DOI:
10.1039/D5SC03190G
(Edge Article)
Chem. Sci., 2025,
16, 12804-12811
Enhanced recyclability of methacrylic resins by copolymerization or pendant modification using trityl esters†
Received
1st May 2025
, Accepted 9th June 2025
First published on 10th June 2025
Abstract
The conversion of pendant groups into poly(methyl methacrylate) (PMMA) to triphenylmethyl (trityl) esters facilitates thermal depolymerization, enabling the recovery of the monomer, methyl methacrylate (MMA). While PMMA offers potential for chemical recycling through depolymerization, its complete degradation necessitates extreme heating conditions exceeding 400 °C. Conversely, a copolymer consisting of MMA (95 mol%) and trityl methacrylate (TMA; 5 mol%), synthesized via free radical copolymerization, undergoes depolymerization at 270 °C, yielding pure MMA with 94.5% efficiency. Additionally, commercially available PMMA sheets and modified acrylic resins incorporating n-butyl acrylate as a comonomer were also successfully depolymerized at 270 °C through pendant conversion to trityl esters, achieving high yields of pure MMA.
Introduction
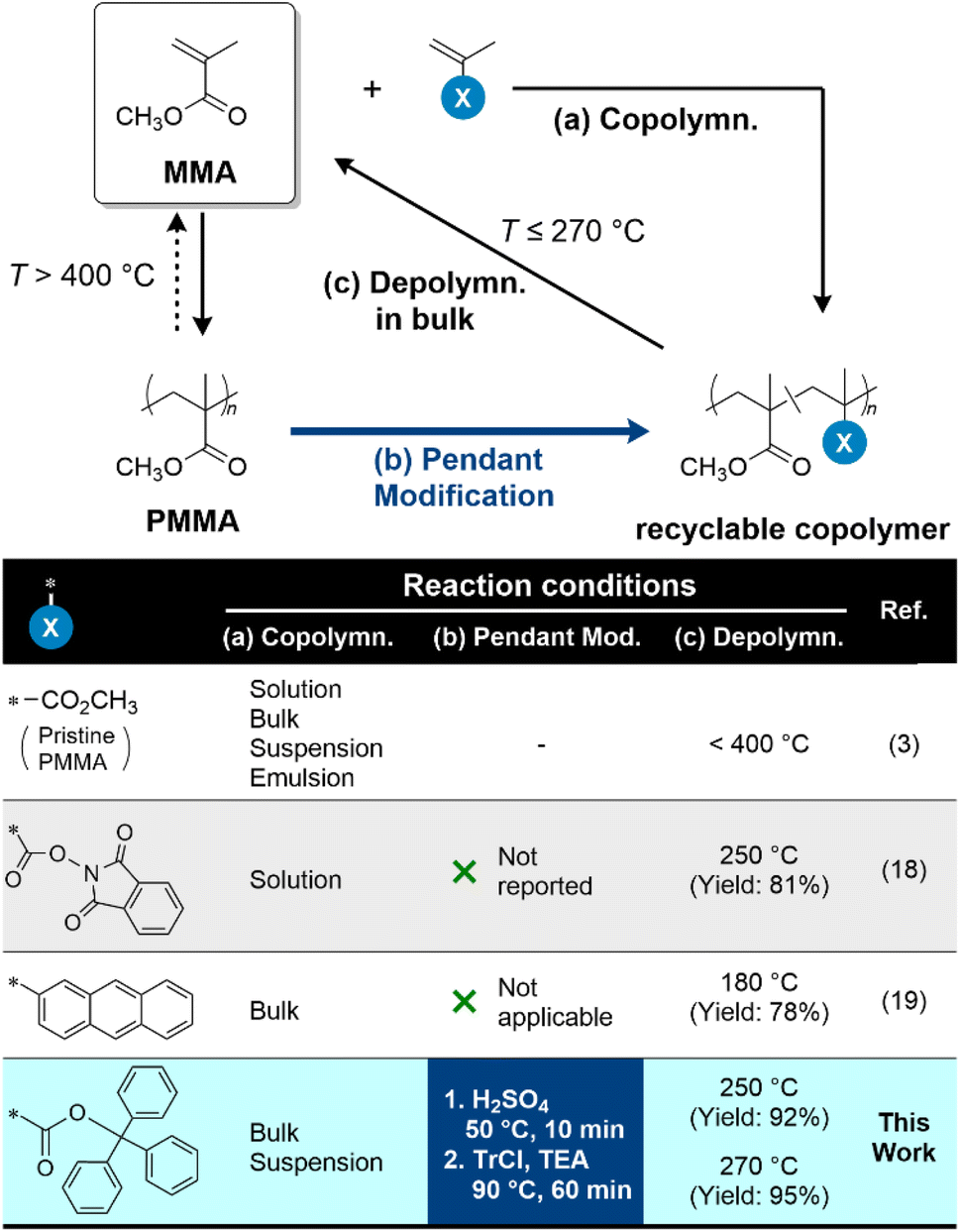
Poly(methyl methacrylate) (PMMA) is industrially produced at approximately 4 million metric tons per year1via free radical polymerization (FRP) under bulk, suspension, or emulsion conditions.2 Since harsh heating conditions (>400 °C) lead to complete degradation to recover the monomer, methyl methacrylate (MMA), via radical depolymerization (Fig. 1),3 PMMA has attracted attention as a promising vinyl polymer for resource circulation.4 However, depolymerization under severe conditions resulted in the generation of byproducts3 and high energy costs.5
 |
| | Fig. 1 Partial replacement approach for PMMA to recover the monomer, MMA, under mild heating conditions. | |
In general, depolymerization requires both the generation of active species and an equilibrium shift to depolymerization. The conditions for generating active species depend on the polymer chain-end structures,6–9 which are classified into several types in FRP. Most heat-resistant chains of PMMA have saturated ends, requiring heating above 300 °C to generate radical active species.6 On the other hand, the ceiling temperature (Tc) of MMA is 202 °C.10 These facts suggest that the active species generation is the energy barrier in inducing depolymerization, and its removal is critical for achieving chemical recycling under mild conditions. To address this issue, the incorporation of chain ends that can easily generate active radical species has been proposed using controlled radical polymerization.11–17 However, depolymerization initiated from the chain ends may not always be effective for PMMA, particularly those with a high degree of polymerization (DP > 103),16 because the active species at the chain ends undergo termination and chain-transfer reactions to lose their reactivity under severe conditions, such as bulk depolymerization. Recently, Sumerlin et al. reported the depolymerization of copolymers consisting of MMA and N-(methacryloxy)phthalimide, a methacryl monomer bearing an active ester.18 The comonomer units generate radicals at the main chain, leading to quantitative depolymerization. Because multiple radical active species are generated per polymer chain, the length of depolymerization per radicals required for quantitative degradation becomes shorter with the copolymerization approach than with the chain-terminating approach. Therefore, this approach was applied to high-DP polymers (DP ∼ 104). Diao et al. reported a similar approach using propenylarenes as comonomers.19 These studies suggest that the incorporation of depolymerization-initiating points in PMMA skeletons effectively imparts facile recyclability.
