
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmployment of a C(sp3)-based nucleophile for photoinduced palladium-catalysed cross-coupling†
Beom Ho
Kim‡
 ,
Byeongdo
Roh‡
,
Dong Jun
Kim
and
Hong Geun
Lee
*
,
Byeongdo
Roh‡
,
Dong Jun
Kim
and
Hong Geun
Lee
*
Department of Chemistry, Seoul National University, Seoul 08826, Republic of Korea. E-mail: hgleee@snu.ac.kr
First published on 11th June 2025
Abstract
The development of efficient methods for C(sp3)–C(sp3) bond formation remains a longstanding challenge in synthetic chemistry, especially in palladium catalysis employing sterically bulky electrophiles. In this study, we present a novel approach for achieving C(sp3)–C(sp3) cross-coupling via photoinduced palladium catalysis, employing cyclopropanols as masked C(sp3)-nucleophiles. Leveraging the unique reactivity of photoexcited palladium, this protocol enables radical-mediated C(sp3)–C(sp3) coupling across a broad range of substrates including sterically hindered and functionally diverse alkyl halides under mild conditions. This method significantly expands the extent of palladium-catalysed cross-coupling for bond construction between sp3-hybridized carbon units, providing streamlined access to structurally complex C(sp3)-rich frameworks that are crucial for medicinal chemistry.
Introduction
The strategic formation of carbon–carbon bonds is one of the most important processes in the synthesis of valuable organic molecules. In this sense, transition metal-catalysed processes have gained considerable attention due to their convergence in nature and high efficiency.1 Among many types, palladium-catalysed cross-coupling has been one of the most popularized methods in forging various types of carbon–carbon bonds due to the facile generation of the reactive palladium(0) catalyst, redox-neutrality, and synthetic robustness.2 Conventional catalytic methods of this kind have predominantly relied on the use of C(sp2)-based coupling partners as nucleophiles and/or electrophiles.3 However, palladium-catalysed C(sp3)–C(sp3) coupling remains severely limited, especially in terms of employing sterically demanding electrophiles with sp3 hybridization (Fig. 1A).3c,f This is mainly due to the