
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atom-economical insertion of hydrogen and sulfur into carbon–nitrogen triple bonds using H2S via synergistic C–N sites†
Ganchang Lei a,
Jiayin Wanga,
Xinhui Liub,
Shiping Wanga,
Shijing Lianga,
Lijuan Shen*b,
Yingying Zhan*a and
Lilong Jiang*a
a,
Jiayin Wanga,
Xinhui Liub,
Shiping Wanga,
Shijing Lianga,
Lijuan Shen*b,
Yingying Zhan*a and
Lilong Jiang*a
aNational Engineering Research Center of Chemical Fertilizer Catalyst, State Key Laboratory of Fluorine & Nitrogen Chemicals, School of Chemical Engineering, Fuzhou University, Gongye Road 523, Gulou District, Fuzhou, Fujian 350002, P. R. China. E-mail: jll@fzu.edu.cn; zhanyingying@fzu.edu.cn
bCollege of Environmental and Resource Science, College of Carbon Neutral Modern Industry, Fujian Key Laboratory of Pollution Control & Resource Reuse, Fujian Normal University, 350007, Fuzhou, Fujian, People's Republic of China. E-mail: ljshen@fjnu.edu.cn
First published on 16th June 2025
Abstract
Developing efficient strategies that convert industrial waste hydrogen sulfide (H2S) into value-added products is meaningful for both applied environmental science and industrial chemistry. Here we report a series of heterogeneous N-doped carbon catalysts with synergistic C–N sites that enable the nucleophilic addition of H2S into aromatic nitrile compounds (PhCN) under mild conditions to produce thiobenzamide (PhCSNH2). The as-designed C–N sites achieve a high thioamide production rate of 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 μmolPhCSNH2 L−1 h−1 and a notable selectivity of ca. 80% at 60 °C within a short 2-hour timeframe. Additionally, the catalyst exhibits easy recyclability and maintains high stability over ten cycles during a 6-month period. Systematic microscopic and in situ spectroscopic characterization, combined with theoretical calculations, reveal that C-pyridinic N coordination sites effectively lower the adsorption energy barrier of the crucial intermediate *PhCSHNH, offering a dynamically favorable pathway for PhCSNH2 production. Furthermore, the protocol demonstrates excellent compatibility with various substituted substrates, providing access to a diverse range of thioamides.
400 μmolPhCSNH2 L−1 h−1 and a notable selectivity of ca. 80% at 60 °C within a short 2-hour timeframe. Additionally, the catalyst exhibits easy recyclability and maintains high stability over ten cycles during a 6-month period. Systematic microscopic and in situ spectroscopic characterization, combined with theoretical calculations, reveal that C-pyridinic N coordination sites effectively lower the adsorption energy barrier of the crucial intermediate *PhCSHNH, offering a dynamically favorable pathway for PhCSNH2 production. Furthermore, the protocol demonstrates excellent compatibility with various substituted substrates, providing access to a diverse range of thioamides.
Broader contextCatalytic conversion of H2S to high value-added organic sulfur-containing compounds is more appealing for the resource utilization of H2S. Herein, we propose an efficient method for synthesizing thiobenzamide (PhCSNH2) through the nucleophilic addition of the cyano group into aromatic nitrile compounds (PhCN) using industrial waste H2S. Heterogeneous N-doped carbon catalysts with synergistic C–N sites were designed for this process. As a result, the C-pyridinic N sites are responsible for the excellent performance of N–GC-X catalysts in PhCSNH2 synthesis under mild reaction conditions, exhibiting a high PhCSNH2 synthesis rate of 26400 μmolPhCSNH2 L−1 h−1. Multiple in situ spectroscopic and microscopic characterization methods combined with theoretical calculations disclose that the pyridinic nitrogen in the carbon support enhances the adsorption and activation of H2S and nitrile reactants and offers sufficient structural N active sites to reduce the energy barrier of the rate-determining step for the nucleophilic addition of H2S to synthesize thioamides. This work unveils the catalytic nature of nitrogen-doped carbon and provides guidance for designing metal-free carbon catalysts for the high-economic utilization of H2S.
|
Introduction
Over the past hundred years, commercial production of sulfur-containing amides has continuously increased and now exceeds 5 million metric tons per year of valuable fine and bulk chemicals, with an annual output value of nearly 49 billion RMB. At present, thioamides are widely applied in the synthesis of biomedicines, cosmetics and agrochemicals.1–3 For instance, nearly 50% of agrochemicals and over 20% of the top 200 retail sale pharmaceuticals are globally derived from the functionalization of thioamide groups.4–6 Traditionally, thioamides are synthesized through the conversion of oxoamides via O–S exchange reactions using unstable and costly Lawesson's reagent and P2S5, or the Willgerodt–Kindler reaction that employs expensive amines and carbonyl compounds in the presence of elemental sulfur.7,8 These routes are generally complicated and uneconomical and suffer from poor atom-utilization efficiency and functional-group compatibility.9 In this regard, green and efficient reactions for chemically selective synthesis of thioamides with atomic economy are highly desirable. Hydrogen sulfide (H2S) molecule is a rich resource but is generally regarded as a corrosive and toxic gas that is produced in large quantities from both industrial processes and natural sources.10–14 The current industrial practices for H2S elimination predominantly rely on the Claus process, wherein elemental sulfur is recovered, while hydrogen is consumed and discharged as wastewater.15–18 There is increasing learning that H2S could become a potentially valuable chemical if a process that can simultaneously utilize H and S elements in H2S to produce high value-added products was developed. Ideally, the direct insertion of toxic yet cheap H2S molecules into readily available industrial nitrile building blocks through the additive reaction provides a compelling strategy for producing functionalized thioamides. This approach offers unique environmental and economic benefits (Fig. 1), with the potential for 100% atom utilization efficiency without waste discharge. Nevertheless, little attention has been paid to the reaction in the open literature. In particular, no efforts have been dedicated to the design and synthesis of active and stable catalysts to operate under sour and corrosive conditions, which greatly hinders the development and optimization of the protocol.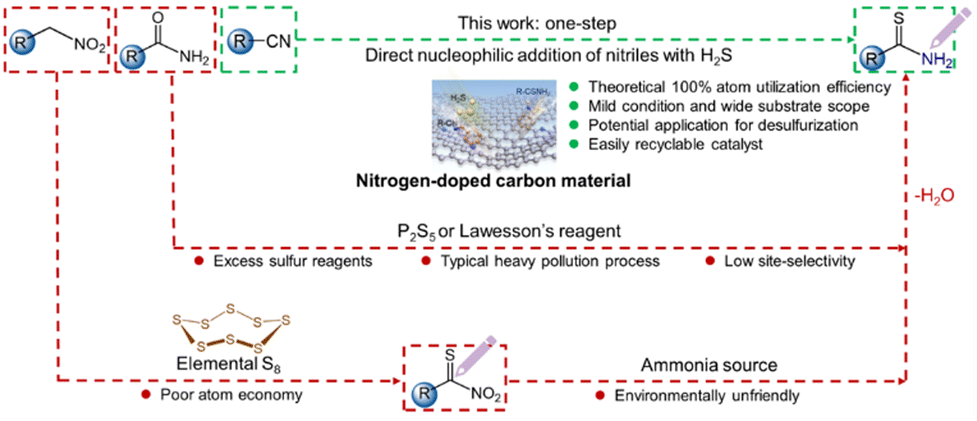 | ||