In addition to producing modified PMMA with improved recyclability, it is also necessary to improve the recyclability of pristine PMMA, already available in the market, to establish a circular society. The primary technical focus is on the mechanisms by which radicals, the active species involved in depolymerization, are generated. Recently, Anastasaki et al. reported the light-induced depolymerization of commercial PMMA in chlorinated solvents.20 In their study, the reaction mechanism was proposed to involve the formation of chlorine radicals from chlorinated solvents upon violet light irradiation, followed by hydrogen-atom transfer from PMMA, leading to the formation of active radical species on the main chains. Consequently, chlorinated solvents act as external radical initiators for depolymerization. Conversely, Sumerlin et al. introduced an internal radical initiator, specifically depolymerization initiation points at chain ends via mechanical main-chain scission and end functionalization.21 As previously mentioned, depolymerization initiated from chain ends may have limitations in applicable DP. Therefore, end-functionalization following main-chain scission is effective for addressing these limitations.
In this study, we focused on triphenylmethyl (trityl) ester pendants, which are promising candidates as depolymerization initiation points. The incorporation of trityl esters into polymethacrylates can be achieved by copolymerization with trityl methacrylate (TMA), a monomer commercially applied for the preparation of a chiral stationary phase for high-performance liquid chromatography (HPLC),22 whereas post-polymerization modification of pristine PMMA also effectively incorporates trityl ester pendants (Fig. 1). Trityl esters cleave radically to carboxylic and trityl radicals above 300 °C.23 In particular, trityl esters of side groups in polyTMA decompose at a temperature of at least 230 °C.24 We applied the thermal decomposition of TMA units to initiate bulk depolymerization, recovering MMA. The incorporation of trityl esters pendants via the classical post-polymerization reaction of PMMA significantly improved their recyclability.
Results and discussion
Enhancement of depolymerization by trityl pendants
To evaluate the effects of trityl esters on the thermal stability, depolymerization behavior, and other physical polymer properties, a series of copolymers with varying TMA content were prepared via bulk free-radical copolymerization (Table 1 and Fig. S1†). The copolymers were coded CP-# with numbers indicating the contents of TMA units in mol%; for example, CP-1 was composed of 1 mol% TMA units and 99 mol% MMA units. Prior to molar mass evaluation by size-exclusion chromatography (SEC), the pendant trityl esters were converted into methyl esters. This approach was necessary because poly(MMA-co-TMA) displayed bimodal peaks in the SEC curves owing to self-aggregation (Fig. S2†).25 The DPs were higher than 103, which were above the limits to allow quantitative depolymerization from the chain ends.16 In other words, the internal generation of radical active species is necessary to achieve quantitative depolymerization from these copolymers.18
Table 1 Synthesis and properties of polymers
| Entry |
Method |
Feed [%] |
Yield [%] |
Comp.e [%] |
M
n
[g mol−1] |
Đ
[°C] |
T
g
[°C] |
T
d95
[°C] |
%Ti |
E
[GPa] |
| MMA |
TMA |
MMA |
TMA |
|
Purchased from Tokyo Chemical Industry Co., Ltd.
Prepared using AIBN (1.7 mol%) at 65 °C for 18 h in bulk.
Prepared using AIBN (0.3 mol%) at 65 °C in a suspension with water.
Prepared from PMMA by pendant modification.
Determined by 1H NMR spectrometry (400 MHz, CDCl3, 25 °C).
Determined by SEC (40 °C, CHCl3, PMMA standard) after the conversion to PMMA.
Determined by DSC.
Determined by TG (under N2, 10 °C min−1).
Determined from the transmittance at 450 nm by UV-vis spectrometry.