inherent mechanistic restrictions of C(sp3)-based electrophiles, which undergo inefficient oxidative addition and compete with rapid β-hydride elimination (Fig. 1B, left).3c,f,4,5 Therefore, extending cross-coupling to the functionalization of C(sp3)-based building blocks has relied on the use of other reactivity categories, such as early transition metal catalysis, photoredox catalysis or electrochemical methods.6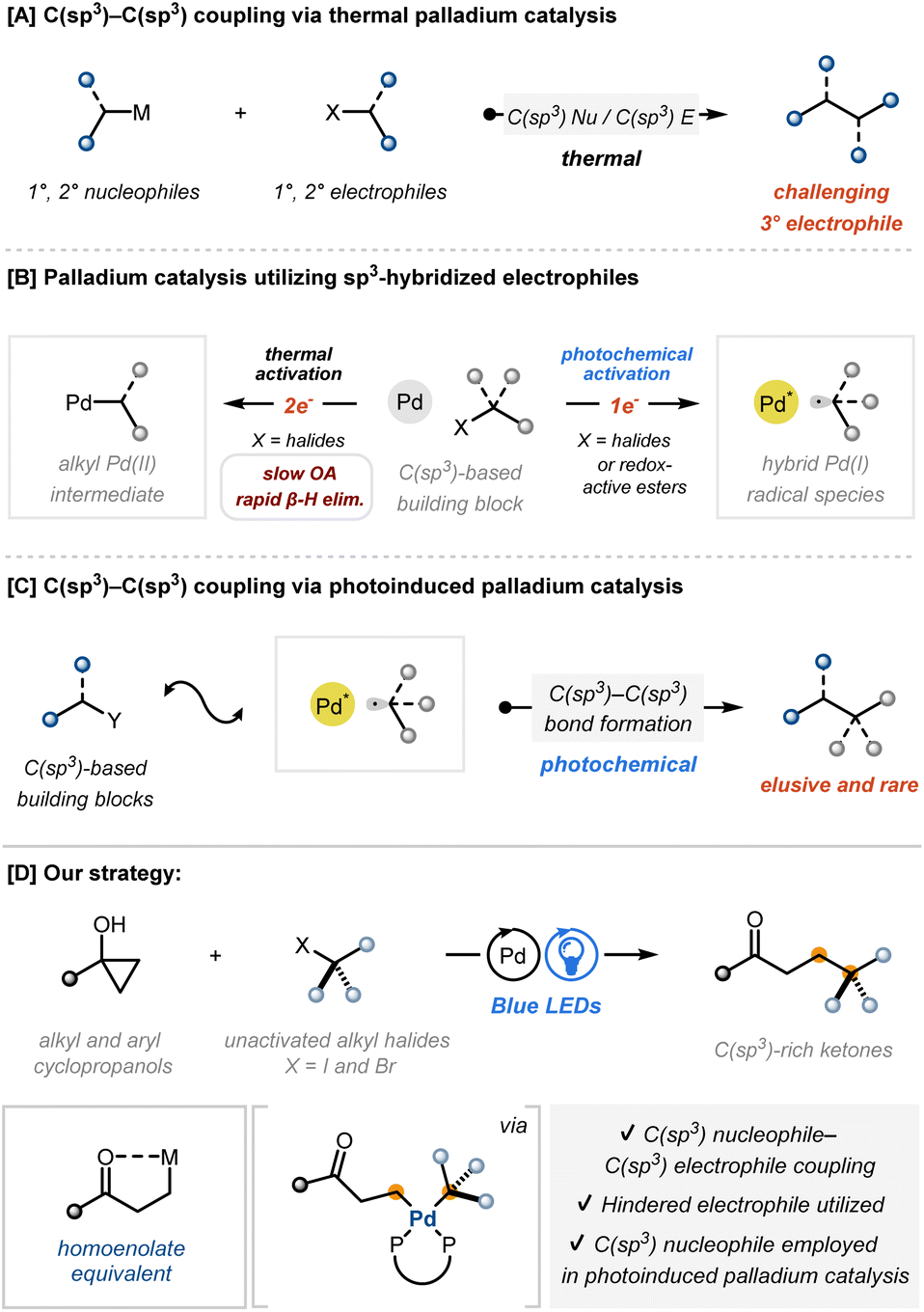 | ||
| Fig. 1 State of the art for palladium-catalysed C(sp3)–C(sp3) cross-coupling. | ||
Photoinduced palladium catalysis has recently emerged as a powerful tool to overcome the limitations of traditional methods that require the activation of C(sp3)-based electrophiles.7 Excited-state palladium complexes, generated by visible light irradiation, can activate C(sp3)-based precursors to form hybrid alkyl radical palladium species via single-electron oxidative addition (Fig. 1B, right). Subsequently, it serves as a valuable radical-based reaction partner for challenging bond-forming transformations.8–11 Nonetheless, the applications of photoinduced palladium catalysis for the formation of C(sp3)–C(sp3) bonds are still limited and the reactivity of hybrid alkyl radical palladium with C(sp3)-based nucleophilic coupling partners remains rare and elusive (Fig. 1C).12
Recognizing the critical importance of introducing sp3-hybridized carbon nucleophiles, the potential of cyclopropanols as latent homoenolate equivalents was investigated in the context of photoinduced palladium catalysis. Cyclopropanols serve as three-carbon synthons by undergoing ring-opening processes and have been utilized as valuable synthetic building blocks to accomplish the formation of various carbon–carbon bonds.13,14 In the present study, cyclopropanols were utilized as masked C(sp3)-based nucleophiles, facilitating the efficient formation of C(sp3)–C(sp3) bonds in conjunction with photoinduced palladium catalysis to achieve the activation of challenging electrophiles under mild reaction conditions (Fig. 1D). This approach underscores the potential of photoinduced palladium catalysis to induce an unprecedented bond formation event with a formally nucleophilic counterpart.