| Fig. 1 Comparison of the different approaches for the synthesis of thioamides. | ||
From a mechanistic standpoint, the nucleophilic addition of H2S to nitrile compounds primarily involves the dissociation of H2S molecules at active sites, yielding HS* and H*, which further react with the –CN group in nitrile compounds to form thioamides. Research studies have revealed that N-doped carbon catalysts with abundant structural base sites exhibit substantial catalytic activity and remarkable sulfur resistance during the H2S catalytic reaction.19–21 The findings motivate us to undertake the first heterogeneous catalytic synthesis of thioamides from nitriles and H2S. In doing so, we herein fabricate a series of N-doped carbon catalysts with different configurations of N (graphitic N, pyrrolic N and pyridinic N) decorated on two-dimensional porous carbon materials. The catalyst with rich pyridinic N exhibits an optimal PhCSNH2 yield of up to approximately 26400 μmolPhCSNH2 mL−1 h−1 with good product selectivity (>80%) and high durability through 10 runs of cyclic testing over 6 months. A wide variety of substituted substrates are viable in this reaction, enabling access to diverse thioamides. Using detailed in situ spectroscopic characterization, combined with theoretical calculations, the common mechanistic pathway is elucidated. The progress is anticipated to inspire more research attention on the cost-efficient synthesis of thioamides with high atom economy, as well as open the way for highly efficient resource utilization of the H2S waste.
Results and discussion
Synthesis and structural analysis of nitrogen-doped carbon catalysts
To realize the high-efficiency synthesis of thioamides, we developed a series of nitrogen-doped carbon catalysts composed of hierarchically porous carbon nanosheets with controllable basic nitrogen sites, as schematically illustrated in Fig. S1 (ESI†). Typically, glucose and dicyandiamide were used as precursors and subjected to pyrolysis at 600 °C, which resulted in the formation of intermediates of carbon polymers and layered carbon nitride (g-C3N4). Subsequently, the mixed intermediates were further carbonized by instant heating at 780 °C for 60 min to yield nitrogen-doped carbon catalysts (N–GC-X). During this process, the formed g-C3N4 can bind to the as-generated carbon intermediates via donor–acceptor interactions and function as a soft template for the formation of hierarchically porous N–GC-X nanosheets.Fig. S2 (ESI†) shows the SEM images of the as-prepared samples. Pristine GC presents a layered block-like structure (Fig. S2a, ESI†). As the dicyandiamide content increases, the obtained N–GC-X catalysts gradually decrease in size, showing thin-layer morphologies with holey structures rather than stacked layers (Fig. S2b–e, ESI†). Further insight from TEM characterization highlights the unique tulle-like morphologies of the N–GC-6 catalysts (Fig. 2a and b). EDS mapping demonstrates uniform distribution of carbon, nitrogen, and oxygen elements in the layered structure (Fig. S3, ESI†). In addition, the TEM analysis uncovers the presence of mesopores, revealing the porous nature of N–GC-6. The layered structure of N–GC-6 with abundant nanoscale pores is also corroborated by the AFM image, as illustrated in Fig. 2c. The randomly measured porous nanosheets have a similar thickness of about 1.8–2.3 nm. Furthermore, Fig. S4, S5 and Table S1 (ESI†) present the nitrogen adsorption–desorption isotherms and the corresponding BJH pore size distributions for the N–GC-X samples. All N–GC-X catalysts exhibit type IV isotherms with narrow H2 hysteresis loops, confirming mesoporous properties according to the IUPAC definition.
 | ||
| Fig. 2 (a) and (b) TEM and (c) AFM images of N–GC-6; high-resolution N 1s XPS profiles of (d) N–GC-2, (e) N–GC-4, (f) N–GC-6, and (g) N–GC-8 catalysts; (h) content of different N species of N–GC-X catalysts from XPS fitting results; and (i) the percentages of pyridinic N, pyrrolic N, and graphitic N over the N–GC-X catalysts. | ||
To obtain more microstructural information of the catalysts, multiple spectroscopic characterization studies have been conducted. Fig. S6 (ESI†) displays the XRD patterns of the as-prepared samples, which all show two characteristic peaks at around 26° and 44° that are assigned to the (002) and (101) crystal planes of graphitic carbon, respectively.22,23 The finding indicates the formation of amorphous carbon networks in the N–GC-X catalysts. Additionally, N–GC-X exhibits a relatively weak peak intensity and a high angular shift in the (002) plane, indicating the presence of numerous structural defects in its amorphous carbon network. The chemical functional groups in the samples have been investigated by FT-IR characterization (Fig. S7, ESI†). The N–GC-X samples exhibit three bands at around 1320–1197 cm−1, 1608–1515 cm−1, and 2200 cm−1, corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, C–N, and OCN groups, respectively.24 This result confirms the successful introduction of functional N species into the N–GC-X framework.
N, C–N, and OCN groups, respectively.24 This result confirms the successful introduction of functional N species into the N–GC-X framework.
Moreover, in the Raman spectra, both pristine GC and N–GC-X display peaks at around 1350 cm−1 and 1590 cm−1, which are attributable to the disorder-induced D-band and the in-plane vibrational G-band, respectively (Fig. S8, ESI†). Notably, the intensity ratio of the D-band to the G-band (ID/IG) increases from 0.98 to 1.07 for the N–GC-X samples, indicating an increased graphitization degree and elevated topological defects.25,26 This deduction is consistent with the XRD analysis results, implying that the incorporation of N species benefits the formation of structural base sites. Fig. S9 (ESI†) displays the 13C NMR spectrum of N–GC-6, which is obtained to unveil the C and N chemical bonding structure of the sample. The bands at 98 ppm and 129 ppm are attributed to aromatic carbon in the pyridine ring. The band at 170 ppm could be attributed to carbon substituted by the CN cross-linker in the pyridine ring.27–29 The results agree well with the results of the following XPS analysis, that is, the doped N in the N–GC-X catalyst is mainly pyridinic N. As depicted in Fig. S10 (ESI†), the C 1s XPS spectra of all N–GC-X catalysts can be deconvoluted into three peaks at around 284.6, 285.9, and 287.8 eV, attributable to the CC/C–C, C–N, and NC signals, respectively.30 In contrast, pristine GC displays only one predominant peak located at approximately 284.6 eV, corresponding to the CC/C–C unit. The N 1s spectra in Fig. 2d–g and Fig. S11 (ESI†) are fitted into four peaks with binding energies of 398.6 eV, 399.7 eV, 400.7 eV and 401.8 eV, which are assigned to pyridinic N, pyrrolic N, graphitic N, and quaternary N, respectively.31,32 The above findings indicate that the structural N sites are successfully doped into the carbon skeleton. Fig. 2h depicts the total N content and the distribution of N species in the catalysts. It can be found that the total N content in the samples follows the order N–GC-6 (39.7 at%) > N–GC-8 (39.3 at%) > N–GC-4 (33.9 at%) > N–GC-2 (26.8 at%). N–GC-6 possesses the highest N content. The results are also validated by the elemental analysis (EA) (Table S1, ESI†). Notably, with the variation of the dicyandiamide content, the percentage of pyridinic N in the series of N–GC-X samples changes from 43.6% to 48.0%, surpassing that of pyrrolic N (from 24.8% to 26.4%) and graphitic N (from 27.2% to 30.0%) (Fig. 2i). The different N species in all N–GC-X samples follow the order pyridinic N > pyrrolic N ≈ graphitic N > quaternary N. It should be noted that quaternary N usually appears inert in heterogeneous catalytic reactions. Thus, we mainly investigate the most adequate pyridinic N, pyrrolic N and graphitic N in the present work. The percentages of pyridinic N in all N–GC-X samples are higher than 43%, which plays a key role in the H2S addition reaction (as discussed later). Collectively, the above characterization methods faithfully validate that we have successfully synthesized a series of N–GC-X catalysts with different weight contents of N doping by varying the amounts of precursors. Importantly, the distribution of N species in these samples has also been modulated, which establishes a foundational framework for identifying active sites and conducting in-depth mechanism studies.
Catalytic performance of N–GC-X
The catalytic activities of the series of N–GC-X samples were initially investigated by the nucleophilic addition of benzonitrile (PhCN) with H2S to yield thiobenzamide (PhCSNH2). This procedure was carried out under an ambient pressure of 5 wt%/95% H2S/N2 at 60 °C in dimethylacetamide (DMAC) solvent. As shown in Fig. 3a and Fig. S12 (ESI†), all N–GC-X catalysts exhibit enhanced activity compared to the pristine GC catalyst, highlighting the promotion role of N sites in the catalysts. The PhCN conversion and PhCSNH2 selectivity of the catalysts increase with the increase of the reaction temperature from 40 to 60 °C, but decline when the reaction temperature exceeds 70 °C (Fig. 3b), plausibly because of side reactions that occur at high temperatures. The influence of reaction time on the nucleophilic addition reaction has also been studied (Table S2 and Fig. S13, ESI†). N–GC-6 achieves a PhCN conversion of 99% with a PhCSNH2 selectivity of 59% after reaction for 1 hour. With the extension of the reaction time to 2 hours, the catalyst shows optimal catalytic activity, reaching a PhCSNH2 selectivity as high as ca. 80%. Notably, the activity of N–GC-6 increases with prolonged reaction time from 1 hour to 3 hours. However, there is no significant improvement in catalytic performance when the reaction time is further increased to 4 hours. Additionally, the solvent effect on catalytic performance has also been studied due to its impact on stabilizing intermediates, affecting the solubility of substrates, or tuning the wettability and dispersion of catalysts. As demonstrated in Fig. S14 (ESI†), the reaction proceeds most effectively in DMAC or DMF. Other polar or medium-polar solvents are either ineffective or produce PhCSNH2 in low yields. | ||
| Fig. 3 (a) Catalytic conversion of PhCN to PhCSNH2 in the presence of N–GC-X catalysts; (b) catalytic results of the nucleophilic addition of H2S to PhCSNH2 by N–GC-6 at different temperatures; the plot for the concentration of PhCSNH2 vs. reaction time under different temperatures over (c) pristine GC and (d) N–GC-6; (e) PhCSNH2 synthesis rate over pristine GC and N–GC-6 (the catalysts were tested for PhCSNH2 synthesis in a feed with a composition of 5%H2S–95%N2 and 10 mmol PhCN); (f) Arrhenius plots of the rate constants for nucleophilic addition of H2S to PhCSNH2 over the catalysts; (g) relationship between the pyridinic N content and the yield of PhCSNH2; and (h) catalytic results of fresh N–GC-6 and that after storing under ambient conditions for six months. | ||
To further elucidate the relationship between the structure of the catalysts and their catalytic properties, kinetic experiments have been performed at low conversions of <20% to achieve accurate kinetic parameters. The ordinary rate formula for the nucleophilic addition of PhCN with H2S can be derived using the following eqn (1). Since the amount of catalyst is constant and the concentration of H2S is significantly higher than the PhCN concentration, eqn (1) can be simplified to eqn (2). As shown in Fig. 3c and d, the curves that depict the decrease in the concentration of PhCN (CPhCN) as a function of reaction time (t) give a straight line from the starting point, denoting a pseudo-zero-order reaction (γ = 0) with respect to PhCN. Thus, eqn (2) can be further expressed as eqn (3) or (4). The experimental results suggest that the reaction rate is in agreement with eqn (4), and there is a linear relationship between CPhCN and t. Based on the Arrhenius equation: k = A e−Ea/RT, the apparent activation energy Ea can be calculated using eqn (5). The PhCSNH2 production rate over N–GC-6 displays an approximately linear enhancement with increasing temperature, ranging from 8600 to 26400 μmolPhCSNH2 L−1 h−1 when the reaction temperature increases from 40 to 70 °C (Fig. 3e). The results indicate that the reaction rate of catalysts is enhanced with the incorporation of structural N base sites. As depicted in Fig. 3f, the extraction of the activation energy Ea from the Arrhenius curve leads to Ea = 35.8 kJ mol−1 and 36.4 kJ mol−1 for N–GC-6 and pristine GC, respectively, in line with the observed trend in catalytic activity. The findings prove that the synergistic effect between C and N sites can significantly lower the Ea and enhance the catalytic performance. By comparing the relationship between the pyridinic N content and the yield of PhCSNH2 for N–GC-X materials (Fig. 3g), it is obvious that the increase of pyridinic N content of the catalysts enhances the PhCSNH2 yield. Furthermore, the optimized N–GC-6 displays better reactivity than the traditional metal-based catalysts, including Fe2O3, CoO, CeO2 and CaO (Fig. S15, ESI†). More importantly, N–GC-6 exhibits stable activity in H2S nucleophilic addition, with no obvious decline of PhCSNH2 selectivity even with the introduction of 1% CO2 (Fig. S16, ESI†). The above test findings clearly reveal that N–GC-6 exhibits high activity and outstanding PhCSNH2 selectivity, which is positively correlated with its high pyridinic N content.
 | (1) |
 | (2) |
 | (3) |
| CBN = −k·t | (4) |
|
lnk = lnA − Ea/RT
| (5) |
Given the high catalytic performance, the catalytic stability of N–GC is tested as it is crucial for practical application. The N–GC-6 catalysts were reused for five cycles via facile centrifugation separation without any other treatment. As exhibited in Fig. 3h, there is no significant decrease in both activity and PhCSNH2 selectivity during the cycle test. N–GC-6 achieves a PhCN conversion of 97% with a PhCSNH2 selectivity of 78% after five runs of the reaction. In addition, after storing under ambient conditions for 6 months, N–GC-6 maintains a high catalytic activity and structural stability comparable to those of a fresh catalyst (Fig. 3h and Fig. S17–S20, ESI†), demonstrating its excellent stability. Besides, the slight decrease in reactivity over the catalysts may be caused by the generation of sulfur species that can cover the active sites or block the pores of the catalysts.
With the confirmed optimal reaction parameters, we then explored the reaction scope for producing different thioamides by nucleophilic addition of the cyano group into nitrile compounds using H2S over an N–GC-6 catalyst. As displayed in Table 1 and Fig. S21 (ESI†), the reaction proceeds smoothly in all cases, no matter for strongly electron-withdrawing or electron-donating substrates. For instance, following a reaction time of 2 h at 60 °C, p-chlorobenzonitrile exhibits a 92% conversion and a good regioselectivity of 80% for the p-chlorothiobenzamide product. Substrates bearing a hydroxyl group also display nearly 80% selectivity for the generation of 4-hydroxythiobenzamide. In brief, N–GC-6 exhibits excellent tolerance to functional groups including chlorine, bromine, fluorine, and methyl. Additionally, the approach is found to be effective in the production of aliphatic thioamides with high yields using acetonitrile and 2-methylpropanenitrile as substrates (thioacetamide and 2-methylpropanethioamide). Furthermore, even upon switching the reaction solvent to DMF, N–GC-6 still demonstrates comparable catalytic performance in transforming the nitrile-based compounds into their corresponding thioamides, highlighting the unique role of the pyridinic N sites in promoting nucleophilic addition reactions.
| Reaction conditions: 4 mmol nitriles in 40 mL of solvents (DMAC and DMF) using 40 mg of a catalyst, reaction for 2 h at 60 °C under atmospheric pressure H2S (5 wt%, 20 mL min−1). |
|---|
 |
Reaction mechanism
According to the characterization and catalytic performance results, the catalytic activity of N–GC-X is improved with an increase in pyridinic N content. To gain a deeper understanding of the reaction mechanism, theoretical calculations were performed to determine the adsorption free energy of key intermediates in the H2S nucleophilic addition reaction. Simultaneously, charge density differences and Bader charge transfer for the key intermediates were explored on various N sites to elucidate the adsorption and desorption abilities. Firstly, the reaction mechanisms and calculated energy distributions for CS and N–H bond formation mediated by C–N centers are presented. The H2S nucleophilic addition reaction involves five major steps: (1) adsorption of PhCN and H2S, (2) H2S dissociation, (3) formation of *PhCNH and *HS, (4) formation of *PhCSHNH, and (5) formation and desorption of PhCSNH2 (Fig. 4a). In theory, an ideal active site should adsorb H2S molecules strongly while concurrently preventing sulfur poisoning. Notably, the C atoms adjacent to the N dopants are usually triggered and thus serve as one of the reaction sites for catalytic reactions.
 | ||
| Fig. 4 (a) The proposed reaction mechanism for CS and N–H bond formation mediated by nitrogen-doped carbon catalysts; theoretical calculations of the catalysts. (b) PhCN adsorption energy over C-graphitic N, C-pyrrolic N and C-pyridinic N; (c) free energy of H2S nucleophilic addition of PhCN to PhCSNH2 on graphitic N, pyrrolic N and pyridinic N; (d) free energy of desorption for the S-containing product on graphitic N, pyrrolic N and pyridinic N; and (e) free energy diagram of the transformation of *PhCNH and *HS to *PhCSHNH key intermediates on graphitic N, pyrrolic N and pyridinic N. | ||
The calculation results reveal that the PhCN molecule can be intensely adsorbed on the C-pyridinic N site and facilely activated to *PhCN with a low activation energy of −0.49 eV. In contrast, adsorption of the PhCN molecule on the C-graphitic N site and the C-pyrrolic N site is less favorable, requiring higher activation energies of −0.21 eV and −0.32 eV, respectively (Fig. 4b and Fig. S22–S24, ESI†). Additionally, the dissociation of *H2S into *HS and *H on the N sites is found to be endothermic, with free energies of −0.41 eV, −0.62 eV and −0.81 eV for graphitic N, pyrrolic N and pyridinic N, respectively (Fig. 4b). A transition barrier energy (TS1) of 0.91 eV is required for the pyridinic N site, significantly lower than the values of 1.54 eV for graphitic N and 1.23 eV for pyrrolic N (Fig. 4c). The higher dissociation energies and TS1 energies of the graphitic N and pyrrolic N sites suggest that the dissociation of H2S on these sites is more challenging, which would result in relatively low activity.
Following dissociation (Fig. 4c), the *H reacts with *PhCN to form the *PhCNH intermediate, with TS2 values of 0.60 eV, 0.44 eV and 0.19 eV for graphitic N, pyrrolic N and pyridinic N, respectively. The formation energies of *PhCNH on graphitic N and pyrrolic N are −0.62 eV and −1.03 eV, compared to −1.25 eV on the pyridinic N, indicating the high activity of the pyridinic N site for the H2S nucleophilic addition reaction. Subsequently, *HS reacts with *PhCNH to form the key intermediate *PhCSHNH, with a TS3 of 0.85 eV for pyridinic N. This value is notably lower than those of graphitic N (1.75 eV) and pyrrolic N (1.18 eV). Importantly, the pyridinic N site can facilitate the –HS spillover on the catalyst surface, leading to the formation of *PhCSHNH species with a free energy of −1.60 eV. In contrast, graphitic N and pyrrolic N exhibit higher formation energies. The *PhCSHNH intermediate then undergoes intramolecular rearrangement of the –SH group to further convert into *PhCSNH2, with a TS4 of 0.56 eV for pyridinic N. Furthermore, the formation energy barrier for PhCSNH2 formation on pyridinic N (−1.56 eV) is much lower compared to that on graphitic N (−1.32 eV) and pyrrolic N (−1.40 eV), indicating that the PhCSNH2 product is more readily generated at the pyridinic N site. Additionally, the PhCSNH2 desorption free energies at the N species follow the order pyridinic N > graphitic N > pyrrolic N, aligning with the activity trend (Fig. 4d). These findings suggest that the pyridinic N centers can serve as efficient sites for the H2S nucleophilic addition. According to the calculations, the rate-determining step of the H2S nucleophilic addition reaction is the nucleophilic attack of *PhCNH by *HS to form *PhCSHNH (Fig. S25a, ESI†). The lower activation energies for the formation of the *PhCSHNH intermediate on pyridinic N consolidate that a higher proportion of pyridinic N facilitates the nucleophilic reaction (Fig. 4e). Moreover, the *PhCSHNH key intermediate serves as a probe to determine the formation enthalpies of the C–S bond (ΔHC–S) on different N sites. The ΔHC–S is calculated to be −33.6 kJ mol−1 on pyridinic N. In contrast, on pyrrolic N and graphitic N, the ΔHC–S is substantially enhanced to −0.29 kJ mol−1 and −0.58 kJ mol−1, respectively. The lower ΔHC–S on pyridinic N signifies that the formation of the C–S bond to generate *PhCSHNH becomes more beneficial, leading to a markedly distinct selectivity in the production of PhCSNH2 on pyridinic N.
The charge density difference and Bader charge analyses were also performed to evaluate the affinity of N sites over reactive atoms (Fig. S25b, ESI†). The variation in charge difference among the three N site-PhCSHNH intermediates is 0.48–0.57 eV, verifying the hypothesis that introducing N atoms enhances the affinity of C sites on the carbon matrix by creating basic sites. The results are consistent with the CO2-TPD characterization results that the reactants are strongly adsorbed on the surface of N–GC-6 due to its plentiful structural base sites (Fig. S26, ESI†). In addition, the pyridinic N sites show weak interaction with PhCSNH2, and the charge shift from the pyridinic N site to PhCSNH2 is 0.45 eV, slightly exceeding the values for graphitic N (0.39 eV) and pyrrolic N (0.35 eV). The comprehensive theoretical calculation suggests that *PhCSHNH is favorable for addition rearrangement to *PhCSNH2 at the pyridinic N-based basic nitrogen sites.
To further explore the reaction process of nucleophilic addition of PhCN with H2S on the catalyst, Quasi-in situ IR spectroscopy of PhCN and H2S co-adsorption were conducted to investigate the real-time intermediates of the reaction (Fig. 5a). The recorded IR spectra display a characteristic band at around 2232 cm−1, which is assigned to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N group.33 Concurrently, a new band at around 1390 cm−1 assigned to ν(H–S) is detected.34 The intensity of the CN group gradually weakens with reaction time, while two vibration bands emerge at around 1758 cm−1 and 1630 cm−1, which can be attributed to the ν(CN) and δ(N–H) groups, respectively.35,36 Notably, the bands attributable to the CS and CS–H groups are detected at around 2188 cm−1 and 990–1210 cm−1, respectively, confirming the generation of *PhCSHNH as a key intermediate in the H2S nucleophilic addition reaction.37,38 This observation aligns with the DFT calculation results. Furthermore, the intensities of the N–H and CS groups enhance with time, revealing the formation of Ph-CSNH2 during the reaction.
N group.33 Concurrently, a new band at around 1390 cm−1 assigned to ν(H–S) is detected.34 The intensity of the CN group gradually weakens with reaction time, while two vibration bands emerge at around 1758 cm−1 and 1630 cm−1, which can be attributed to the ν(CN) and δ(N–H) groups, respectively.35,36 Notably, the bands attributable to the CS and CS–H groups are detected at around 2188 cm−1 and 990–1210 cm−1, respectively, confirming the generation of *PhCSHNH as a key intermediate in the H2S nucleophilic addition reaction.37,38 This observation aligns with the DFT calculation results. Furthermore, the intensities of the N–H and CS groups enhance with time, revealing the formation of Ph-CSNH2 during the reaction.
 | ||
| Fig. 5 (a) Quasi-in situ IR and (b and c) Quasi-in situ Raman spectra of PhCN hydrogenation to PhCSNH2 in the presence of N–GC-6 recorded within 3500–600 cm−1 by flowing H2S (5 wt%) as a reaction gas; (d) free energy diagram for the *PhCNH, *PhCSHNH and *PhCSNH2 formation process on pyridinic N; and (e) catalytic reaction mechanism of H2S nucleophilic addition of PhCN to PhCSNH2 over N–GC-6. | ||
Quasi-in situ Raman spectra of H2S and PhCN co-adsorption on N–GC-X have also been recorded to shed light on the reaction process (Fig. 5b and c). The characterization was conducted under experimental conditions, i.e., atmospheric pressure, T = 60 °C, using 10 mL PhCN + 5 wt% H2S (balance N2). Before evaluation, 10 mL of PhCN and 40 mg of the catalyst were carefully added into a reaction tank, followed by the continuous introduction of 5 wt% H2S into the reaction system. It is observed that the intensity of the characteristic peak at 2230 cm−1 associated with ν(CN) is decreased with reaction time,39 while the intensity of the –NH2 group (3195 cm−1) is enhanced. The phenomenon indicates that H atoms in H2S are easily added to PhCN to form NH/NH2 groups, demonstrating a pronounced nucleophilic addition of H2S. Additionally, a band at 2576 cm−1 attributed to the generation of the *HS intermediate is detected,40 further validating the nucleophilic addition between H2S and PhCN.
Furthermore, four bands located at 377 cm−1, 459 cm−1, 550 cm−1 and 1490 cm−1 can be attributed to the ν(CS) and C–S unit vibrations,41,42 respectively. The intensity of these peaks gradually enhances with reaction time, indicating the enrichment of CS species on the catalyst surface. The above results suggest that PhCN is first reduced to *PhCNH by dissociated *H in H2S. *PhCNH is then attacked by *HS to generate *PhCSNHNH, which finally rearranges into *PhCSNH2. The results of Quasi-in situ IR and Quasi-in situ Raman characterization are consistent with the calculated activation barriers for the formation of *PhCNH, *PhCSHNH and *PhCSNH2 intermediates on the pyridinic N (Fig. 5d), which reveals that *PhCSHNH is the rate-limiting intermediate in this reaction. Overall, the formation of structural base sites is the main contributor to high activity and product selectivity in the H2S nucleophilic addition reaction (Fig. 5e).
Conclusions
In this work, holey nitrogen-doped carbons with various coordination configurations and contents of N have been synthesized to explore the active centers for the H2S-nucleophilic addition reaction. Results of in situ spectroscopic characterization and theoretical calculations reveal that C-pyridinic N coordination sites can effectively reduce the adsorption energy barrier of the key intermediate *PhCSHNH. As a result, the C-pyridinic N sites are responsible for the excellent performance of N–GC-X catalysts in PhCSNH2 synthesis under mild reaction conditions, exhibiting a high PhCSNH2 synthesis rate of 26400 μmolPhCSNH2 L−1 h−1. In addition, the as-designed catalyst shows easy recyclability with sustained high stability over ten cycles over a period of 6 months. The present work unveils the catalytic nature of nitrogen-doped carbon and provides guidance for designing metal-free carbon catalysts for the high-economic utilization of H2S.
Experimental section
Chemicals and reagents
Dicyandiamide (≥98.0%) and glucose (≥99.0%) were purchased from Sinopharm Chemical Reagent (Shanghai, China). Ethanol (>99.5%), N,N-dimethylformamide (DMF, ≥99.9%), dimethyl sulfoxide (DMSO, >99.8%) and dimethylacetamide (DMAC, ≥99.9%) were collected from Sinopharm Chemical Reagents, China. Isopropylamine (C3H9N, ≥99.0%), benzonitrile (C7H5N, ≥99.0%), 4-methoxybenzonitrile (C8H7NO, ≥99.0%), 4-chlorobenzonitrile (C7H4ClN, ≥98.0%), 4-hydroxybenzonitrile (C7H5NO, ≥98.0%), 4-ethylbenzonitrile (C9H9N, ≥98.0%), 4-bromobenzonitrile (C7H4BrN, ≥97.0%), 4-fluorobenzonitrile (C7H4FN, 99.0%), 2-fluorobenzonitrile (C7H4FN, ≥99.0%), 4-phenoxybenzonitrile (C13H9NO, ≥98.0%), 4-amino-2-chlorobenzonitrile (C7H5ClN2, 99.0%), acetonitrile (C2H3N, 99.8%), 2-methylpropanenitrile ((CH3)2CHCN, 99.6%) and ethyl cyanoformate (C3H5NO2, 98%) were collected from Aladdin (Shanghai) Chemistry Co., Ltd. Deionized water was provided from local sources. N2 gas (≥99.9%) was purchased from Linde Industrial Gases (Xiamen, China). A reaction gas containing H2S (50000 ppm) and N2 (balance) was supplied from Dalian Special Gases (Dalian, China). All chemicals were used as received without further treatment.
Synthesis of samples
N–GC-X samples were prepared by carbonizing the mixture of dicyandiamide and glucose. Typically, 1 g of glucose with designed amounts of dicyandiamide (i.e., 0, 2, 4, 6 and 8 g) was mixed well, and then the mixture was transferred into a tube furnace under flowing N2. The temperature of the tube furnace experienced the following heating procedures: from room temperature to 600 °C (2.3 °C min−1), from 600 °C to 780 °C (2 °C min−1), and maintained at 780 °C for 60 min. The collected N–GC-X are herein denoted as pristine GC, N–GC-2, N–GC-4, N–GC-6, and N–GC-8 for dicyandiamide = 0, 2, 4, 6, and 8 g, respectively.Characterization
The details of scanning electron microscopy (SEM), transmission electron microscopy (TEM), BET surface area analysis, X-ray diffraction (XRD), Raman, FT-IR, 13C-NMR, and X-ray photoelectron spectroscopy (XPS) methods, elemental analysis (EA), and CO2 temperature-programmed desorption (CO2/O2-TPD) are shown in the ESI.†In situ characterization
Quasi-in situ diffuse reflection infrared Fourier transform spectroscopy (Quasi-in situ DRIFTS): Quasi-in situ DRIFTS were recorded using an ALPHA II spectrometer in the range of 4000–650 cm−1 with 16 scans at a spectral resolution of 2 cm−1. Firstly, benzonitrile (10 mL) and catalyst (40 mg) were carefully added into a 100 mL three-necked flask. Then, the reaction system was purged with 5 wt% H2S for three times. A gas mixture containing 5 wt% H2S and balance N2 was fed into the reactor under a flow rate of 20 mL min−1 at 60 °C for 30 min. Afterward, continuous sampling was conducted every two minutes and the samples were quickly transferred to an ALPHA II spectrometer for testing. Finally, DRIFTS spectra were collected. Quasi-in situ Raman spectra were collected using a Renishaw spectrometer laser beam (λ = 532 nm). Accordingly, the reaction studies were carried out at 60 °C for 16 min.DFT calculations
We have employed the Vienna Ab initio Simulation Package (VASP)43,44 to perform all the density functional theory (DFT) calculations within the generalized gradient approximation (GGA) using the PBE45 formulation. We have chosen the projected augmented wave (PAW) potentials46,47 to describe the ionic cores and take valence electrons into account using a plane wave basis set with a kinetic energy cutoff of 520 eV. Partial occupancies of the Kohn–Sham orbitals were allowed using the Gaussian smearing method and a width of 0.05 eV. The electronic energy was considered self-consistent when the energy change was smaller than 10−5 eV. Geometry optimization was considered convergent when the force change was smaller than 0.02 eV Å−1. Grimme's DFT-D3 methodology48,49 was used to describe the dispersion interactions. The free energy of a gas phase molecule or an adsorbate on the surface was calculated using the equation G = E + ZPE − TS, where E is the total energy, ZPE is the zero-point energy, T is the temperature in kelvin (298.15 K is set here), and S is the entropy.Catalyst measurement
Typical procedure for the synthesis of thiobenzamide: firstly, the substrate (benzonitrile and its derivatives, 4 mmol), the solvent (DMAC, 40 mL), isopropylamine (1 mL) and the catalyst (40 mg) were carefully added into a 100 mL three-necked flask. Then, the reaction system was purged with 5 wt% H2S for three times. A gas mixture containing 5 wt% H2S and balance N2 was fed into the reactor at a flow rate of 20 mL min−1. Subsequently, the reaction was allowed to occur at 30–100 °C with a constant stirring speed of 800 rpm and maintained for 0.5–2 h. After the reaction, the liquid phase of the reaction mixture was analyzed by gas chromatography with a flame ionization detector (Shimadzu, GC-2010Plus). The conversion (Con.) and selectivity (Sel.) are defined as follows:

For the reusability evaluation, the catalysts were separated by centrifugation after the end of the reaction and washed with ethanol three times, followed by drying in vacuo before the next catalytic cycle.
Author contributions
G. Lei, L. Shen and L. Jiang conceived the project and designed the experiment. G. Lei, J. Wang and X. Liu carried out material characterization and tests. G. Lei wrote the initial manuscript with the input of all authors. S. Wang, S. Liang and Y. Zhan participated in the data analysis. All authors participated in the discussion of the data. L. Shen, Y. Zhan and L. Jiang conducted supervision, formal analysis, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.† Experimental procedures, catalyst characterization, reaction activity, and DFT calculations (Fig. S1–S26 and Tables S1 and S2) are provided in the ESI.†Acknowledgements
This work was funded by the National Natural Science Foundation of China (22208053, 22178057, 22278073, and 22208055), the Natural Science Foundation of Fujian Province (2023J05113 and 2022J05131), the Natural Science Foundation for Distinguished Young Scholars of Fujian Province (2024J010026), and the Fuzhou University Undergraduate Research Training Program (SRTP).Notes and references
- Z. Liu, F. Huang, M. Peng, Y. Chen, X. Cai, L. Wang, Z. Hu, X. Wen, N. Wang, D. Xiao, H. Jiang, H. Sun, H. Liu and D. Ma, Nat. Commun., 2021, 12, 6194 CrossRef PubMed.
- J. Zhang, Y. Liu, Q. Jia, Y. Wang, Y. Ma and M. Szostak, Org. Lett., 2022, 22, 6884–6890 CrossRef.
- X. Wang, S. Xu, Y. Tang, M. J. Lear, W. He and J. Li, Nat. Commun., 2023, 14, 4626 CrossRef CAS PubMed.
- P. Devendar and G.-F. Yang, Top. Curr. Chem., 2017, 375, 82 CrossRef PubMed.
- K. A. Scott and J. T. Nijardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef.
- N. Wang, P. Saidhareddy and X. Jiang, Nat. Prod. Rep., 2020, 37, 246–275 RSC.
- K. Xu, Z. Li, F. Cheng, Z. Zou, T. Wang, M. Wang and L. Liu, Org. Lett., 2018, 20, 2228–2231 CrossRef CAS.
- R. Maini, H. Kimura, R. Takatsuji, T. Katoh, Y. Goto and H. Suga, J. Am. Chem. Soc., 2019, 141, 20004–20008 Search PubMed.
- M. Saito, S. Murakami, T. Nanjo, Y. Kobayashi and Y. Takemoto, J. Am. Chem. Soc., 2020, 142, 8130–8135 CrossRef CAS PubMed.
- Y. Wang, X. Chen, H. Shi and J. A. Lercher, Nat. Catal., 2023, 6, 204–214 Search PubMed.
- M. S. Shah, M. Tsapatsis and J. I. Siepmann, Chem. Rev., 2017, 117, 9755–9803 Search PubMed.
- W. Xiong, M. Shi, X. Zhang, Z. Tu, X. Hu and Y. Wu, Green, Chem., 2021, 23, 7969–7975 RSC.
- X. Zheng, G. Lei, S. Wang, L. Shen, Y. Zhan and L. Jiang, ACS Catal., 2023, 13, 11723–11752 CrossRef CAS.
- M. Zhang, J. Guan, Y. Tu, S. Chen, Y. Wang, S. Wang, L. Yu, C. Ma, D. Deng and X. Bao, Energy Environ. Sci., 2020, 13, 119–126 RSC.
- F. Zhao, C. Wang, R. Xiong, X. Dai, B. Sa, C. Yang, G. Xu and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202314891 CrossRef CAS PubMed.
- G. Lei, Z. Yao, J. Qu, J. Chen, L. Shen, X. Zheng, S. Wang, Y. Cao and Y. Zhan, Chem. Eng. J., 2024, 485, 150007 CrossRef CAS.
- G. Lei, S. Qi, H. Li, Y. Xue, L. Shen, X. Zheng, S. Wang, Y. Cao and Y. Zhan, Phys. Chem. Chem. Phys., 2023, 25, 32317–32322 RSC.
- W. Ma, H. Wang, W. Yu, X. Wang, Z. Xu, X. Zong and C. Li, Angew. Chem., Int. Ed., 2018, 57, 3473–3477 CrossRef CAS.
- C. Xu, Q. Gu, S. Li, J. Ma, Y. Zhou, X. Zhang, C. Jiang, C. Pham-Huu and Y. Liu, ACS Catal., 2021, 11, 8591–8604 CrossRef CAS.
- B. Yang, D. Guo, P. Lin, L. Zhou, J. Li, G. Fang, J. Wang, H. Jin, X. Chen and S. Wang, Angew. Chem., Int. Ed., 2022, 61, e202204327 CrossRef CAS PubMed.
- Y. Sun, J. Wang, T. Shang, Z. Li, K. Li, X. Wang, H. Luo, W. Lv, L. Jiang and Y. Wan, Angew. Chem., Int. Ed., 2023, 62, e202306791 CrossRef CAS PubMed.
- J. Li, N. Huang, M. Lv, N. Su, C. Li, Y. Huang, Y. Wang, Y. Zheng, W. Liu, T. Ma and L. Ye, EES Catal., 2023, 1, 987–997 RSC.
- A. Ly, E. Murphy, H. Wang, Y. Huang, G. Ferro, S. Guo, T. Asset, Y. Liu, I. V. Zenyuk and P. Atanassov, EES Catal., 2024, 2, 624–637 RSC.
- W. Zhang, J. Yin, C. Chen and X. Qiu, Chem. Eng. Sci., 2021, 241, 116709 CrossRef CAS.
- B. Guo, W. Du, T. Yang, J. Deng, D. Liu, Y. Qi, J. Jiang, S.-J. Bao and M. Xu, Adv. Sci., 2020, 7, 1902617 CrossRef CAS PubMed.
- J.-H. Feng, F. Lu, Z. Chen, M.-M. Jia, Y.-L. Chen, W.-H. Lin, Q.-Y. Wu, Y. Li, M. Xue and X.-M. Chen, Chem. Sci., 2024, 15, 9057–9565 Search PubMed.
- F. Liu, K. Huang, A. Zheng, F.-S. Xiao and S. Dai, ACS Catal., 2018, 8, 372–391 CrossRef CAS.
- F. Liu, K. Huang, Q. Wu and S. Dai, Adv. Mater., 2017, 29, 1700445 CrossRef PubMed.
- S. Ali, A. Wisal, M. N. Tahir, A. D. Abdullah, A. Ali, S. Hameed and M. N. Ahmed, J. Mol. Struct., 2020, 1210, 128035 CrossRef CAS.
- Y. Pei, A.-Y. Chang, X. Liu, H. Wang, H. Zhang, A. Radadia, Y. Wang, W. W. Yu and S. Wang, AIChE J., 2021, 67, e17132 CrossRef CAS.
- X. Yu, C. Zhao, L. Yang, J. Zhang and C. Chen, EES Catal., 2024, 2, 811–822 RSC.
- G. Lei, X. Lin, H. Yan, L. Shen, S. Wang, S. Liang, Z.-J. Zhao, F. Liu, Y. Zhan and L. Jiang, ACS Catal., 2024, 14, 17103–17112 CrossRef CAS.
- Z.-Y. Li, F. Horn, Y. Li, L.-H. Mou, W. Schollkopf, H. Chen, S.-G. He and K. R. Asmis, Chem. – Eur. J., 2023, 29, e202203384 CrossRef CAS PubMed.
- X. Zheng, Y. Li, W. You, G. Lei, Y. Cao, Y. Zhang and L. Jiang, Chem. Eng. J., 2022, 430, 132917 CrossRef CAS.
- P. Pietrzyk, K. Gora-Marek, T. Mazur, B. Mozgawa, M. Radon, M. Chiesa, Z. Zhao and Z. Sojka, J. Catal., 2021, 394, 206–219 CrossRef CAS.
- T. Han, X. Cao, H.-C. Chen, J. Ma, Y. Yu, Y. Li, W. Xu, K. Sun, A. Huang, Z. Chen, C. Chen, H. Zhang, B. Ye, Q. Peng and Y. Li, Angew. Chem., Int. Ed., 2023, 135, e202313325 CrossRef.
- J. Madarász and G. Poko, J. Therm. Anal. Calorim., 2007, 88, 329–336 CrossRef.
- A. Das, P. Sarkar, S. Maji, S. K. Pati and S. K. Mandal, Angew. Chem., Int. Ed., 2022, 61, e202213614 CrossRef CAS PubMed.
- P. Marabotti, M. Tommasini, C. Castiglioni, S. Peggiani, P. Serafini, B. Rossi, A. L. Bassi, V. Rosso and C. S. Casari, Carbon, 2024, 216, 118503 CrossRef CAS.
- E. T. Peltzer, X. Zhang, P. M. Walz, M. Luna and P. G. Brewer, Mar. Chem., 2016, 184, 32–42 CrossRef CAS.
- K. Sambathkumar, Spectrochim. Acta, Part A, 2015, 147, 51–66 CrossRef CAS PubMed.
- S. Lyu, J. Wang, Y. Zhou, C. Wei, X. Liang, Z. Yu, W. Lin, Y. Hou and C. Yang, Adv. Funct. Mater., 2023, 34, 2310286 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 Search PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvst, J. R. Kitchin, T. Bligaard and H. Jønsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ey00110b |
| This journal is © The Royal Society of Chemistry 2025 |