Determined by tensile testing.
|
|
HP
|
Purchased PMMAa |
— |
— |
— |
100 |
— |
340![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
2.07 |
99 |
395 |
91.9 |
1.03 ± 0.11 |
|
CP-1
|
Bulk polymn.b |
99 |
1 |
84 |
99 |
1 |
110300 |
6.07 |
120 |
374 |
n.d. |
n.d. |
|
CP-3
|
|
97 |
3 |
87 |
97 |
3 |
108700 |
4.34 |
124 |
316 |
91.7 |
1.39 ± 0.68 |
|
CP-5
|
|
95 |
5 |
96 |
95 |
5 |
127600 |
4.59 |
126 |
309 |
92.3 |
1.36 ± 0.09 |
|
CP-10
|
|
90 |
10 |
92 |
90 |
10 |
154500 |
3.29 |
130 |
297 |
85.2 |
1.39 ± 0.11 |
|
CP-S4
|
Suspension polymn.c |
95 |
5 |
84 |
96 |
4 |
279900 |
5.51 |
126 |
351 |
n.d. |
n.d. |
|
CP-S10
|
|
90 |
10 |
95 |
90 |
10 |
235000 |
4.51 |
130 |
309 |
n.d. |
n.d. |
|
CP-Ma
|
Pendant modificationd |
— |
— |
— |
93 |
7 |
— |
— |
— |
354 |
n.d. |
n.d. |
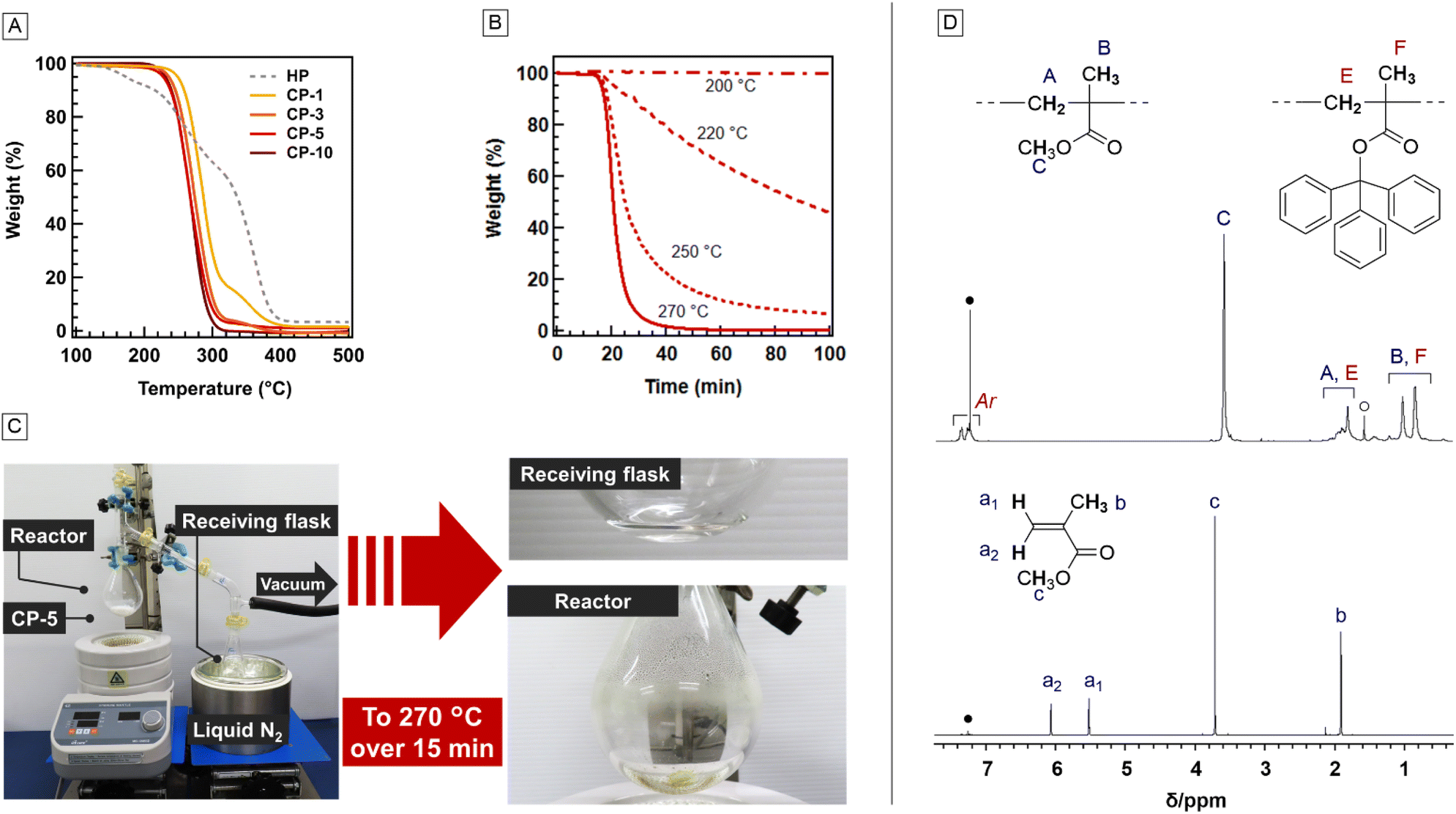
To understand the effects of TMA units, thermogravimetry (TG) analysis was performed (Fig. 2A). The purchased PMMA, HP, exhibited a 36% weight loss at 300 °C, whereas that of CP-1 was 71%. These differences imply the significant impact of the TMA units on thermal decomposition, even when the composition was only 1 mol%. Nevertheless, the 95% weight loss temperature (Td95) of CP-1 was 374 °C, which was close to that mass (Đ = 6.07), the copolymer should contain polymer chains with few TMA units that do not exhibit superior degradability.26 Thus, 1 mol% TMA units seemed insufficient to facilitate complete depolymerization. On the other hand, Td95 were observed for CP-3, CP-5, and CP-10 at 80–100 °C lower than that of HP, suggesting sufficient TMA unit content for depolymerization. Notably, PMMA derived from CP-5 showed only 7.0% weight loss at 300 °C and Td95 at 392 °C (Fig. S4†), highlighting the effects of the TMA units on thermal decomposition. The activation energies (Ea) for the decomposition of HP and copolymers were calculated using the Flynn–Wall–Ozawa plot (Fig. S5 and Table S1†).27,28 The thermal decomposition of HP can be roughly divided into three stages: 150–220, 220–320 and 320–400 °C, which are assignable to the degradation initiated by the radical generation from head-to-head linkages,8 unsaturated chain end,7 and random chain scission.6,9 The Ea for the degradation at the third stage corresponding to the 80–95% weight loss was 223 kJ mol−1, which was similar to the reported value.28 On the other hand, CP-3 and CP-5 indicated a single-step decomposition, and the Ea estimated from the range of 15–95% wight loss was 159 kJ mol−1. Thus, the incorporation of TMA units was effective in reducing the energy barrier for thermal decomposition. However, the Ea of CP-10 was estimated to be 177 kJ mol−1, which was slightly higher than those of CP-3 and CP-5. This value might have been overestimated because the slow evaporation of the decomposed TMA unit, involving the depolymerized TMA monomer (Fig. S6†), might have affected the observed weight loss. The details of the degradation of the TMA units are discussed later. Although Ea was evaluated from the weight loss during the heating process, the actual monomer recovery via thermal depolymerization should be conducted at a constant temperature. Therefore, TG analysis was performed again at a lower heating rate (1.0 °C min−1) to detect the initiation temperature of thermal decomposition. The 5% weight-loss temperature (Td5) of CP-5 was 204 °C (Fig. S7†). Thus, the weight loss of CP-5 at constant temperatures of 200, 220, 250, and 270 °C was investigated (Fig. 2B). CP-5 was stable at 200 °C, while 46% and 93% weight losses were observed at 220 and 250 °C after heating for 100 min. This was a sharp contrast to HP, of which weight