Results and discussion
We commenced our investigations by cross-coupling of phenylcyclopropanol 1a with commercially available tert-butyl bromide 2a in the presence of a palladium catalyst and visible light irradiation (456 nm) (Table 1).15 It has been shown that rac-BINAP is the optimal supporting ligand for the palladium catalyst (entries 2–5). In addition, the use of other palladium sources decreased the reaction efficiency, regardless of the initial oxidation state of palladium precatalysts (entry 6). In the case of the base additive, other inorganic bases were not as effective as cesium carbonate (entry 7), and similar observations were made with amine-based organic bases (entry 8). Additional control experiments demonstrated that all reaction components, including palladium precatalyst, base, and the irradiation of light are essential for optimal reactivity (entries 9–11). Moreover, it was found that the use of 10 mol% of palladium catalyst and 20 mol% of ligand is required for the best performance of the system (entry 12). Finally, the protocol was smoothly performed in a larger scale (entry 13).|
|
||
|---|---|---|
| Entrya | Deviation from standard conditions | Yieldb |
| a Reaction conditions: 1a (0.1 mmol), 2a (2.0 equiv.), Pd(OAc)2 (10 mol%), rac-BINAP (20 mol%), Cs2CO3 (2.0 equiv.) in DCM (0.6 mL), rt, N2, 18 h, irradiated with blue LEDs (456 nm). b Determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard; isolated yields given in parentheses. | ||
| 1 | None | 90 (85) |
| 2 | No rac-BINAP | 45 |
| 3 | XantPhos instead of rac-BINAP | 41 |
| 4 | t-Bu XantPhos instead of rac-BINAP | 15 |
| 5 | DPEPhos instead of rac-BINAP | 28 |
| 6 | PdI2, PdCl2(PhCN)2, PdCl2, Pd2dba3 | 74, 60, 39, 6 |
| 7 | Li2CO3, Na2CO3, K2CO3, CsOAc, CsOPiv | 30, 7, 7, 54, 36 |
| 8 | TEA, DIPEA | 28, 28 |
| 9 | No Pd(OAc)2 | <5 |
| 10 | No Cs2CO3 | 8 |
| 11 | No light irradiation | <5 |
| 12 | Pd/L = 5/10 mol% | 53 |
| 13 | 2.0 mmol scale | (88) |

|
||

After establishing the optimal reaction conditions, we began to evaluate the applicability of the developed method (Table 2). Initially, we investigated the functional group tolerance of our system with a variety of substituted cyclopropanols. Cyclopropanols containing an electron-donating group, such as methyl (3ab), methoxy (3ac), or dimethylamino groups (3ad), were found to be effective reaction partners to afford the cross-coupling products. Moreover, cyclopropanols with an electron-withdrawing group (3ae) also underwent the desired transformation smoothly. Halogen-substituted substrates (3af, 3ag) were also compatible with the reaction conditions, indicating the orthogonal characteristics of the protocol with aryl halides. Cyclopropanol pronucleophiles with an extended π-system, such as naphthyl (3ah) or vinyl (3ai) moieties, also participated in the reaction with high efficiency. Notably, further addition reaction did not occur to the corresponding α,β-unsaturated carbonyl product, demonstrating the mildness of the developed protocol (3ai). Moreover, medicinally relevant heterocycles, including indole (3aj), pyrrole (3ak) or thiophene (3al), were well-tolerated during the transformations. Finally, various aliphatic cyclopropanols bearing cyclopentyl (3am), tetrahydropyranyl (3an), n-pentyl (3ao) or benzylic (3ap) substituents were also efficiently coupled with electrophiles, affording C(sp3)-rich ketones. In contrast, β-substituted cyclopropanols failed to participate effectively, presumably due to increased steric hindrance (ESI Table S5†).
