de Nantes Jean Rouxel, IMN, F-44000, France. E-mail: houria.kabbour@cnrs-imn.fr
First published on 25th July 2025
Abstract
The emerging field of spintro-catalysis, at the interface of catalysis and spintronics, offers a compelling strategy to overcome long-standing limitations in catalytic reactions involving spin-sensitive intermediates. In particular, the redox chemistry of molecular oxygen, central to the oxygen evolution and reduction reactions (OER and ORR), is subject to spin-selection rules due to the triplet ground state of O2. Traditional catalysts, often in singlet spin states, suffer from inefficient spin-incoherent electron transfer, which hampers reaction kinetics. Recent advances demonstrate that ferromagnetic materials, through spin-polarized charge transport and coherent spin alignment, can facilitate these challenging transformations more efficiently than antiferromagnetic analogues. This frontier article examines how magnetic ordering, superexchange interactions, and spin state alignment influence catalytic performance, with a focus on perovskite and spinel-type oxides and their emerging chalcogenide analogues. The inclusion of mixed-anion chemistry further enhances the tunability of electronic and magnetic structures. Looking ahead, ab initio simulations such as spin-polarized density functional theory (DFT) will be critical for unveiling spin-related mechanisms and guiding the rational design of next-generation spin-active catalysts. By integrating spin dynamics into catalyst design, spintro-catalysis offers a transformative approach for energy conversion and sustainable chemical transformations.
Introduction
Catalysis plays a pivotal role in modern society, driving chemical processes essential for energy production, environmental protection, and the synthesis of vital chemicals.1 A notable example is the Haber–Bosch process, which enables the large-scale synthesis of ammonia from nitrogen and hydrogen. This process is fundamental to global agriculture, as ammonia serves as a key precursor for fertilizers that sustain food production for billions of people. Despite its importance, the Haber–Bosch process remains energy-intensive, consuming over 1% of the world's total energy supply, highlighting the ongoing need for more efficient catalytic solutions.2 Catalysis is also an integral part of sustainable fuel production, playing a key role in the generation of biofuels and hydrogen. Electrocatalysis and photocatalysis are crucial for reactions such as CO2 reduction towards products such as methane or ethanol3 or water splitting to produce green hydrogen fuel,4 which are promising alternatives to conventional fuels derived primarily from fossil sources.Notably, many catalysts incorporate magnetic transition-metal elements such as iron, cobalt, and nickel.5–10 Recent studies have illuminated the significant influence of magnetism on catalytic properties. For instance, research has demonstrated that applying a magnetic field can enhance the oxygen evolution reaction (OER) activity of CoFe2O4 nanosheets by modulating their intrinsic magnetism, reducing the overpotential required for the reaction.11 Additionally, investigations into the hydrogen evolution reaction (HER) have revealed that magnetic fields can alter catalytic efficiency across various materials, including ferromagnetic and paramagnetic catalysts, by affecting the binding energy between reaction intermediates and the catalyst surface.12 An external magnetic field can further enhance the catalytic performance by reducing the volume fraction of domain walls, the resulting large ferromagnetic (FM) domains promoting spin-facilitated pathways for the OER.13–15
The intrinsic magnetism and spontaneous magnetic interactions, especially for room temperature ordered systems, are, however, already consequential for catalysis without an applied external magnetic field. Quantum-spin exchange interactions that reduce the repulsion between same-spin electrons were predicted to be at the origin of these effects.16,17 Ferromagnetism was identified in general to be beneficial for high catalytic activity of reactions such as the oxygen reduction reaction (ORR), the OER, or ammonia synthesis.17,18 These insights underscore the potential of leveraging magnetism to design more efficient catalysts for applications in electrocatalysis and photocatalysis. Due to the effect of the spin on catalysis and in analogy to the field of spintronics,19 the term spintro-catalysis was coined20 to describe the direct effect of the magnetic structure on the catalytic activity.
The O2 redox chemistry and its relation to spintro-catalysis
This spin-dependent activity seems especially pronounced for the O2 redox chemistry, which stems from O2's particular electronic structure. In its ground state, the O2 molecule exists in a triplet (3Σg−) state due to the preferential occupation of its two-degenerate antibonding π* molecular orbitals by electrons with parallel spins, as shown in Fig. 1. These antibonding π* orbitals play a crucial role in the electrochemistry of O2, particularly in the oxygen reduction reaction (ORR) and the (OER).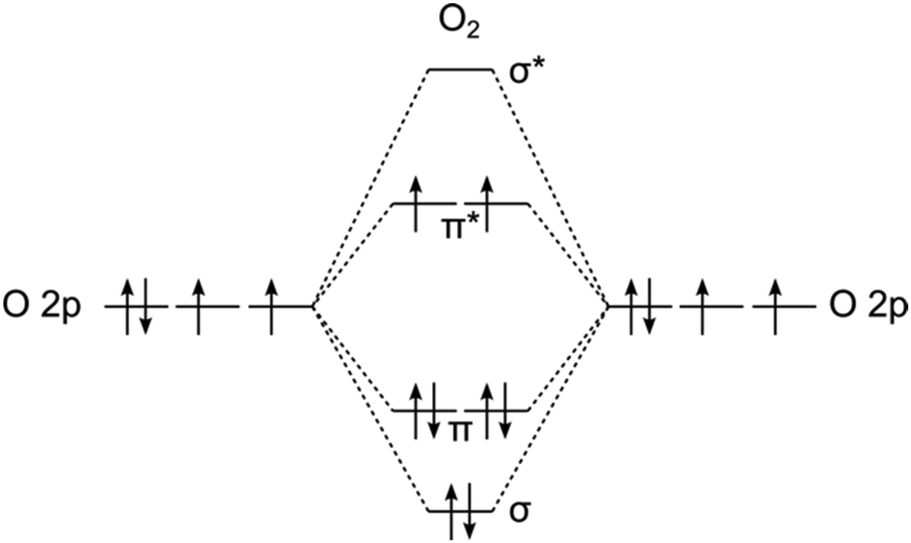 | ||
| Fig. 1 Simplified O2 molecular orbital diagram considering only the O 2p states. | ||
During the ORR, electrons are first transferred into these π* antibonding orbitals, progressively weakening the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond.21 As reduction continues, the occupation of the higher-energy σ* antibonding orbital further destabilizes the bond, leading to O2 dissociation and the formation of either water in acidic conditions or hydroxide (OH−) in alkaline conditions.22 Conversely, during the OER, the process is reversed: electrons are successively removed from oxygen-containing intermediates such as *OH, *O, and *OOH until the OO bond becomes stable, allowing molecular oxygen to form and be released.
O bond.21 As reduction continues, the occupation of the higher-energy σ* antibonding orbital further destabilizes the bond, leading to O2 dissociation and the formation of either water in acidic conditions or hydroxide (OH−) in alkaline conditions.22 Conversely, during the OER, the process is reversed: electrons are successively removed from oxygen-containing intermediates such as *OH, *O, and *OOH until the OO bond becomes stable, allowing molecular oxygen to form and be released.
A key challenge in both reactions arises from this intrinsic triplet spin state of molecular oxygen, which imposes spin-selection constraints on electron transfer.17,20 In the ORR, for example, the first two electrons transferred must be of opposite spin to those already occupying the π* orbitals. In the OER, on the other hand, the last two electrons removed from the molecule must have the same spin. However, if the catalytic metal site is in a singlet state, as is common for most elemental transition metals, there is no intrinsic preference for spin orientation. As a result, approximately half of the electron transfer events to or from these π* orbitals will be spin-forbidden, leading to slow reaction kinetics via incoherent transport mechanisms such as hopping.17,20 This spin restriction necessitates catalytic strategies that promote spin-polarized electron transfer (Fig. 2).
 | ||
| Fig. 2 Reproduced with permission from ref. 20, Gracia et al., J. Catal., 2018, 361, 331–338. © 2018 Elsevier. Illustration of the spin-selection constraints on electron transfer for the ORR. The schematic representation showing the density of states (left) of FM La1−xSrxMnO3 oxides and (right) preferential antiparallel spin orientation of an adsorbed O2 molecule. Half-metals are materials that conduct electrons of one spin orientation while blocking the other, making them highly interesting for spin-polarized charge transfer in catalysis. | ||
Magnetic materials to promote the O2 redox chemistry
The magnetic order of the catalyst is one route to promote spin-polarized electron transfer.21 While ferromagnetically (FM) ordered catalysts have high-lying transition-metal d-electrons of one preferred spin-polarization in each ferromagnetic domain, the local spin order of a ferrimagnetic (FiM) or antiferromagnetic (AFM) material can also promote spin-polarized charge transfer if the reactant adsorbs to two same-spin transition-metals sites. It was also stated that the FM order must persist along the electron transfer pathway and that a covalent bond of intermediate strength between the active site and the reactant simultaneously optimizes the charge transfer and the desorption rate according to the Sabatier principle.Gracia et al.17 propose an extension of the Kanamori-Goodenough rules23,24 to (electro)catalytic interfaces and in particular the OER and ORR reactions. In the case of spintro-catalysis, FM interactions are expected to enhance electronic conduction to/from the O2 triplet state in the OER and ORR as they provide enhanced spin conductivity while AFM provides localization leading to higher energy barriers, limited charge conductivity, and weak chemisorption for electron donors. Based on the above, Gracia et al. have analyzed transition metal oxidation states in some catalysts active for the OER and ORR. They found that higher oxidation states are performing better for the OER, because they provide electrophile FM surfaces capable of forming spin-polarized O2* easily. For the ORR, low-intermediate oxidation states are preferred because they provide nucleophile FM surfaces that can supply electrons to actively adsorb O2 → O− (such as Mn3+, Co3+, Ni3+, and Ru4+ for instance). This leads to the trend that good catalysts for the ORR are less performant for OER and vice versa.17
While FM order can exist in metallic alloys,25 we focus here on this magnetic order in oxides and mixed-anion materials that are emerging in various fields. In oxides, the stable magnetic order is primarily driven by superexchange, that is the alignment of spins is determined by their interaction through an oxygen ligand.23,24 As such AFM order is favored when two empty or two filled d orbitals are connected by a 180° bond via an oxygen 2p orbital. FM can emerge when the bond angle approaches 90° or when a filled d orbital is connected to an empty one via the O 2p orbital. Since most ABO3 perovskites have bond angles close to 180° and oxygen connects the same (empty or filled) B-site d orbitals, most of them display an AFM order. Notable exceptions are doped i.e. (La,Sr)MnO3, or defective perovskite oxides in which the altered bond geometry or the orbital occupations can favor FM.23,26,27 In La1−xSrxMnO3, the dominant magnetic interaction is double exchange, involving electron hopping between Mn3+ and Mn4+ ions through eg orbitals. This interaction leads to FM alignment and differs from superexchange, which occurs between same-valence cations and typically favors AFM coupling. FM can also occur in mixed B-site double perovskites,28 where transition metal d-orbitals of different occupations are linked by oxygen.
Extension of the mechanistic principles of the O2 triplet state from perovskites to other magnetic catalysts
As shown above, a huge literature (both experimental and theoretical) is now available for perovskite-type catalysts. On the other hand (thio)spinels are emerging with great promise and we focus here on the prospect of their magnetic properties in (photo)electrocatalytic applications. They have the general formula AB2X4 (X = O2− or S2−), featuring A cations in tetrahedral sites and B cations in octahedral sites within a cubic close-packed anion lattice as shown in Fig. 3. In inverse spinels, half of the B cations swap with A cations, forming B(AB)X4, as in magnetite (Fe3O4). | ||
| Fig. 3 Spinel crystal structure shown for the example of NiCo2O4. | ||
The diverse magnetic behavior of spinels is due to the intricate cation distribution within their face-centered cubic oxygen lattice. The magnetic properties of spinels also arise primarily from superexchange interactions between metal cations occupying tetrahedral (A) and octahedral (B) sites. Depending on the nature of these interactions, spinels can exhibit FM, FiM, or AFM. FM spinels arise when direct exchange (overlap of neighboring metal cation orbitals) or double exchange (electron hopping between mixed-valence cations, favoring parallel spin alignment) dominates. FiM spinels result from unequal antiparallel spin sublattices, leading to a net magnetization, while AFM spinels typically occur when one of the two metal elements is non-magnetic, leading to strong superexchange coupling. Like in perovskites, defects such as cation or oxygen vacancies can alter orbital occupations, or alter the exchange pathways, leading to different magnetic behavior.
Spinel materials are crucial for catalytic applications, magnetic storage, spintronics for low-energy computing,29–31 and so on. These applications highlight their significance in advancing sustainable energy and materials science. For instance, lithium manganese oxide (LiMn2O4) is a key cathode material in lithium-ion batteries.32 Spinel-based catalysts like Co3O4 and NiCo2O4 drive oxygen evolution and reduction reactions in fuel cells and water splitting.33,34 On the other hand, thiospinels, with sulfur replacing oxygen, exhibit unique electronic and magnetic properties35 due to sulfur's larger size and polarizability. For instance, CuCo2S4, is promising for supercapacitors and batteries due to its high conductivity and redox activity.36 While oxide spinels have been extensively explored for their catalytic properties,37 their conductivity and the number of active sites can be improved in their thiospinel counterparts, the redox activity of which is better than that of the corresponding oxides.38 Therefore, thiospinels are promising emergent counterparts to spinels in the catalysis field to promote efficient electron transfer and to stabilize reaction intermediates for highly effective energy conversion technologies. Room-temperature FM exists in spinels such as magnetite (Fe3O4), which exhibits a Verwey transition at ∼120 K but remains ferrimagnetic up to 860 K,39 and Li0.5Fe2.5O4,40 which shows ferrimagnetism above room temperature. Ferromagnetic spinels, such as Co3O4 and NiFe2O4, are particularly notable for their catalytic properties in OER and ORR. For thiospinels, CuCr2S4 and CuCr2Se4 are also ferromagnetic near room temperature, with Curie temperatures (TC) of 376 K and 430 K.41 On the other hand, FeCr2S4 exhibits ferrimagnetism (magnetic LRO) but below 170 K and is subject to orbital ordering.42
Ferromagnetic thio(spinels) and their catalytic behavior
If we consider first FM spinel oxides, a notable comparative mechanistic study of the OER reaction pathways for NiFe2O4 and CoFe2O4 (inverse spinels) concluded that CoFe2O4 exhibits lower barriers compared to NiFe2O4, especially in the Co-site-assisted OER mechanism.43 This study provides also a valuable methodology (see Fig. 4) based on O2 formation transition-state structures to determine barriers, which may enhance the comprehension of such catalytic reactions. As seen for numerous spinel-type materials, the later Co–Fe based system demonstrates a great anionic flexibility with the existence of the oxide CoFe2O4 for which theoretical investigations showed lower barriers and increased TOF compared to other spinel oxides, of the fluorine doped CoFe2O4:F![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 44 and of the thiospinel counterpart CoFe2S4 which exhibit water oxidation activity.45
44 and of the thiospinel counterpart CoFe2S4 which exhibit water oxidation activity.45
 | ||
| Fig. 4 Reproduced from ref. 43 under CC-BY 4.0 (https://creativecommons.org/licences/by/4.0). DFT modeling of the mechanism and energetics of OER on the (001) facets of NiFe2O4 and of CoFe2O4 spinels. The mechanistic pathways investigated include the lattice oxygen mechanism (LOM) and the adsorbate evolution mechanism (AEM). In the case of NiFe2O4, a Fe-site-assisted LOM pathway is found to be preferred. In the case of CoFe2O4, the authors found possible coexistence of active sites: with a Fe-site-assisted LOM pathway and a Co-site-assisted AEM pathway. The turnover frequencies (TOF) suggest higher efficiency for CoFe2O4. | ||
CuCr2O4 oxide spinel is among the most efficient catalyst for many reactions such as CO oxidation.46 Many synthesis methods (e.g. solvothermal, co-precipitation, sol–gel) showed a versatile system with controllable nanostructures that impact several catalytic or photocatalytic reactions (e.g. depollution, photodegradation of organic dyes, H2 evolution). Theoretical investigations have been carried out on various MCr2O4 materials including CuCr2O4: stable surface terminations were established, and CO oxidation activity could be related to the oxygen vacancy formation energy, with a dependence related to the M transition metal electronic structure.47 Considering advances on this particular Cu–Cr system with the existence of the pure oxide and the pure chalcogenides (S and Se), it is interesting to mention that recently sulfur doped CuCr2O4:S with different doping levels was reported, therefore showing the possibility of mixing anions. Without doping, CuCr2O4 is a visible light photocatalyst with a 2.4 eV band gap.48 When O, S and Se occupy the anionic sites, this system should be a rich playground for mixed-anion tuning of the catalytic properties. In the S-doped sample, a certain level of sulfur incorporation induces a morphological change of the particles toward well-defined nanosheets with improved catalytic performance, showing a dual effect of anionic doping that impacts both the electronic structure and the nanostructure.49 Other recent studies on spinel catalysts suggest that nanostructuring50 and defect engineering51 can be applied to thiospinels to enhance their catalytic potential. Further research into these materials and the relationship between their magnetic and catalytic properties could open new avenues for sustainable energy technologies.
Further prospects of mixed-anion spinels as spintro-catalysis materials
The above lays out that the intrinsic magnetism of a catalyst can have a marked effect on the catalytic activity, FM generally leading to higher activities than AFM. While perovskite complex oxides have been intensively studied as catalysts, the spinel family of compounds is advantageous compared to perovskites as they offer a richer tunability of their magnetic order in addition to being composed primarily of earth-abundant elements and being accessible via various synthetic routes. While the role of the transition metal on the catalytic activity was previously investigated for spinels, the role of the intrinsic magnetism was not systematically studied. Moreover, the versatile anion chemistry of spinels (i.e. the formation of thiospinels) and their potential to form mixed-anion phases such as oxysulfides, oxynitrides and oxyfluorides, offers additional tunability of the transition-metal d orbital occupation (fluorine will reduce transition metals, which is beneficial for the ORR, nitrogen will oxidize, which favors the OER and sulfur enhances the covalency) and hence the magnetic structure and catalytic activity.On the other hand, the magnetic ordering temperature of catalysts is a key consideration for practical applications. Catalysts which exhibit ferromagnetic order above room temperature, allow spintronic effects to be exploited under ambient conditions. For materials with lower ferromagnetic ordering temperatures, external magnetic fields may be applied to induce spin alignment. It is also interesting to mention the study of NiFeN@NiFeOOH core–shell catalyst in which the shell is antiferromagnetic until 70 °C.52 The authors could accelerate the OER reaction by heating up to 70 °C where the entire sample is in its paramagnetic state. This heat-induced magnetic transition allowed to diminish the spin-related electron transfer barrier in this particular case without stability issues reported. Furthermore, assessing environmental stability beyond temperature (e.g. via Pourbaix diagrams) is essential to ensure sustained catalytic activity in realistic electrochemical environments. In particular, the stability of thiospinels under aqueous conditions should be considered. For instance, CoFe2S4 mentioned above (ref. 45), undergoes surface oxidation and forms oxohydroxide layers during OER. In this case, the transformation is not detrimental but forms part of the activation mechanism, resulting in a catalytically active interface. In the later, both Fe–S component and active oxyhydroxide (Co(Fe)OxHy) suggest that mixed-anion systems can enhance both stability and reactivity through dynamic surface restructuring.
Conclusions and future prospects
The emerging field of spintro-catalysis highlights the importance of intrinsic magnetism and spin-polarized charge transport in governing the kinetics and efficiency of catalytic reactions, particularly those involving spin-sensitive species such as molecular oxygen. Ferromagnetic materials, by enabling coherent spin alignment and reducing spin-forbidden transitions, offer clear advantages over their antiferromagnetic counterparts in key processes like the oxygen evolution and reduction reactions (OER and ORR). Spinels and thiospinels, due to their compositional flexibility and diverse magnetic behaviour, are promising candidates for realizing these effects in practical catalytic systems.It is also worth mentioning that beyond spinels and the mentioned perovskites, several classes of magnetic oxides and intermetallics have been explored for spin-influenced catalysis, including layered double hydroxides53 and Heusler-type54 compounds. These materials exhibit varied magnetic behaviors and tunable electronic structures suitable for OER and ORR. Besides, spin-dependent effects are also crucial in other reactions such as the ones involving NOx chemistry for instance. Here again, magnetic catalysts may influence selectivity and efficiency in these reactions through spin polarization.55
A major limitation in electrocatalysis is the presence of linear scaling relationships between the adsorption energies of intermediates (e.g., *OH, *O, *OOH), which restrict optimization. Here, multiferroic materials, exhibiting coupled ferroelectric and magnetic ordering that can be externally manipulated, offer additional prospects. This tunability may enable selective modification of surface reactivity and offer a pathway to break scaling relationships.56–59
In addition to experimental advances, theoretical approaches are also crucial in shaping the future of spintro-catalyst design. Accurately modeling spintronic effects in catalysis requires methods capable of treating magnetic ordering, spin–orbit coupling, and strong electronic correlations. Spin-polarized DFT enables access to key descriptors such as the projected density of states and Bader charges, and can be combined with reaction path methods like nudged-elastic band (NEB) calculations. However, more advanced methods such as Hubbard-corrected functionals (e.g DFT+U or DFT+U+V, potentially in a self-consistent and/or site-dependent formalism), computationally intensive metaGGA or hybrid functionals or other strong correlation methods such as dynamical mean-field theory (DMFT) would be required. Furthermore, modeling catalysis at solid–liquid interfaces remains highly nontrivial, necessitating techniques such as implicit solvation models, joint DFT, ab initio molecular dynamics, metadynamics, or thermodynamic integration.
Ultimately, spintro-catalysis represents a multidisciplinary framework integrating solid-state physics, surface science, and theoretical chemistry, for the rational development of next-generation catalysts that leverage spin to achieve both activity and selectivity in sustainable energy technologies.
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this Frontiers short review.References
- C. M. Friend, et al., Acc. Chem. Res., 2017, 50(3), 517–521, DOI:10.1021/acs.accounts.6b00510.
- L. Torrente-Murciano, et al., Nat. Synth., 2023, 2, 587–588, DOI:10.1038/s44160-023-00339-x.
- C. Yang, et al., ACS Appl. Energy Mater., 2021, 4(2), 1034–1044, DOI:10.1021/acsaem.0c02648.
- B. You, et al., Acc. Chem. Res., 2018, 51(7), 1571–1580, DOI:10.1021/acs.accounts.8b00002.
- B. Su, et al., Acc. Chem. Res., 2015, 48(3), 886–896, DOI:10.1021/ar500345f.
- D. Wang, et al., Chem. Soc. Rev., 2017, 46, 816–854, 10.1039/C6CS00629A.
- K. E. Dalle, et al., Chem. Rev., 2019, 119(4), 2752–2875, DOI:10.1021/acs.chemrev.8b00392.
- J. I. van de Vlugt, Eur. J. Inorg. Chem., 2012, 363–375, DOI:10.1002/ejic.201100752.
- J. C. Védrine, Catalysts, 2017, 7(11), 341, DOI:10.3390/catal7110341.
- J. Wang, et al., Adv. Mater., 2016, 28, 215–230, DOI:10.1002/adma.201502696.
- Y. Ma, et al., ACS Appl. Mater. Interfaces, 2023, 15(6), 7978–7986, DOI:10.1021/acsami.2c19396.
- G. Li, et al., CCS Chem., 2021, 3, 2259–2267, DOI:10.31635/ccschem.021.202100991.
- X. Ren, et al., Nat. Commun., 2023, 14, 2482, DOI:10.1038/s41467-023-38212-2.
- Q. Wang, et al., J. Am. Chem. Soc., 2024, 146(50), 34681–34689, DOI:10.1021/jacs.4c13017.
- L. L. Hao, et al., ACS Catal., 2025, 15, 5640–5650, DOI:10.1021/acscatal.5c00081.
- J. Gracia, Phys. Chem. Chem. Phys., 2017, 19, 20451–20456, 10.1039/C7CP04289B.
- C. Biz, et al., ACS Catal., 2021, 11(22), 14249–14261, DOI:10.1021/acscatal.1c03135.
- J. Munarriz, et al., ChemPhysChem, 2018, 19, 2843–2847, DOI:10.1002/cphc.201800633.
- A. Hirohata, et al., J. Magn. Magn. Mater., 2020, 509, 16671, DOI:10.1016/j.jmmm.2020.166711.
- J. Gracia, et al., J. Catal., 2018, 361, 331–338, DOI:10.1016/j.jcat.2018.03.012.
- J. E. McGrady, et al., Inorg. Chem., 1999, 38(3), 550–558, DOI:10.1021/ic981253k.
- H. Li, et al., Adv. Energy Sustainability Res. DOI:10.1002/aesr.202400326.
- J. B. Goodenough, Phys. Rev., 1955, 100, 564, DOI:10.1103/PhysRev.100.564.
- J. Kanamori, J. Phys. Chem. Solids, 1959, 10(2–3), 87–98, DOI:10.1016/0022-3697(59)90061-7.
- H. Li, et al., Adv. Energy Sustainability Res., 2024, 2400326, DOI:10.1002/aesr.202400326.
- I. R. Shein, et al., Phys. Lett. A, 2007, 371(1–2), 155–159, DOI:10.1016/j.physleta.2007.06.013.
- N. Biškup, et al., Phys. Rev. Lett., 2014, 112, 087202, DOI:10.1103/physrevlett.112.087202.
- N. S. Rogado, et al., Adv. Mater., 2005, 17, 2225–2227, DOI:10.1002/adma.200500737.
- S. Yang, et al., ChemSusChem, 2025, 18, e202401115, DOI:10.1002/cssc.202401115.
- U. Lüders, et al., Adv. Mater., 2006, 18, 1733–1736, DOI:10.1002/adma.200500972.
- Y. Li, et al., Sensors, 2020, 20(18), 5413, DOI:10.3390/s20185413.
- W. Zeng, et al., Nat. Commun., 2024, 15, 7371, DOI:10.1038/s41467-024-51168-1.
- A. Ashok, et al., ACS Omega, 2018, 3(7), 7745–7756, DOI:10.1021/acsomega.8b00799.
- Y. Wang, et al., Green Chem., 2023, 25, 8181–8195, 10.1039/D3GC01828H.
- F. Ozel, et al., Mater. Today Energy, 2021, 21, 100822, DOI:10.1016/j.mtener.2021.100822.
- J. Sun, et al., J. Materiomics, 2021, 7(1), 98–126, DOI:10.1016/j.jmat.2020.07.013.
- V. Jeyavani, et al., ACS Appl. Nano Mater., 2024, 7(15), 17776–17785, DOI:10.1021/acsanm.4c03007.
- S. Li, et al., Catalysts, 2023, 13(5), 881, DOI:10.3390/catal13050881.
- J. P. Wright, et al., Solid State Sci., 2000, 2, 747–753, DOI:10.1016/S1293-2558(00)01107-9.
- M. Ahmad, et al., J. Mater. Res. Technol., 2022, 18, 3386–3395, DOI:10.1016/j.jmrt.2022.03.113.
- F. K. Lotgering, Solid State Commun., 1964, 2, 55–56, DOI:10.1016/0038-1098(64)90573-3.
- N. Büttgen, et al., New J. Phys., 2004, 6, 191, DOI:10.1088/1367-2630/6/1/191.
- Ö. N. Avcı, et al., ACS Catal., 2022, 12(15), 9058–9073, DOI:10.1021/acscatal.2c01534.
- Y. G. Ji, et al., Chem. Eng. J., 2024, 154211, DOI:10.1016/j.cej.2024.154211.
- T. Wu, et al., Adv. Mater., 2023, 35, 2207041, DOI:10.1002/adma.202207041.
- S. Mobini, F. Meshkani and M. Rezaei, J. Environ. Chem. Eng., 2017, 5, 4906–4916, DOI:10.1016/j.jece.2017.09.027.
- P. Zhao, et al., Theoretical insight into oxidation catalysis of chromite spinel MCr2O4 (M = Mg, Co, Cu, and Zn): Volcano plot for oxygen-vacancy formation and catalytic activity, J. Catal., 2021, 393, 30–41, DOI:10.1016/j.jcat.2020.11.006.
- R. Peymanfar and H. Ramezanalizadeh, Sol-gel assisted synthesis of CuCr2O4 nanoparticles: An efficient visible-light driven photocatalyst for the degradation of water pollutions, Optik, 2018, 169, 424–431, DOI:10.1016/j.ijleo.2018.05.072.
- A. R. Rashid, et al., Facile fabrication of Sulfur-Doped CuCr2O4 Nanocatalysts: For enhanced bifunctional oxygen and hydrogen evolution reactions, Inorg. Chem. Commun., 2025, 178, 114492, DOI:10.1016/j.inoche.2025.114492.
- T. E. Meyer, et al., Nano Lett., 2025, 25(11), 4234–4241, DOI:10.1021/acs.nanolett.4c05699.
- H. Xu, et al., J. Colloid Interface Sci., 2023, 650, 1500–1508, DOI:10.1016/j.jcis.2023.07.109.
- M. Lu, et al., Nano Lett., 2022, 22(22), 9131–9137, DOI:10.1021/acs.nanolett.2c03634.
- L. Lin, et al., ACS Catal., 2023, 13, 1431–1440, DOI:10.1021/acscatal.2c04983.
- M. Yu, et al., Angew. Chem., Int. Ed., 2021, 60, 5800–5805, DOI:10.1002/anie.202013610.
- J. Hwang, R. R. Rao and L. Giordano, et al., Regulating oxygen activity of perovskites to promote NOx oxidation and reduction kinetics, Nat. Catal., 2021, 4, 663–673, DOI:10.1038/s41929-021-00656-4.
- X. Li, et al., Nat. Commun., 2019, 10, 1409, DOI:10.1038/s41467-019-09191-0.
- A. Kakekhani, et al., ACS Catal., 2015, 5(8), 4537–4545, DOI:10.1021/acscatal.5b00507.
- A. Kakekhani, et al., J. Mater. Chem. A, 2016, 4, 5235–5246, 10.1039/C6TA00513F.
- A. Kakekhani, et al., Phys. Chem. Chem. Phys., 2016, 18, 19676–19695, 10.1039/C6CP03170F.
| This journal is © The Royal Society of Chemistry 2025 |