loss was only 60.9% even after 300 min (Fig. S8†). Moreover, 40 min was sufficient to achieve a 95% weight loss of CP-5 at 270 °C. Therefore, the temperature for the monomer recovery from CP-5via bulk depolymerization should be higher than 220 °C and appropriate at 270 °C. Note that CP-5 exhibited a gradual decrease in molar mass during decomposition at 250 °C (Fig. S9†). This suggests that a single polymer chain did not fully depolymerize from one radical species generated from a trityl pendant, probably because of its high DP. In other words, several radical species are necessary for complete depolymerization.
 |
| | Fig. 2 (A) TG curves of the (co)polymers (heating rate: 10 °C min−1, N2 atmosphere). (B) TG curves of CP-5 at constant temperature conditions of 270, 250, 220, and 200 °C. (C) Photographs of the monomer recovery test for CP-5. (D) 1H NMR spectra of CP-5 (top) and the liquid collected in the receiving flask (bottom) (400 MHz, CDCl3, 25 °C). ●: CHCl3, ○: H2O. | |
The copolymers of MMA and TMA were also prepared via suspension polymerization, another practical method for the industrial production of PMMA.29,30 Mixtures of TMA (5 or 10 mol%), MMA (95 or 90 mol%), and 2,2′-azobis(isobutyronitrile) (AIBN) were suspended in pure water at 65 °C, resulting in polymers CP-S4 and CP-S10 (Table 1). Although CP-S4 has a high Mn (>2.0 × 106 g mol−1) and high Td95 at 351 °C, heating at 270 °C resulted in complete weight loss after 150 min (Fig. S10†). CP-S10 exhibited a lower Td95 at 309 °C and 50 min was sufficient for complete thermal degradation at 270 °C (Fig. S10†).
Monomer recovery from poly(MMA-co-TMA)
Based on the above results, monomer recovery via thermal decomposition was investigated. CP-5 (1.0 g) was heated under a reduced pressure of 27 hPa (Fig. 2C and Table 2, Entry 1). The reactor was heated to 270 °C for 15 min, although most of CP-5 had already disappeared before the temperature reached the target. A slightly yellow residue at the bottom and a transparent liquid on the wall remained in the reactor, whereas a white solid, which turned into a colorless transparent liquid at 25 °C, was collected in the receiving flask cooled by liquid N2. The collected product was identified as pure MMA using 1H NMR spectroscopy (Fig. 2D) and gas chromatography (GC, purity >99%). The MMA recovery yield was 94.5%, as calculated from the number of MMA units contained in CP-5. The residue in the reactor was analyzed using 1H NMR spectroscopy (Fig. 3B),which suggested the existence of MMA, TMA, the polymer fragment with an olefin end, and triphenylmethane (TPM) (Fig. 3A). The detection of TPM and olefinic polymer ends suggested a radical cleavage mechanism of the trityl ester pendant that induced depolymerization.18,23,24 Expansion of the reaction time to 60 min resulted in 92.8% MMA recovery in the receiving flask, whereas a slight residue consisting of PMMA with olefin ends remained at the bottom of the reactor (Fig. S11†). The incorporation of the olefin end into the polymer fragment was supported by the results of DOSY NMR spectroscopy (Fig. S12†). Notably, the colorless transparent liquid remaining on the upper part of the wall of the reactor was identified as a mixture of mainly TMA (89%) and a small amount of TPM (11%) using the 1H NMR spectrum (Fig. S11†). The weight of the remaining liquid corresponds to 97% of the weight of the TMA units in the initial polymer. This suggests that TMA units that do not function as the depolymerization initiation points may be recyclable, although an improvement in the reaction system is required to collect TMA because of its high boiling point.