| a All reactions were performed on a 0.2 mmol scale. |
|---|

|
Subsequently, we surveyed the generality of the method in terms of the electrophilic reaction partner to find that a wide range of alkyl halides — primary, secondary, or tertiary — can readily undergo the desired transformation. Various sterically hindered tertiary halides performed well in this transformation (3bb–3bd). Additionally, cyclic secondary alkyl halides containing cyclopentyl (3be), cyclohexyl (3bf), tetrahydropyranyl (3bg) or piperidinyl (3bh) moieties provided the cross-coupling products with good to excellent yields. Additionally, an acyclic secondary alkyl halide (3bi) was shown to be compatible with the protocol. Lastly, primary alkyl halides were also successfully engaged in the photoinduced palladium catalysis, demonstrating the generality of the method. Primary alkyl halides substituted with arene (3bj), tetrahydrofuran (3bk) or silyl ether (3bl) were viable substrates for the reaction. Also, synthetically useful functional groups such as carboethoxy (3bm), cyano (3bn) or boronic acid pinacol ester (3bo) groups were shown to be compatible with the reaction conditions. Notably, alkyl substrates containing multiple carbon–halogen bonds afforded the desired product in a chemo-selective manner (3bp). Additionally, alkyl halides bearing α-heteroatom (3bq) or α-carbonyl (3br) substituents were efficiently coupled with cyclopropanols.
The applicability of the developed method toward complex bioactive architectures was also evaluated (Scheme 1A). A series of drug molecules and natural products, such as naproxen (4a), adapalene (4b) and cholic acid (4c), could be conveniently converted to the corresponding cyclopropanols. By subjecting the complex cyclopropanol pronucleophiles to the developed reaction protocol, the extended ketone derivatives of the parent precursors could be successfully prepared. Additionally, complex alkyl halides derived from bioactive molecules, such as indomethacin (4d), menthol (4e), or cholesterol (4f), were also proved to be competent coupling partners in this transformation, affording C(sp3)-rich architectures.
 | ||
| Scheme 1 Synthetic utility. | ||
To further highlight the synthetic utility of the protocol, various downstream transformations of the β-alkylated ketone products were attempted (Scheme 1B). A substituted δ-keto ester (5aa), which could be conveniently prepared using the developed method, was subjected to either enantioselective carbonyl or imine reduction conditions to yield chiral δ-hydroxy (5ab) or δ-amino ester products (5ae), respectively (paths a and b). Subsequent acid- or base-mediated cyclization afforded lactone (5ac) and lactam (5af) products in enantiopure form. Through this divergent synthetic sequence, pharmaceutically important scaffolds could be readily synthesized, which would otherwise be difficult to access with conventional reactivity of ester enolates. Moreover, the carboxyl group of the product could be further exploited as a functional handle to furnish more complex molecular architectures. Another substituted δ-keto ester product (5ba) underwent straightforward hydrolysis to give the corresponding δ-keto acid (5bb). The reactive intermediate could be advanced to the amino ketone product (5bd) by Curtius rearrangement (path c). Alternatively, the carboxylic acid intermediate could be converted to its redox-active ester form (5be) to undergo a further electron-donor-acceptor (EDA) complex-mediated Giese addition (5bf, 5bg) (path d).16 Importantly, through the use of carboxylate as a secondary functional handle, it was possible to construct product structures that are either difficult to access using a halide-based precursor, due to their susceptibility to elimination (5bd), or resistant to undergo the desired transformation under the developed conditions (5bg).
The synthetic versatility of the developed method was further examined by evaluating its chemical orthogonality (Scheme 1C). Cyclopropanols bearing either a bromine (1q) or a boronic acid ester (1r) on its aryl substituent were subjected to the developed reaction conditions to furnish the cross-coupling products (6aa, 6ba) (Scheme 1C, a). The coupling products with an existing functional handle could be successfully converted into more complex biaryl ketones (6ab, 6bb) by the thermally-induced palladium-catalysed C(sp2)–C(sp2) cross-coupling. Analogously, alkyl halide electrophiles containing identical functional handles could be further elaborated in a similar manner (Scheme 1C, b). These examples demonstrate that the developed method is an orthogonal strategy that can be coupled with the conventional cross-coupling reactions based on the Pd(0)/Pd(II) cycle.