Table 2 Thermal depolymerization of MMA copolymers
| Entrya |
Copolymer |
Weight [g] |
Temp. [°C] |
Timeb [min] |
Pressure [hPa] |
Yieldc [%] |
|
The evaporated MMA was collected using a flask cooled in a liquid nitrogen bath as described in Fig. 2C.
Time from the start of heating.
calculated from the weight of MMA units contained in initial poly(MMA-co-TMA).
Most of the copolymers disappeared when the temperature reached 270 °C.
The evaporated MMA was collected using a flask cooled in an ice bath.
|
| 1 |
CP-5
|
1.0 |
≤270d |
15 |
27 |
94.5 |
| 2 |
CP-5
|
1.0 |
250 |
40 |
27 |
92.3 |
| 3e |
CP-5
|
1.0 |
270 |
120 |
800 |
82.2 |
| 4 |
CP-S10
|
10.0 |
270 |
40 |
27 |
80.9 |
| 5 |
CP-Ma
|
9.7 |
270 |
40 |
27 |
91.3 |
| 6 |
CP-Mb
|
13.0 |
270 |
60 |
27 |
87.6 |
| 7 |
CP-BA1-M
|
1.0 |
270 |
60 |
27 |
92.6 |
 |
| | Fig. 3 (A) Proposal depolymerization-initiation mechanism for CPs with (B) 1H NMR spectra (400 MHz, DMSO-d6, 25 °C) of the residue in the reactor after the monomer recovery test from CP-5 at 270 °C within 15 min. | |
The effects of temperature and pressure were investigated to achieve MMA recovery under milder conditions. As indicated by the TG analysis (Fig. 2B), almost complete thermal degradation proceeded even at 250 °C. In fact, heating CP-5 at 250 °C under reduced pressure (27 hPa) for 40 min afforded MMA in 92.3% yield (Table 2, Entry 2). On the other hand, high vacuum did not appear to be essential for the recovery of MMA by depolymerization because TG analysis was performed under atmospheric pressure. Therefore, CP-5 was heated to 270 °C under slightly reduced pressure (800 hPa) with a receiving flask cooled in an ice bath (Entry 3). This approach also yielded MMA (purity >99%), with a recovery yield of 82.2%, although a prolonged reaction time of 120 min was required. These results suggest that depolymerization and almost quantitative recovery of MMA can be achieved even at 250 °C or under 800 hPa pressure although a long heating is required. In other words, a higher temperature (270 °C) and stronger reduced pressure (27 hPa) are effective in enhancing the depolymerization by shifting the depolymerization/polymerization equilibrium through the rapid removal of MMA from the reactor. Therefore, a large-scale monomer recovery test (10.0 g-scale) was conducted at 270 °C under a pressure of 27 hPa using CP-S10 (Entry 4). CP-S10 was used instead of CP-5, because it was prepared on a larger scale. Even at this scale, MMA was recovered in a high yield (80.9%).
Effects of TMA units on resin performances
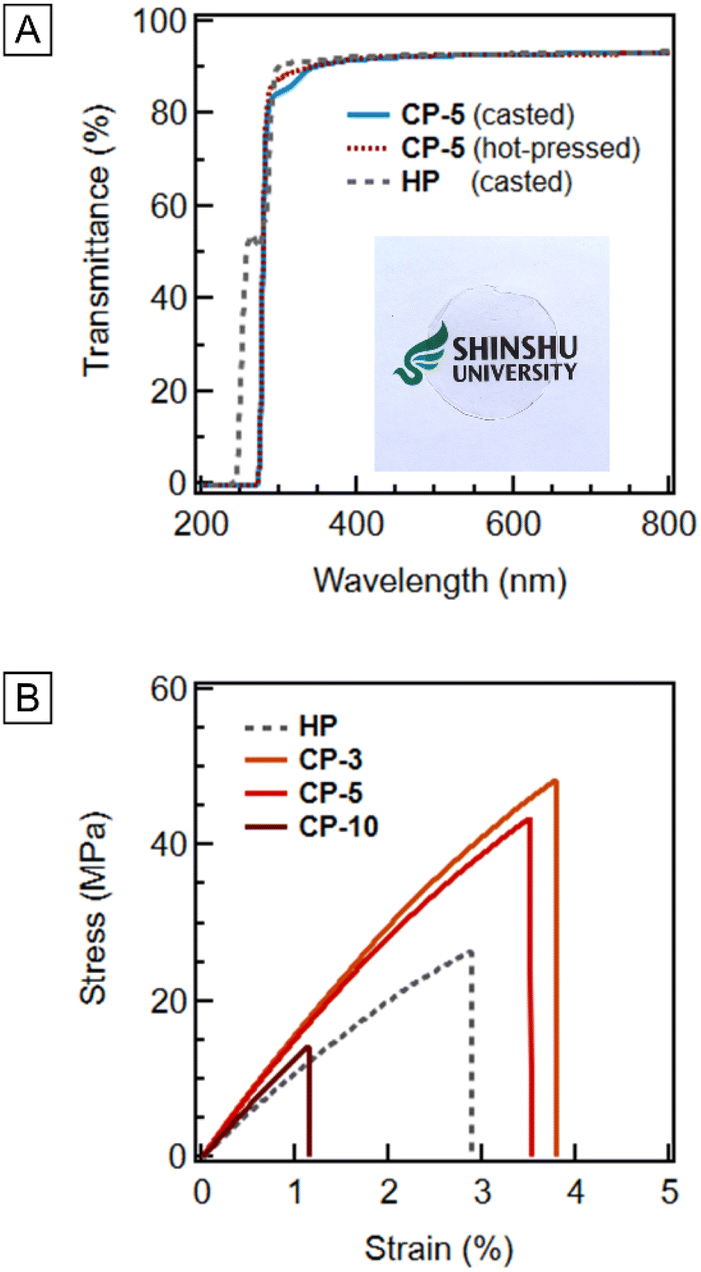
The results described above suggest that the incorporation of TMA units improves the thermal degradability of acrylic resins. Herein, the effects of TMA units on the resin performances, such as optical and mechanical properties, were investigated because the incorporation of TMA might deteriorate resin properties. The cast film of CP-5 exhibited high transparency comparable to that of PMMA in the visible light range (Fig. 4A). The low depolymerization temperatures of the copolymers may cause problems during thermal processing. Therefore, a film of CP-5 was formed by hot-pressing at 200 °C, which is within the range of general processing temperature for molding PMMA (180–280 °C;30 Fig. S13†). The transmittance of the film CP-S10 pressed at 180 °C also exhibited a high transparency, although slight yellowing was observed at 200 °C (Fig. S14†). The Young's modulus (E′) of CPs was approximately 1.4 GPa, which was slightly higher than that of HP (1.0 GPa; Table 1 and Fig. S15†). The toughness of CP-3 and CP-5 was 5.74 ± 3.71 and 4.16 ± 1.39 kJ comparable to 4.35 ± 1.35 kJ of HP, whereas CP-10 showed fragile behavior (0.53 ± 0.22 kJ; Fig. 4B and Table S2†). Therefore, to ensure the preservation of a comparable Young's modulus and prevent a reduction in mechanical toughness from PMMA, the composition of TMA should be maintained at 5 mol% or less. PMMA sheets are commercially produced by cell casting and continuous methods,30 which are based on bulk polymerization. Therefore, the copolymerization approach described above is promising for such resin production.
 |
| | Fig. 4 (A) Transmittance of CP-5 and PMMA films. A photograph in the frame shows the cast film prepared from CP-5. (B) Stress–strain curves of the samples of poly(MMA-co-TMA)s and PMMA cut from their films pressed at 200 °C. | |
MMA recovery via pendant modification
The advantage of our strategy using trityl esters is its ability to convert pristine PMMA into easily recyclable polymers via a post-polymerization modification process (Fig. 5A). Among the reported procedures for the partial hydrolysis of PMMA,31,32 using H2SO4 was convenient for the shortest reaction time.33HP (10.0 g), a purchased PMMA in a reagent grade, was treated with conc. H2SO4 at 50 °C for 10 min. The degree of hydrolysis was estimated to be 10%, based on the 1H NMR spectrum (Fig. S16†). The product was reacted with trityl chloride (TrCl) in the presence of triethylamine (TEA) at 90 °C for 60 min, resulting in 9.7 g of the modified polymer CP-Ma with 7% of TMA unit composition (Fig. S17†). The TG curve of CP-Ma showed Td95 at 354 °C (Table 1 and Fig. 5B). Heating 9.7 g of CP-Ma at 270 °C under 27 hPa pressure resulted in a 91.3% yield of MMA. The total MMA recovery calculated from the initial amount of HP was 69%. The pendant-modification method was also applied to commercial PMMA sheets (Fig. 5A and S18†). The debris from the PMMA sheets (10.0 g) was powdered by ball milling at 30 Hz for 20 min to improve its solubility in H2SO4. Although main-chain scission of PMMA by mechanical stimuli has been reported,20 the molar mass did not change after ball-milling (Fig. S17†). Pendant modification afforded a copolymer containing 17% TMA units (CP-Mb), which recovered MMA in 67.4% yield by heating at 270 °C under 27 hPa for 60 min.