To gain mechanistic insight into the developed synthetic strategy, a number of control experiments were conducted (Scheme 2). The presence of 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO), a radical scavenger, significantly inhibited reactivity, and the cross-coupled product (3ap) was not formed at all (Scheme 2A). Instead, TEMPO adducts derived from both cyclopropanol and alkyl halide were observed. At this point, it can be speculated that both the formally nucleophilic homoenolate, originating from the cyclopropanol precursor, and the alkyl counterpart participate in the catalytic cycle in the form of radical species. Furthermore, the incorporation of electron-rich or -deficient olefins, such as p-methoxystyrene or 2-benzylidenemalononitrile, as radical acceptors led to the formation of radical addition products from the alkyl halide precursors (7a and 7b) (Scheme 2B). In addition, competition experiments based on the simultaneous usage of the tertiary and secondary alkyl bromide substrates yielded only the cross-coupled product derived from the tertiary halide (3ab), suggesting the involvement of a thermodynamically more stable tertiary radical (Scheme 2C). These results reinforced the involvement of intermediary alkyl radical species.
 | ||
| Scheme 2 Mechanistic studies and proposed mechanism. | ||
Based on the mechanistic experiments and existing literature precedents,14c,d,17 a plausible reaction mechanism is depicted (Scheme 2D). Initially, alkyl halides undergo a single electron transfer mediated by a photoexcited Pd(0) complex A to generate hybrid alkyl Pd(I) radical B1, which is in equilibrium with ionic oxidative addition intermediate B2. Then, ligand exchange of the halide ligand with cyclopropanol results in the formation of a Pd(I) cyclopropoxide species C. The subsequent β-carbon elimination step produces palladium(I) homoenolate intermediate D. Finally, C(sp3)–C(sp3) bond-forming radical-induced reductive elimination occurs, yielding the β-alkylated ketone product and regenerating the Pd(0) catalyst. Indeed, alkyl palladium(I) complex D leads to the formation of alkyl palladium(II) complex D2 and hybrid β-keto radical palladium(I) species D1, which are in equilibrium with the corresponding palladium(I) intermediate, as supported by mechanistic investigations (Scheme 2A). The presence of the coupled product was detected when a bicyclic cyclopropanol was used as a mechanistic probe; nevertheless, the possibility of a radical ring-opening pathway cannot be completely ruled out at this stage (ESI Table S5†).
Conclusions
In conclusion, we have developed a photoinduced palladium-catalysed C(sp3)–C(sp3) cross-coupling reaction using cyclopropanols as masked C(sp3) nucleophiles. By strategically utilizing a photoinduced hybrid palladium alkyl radical, the challenging palladium-catalysed C(sp3)–C(sp3) coupling could be accomplished under mild conditions using a diverse range of cyclopropanols and alkyl halides, including sterically hindered tertiary halides. Notably, the method offers a streamlined route to the synthesis of structurally diverse β-alkylated ketones, including the derivatives of complex natural products and drug molecules. We anticipate that this strategy would be a milestone for further advancement in C(sp3)–C(sp3) cross-coupling in palladium catalysis.Data availability
All data supporting this article have been included in the ESI.†Author contributions
H. G. L. supervised and provided guidance to the project. B. R. initiated research project and B. H. K. performed the reaction optimisation. B. H. K. and D. J. K. carried out the synthetic experiments and B. R. and B. H. K. conducted the mechanistic investigations. All the authors analysed and discussed the experimental data. H. G. L., B. H. K. and B. R. prepared the manuscript, and B. H. K. and B. R. prepared the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. RS-2022-NR069773), and by the Global-LAMP Program of the National Research Foundation of Korea (NRF) grant funded by the Ministry of Education (No. RS-2023-00301976).Notes and references
- For reviews, see: (a) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed; (b) W. Shi, C. Liu and A. Lei, Chem. Soc. Rev., 2011, 40, 2761–2776 RSC; (c) F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC; (d) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed; (e) J. H. Docherty, T. M. Lister, G. Mcarthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Chem. Rev., 2023, 123, 7692–7760 CrossRef CAS PubMed.
- V. M. Chernyshev and V. P. Ananikov, ACS Catal., 2022, 12, 1180–1200 CrossRef CAS.
- For reviews, see: (a) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656–2670 CrossRef CAS PubMed; (b) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed; (c) N. Kambe, T. Iwasaki and J. Terao, Chem. Soc. Rev., 2011, 40, 4937–4947 RSC; (d) C.-Y. Wang, J. Derosa and M. R. Biscoe, Chem. Sci., 2015, 6, 5105–5113 RSC; (e) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587–9652 CrossRef CAS PubMed; (f) Z. Qureshi, C. Toker and M. Lautens, Synthesis, 2017, 49, 1–16 CAS; (g) X. Ma, B. Murray and M. R. Biscoe, Nat. Rev. Chem., 2020, 4, 584–599 CrossRef CAS PubMed; (h) B. Roh and H. G. Lee, Synthesis, 2024, 56, 2614–2626 CrossRef CAS.
- (a) H. Chen and M. Z. Deng, J. Org. Chem., 2000, 65, 4444–4446 CrossRef CAS PubMed; (b) T. Brenstrum, D. A. Gerristma, G. M. Adjabeng, C. S. Frampton, J. Britten, A. J. Robertson, J. McNulty and A. Capretta, J. Org. Chem., 2004, 69, 7635–7639 CrossRef CAS PubMed; (c) N. Hadei, G. T. Achonduh, C. Valente, C. J. O'Brien and M. G. Organ, Angew. Chem., Int. Ed., 2011, 50, 3896–3899 CrossRef CAS PubMed; (d) L. C. McCann, H. N. Hunter, J. A. Clyburne and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 7024–7027 CrossRef CAS PubMed; (e) R. A. Lippa, D. J. Battersby, J. A. Murphy and T. N. Barrett, J. Org. Chem., 2021, 86, 3583–3604 CrossRef CAS PubMed.
- For review, see: J. Choi and G. C. Fu, Science, 2017, 356, eaaf7230 CrossRef PubMed.
- For reviews, see: (a) A. C. Frisch and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674–688 CrossRef CAS PubMed; (b) D. A. Everson and D. J. Weix, J. Org. Chem., 2014, 79, 4793–4798 CrossRef CAS PubMed; (c) J. Gu, X. Wang, W. Xue and H. Gong, Org. Chem. Front., 2015, 2, 1411–1421 RSC; (d) E. L. Lucas and E. R. Jarvo, Nat. Rev. Chem., 2017, 1, 0065 CrossRef; (e) A. Claraz and G. Masson, ACS Org. Inorg. Au, 2022, 2, 126–147 CrossRef CAS PubMed; (f) R. Kranthikumar, Organometallics, 2022, 41, 667–679 CrossRef CAS; (g) T. Iwasaki and N. Kambe, Org. React., 2023, 113, 1–456 Search PubMed; (h) A. L. Gabbey, K. Scotchburn and S. A. Rousseaux, Nat. Rev. Chem., 2023, 7, 548–560 CrossRef CAS PubMed; (i) D. Sun, Organometallics, 2024, 43, 1662–1681 CrossRef.
- For reviews, see: (a) M. Parasram and V. Gevorgyan, Chem. Soc. Rev., 2017, 46, 6227–6240 RSC; (b) P. Chuentragool, D. Kurandina and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 11586–11598 CrossRef CAS PubMed; (c) W. J. Zhou, G. M. Cao, Z. P. Zhang and D. G. Yu, Chem. Lett., 2019, 48, 181–191 CrossRef CAS; (d) R. Kancherla, K. Muralirajan, A. Sagadevan and M. Rueping, Trends Chem., 2019, 1, 510–523 CrossRef CAS; (e) W. M. Cheng and R. Shang, ACS Catal., 2020, 10, 9170–9196 CrossRef CAS; (f) K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543–1625 CrossRef PubMed; (g) S. Sarkar, K. P. S. Cheung and V. Gevorgyan, Angew. Chem., Int. Ed., 2024, 63, e202311972 CrossRef CAS PubMed.
- (a) M. Parasram, P. Chuentragool, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2016, 138, 6340–6343 CrossRef CAS PubMed; (b) M. Parasram, P. Chuentragool, Y. Wang, Y. Shi and V. Gevorgyan, J. Am. Chem. Soc., 2017, 139, 14857–14860 CrossRef CAS PubMed; (c) P. Chuentragool, M. Parasram, Y. Shi and V. Gevorgyan, J. Am. Chem. Soc., 2018, 140, 2465–2468 CrossRef CAS PubMed; (d) W. M. Cheng, R. Shang and Y. Fu, Nat. Commun., 2018, 9, 5215 CrossRef PubMed; (e) W. L. Xing, R. Shang, G. Z. Wang and Y. Fu, Chem. Commun., 2019, 55, 14291–14294 RSC; (f) S. Yang, H. Fan, L. Xie, G. Dong and M. Chen, Org. Lett., 2022, 24, 6460–6465 CrossRef CAS PubMed; (g) W. Jin and S. Yu, J. Org. Chem., 2022, 87, 14715–14722 CrossRef CAS PubMed.
- (a) X. Y. Ruan, T. Zhang, W. A. Li, Y. Z. Yin, Z. Y. Han and L. Z. Gong, Sci. China Chem., 2022, 65, 863–869 CrossRef CAS; (b) G. Zhao, U. Mukherjee, L. Zhou, J. N. Mauro, Y. Wu, P. Liu and M. Y. Ngai, CCS Chem., 2023, 5, 106–116 CrossRef CAS PubMed; (c) H. Fang, C. Empel, I. Atodiresei and R. M. Koenigs, ACS Catal., 2023, 13, 6445–6451 CrossRef CAS; (d) N. Kvasovs, J. Fang, F. Kliuev and V. Gevorgyan, J. Am. Chem. Soc., 2023, 145, 18497–18505 CrossRef CAS PubMed; (e) Z. Zhang and V. Gevorgyan, Angew. Chem., Int. Ed., 2023, 62, e202311848 CrossRef CAS PubMed; (f) K. Muralirajan, R. Kancherla, B. Maity, S. Karuthedath, F. Laquai, L. Cavallo and M. Rueping, Nat. Commun., 2023, 14, 6622 CrossRef CAS PubMed; (g) Y. Cai, G. Gaurav and T. Ritter, Angew. Chem., Int. Ed., 2024, 63, e202311250 CrossRef CAS PubMed; (h) Z. L. Liu, Z. P. Ye, Z. H. Liao, W. D. Lu, J. P. Guan, Z. Y. Gao, K. Chen, X. Q. Chen, H. Y. Xiang and H. Yang, ACS Catal., 2024, 14, 3725–3732 CrossRef CAS; (i) G. X. Liu, X. T. Jie, G. J. Niu, L. S. Yang, X. L. Li, J. Luo and W. H. Hu, Beilstein J. Org. Chem., 2024, 20, 661–671 CrossRef CAS PubMed; (j) Y. Liang, T. Bian, K. Yadav, Q. Zhou, L. Zhou, R. Sun and Z. Zhang, ACS Cent. Sci., 2024, 10, 1191–1200 CrossRef CAS PubMed; (k) J. Yang, C. R. Li, X. Guo, Z. Chen, K. Hu and L. X. Li, Org. Lett., 2024, 26, 5110–5114 CrossRef CAS PubMed; (l) H. Han, W. Yi, S. Ding, X. Ren and B. Zhao, Angew. Chem., Int. Ed., 2024, e202418910 Search PubMed.
- (a) Q. Fan, K. Jiang, B. Liu, H. Jiang, X. Cao and B. Yin, Adv. Sci., 2024, 11, 2307074 CrossRef CAS PubMed; (b) L. Liu, J. Huang, Y. Li, Q. Wang, M. Liu, B. Liu and K. Feng, J. Organomet. Chem., 2024, 1008, 123066 CrossRef CAS; (c) S. Senapati, S. K. Hota, L. Kloene, C. Empel, S. Murarka and R. M. Koenigs, Angew. Chem., Int. Ed., 2024, e202417107 Search PubMed.
- (a) I. Abdiaj, L. Huck, J. M. Mateo, A. de la Hoz, M. V. Gomez, A. Díaz-Ortiz and J. Alcázar, Angew. Chem., Int. Ed., 2018, 57, 13231–13236 CrossRef CAS PubMed; (b) Y.-C. Luo, F.-F. Tong, Y. Zhang, C.-Y. He and X. Zhang, J. Am. Chem. Soc., 2021, 143, 13971–13979 CrossRef CAS PubMed; (c) N. Oku, M. Murakami and T. Miura, Org. Lett., 2022, 24, 1616–1619 CrossRef CAS PubMed; (d) J. Lai, Y. Zhang, Y. Zhan, Z. Zhou, Z. Wang, H. Liu, Q. Zhang, J.-S. Sun and L. Wang, Org. Chem. Front., 2024, 11, 5044–5053 RSC.
- (a) W. J. Zhou, G. M. Cao, G. Shen, X. Y. Zhu, Y. Y. Gui, J. H. Ye, L. Sun, L. Liao, J. Li and D. G. Yu, Angew. Chem., Int. Ed., 2017, 56, 15683–15687 CrossRef CAS PubMed; (b) H. Xin, L.-N. Guo, M. Yang, C. Guan, Z.-H. Yuan and X.-H. Duan, Org. Chem. Front., 2023, 10, 1147–1152 RSC.
- (a) D. Rosa, A. Nikolaev, N. Nithiy and A. Orellana, Synlett, 2015, 26, 441–448 CrossRef CAS; (b) A. Nikolaev and A. Orellana, Synthesis, 2016, 48, 1741–1768 CrossRef CAS; (c) L. R. Mills and S. A. L. Rousseaux, Eur. J. Org Chem., 2019, 2019, 8–26 CrossRef CAS; (d) T. R. McDonald, L. R. Mills, M. S. West and S. A. L. Rousseaux, Chem. Rev., 2021, 121, 3–79 CrossRef CAS PubMed; (e) Y. Sekiguchi and N. Yoshikai, Bull. Chem. Soc. Jpn., 2021, 94, 265–280 CrossRef CAS; (f) Y. Sekiguchi and N. Yoshikai, J. Am. Chem. Soc., 2021, 143, 18400–18405 CrossRef CAS PubMed; (g) F. Doraghi, S. Pegah Aledavoud, A. Fakhrioliaei, B. Larijani and M. Mahdavi, ChemistrySelect, 2023, 8, e202301438 CrossRef CAS.
- (a) N. Nithiy and A. Orellana, Org. Lett., 2014, 16, 5854–5857 CrossRef CAS PubMed; (b) Z. Ye, K. E. Gettys, X. Shen and M. Dai, Org. Lett., 2015, 17, 6074–6077 CrossRef CAS PubMed; (c) Z. Ye, X. Cai, J. Li and M. Dai, ACS Catal., 2018, 8, 5907–5914 CrossRef CAS PubMed; (d) L. R. Mills, C. Zhou, E. Fung and S. A. L. Rousseaux, Org. Lett., 2019, 21, 8805–8809 CrossRef CAS PubMed; (e) X. Zhang, S. Cui, S. Wei, M. Zhao, X. Liu and G. Zhang, Org. Lett., 2024, 26, 2114–2118 CrossRef CAS PubMed.
- See the ESI† for details..
- C. Zheng, G. Z. Wang and R. Shang, Adv. Synth. Catal., 2019, 361, 4500–4505 CrossRef CAS.
- G. M. Torres, Y. Liu and B. A. Arndtsen, Science, 2020, 368, 318–323 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data for newly synthesized compounds and other experimental details. See DOI: https://doi.org/10.1039/d5sc02325d |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2025 |