 |
| | Fig. 5 (A) Pendant modification from PMMA to poly(MMA-co-TMA). (B) TG curves of HP and CP-Ma prepared from HP (under N2, 10 °C min−1). | |
MMA recovery from copolymers of MMA and acrylates
Acrylic resins for molding are generally synthesized through the copolymerization of MMA with minor quantities of acrylates, such as butyl acrylate (BA), to enhance thermal stability and melt viscosity at the molding temperature.28 In this context, copolymerization with TMA or other monomers to promote depolymerization is inherently contradictory, as BA is copolymerized to inhibit depolymerization. Consequently, pendant modification emerges as a promising strategy for such copolymers. To investigate this potential, copolymers of MMA and BA with varying compositions were prepared (Fig. S20–22†). These copolymers are designated as CP-BA#, where # represents the composition of the BA units in mol%. CP-BA1, CP-BA2, and CP-BA4 exhibited similar Td5 values of approximately 260 °C, which were significantly higher than that of HP, observed at 173 °C (Fig. S23†). These findings indicate that 1 mol% BA units are sufficient to suppress depolymerization at low temperatures. Pendant modification of CP-BA# through hydrolysis and tritylation yielded the corresponding copolymers CP-BA#-M, comprising 7–10 mol% trityl units (Fig. S20–22†). This pendant modification is significant in facilitating thermal (co)polymers are summarized in Fig. 6C, clearly indicating the enhancement of thermal degradation by trityl ester pendants. During thermal decomposition, the tritylated units function as depolymerization-initiating sites, and the acrylate units function as barriers to inhibit depolymerization (Fig. 6A). Depolymerization progressed until the active species reached the acrylate unit because the DP between the two acrylate units was sufficiently low. However, one fragment between the acrylate and tritylated units or two acrylate units should remain without depolymerization because the depolymerization progresses in only one direction from one tritylated unit. Based on this anticipated mechanism, the maximum expected value (MEV) of weight loss was calculated as follows (Table S3†): The number-average DP of the pristine (co)polymers (Xn) was calculated using eqn (1).Here, M1 is the average molar mass of one unit, calculated using eqn (2).| | | M1 = MMMA × FMMA + MBA × FBA | (2) |
MMMA and MBA are the molar masses of MMA and BA, respectively, whereas FMMA and FBA are the molar fractions of the MMA and BA units in the pristine (co)polymers, respectively. After tritylation, the depolymerization was terminated at the acrylate units. Therefore, we focused on the segment between the two acrylate units. The number-average DP between two acrylate units (Yn) was calculated as follows:| | | Yn = Xn/(Xn × FBA + 1) | (3) |
 |
| | Fig. 6 (A) Pendant modification of poly(MMA-co-BA) and the monomer recovery via depolymerization. (B) TG curves of CP-BA1 and CP-BA1-M (heating ratio of 10 °C min−1). (C) Weight losses of pristine and modified (co)polymers at 320 °C in TG curves and their maximum expected values. (D) TG curves of CP-BA1-M–CP-BA4-M under constant temperature conditions of 270 °C. | |
The number-averaged DP between the two tritylated units (Zn) in this segment is expressed as follows:
FT is the molar fraction of tritylated units. As described in
Fig. 6A, the depolymerization initiated at the tritylated units and terminated at the acrylate units, and an oligomer fragment between an acrylate and a tritylated unit or between two tritylated units remained. Therefore, the weights of the remaining fragments (
W) were estimated as follows:
Here,
M1m is the average molar mass of one unit of the tritylated (co)polymer. The initial weight of the segment between the two acrylate units (
W0) was expressed as follows:
Therefore, the maximum expected value (MEV) of weight loss can be estimated as follows:
eqn (5)–(7) can be rewritten as
The aforementioned model does not account for the scenario in which tritylation occurs in two adjacent units. Consequently, the values it produces represent merely the ‘maximum’ expected value for weight loss. The calculated MEVs are presented in Fig. 6C, indicating that the MEV associated with weight loss, which correlates with the yield of MMA recoverable through depolymerization, is substantially reduced by the copolymerization of BA with a minimal molar fraction. Notably, the MEV for the weight loss of CP-BA4-M was merely 60%, while the observed value was 60.3%. This indicates that it is fundamentally unfeasible to achieve high-yield recycling of MMA from acrylic resins with some proportion of BA units.
On the other hand, as illustrated by the TG curve of CP-BA1 (Fig. 6B), the incorporation of 1 mol% BA was adequate to enhance the thermal stability of the acrylic resins by inhibiting depolymerization. This indicates that 1 mol% BA units are sufficient if copolymerization is performed only to improve thermal stability, and such copolymers promise the chemical recycling of MMA in a moderate yield. Thus, the recovery of MMA from CP-BA1 was investigated. The weight loss of CP-BA1 during heating at a constant temperature of 270 °C, observed by TG analysis, was 37.1%, whereas that of CP-BA1-M was 82.9% (Fig. 6D and S25†). Thus, tritylation was effective in enhancing depolymerization at 270 °C. In fact, MMA was obtained in 71.1% yield from 1.0 g of CP-BA1-M by heating at 270 °C for 60 min (Fig. S26†).
Conclusions
The partial replacement of MMA units in PMMA with TMA units through bulk/suspension copolymerization or post-polymerization modification enhances the chemical recyclability of the acrylic resins. MMA was recovered with high purity and yield via depolymerization under milder thermal conditions than those required for pristine PMMA. The improvement in chemical recyclability through copolymerization is effective for acrylic resins produced by cell casting and continuous casting methods, which utilize bulk polymerization techniques and are not designed for molding. The optimal composition of the TMA unit (∼5%) resulted in slightly improved mechanical toughness, comparable transparency, and adequate chemical recyclability. The appeal of TMA in this copolymerization approach lies in its availability through a single-step reaction using commercial reagents, and its solubility in MMA.
Additionally, TMA or trityl esters offer the advantage of incorporation via post-polymerization pendant conversion. This pendant modification effectively enhanced the thermal decomposition of methacrylic resins intended for molding, where thermal depolymerization was inhibited by copolymerization with comonomers such as BA. However, the composition of the BA copolymers should be considered because the anticipated MMA yields were significantly reduced with a slight increase in BA content. The multistep modification process presents another challenge from an economic cost perspective. Therefore, optimization of the copolymer composition and pendant modification procedures are necessary for industrial applications.
Data availability
The data supporting this manuscript is available as part of the ESI.†
Author contributions
Y. C.: conceptualization, data curation, funding acquisition, investigation, methodology, validation, visualization, writing – original draft. S. H.: investigation, and writing – review & editing. Y. K.: funding acquisition, project administration, resources, supervision, and writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by JST, PRESTO, Grant Number JPMJPR22N4 (for Y. K.), and JSPS KAKENHI, Grant Numbers JP 22H02129 and JP 23K23397 (for Y. K.) and JP 23KJ1042 (for Y. C.).
Notes and references
-
M. Sponchioni and S. Altinok, in Advanced Chemical Engineering, ed. D. Moscatelli and M. Pelucchi, Academic Press, 2022, vol. 60, pp. 269–287 Search PubMed.
- U. Ali, K. J. B. A. Karim and N. A. A. Buang, Polym. Rev., 2015, 55, 678–705 CrossRef CAS.
- K. Gkaliou, L. Benedini, Z. Sárossy, C. D. Jensen, U. B. Henriksen and A. E. Daugaard, Waste Manage., 2023, 164, 191–199 CrossRef CAS PubMed.
- J. C. Worch and A. P. Dove, ACS Macro Lett., 2020, 9, 1494–1506 CrossRef CAS PubMed.
- J. De Tommaso and J.-L. Dubois, Polymers, 2021, 13, 2724 CrossRef CAS PubMed.
- L. E. Manring, Macromolecules, 1988, 21, 528–530 CrossRef CAS.
- L. E. Manring, Macromolecules, 1989, 22, 2673–2677 CrossRef CAS.
- L. E. Manring, D. Y. Sogah and G. M. Cohen, Macromolecules, 1989, 22, 4652–4654 CrossRef CAS.
- L. E. Manring, Macromolecules, 1991, 24, 3304–3309 CrossRef CAS.
-
W. K. Busfield, in Polymer Handbook, ed. J. Brandrup, R. H. Immergut and E. A. Grulke, Wiley, 1999, vol. 1 Search PubMed.
- Y. Sano, T. Konishi, M. Sawamoto and M. Ouchi, Eur. Polym. J., 2019, 120, 109181 CrossRef CAS.
- H. S. Wang, N. P. Truong, Z. Pei, M. L. Coote and A. Anastasaki, J. Am. Chem. Soc., 2022, 144, 4678–4684 CrossRef CAS.
- R. Whitfield, G. R. Jones, N. P. Truong, L. E. Manring and A. Anastasaki, Angew. Chem., Int. Ed., 2023, 62, e202309116 CrossRef CAS PubMed.
- M. R. Martinez, D. Schild, F. De Luca Bossa and K. Matyjaszewski, Macromolecules, 2022, 55, 10590–10599 CrossRef CAS.
- F. De Luca Bossa, G. Yilmaz and K. Matyjaszewski, ACS Macro Lett., 2023, 12, 1173–1178 CrossRef CAS.
- J. B. Young, R. W. Hughes, A. M. Tamura, L. S. Bailey, K. A. Stewart and B. S. Sumerlin, Chem, 2023, 9, 2669–2682 CAS.
- N. De Alwis Watuthanthrige, R. Whitfield, S. Harrisson, N. P. Truong and A. Anastasaki, ACS Macro Lett., 2024, 13, 806–811 CrossRef CAS PubMed.
- R. W. Hughes, M. E. Lott, I. S. Zastrow, J. B. Young, T. Maity and B. S. Sumerlin, J. Am. Chem. Soc., 2024, 146, 6217–6224 CrossRef CAS.
- M. T. Chin, T. Yang, K. P. Quirion, C. Lian, P. Liu, J. He and T. Diao, J. Am. Chem. Soc., 2024, 146, 5786–5792 CrossRef CAS.
- H. S. Wang, M. Agrachev, H. Kim, N. P. Truong, T.-L. Choi, G. Jeschke and A. Anastasaki, Science, 2025, 387, 874–880 CrossRef CAS.
- J. B. Young, S. L. Goodrich, J. A. Lovely, M. E. Ross, J. I. Bowman, R. W. Hughes and B. S. Sumerlin, Angew. Chem., Int. Ed., 2024, e202408592 CAS.
- Y. Okamoto, Proc. Jpn. Acad., Ser. B, 2015, 91, 246–261 CrossRef CAS.
- K. D. Berlin, L. H. Gower, B. S. Rathore, G. P. Sturm, J. W. White, J. B. Richards and M. Peterson, J. Org. Chem., 1963, 28, 2039 CrossRef.
- Y. Wang, M. Ding, Y. Luo and F. Wang, J. Polym. Sci., Part C, 1989, 27, 475–479 CrossRef CAS.
- K. Ishitake, K. Satoh, M. Kamigaito and Y. Okamoto, Angew. Chem., Int. Ed., 2009, 48, 1991–1994 CrossRef CAS PubMed.
- Thermal degradation below 200 °C was not observed for CP-1, suggesting the absence of head-to-head linkages. The detailed discussion is available in the ESI (Fig. S3 and accompanying discussion)†.
- T. Ozawa, Bull. Chem. Soc. Jpn., 1965, 38, 1881–1886 CrossRef CAS.
- Y.-H. Hu and C.-Y. Chen, Polym. Degrad. Stab., 2003, 82, 81–88 CrossRef CAS.
- B. Brooks, Chem. Eng. Technol., 2010, 33, 1737–1744 CrossRef CAS.
-
K. Albrecht, M. Stickler, T. Rhein, in Ullmann's Polymers and Plastics: Products and Processes, Wiley-VCH, 2016, vol. 2, pp. 885–898 Search PubMed.
- B. Sandner and C. Bischof, Angew. Makromol. Chem., 1983, 115, 207–219 CrossRef CAS.
- G. Jiang, C. Guhrenz, A. Kirch, L. Sonntag, C. Bauer, X. Fan, J. Wang, S. Reineke, N. Gaponik and A. Eychmüller, ACS Nano, 2019, 13, 10386–10396 CrossRef CAS.
- P. Smith and L. Goulet, J. Polym. Sci., Part B, 1993, 31, 327–338 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra, SEC traces, TG curves, UV spectra, and additional tables. See DOI: https://doi.org/10.1039/d5sc03190g |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab