
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A mechanism review of metal phthalocyanines as single-atomic catalysts in electrochemical energy conversion
Zikang
Li
,
Ziqi
Zhou
,
Mingzi
Sun
,
Tong
Wu
,
Qiuyang
Lu
,
Lu
Lu
,
Baian
Chen
,
Cheuk Hei
Chan
,
Hon Ho
Wong
and
Bolong
Huang
 *
*
Department of Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon, 999077, Hong Kong SAR. E-mail: b.h@cityu.edu.hk
First published on 14th July 2025
Abstract
Metal phthalocyanines (MPcs) are emerging as model single-atom catalysts (SACs) with atomically defined MN4 cores and tailorable peripheries, enabling precise mechanistic explorations and rational performance tuning. Herein, we review recent progress on carbon-supported MPc catalysts for key electrochemical energy-conversion reactions, including the oxygen reduction/evolution (ORR/OER), hydrogen evolution (HER), CO2 reduction (CO2RR) and nitrogen/nitrate reduction (N2RR/NOxRR) reactions. We emphasize mechanistic insights obtained from density-functional theory (DFT) and how π–π stacking, defect engineering, and curvature in graphene or carbon nanotubes modulate the electronic structure of MPcs, optimize intermediate adsorption, and suppress competing pathways. Meanwhile, we focus on specific computational methods like grand-canonical DFT (GC-DFT) and ab initio molecular dynamics (AIMD), which provide potential- and solvent-explicit descriptions of reaction energetics, bridging gaps between conventional constant-charge calculations and experimental observations. Besides, the machine-learning (ML) applications in MPc screening and identification based on metal centers, axial ligands, and dual-site motifs are discussed, followed by a future outlook of remaining challenges and further development of next-generation MPc-based catalysts for sustainable energy technologies.
 Bolong Huang | Bolong Huang is a full Professor at the Department of Chemistry of City University of Hong Kong. His main research fields are electronic structures of nanomaterials, energy materials, solid functional materials, and rare earth materials, as well as their applications in multi-scale energy conversion and supply systems. Dr Huang has published 364 research papers and received citations over 32 |
1. Introduction
Fossil fuels, which currently dominate the energy sector, not only contribute to greenhouse gas emissions but also face finite availability. The environmental issues brought about by the overuse of fossil fuels, like climate change, air pollution, and ecosystem degradation, have increased global attention to address sustainable energy development. To this end, electrochemical technologies, such as water-splitting electrolyzers, CO2 reduction reactors, and fuel cells, have emerged as promising solutions for converting sustainable energy sources into storable chemical fuels like hydrogen or hydrocarbons. These systems bridge the gap between intermittent renewable energy generation and consistent industrial demand, positioning clean energy conversion as a cornerstone of both environmental sustainability and economic resilience.A wide range of catalysts have been developed for clean energy conversion, including metal-based catalysts,1 single-atom catalysts (SACs),2 carbon-based metal-free catalysts,3etc. Among them, metal phthalocyanines (MPcs) as some of the M–N–C type SACs have attracted increasing research interest due to their well-defined molecular structures and good catalytic activities.4–14 Compared to normal M–N–C type SACs, in spite of disadvantages such as low dispersion of metal active sites that leads to lower activity, MPcs offer the advantages of straightforward and scalable synthesis and diverse ligand derivatives that can be prepared for various applications, and their precise structural composition facilitates a deeper understanding of reaction mechanisms at the molecular level and enables thorough kinetic evaluations of catalytic performance.15 Moreover, MPcs have been used for different types of catalysts based on their inherent properties and structure modifications, such as acting as photosensitizers in photocatalysts,16 forming porous organic polymers via polymerization,17 building metal organic frameworks (MOFs)18,19 or covalent organic frameworks (COFs)20,21 and pyrolyzing into carbon-based SACs.22–24 These modifications improve catalytic efficiency and enable precise control over product selectivity in energy conversion.
To date, MPcs have been applied in various electrocatalytic energy conversions, which have been discussed regarding the characteristics, performances, and developments of FePc, CoPc, and NiPc in energy conversions, and have elucidated structure–performance relationships.25–31 However, plenty of the experimental studies on electrocatalysts rely on traditional trial-and-error approaches. The conventional methods, whether through experimental synthesis and evaluation or numerical simulations, are heavily dependent on the expertise and intuition of researchers, often leading to suboptimal results. The inherent limitations of this approach hinder the improvements in catalytic performance both in time and space. Besides, traditional research methodologies cannot efficiently manage the complexity of systems involving numerous variables with a heavy workload.32 In the era of rapid advancements in data science and computational technology, there is an urgent need for more effective, data-driven strategies to accelerate the discovery of novel electrocatalyst candidates and identify optimal material combinations, ultimately enhancing efficiency and innovation in this labor-intensive field.33–35
In this review, we provide a comprehensive overview of recent advancements in carbon-supported metal phthalocyanine (MPc) catalysts for energy conversion (Fig. 1). Specifically, we focus on research conducted using density functional theory (DFT) calculations, along with other computational methods that offer deeper insights into electrocatalytic systems. First, we summarize the catalytic mechanisms of MPc catalysts that are used in various electrochemical reactions, including the hydrogen evolution reaction (HER), oxygen evolution reaction (OER), oxygen reduction reaction (ORR), carbon dioxide reduction reaction (CO2RR) and nitrogen/nitrite/nitrate reduction reaction (N2RR/NOxRR). Given the critical role of carbon-based substrates in MPc catalysts, we also explore the structure–activity relationship between the molecular catalyst and its substrate, as revealed by theoretical studies. Furthermore, we introduce computational methods, such as grand-canonical DFT (GC-DFT) and ab initio molecular dynamics (AIMD) simulations, which aim to simulate a more explicit representation of the electrocatalytic environment and reveal the details of the mechanism. Additionally, we discuss the integration of machine learning (ML) approaches for molecular screening and theoretical guidance in MPc catalyst design. Finally, we outline the key challenges and future perspectives for MPc-based catalysts in this rapidly evolving field. This review highlights the significance of theoretical studies in advancing MPc-based catalysis and lays the groundwork for structural engineering in SACs for energy conversion applications.
 | ||
| Fig. 1 An overview of MPc electrochemical energy conversions and applied computational methods in this review. | ||
2. Electrochemical energy conversions of MPcs
2.1 ORR/OER/HER
Hydrogen and oxygen related energy conversion reactions have been studied for decades, and are the fundamental reactions of water electrolysis and fuel cells.36,37 The mechanisms for these reactions are well investigated in different possible pathways as shown in Table 1, based on acidic or alkaline media in the reduction system.| Acidic media | Alkaline media |
|---|---|
| ORR | |
| * + O2 → *O2 | |
| *O2 + H+ + e− → *OOH | *O2 + H2O + e− → *OOH + OH− |
| *OOH + H+ + e− → *O + H2O | *OOH + e− → *O + OH− |
| *O + H+ + e− → *OH | *O + H2O + e− → *OH + OH− |
| *OH + H+ + e− → * + H2O | *OH + e− → * + OH− |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
| OER | |
| * + H2O → *OH + H+ + e− | * + OH− → *OH + e− |
| *OH → *O + H+ + e− | *OH + OH− → *O + H2O + e− |
| *O + H2O → *OOH + H+ + e− | *O + OH− → *OOH + e− |
| *OOH → * + O2 + H+ + e− | *OOH + OH− → * + O2 + H2O + e− |
|
|
| HER | |
| * + H+ + e− → *H | * + H2O + e− → *H + OH− |
| *H + H+ + e− → * + H2 | *H + H2O + e− → * + H2 + OH− |
| 2 *H → 2 * + H2 | |
The ORR can be categorized into two main pathways based on the number of electrons transferred: the four-electron and two-electron pathways.38,39 In the four-electron pathway, molecular oxygen (O2) is directly reduced to either water (H2O) in acidic media (O2 + 4H+ + 4e− → 2H2O) or hydroxide ions (OH−) in alkaline media (O2 + 2H2O + 4e− → 4OH−), without the formation of H2O2 and can follow either dissociative or associative mechanisms, depending on the oxygen dissociation barrier on the adsorbing atom. In contrast, the two-electron pathway leads to the formation of hydrogen peroxide (H2O2), which affects the fuel cell applications due to its lower efficiency and potential side reactions. For MPc SACs, the associative mechanism is favorable since the O–O bond can be activated through metal coordination.
The OER is the reverse reaction of the ORR that requires a strong water affinity on the adsorbing atom.40 Owing to the high selectivity on the single reaction site, SACs prefer an additional step on the *OOH intermediate rather than generating dual *O for oxygen association. Like the HER, it involves two transferred electrons with two possible mechanisms: the Volmer–Heyrovsky and Volmer–Tafel process.41,42 Unlike the ORR and OER, the easy H binding on the surrounding atoms of the reaction site may offer an additional adsorbed H atom for the Tafel step, which can be attributed to the neighboring effect on the SACs.
Early in 2013, Jiang et al. prepared the graphene-supported FePc (g-FePc) composite with good ORR activity in alkaline media, matching that of the commercial Pt/C catalyst.43 In 2015, Wang et al. demonstrated that the two-dimensional FePc monolayer exhibits high catalytic activity for the ORR in acidic environments, which is another non-platinum alternative for fuel cells.44 They applied GGA with the PBE functional and found that O2 molecules adsorb on the Fe center in an end-on configuration, activating the O–O bond via charge transfer and elongation, which facilitates efficient reduction (Fig. 2(a) and (b)). This demonstration was examined in 2016 when Wang et al. synthesized an FePc polymer sheathed on multiwalled carbon nanotubes (MWCNTs).45 These research studies on MPcs integrated with carbon materials have revealed that the carbon substrates are pivotal in creating a robust, conductive, and durable structure that maximizes the ORR activity of FePc-based SACs. Yang et al. studied FePc with single-walled carbon nanotubes (SWCNTs) and found that when FePc is anchored on SWCNTs, the π–π stacking between the planar FePc molecules and the curved graphitic surface of SWCNTs induces a rehybridization of Fe 3d orbitals, modifying the electronic state of the Fe center.46 This interaction increases the electron density around the Fe atom, facilitating stronger adsorption of O2 molecules and weakening the O–O bond in intermediates like OOH, thereby promoting a more efficient 4-electron ORR pathway. Besides, Yu et al. conducted a DFT modulation on FePc with defective graphene in ORR applications.7 Compared to normal graphene (G) and nitrogen-doped graphene (NG), they found that the defective graphene with specific 585 topology defects (DG-585) significantly enhances ORR performance of FePc through tailored electronic interactions, specifically charge redistribution, transferring electrons to Fe atoms in FePc, and creating an electron-rich environment (Fig. 2(c–e)). This electron transfer upshifts the d-band center of Fe atoms, strengthening O2 adsorption and accelerating reaction kinetics.
 | ||
| Fig. 2 (a) The optimized structures of intermediates (OOH*, O*, OH*, H2O, and H2O2) along the reaction path of the ORR proceeded on the FePc monolayer. (b) Free energy profile for the ORR at zero potential. The solid and dotted lines denote the 4e and 2e reduction pathways, respectively.44 (c) The top views of optimized FePc/G, FePc/NG and FePc/DG-585 based hybrid interfaces. (d) The side views of the charge density difference plot for the interfaces between G, NG and the DG-585 sheet and the FePc layer. Yellow and cyan isosurfaces represent charge accumulation and depletion. (e) The schematic diagram of the probable ORR electrocatalytic mechanism of FePc/DG-585. The red spheres represent electrons.7 Reproduced from ref. 44 and 7 with permission from Royal Society Chemistry and Elsevier, copyright 2015, 2021. | ||
The first research on MPc-based SACs for the OER was proposed by Ladouceur et al., who have pyrolyzed CoPc with carbon black and conducted the OER in acidic environments.47 Based on this, Li and his colleagues reported a sulfonated CoPc/CNT hybrid that had both ORR and OER performance.48 Aralekallu et al. studied CoPc polymer-coated Ni foam as an OER catalyst and they examined the catalytic enhancement from the metal phthalocyanine by mixing it with the benchmark catalyst IrO2.49 Nevertheless, the applications of MPcs in the OER are still limited by energy barriers. To overcome these challenges, researchers proposed some methods to enhance the OER activity of MPcs by studying the substrate effect in the catalyst system. Wan and his colleagues demonstrated that the OER performance of 3d-block transition metal MPcs can be further enhanced by the charge transfer between the catalysts and SWCNTs.50 The carbon substrates reduced the OER energy barrier for almost every step, which is attributed to the weaker interaction between the O atom and H atom of adsorbed water molecules on MPc/SWCNTs than on individual MPcs (Fig. 3(a–c)). Zhang et al. conducted a first-principles investigation to explore the influence of graphene-based substrates on the OER performance of FePc.51 They demonstrated an analogous substrate-driven effect in OER catalysis to those observed in ORR catalysis: the integration of a graphene substrate enhances electron transfer between FePc and oxygen-containing intermediates. Their findings showed that FePc displays varying OER activity depending on the graphene substrate type, with defective graphene exhibiting a significant enhancement in catalytic performance (Fig. 3(d–f)). This improvement was attributed to axial interactions between the iron center in FePc and carbon atoms in the defective graphene substrate, which optimized the adsorption strength of oxygen-containing intermediates, thereby balancing catalytic efficiency and stability.
 | ||
| Fig. 3 (a) The optimized structures of OER reactants and intermediates (H2O, OH*, O*, OOH*, and O2) on the MPc. (b) and (c) The OER pathway of FePc and TiPc with SWNT supports, respectively.50 (d)–(f) The charge density difference plots of the adsorbed O atom on FePc. The charge accumulations (blue color) occur mainly around the adsorbed O atom and the depletion (red color) is centered on the Fe atom. FePc is supported on pure graphene (d), nitrogen doped graphene (e), and defective graphene (f).51 Reproduced from ref. 50 and 51 with permission from Royal Society Chemistry and Elsevier, copyright 2020, 2022. | ||
In addition, the HER is typically considered a competing reaction to other reduction processes such as the CO2RR and N2RR, making it less attractive for energy conversion applications. However, Kwon et al. discovered that FePc/MoS2 two-dimensional composites exhibited enhanced HER catalytic activities, in which FePc promoted the reaction on the surface of MoS2 through electron concentration according to the partial density of states (PDOS) analysis.52
2.2 CO2RR
Converting CO2 into energy-rich molecules presents a dual-benefit strategy that mitigates atmospheric CO2 levels while generating industrially valuable products. For instance, CO2 can be reduced to single-carbon (C1) products like CO, formic acid (HCOOH), formaldehyde (CH2O), methanol (CH3OH), or methane (CH4), depending on the electrode material and applied potential. Researchers have explored diverse methods for this purpose, including thermal,53 chemical54 and photochemical techniques.55–58 However, these approaches face limitations such as high operational costs, suboptimal product selectivity, inefficient energy use, scalability challenges, and stringent reaction conditions. In comparison, the electrocatalytic CO2RR has become a potential solution due to its good selectivity and efficiency in synthesizing C1 or multi-carbon (C2+) products, and adaptability to industrial applications.31,59 Despite these advantages, the CO2RR faces two critical challenges: first, sluggish reaction kinetics caused by the stability of CO2 molecules; and second, competition from the HER at the electrode–electrolyte interface, which reduces the overall process efficiency.60,61The catalytic activity of MPcs in CO2 reduction was first reported by Meshitsuka et al. in 1974.62 Their pioneering work involved compositing various phthalocyanines with graphite as reaction electrodes, revealing that nickel phthalocyanine (NiPc) and cobalt phthalocyanine (CoPc) exhibited promising CO2 reduction performance. In recent years, significant advancements have been achieved in single-atom catalysts based on metal phthalocyanines for CO2 reduction. A methylated nickel phthalocyanine-multiwalled carbon nanotube (NiPc-MWCNTs) composite demonstrated high activity in converting CO2 to CO, achieving 300 mA cm−2 with a faradaic efficiency exceeding 99.5%.63 Similarly, a cobalt phthalocyanine-multiwalled carbon nanotube (CoPc-MWCNTs) composite enabled CH3OH synthesis, attaining a faradaic efficiency of 44% at high overpotentials.64 These breakthroughs highlight the versatility of metal phthalocyanine-based structures in tailoring catalytic pathways for CO2 conversion into value-added products.
The mechanisms governing the CO2RR on MPcs are primarily described through two pathways: the sequential proton-electron transfer (SPET) mechanism and the proton-coupled electron transfer (PCET) mechanism.65–67 These frameworks explain how CO2 molecules adsorb onto catalytic sites and undergo subsequent intermediate transformations. In the two-electron CO2RR process, which generates CO and HCOOH, product selectivity is critically influenced by the electronic structure of the active metal center on the catalyst.68–70 This structural property governs both the reaction energy barriers and the adsorption strength of intermediates. For example, copper phthalocyanine (CuPc) uniquely facilitates HCOOH production in 3d transition metal-based Pc,71 while most MPcs preferentially catalyze CO formation due to their distinct electronic configurations that favor specific intermediate stabilization.
The mechanism of CH3OH formation in CO2 electroreduction has attracted intensive attention since the research of Boutin et al. and Wu et al. which showed that cobalt phthalocyanine anchored on multiwalled carbon nanotubes (CoPc/MWCNT) catalyzes CH3OH production via a CO2–CO–CH3OH cascade reaction in aqueous media, with CO serving as the key intermediate.64,72 As depicted in Fig. 4, the competitive binding dynamics between CO2 and CO intermediates on the CoPc active sites critically govern CH3OH selectivity.73 In this pathway, CO2 absorbs onto CoPc and is reduced to a CO-bound intermediate (Reaction (1)). Due to the weak CoPc–CO interaction, incoming CO2 readily displaces the adsorbed CO (Reaction (3)), favoring further CO2-to-CO conversion rather than CO-to-CH3OH progression (Reaction (2)).
 | ||
| Fig. 4 Schematic illustration of the CO2/CO competitive binding mechanism on CoPc. Reaction (1) represents the reduction from CO2 to CO. Reaction (2) is the further reduction from CO to CH3OH. Reaction (3) is the competitive displacement of CO by CO2.73 Reproduced from ref. 73 with permission from American Chemical Society, copyright 2024. | ||
According to the Sabatier principle, optimal CO binding strength to CoPc is essential to enable efficient CH3OH synthesis.74–77 However, experimental and theoretical studies quantified by Yao et al. through kinetic modeling reveal that CO2 exhibits a binding affinity to CoPc over three times stronger than that of CO, allowing CO2 to dominate active site occupancy.77 This competitive displacement explains the prevalence of CoPc and other metal phthalocyanines as CO catalysts with high selectivity. On the other hand, directly promoting the CO reduction reaction (CORR) to improve CH3OH yields remains challenging, as weak CO binding in CO-rich environments favors the HER over CH3OH production.78,79
Researchers focused on modifying the electronic properties of MPcs to promote the CO-binding affinity on the metal center and thus improve the CH3OH reduction performance. Ding et al. discovered that binuclear cobalt phthalocyanine (B-CoPc) undergoes a spin-state transition from low spin (LS, S = 1/2) to high spin (HS, S = 3/2) in its Co3+ 3d orbitals when thermally treated at 400 °C (Fig. 5).74 This transition is correlated with a loss of square-planar symmetry in B-CoPc, a structural distortion of 15° on the N–Co–N angle linked to enhanced CO2RR activity compared to mononuclear CoPc (M-CoPc). Experimental and DFT analyses revealed that the HS state shifts the rate-determining step (RDS) of the CORR from *CO/*CHxO protonation to CH3OH desorption, reducing the RDS energy barrier from 0.75 eV to 0.43 eV. From the perspective of electron configuration, the HS Co2+ center in B-CoPc features two unpaired electrons in its dxz/dyz orbitals, which strengthen π back-donation to adsorbed CO. This electronic interaction strengthens the Co–C bond while weakening the C–O bond. The stabilized CO intermediate and destabilized C–O bond synergistically accelerate subsequent *CO hydrogenation steps, ultimately boosting CH3OH production. Consequently, the high-spin Co(II) achieves significantly improved methanol selectivity and activity via the CORR compared to its low-spin counterpart.
 | ||
| Fig. 5 (a) and (c) 3d orbital diagrams of M-CoPc and B-CoPc after heat treatments and the corresponding illustration of interactions with CO molecular frontier orbitals (5σ and 2π*). Black dots represent the electrons from electrodes under cathodic bias. (b) and (d) Schematic of σ and π-donation bonds between CO and 3d orbitals of LS-Co2+ and HS-Co2+. (e) Free energy diagram of the CORR over LS-Co2+ and HS-Co2+.74 Reproduced from ref. 74 with permission from Springer Nature, copyright 2023. | ||
Enhancing CH3OH production at metal centers can be achieved through axial modification strategies as well, which tailor the electronic or geometric properties of active sites via axial ligand coordination or engineered catalyst–support interactions. Su et al. systematically investigated how CNT supports influence CO2-to-CH3OH conversion efficiency (Fig. 6).80 Their study revealed that single-walled CNTs (SWCNTs) with smaller diameters (∼1 nm) induce greater curvature in CoPc molecules compared to multi-walled CNTs (MWCNTs), owing to stronger interfacial interactions between CoPc and SWCNTs. This curvature reduces the N–Co–N bond angle within the CoPc macrocycle. The distorted, curved geometry of CoPc on SWCNTs strengthens CO adsorption across a range of electrochemical potentials relative to planar CoPc configurations. This enhanced CO binding facilitates subsequent hydrogenation steps critical for CH3OH formation. The work demonstrates how axial structural modulation can optimize intermediate binding energetics to boost methanol selectivity and activity, providing a blueprint for designing next-generation electrocatalysts through strategic catalyst–support engineering.
 | ||
| Fig. 6 (a) Illustration of the structural distortion of CoPc on different-diameter CNTs, assuming that CoPc is fully elastic. (b) Free energies (eV) at U = −0.93 V versus RHE and pH 7 for CO2 reduction to methanol on curved and flat CoPc. ΔGC and ΔGF are reaction step free energies for the curved and flat CoPc, respectively. (c) The optimized intermediates of the CO2RR towards methanol on CoPc. Green boxes indicate the preferred intermediates, and red boxes indicate the higher-energy intermediates.80 Reproduced from ref. 80 with permission from Springer Nature, copyright 2023. | ||
2.3 N2RR/NOxRR
In 1989, Furuya and Yoshiba reported that MPcs immobilized on gas diffusion electrodes could catalyze the electrochemical reduction of nitrogen (N2) to ammonia (NH3) in aqueous media.81 Their work highlighted that the current efficiency and stability of phthalocyanine-based catalysts for the N2RR are strongly influenced by the identity of the central metal ion. Owing to its catalytic potential, FePc has become a focal point in N2RR studies. In 2019, He et al. developed a hybrid catalyst comprising FePc molecules dispersed on porous carbon (FePc/C), establishing it as a benchmark system for N2RR catalysis by MPcs (Fig. 7).82 Critical control experiments, including thiocyanate (SCN−) ion poisoning, confirmed that the N2RR takes place at the Fe center in FePc rather than on carbon or nitrogen moieties. Adsorption energy analyses further revealed that the Fe site exhibits the lowest energy barrier for N2 adsorption, favoring the spontaneous binding of N2 molecules. Mechanistic investigations into N2RR pathways demonstrated a preference for the alternating pathway (via *NNH → *NHNH intermediates) over the distal pathway (*NNH → *NNH2), as the latter involved higher free energy steps. The rate-determining step was identified as the initial *N2 → *NNH transition, with an energy barrier of 0.85 eV. Subsequent studies synthesized functionalized graphene sheets with finely dispersed FePc for the N2RR, revealing that π–π stacking interactions between FePc and graphene enhance catalytic activity by stabilizing the structure. Additionally, orbital rearrangement promotes end-on adsorption of N2 molecules at the Fe-N4 sites and stabilizes reaction intermediates, enabling selective NH3 production. To date, diverse metals (e.g., Mn, Co, Ni, and Cu) coordinated within phthalocyanine frameworks have demonstrated N2RR potential, with improved catalytic performance through structural modifications to optimize N2 affinity and protonation efficiency.83–86 | ||
| Fig. 7 (a) Free-energy diagrams of N2 adsorption on five active sites of FePc, showing that the Fe site possesses the lowest energy barrier. (b) Different free-energy diagrams for the NRR on an Fe atom through distal and alternating mechanisms at zero potential. (c) Geometric structures of intermediates along the alternating pathway of the NRR proceeded on FePc.82 Reproduced from ref. 82 with permission from American Chemical Society, copyright 2019. | ||
The electroreduction of a nitrite/nitrate ion (NO2−/NO3−) to NH3 (NOxRR) is a multi-step six- or eight-electron transfer process that generates many intermediates and by-products (e.g., N2, NH2OH, N2H4, NO, and N2O). Compared to the N2RR, the NOxRR exhibits significantly faster reaction kinetics for NH3 synthesis, primarily due to the following circumstances: the high water solubility of NOx species facilitates their accessibility to catalytically active sites; the dissociation energy of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (204 kJ mol−1) is much lower compared to that of the N
O bond (204 kJ mol−1) is much lower compared to that of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond (941 kJ mol−1); the higher theoretical redox potential of the NOxRR makes it thermodynamically more favorable than the N2RR. Furthermore, the NOxRR effectively mitigates interference from the competing HER, as the reduction potentials of the NOxRR and HER differ substantially. These advantages have induced research interest in the NOxRR for electrocatalytic NH3 production, with numerous studies exploring catalyst design and mechanistic pathways.
N triple bond (941 kJ mol−1); the higher theoretical redox potential of the NOxRR makes it thermodynamically more favorable than the N2RR. Furthermore, the NOxRR effectively mitigates interference from the competing HER, as the reduction potentials of the NOxRR and HER differ substantially. These advantages have induced research interest in the NOxRR for electrocatalytic NH3 production, with numerous studies exploring catalyst design and mechanistic pathways.
MPc has emerged as a type of highly efficient electrocatalyst for the NOxRR. Pioneering work by Chebotareva et al. demonstrated that MPcs (M = Fe, Co, Ni, Zn, Cu, Mn) immobilized on glassy carbon electrodes effectively catalyze NO2−/NO3− reduction in alkaline media, with NH3 as the primary product.87 Among these, CuPc exhibited the lowest overpotential for the NOxRR. Subsequent studies revealed that dispersing MPcs on conductive carbon substrates (e.g., carbon black and graphene) significantly enhances their electrocatalytic activity. For instance, Adalder et al. designed a heterostructure catalyst by anchoring MnPc on monolayer reduced graphene oxide (RGO), where strong π–π interactions between the graphene substrate and MnPc modulate the electron density of the Mn center, thereby optimizing NO3− adsorption and activation (Fig. 8). The support reduced the energy barrier across all NOxRR steps, accelerating NH3 formation.88
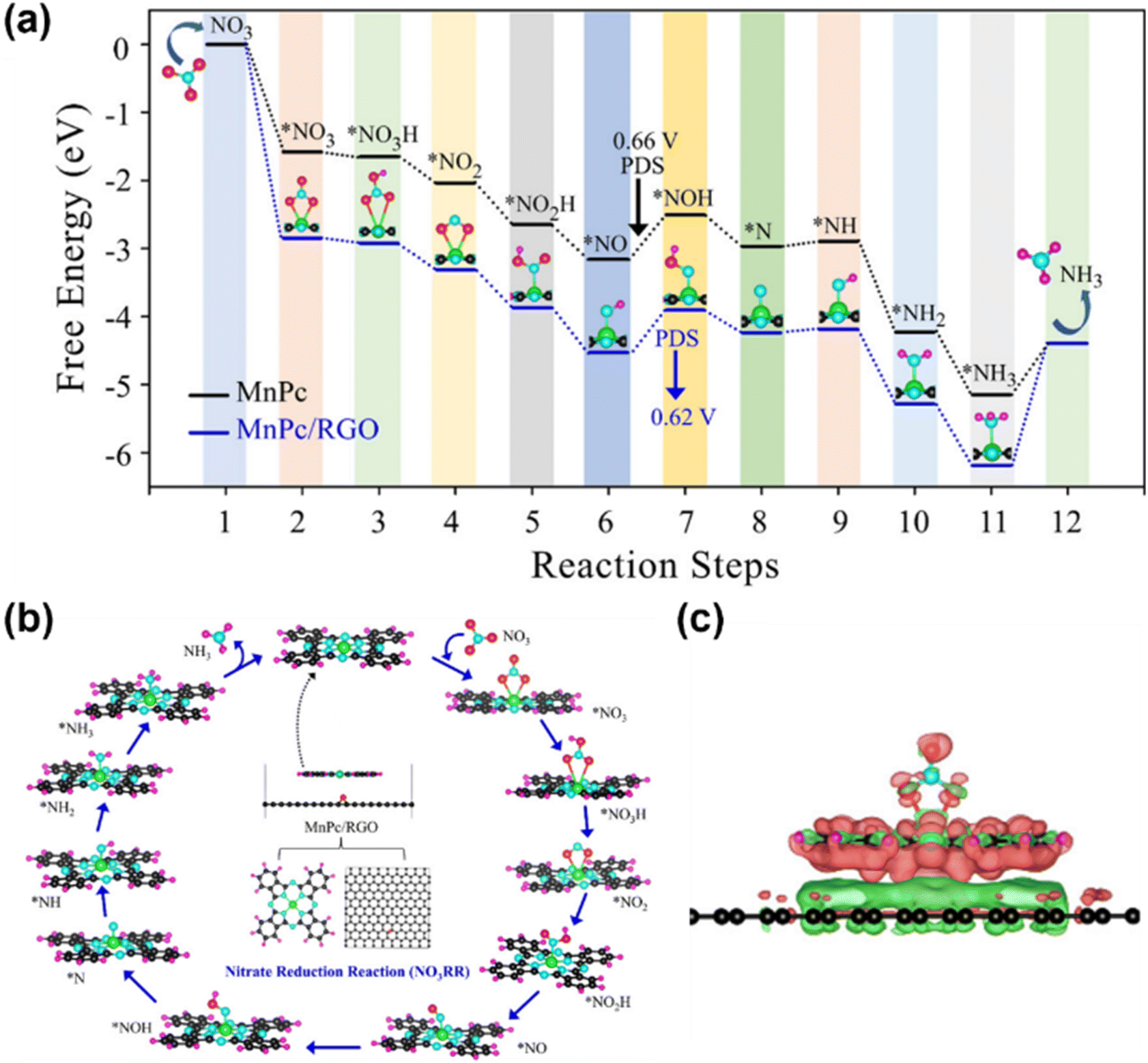 | ||
| Fig. 8 (a) Free energy profiles for both MnPc and MnPc/RGO systems; (b) reaction mechanism of the nitrate reduction reaction on the MnPc/RGO catalyst and the optimized structures of the intermediates. (c) Visualization of the charge density difference during NO3 adsorption on the MnPc/RGO catalyst.88 Reproduced from ref. 88 with permission from American Chemical Society, copyright 2023. | ||
Further mechanistic insights were provided by Harmon et al., who investigated CNT-supported catalysts.89 They found that oxygen-containing groups on CNTs do not directly participate in the rate-determining step of NO3− reduction but instead facilitate proton transfer, promoting competing HER and reducing NH3 selectivity. These findings indicate the importance of tailoring catalyst supports to suppress the HER while maintaining efficient charge/proton transfer. Collectively, these studies highlight how strategic modifications of MPc–support interactions can guide the design of high-performance NOxRR electrocatalysts.
3. Computation methods on MPc catalysts
3.1 Grand-canonical DFT (GC-DFT)
Most computational studies of electrochemical processes rely on conventional DFT, which assumes neutral, zero-potential conditions. These simulations typically incorporate solvation effects via implicit or explicit models while approximating applied potentials using the computational hydrogen electrode (CHE) framework.90–92 The CHE method adjusts DFT-derived energies of adsorbed species by using a term neφ (where φ is the applied potential and n is the formal electron count in the redox step), but it is limited to PCET intermediates and ignores critical interfacial effects of potential.39,93 Specifically, CHE neglects potential-induced electronic structure changes such as electron addition/removal, non-integer electron transfers, electric field polarization and structural relaxations of adsorbates at varying potentials. These simplifications under constant-charge assumptions often yield predictions that are different from experiments. For instance, CHE-based models consider CoPc SACs to be ineffective for the CO2RR and misjudge the priority for hydrogen evolution, yet experiments show high CO2RR activity with minimal hydrogen evolution of CoPc at −0.5 to −0.9 V vs. RHE.20,94 Similar discrepancies are observed for other CO2RR catalysts, underscoring the inadequacy of the CHE in modeling electrochemical interfaces.95,96To address this, researchers have developed constant-potential simulation strategies. Nørskov proposed cell extrapolation and charge extrapolation to manually add different amounts of charge in the system to control the work function, but these methods require high calculation costs.97–99 A breakthrough emerged with grand canonical DFT (GC-DFT) developed by Sundararaman, which integrates joint density functional theory (JDFT)100 and continuum solvation models to explicitly simulate solid–liquid interfaces.101 GC-DFT self-consistently solves the Kohn–Sham equations while varying electron counts to maintain a fixed Fermi level, enabling direct modeling of potential-dependent reaction pathways. Though computationally intensive, when paired with advanced solvation models, GC-DFT captures potential-driven electronic and structural changes that are absent in conventional DFT, providing accurate explanations of experimental results. This method can reasonably describe the electrode–electrolyte interface and has been used to elucidate many electrochemical reactions in metal surfaces and SAC-based electrocatalysis.102,103
Brimley et al. utilized GC-DFT to analyze the influence of applied potential on the CO2RR energetics and electronic properties of 3d-block transition metal MN4C catalysts.104 Their calculations revealed a pronounced sensitivity of the *CO2 adsorption grand free energy to electrode potential across all MN4Cs, emphasizing the critical role of stabilizing the *CO2 intermediate to initiate the CO2RR. PDOS analysis further demonstrated nonlinear shifts in the metal d-orbital and CO2 electronic states at increasingly reductive potentials, showing how applied potential dynamically modulates the electrocatalyst electronic structure. Among the pyridinic MN4Cs studied, Sc-, Ti-, Co-, Cu-, and ZnN4C were identified as active for CO production at moderate to highly reducing potentials (−0.7 to −1.2 V vs. SHE). Notably, ZnN4C exhibited good CO2RR thermodynamics even at lower potentials, positioning it as a highly promising candidate for CO generation. Additionally, CoN4C displayed pH-independent CO2RR activity across all potentials, similar to molecular cobalt protoporphyrin catalysts. In contrast, Cr-, Mn-, and FeN4C suffered from prohibitively high CO desorption free energies, severely limiting their CO2RR rates. This study demonstrates the universality of the CO2RR mechanism in M–N–C type SACs and establishes a foundational framework for understanding MN4C electrocatalysts and provides critical theoretical guidelines for MPc catalyst design.
To elaborate further, Wang et al. employed GC-DFT to unravel the interplay between the applied potential, CO2RR and hydrogen evolution on CoPc SACs.105 Their work systematically resolved how CO2 adsorption behavior evolves with applied potentials. Physical adsorption on CO2 dominates at low potential, while chemical adsorption prevails at higher potential due to enhanced surface charge, which activates CO2 by facilitating electron transfer. Besides, they revealed that protonation steps in the CO2RR exhibit distinct potential dependencies. The initial protonation (*CO2− + H+ → *COOH) emerged as the potential-determining step with an onset potential of −0.46 V vs. RHE, while CO desorption showed negligible sensitivity to potential, as it involves minimal electron redistribution. Meanwhile, the competitive HER shared the same −0.46 V onset as the CO2RR. At a highly reducing potential, CO2RR activity decreases due to competitive surface coverage by adsorbed H and CO2 intermediates (Fig. 9(a) and (b)). These findings highlight the critical role of electron transfer dynamics in mediating intermediate coverage and catalytic selectivity under operational conditions.
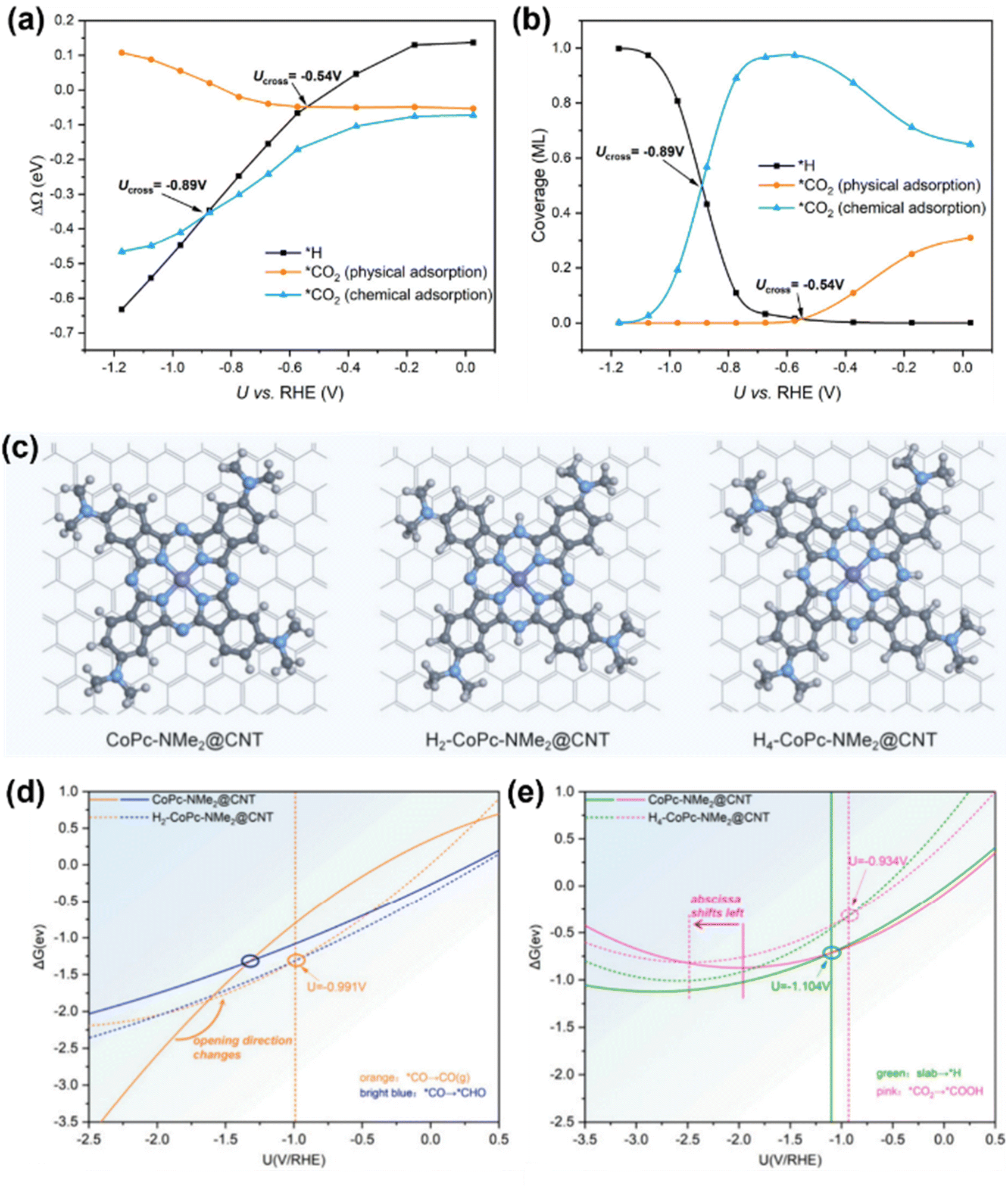 | ||
| Fig. 9 (a) Potential-dependent reaction energetics of physical and chemical adsorption of CO2 and *H formation on CoPc. (b) Potential-dependent competition of coverages among physical and chemical adsorption of CO2 and *H formation on CoPc.105 (c) Structural illustration of CoPc-NMe2@CNT, H2-CoPc-NMe2@CNT and H4-CoPc-NMe2@CNT. (d) and (e) Curves of Gibbs free energy change (ΔG) for the key steps determining the potential for maximum MeOH selectivity for CoPc-NMe2@CNT, H2-CoPc-NMe2@CNT and H4-CoPc-NMe2@CNT as a function of applied potential.106 Reproduced from ref. 105 and 106 with permission from American Chemical Society, copyright 2023, 2024. | ||
Xu et al. developed dynamic product distribution on CoPc derivative catalysts based on the GC-DFT method.106 Their study identified CoPc functionalized with a strong electron-donating dimethylamino (–NMe2) group anchored on carbon nanotubes (CoPc-NMe2@CNT) as a promising catalyst that achieves high MeOH selectivity and good durability. Dynamic product distribution analysis revealed that the hydrogenated forms of the catalyst (H2-CoPc-NMe2@CNT and H4-CoPc-NMe2@CNT) maintain high MeOH selectivity, driven by a –NMe2 induced potential shift that optimizes intermediate stabilization. Besides, HER suppression was attributed to ligand hydrogenation, which weakens charge transfer from the cobalt center to adsorbed hydrogen atoms, thereby hindering H adsorption (Fig. 9(c–e)). This mechanism reveals the critical role of substituent-driven electronic modulation in balancing selectivity and stability. By correlating substituent effects with reaction dynamics, this work highlights the promise of strong electron-donating groups in CO2 electroreduction, offering important insights for advanced heterogeneous CoPc-based systems.
3.2 Ab initio molecular dynamics (AIMD)
AIMD was first developed by Car and Parrinello,107 and is used to model and understand the dynamic behaviors of atoms and electrons at the electrode–electrolyte interface under realistic conditions, providing mechanistic insights for catalyst design. Since AIMD is fundamentally limited by computational cost and timescale, a range of statistical and enhanced sampling methods have been developed to efficiently explore rare events and compute free energy surfaces, which are crucial for understanding reaction mechanisms.Among these approaches, thermodynamic integration (TI) is a rigorous approach for determining free energy differences between states by integrating the ensemble-averaged derivative of the Hamiltonian along a coupling parameter (λ).108 These simulations are typically conducted with temperature control provided by the Nosé–Hoover thermostat, which maintains the system at constant temperature and ensures sampling within the canonical ensemble.109,110 The slow-growth method represents a typical case of TI, in which λ is incrementally varied during a single trajectory, with the accumulated work providing an estimate of the free energy difference.111 For those reaction processes described by a specific reaction coordinate, blue-moon sampling constrains the system along this coordinate, employing algorithms such as SHAKE to maintain the constraint, yielding accurate potential of mean force (PMF) profiles.112,113 Umbrella sampling114 and metadynamics115 are further enhanced sampling strategies: umbrella sampling applies biasing potentials to allow sampling of rarely visited regions along a known reaction coordinate, while metadynamics adds history-dependent bias to collective variables (CVs) to escape local minima and reconstruct multidimensional free energy landscapes. The selection of a suitable method depends on the system, the reaction pathway of interest, and the nature of the process. These methodologies, when combined with AIMD, enable detailed exploration of reaction mechanisms and energetics at electrode–electrolyte interfaces, providing both atomic-level insight and quantitative predictions that guide experimental catalyst development (Table 2).
| Method | Purpose | Key feature | Limitation |
|---|---|---|---|
| Thermo-dynamic integration | Free energy difference | Integrates along λ | Requires multiple simulations |
| Slow-growth | Free energy difference | Gradual change in λ within one trajectory | Sensitive to the rate and may not be reversible |
| Blue-moon sampling | Free energy profile | Constrains a reaction coordinate | Requires an effective constraint algorithm |
| Umbrella sampling | Free energy profile | Applies biasing along the coordinate | Requires overlap between windows |
| Meta-dynamics | Free energy surface | Adds history-dependent bias to CVs | Depends critically on the choice of variables |
In the early years, AIMD was applied for analyzing the CO2RR performance of cobalt porphyrin in water, integrated with DFT calculations with hybrid functionals and dielectric continuum solvation models.116 This advanced method has been used in the investigations of CoPc, which shows a better catalytic performance in CH3OH formation and possible C2 product formation.
Liu et al. employed AIMD simulations to highlight the key role of intermolecular interactions in driving the Eley–Rideal (ER) protonation and Langmuir–Hinshelwood (LH) protonation of *CO during CO hydrogenation on metal–nitrogen–carbon (M–N–C) SACs (Fig. 10(a)), which was on the explicit water bilayer and lasted for 10 ps while maintaining a temperature of 300 K based on the Nosé–Hoover thermostat.117 In the ER pathway, adsorbed *CO directly accepts hydrogen from nearby water molecules, whereas the LH mechanism involves a two-step process with an adsorbed *H species (formed via water dissociation) that couples with *CO at separate active sites.118 Further analysis by Li et al. revealed a unique C⋯H–O hydrogen bond, formed exclusively between water and *CO on charged CoPc, where an antibonding electron pair acted as a proton acceptor.119 They adapted a combination of slow-growth and blue-moon sampling methods with the SHAKE algorithm for the free energy profiles of *CO protonation. Unlike MnPc and FePc systems, the proton transfer to *CO does not involve the diffusion of the water molecule, and the C⋯H–O hydrogen bond additionally reduces the energy barrier for O–H bond dissociation and C–H bond formation. This unique interaction significantly accelerated CO protonation via the ER pathway, explaining the enhanced multielectron CO2RR activity of CoPc. They highlighted the electronic structure of CoPc as critical to this hydrogen bond formation: the half-filled dz2 orbital of the Co ion enabled CO chemisorption, while orbital alignment between CoPc and CO facilitated occupation of the Co–CO σ antibonding orbital at operational potentials (Fig. 10(b–i)). Their work demonstrates hydrogen-bonding interactions that optimize proton transfer in electrochemical systems, offering a strategic route to enhance catalytic efficiency.
 | ||
| Fig. 10 (a) Schematic diagram of the mechanistic pathways of CO reduction to *CHO via the ER and LH mechanisms on M–N–C SACs.117 (b)–(d) Frequency density distribution of dmin(C⋯H) for (b) [MnPc]2−, (c) [FePc]2−, and (d) [CoPc]2− in explicit water, respectively. The red dashed lines indicate a dmin(C⋯H) of 2.84 Å. Representative snapshots from the trajectory are presented, showing instances in which dmin(C⋯H) aligns with the peak of the frequency density. The H2O closest to *CO is highlighted. (e) and (f) The evolution of the Co–C–O angle (θ1) and the C⋯H–O angle (θ2) during the first 2 ps of the molecular dynamics simulation. Snapshots of the Co atom with *CO and the closest H2O are included, corresponding to the simulation time indicated by the arrowheads. (g)–(i) Free energy profile and evolution of average d(O–H) along the reaction coordinate for (g) FePc, (h) MnPc, and (i) CoPc systems. The reaction coordinate is quantified by the collective variable of ξ = d(O–H) − d(C–H).119 Reproduced from ref. 117 and 119 with permission from American Chemical Society, copyright 2023, 2024. | ||
Li and his colleagues investigated the free-energy profiles of the CO2RR on CoPc with graphene at varying electrode potentials using constrained AIMD simulations combined with TI and fully explicit solvation models, which were sampled by the canonical ensemble employing the Nosé–Hoover thermostat with a time step of 1.0 fs at the target temperature of 300 K.120 Their study revealed that the reaction free energy, energy barrier, and transition-state location during CO2 adsorption are strongly influenced by the applied electrode potential. Furthermore, they identified the proton-transfer step as the rate-determining process for the CO2RR on CoPc, elucidating its pH-dependent behavior. The energy barrier of this critical step could be effectively lowered by applying more negative potentials or introducing electron-withdrawing substituents to the phthalocyanine framework, providing mechanistic insights into tailoring catalytic performance through electronic and electrochemical modulation.
3.3 Machine learning (ML)
As a subset of artificial intelligence, machine learning (ML) enables systems to learn from data, identify patterns, and make decisions with minimal human intervention by building analytical models automatically.121 Compared to traditional DFT calculations, ML methods effectively reduce computational costs while achieving high predictive accuracy when paired with appropriate algorithms and feature engineering. This efficiency has promoted the innovative integration of DFT and ML into a hybrid computational framework, combining the accuracy of DFT with the speed of ML by training ML models on DFT-calculated data. In this approach, DFT is used to generate a dataset of key properties, such as total energies, atomic forces, or electronegativities, which are then paired with carefully chosen descriptors of the atomic structure to train an ML model. The primary goal of the ML component is to learn the mapping from structure to property, enabling rapid and accurate predictions of DFT-level properties for new, unknown structures without the need for further costly quantum mechanical calculations. This combined approach nowadays has widely been applied in catalysis research, such as active oxygen evolution catalyst screening, the CO2 reduction network on crystalline solids, and SAC exploration, which allows for the prediction of catalytic performance in complex systems and uncovers intrinsic descriptors governing their underlying activity.122–124 To assess the accuracy of ML models, two metrics—root-mean-square error (RMSE) and the coefficient of determination (R2 score)—are commonly employed. The R2 score, ranging from 0 to 1, reflects prediction quality, where values closer to 1 indicate good performance. RMSE quantifies the deviation between predicted and actual values, where lower values represent better model performance.In recent years, researchers have employed ML algorithms integrated with DFT methods to screen MPcs for electrocatalysis by predicting the catalytic performance and revealing intrinsic descriptors of the catalytic systems.125,126 Wan et al. developed a DFT-based ML method for catalysis program (DMCP) based on ten common algorithms to implement the DFT-ML scheme.127 These ML models can be grouped into several algorithmic families. Tree-based ensemble methods include gradient boosted regression (GBR),128,129 random forest regression (RFR),130 and extra trees regression (ETR).131 All these build collections of decision trees either sequentially, in parallel, or with added randomness to enhance predictive accuracy and robustness. Linear and regularized regression techniques such as kernel ridge regression (KRR),132 least absolute shrinkage and selection operator (LASSO),133 and elastic net regression (ENR)134 extend linear regression by introducing regularization (L1, L2, or both) or kernel methods to capture nonlinearities and select relevant features. Instance-based learning is represented by k-nearest neighbor regression (KNN),135 which bases predictions on the similarity between new samples and known data points. Kernel-based and probabilistic models, including support vector regression (SVR)136 and Gaussian process regression (GPR),137 use kernels to handle nonlinear patterns, with GPR also providing uncertainty estimates for its predictions. Finally, feedforward neural networks (FNNs)138 are a type of deep learning model that uses layered, interconnected nodes to capture complex, nonlinear relationships in data. Using DMCP to investigate the CO2RR on phthalocyanine dual-metal-site catalysts (Pc DMSCs), they applied an 8:2 ratio for the training and test sets through random shuffling and division. The results demonstrated that the GBR model is the best-performing and most well-trained machine learning model, achieving high accuracy with an RMSE of 0.08 eV and an R2 score of 0.96.139 Among 289 Pc DMSC candidates, Ag-MoPc emerged as a highly stable and efficient electrocatalyst, exhibiting a low limiting potential of −0.33 V, and the ML prediction error is only 0.02 V. The DFT-ML hybrid approach proved approximately sevenfold faster than conventional DFT methods (Fig. 11). This methodology bridges computational efficiency and precision, offering a scalable strategy to identify optimal catalytic materials for the CO2RR while drastically reducing resource demands.
 | ||
| Fig. 11 (a) Schematic diagrams of the procedure of the machine-learning-accelerated catalytic activity prediction of Pc DMSCs, and the selected features are listed in the green box. (b) Structures of TMPc and TM1-TM2Pc, respectively, together with all screened transition metal elements. (c) and (d) Comparison of the RMSE and the R2 score for each model on the training set and the test set. (e) The heat map of ML-predicted theoretical limiting potentials of Pc DMSCs. The redder the color, the better the catalytic activity for the CO2RR. (f) Pearson correlation map between each of the 20 input features. Red and blue colors correspond to strong and direct correlation, while white represents no correlation.139 Reproduced from ref. 139 with permission from American Chemical Society, copyright 2021. | ||
Zhang and Wang investigated the CO2RR to CO on NiPc using a combined DFT and ML approach.140 Their DFT calculations revealed that the adsorption properties and structural stability of the Ni–N4 active site depend on the electron density around the Ni center, which can be modulated by attaching electron-withdrawing or electron-donating groups. They also explored the interaction between NiPc and nanocarbon substrates, uncovering a correlation between the π–π stacking interaction energy and charge transfer. Notably, this interaction strength was governed by the polarizability and chemical nature of the substituent rather than electronic effects on the Ni–N4 moiety, suggesting a novel guidance for molecular design. On the other hand, they developed a descriptor-driven ML framework, employing genetic algorithms, semiempirical quantum calculations, and deep neural networks to screen substituted NiPc derivatives. This computational pipeline identified several high-performing candidates, with the optimal molecule surpassing the leading reference catalyst by 100 mV in reduction potential while maintaining good stability. The study provides a general blueprint for designing hybrid catalytic materials, balancing electronic modification and interfacial interactions to optimize performance across diverse applications.
ML has been extended to optimize MPc catalytic systems apart from CO2 reduction. Wang and collaborators systematically assessed 224 carbon-supported SACs, including MPcs, for the HER and OER.141 Their analysis identified CoPc as a HER catalyst candidate, achieving an overpotential below 0.15 V, while Co/Rh/Ir-Pc SACs emerged as competitive OER alternatives to conventional IrO2. Furthermore, CoPc exhibited bifunctional activity and stability for overall water splitting. To decode these trends, they trained the GBR model and highlighted that HER activity in SACs is predominantly determined by the spatial arrangement of atoms around the transition metal site and the electronic characteristics of the metal. This approach not only accelerates catalyst discovery but also provides atomistic insights into tailoring active sites for renewable energy applications. Xia et al. used the RFR method to analyze the influence of physicochemical properties on ORR performances for the carbon materials and discovered the two-dimensional defective nitrogen-doped graphene nanomesh (NGM) as a promising candidate for loading catalysts.142 They successfully synthesized the FePc/NGM catalyst and examined the ORR performance, showing the potential of bridging the ML and synthetic chemistry for the discovery of novel catalysts for practical applications.
4. Summary and outlook
Metal phthalocyanines (MPcs) have emerged as versatile molecular catalysts for electrochemical energy conversion, offering tunable electronic structures, well-defined active sites, and adaptability across diverse reactions such as the oxygen reduction reaction (ORR), oxygen evolution reaction (OER), hydrogen evolution reaction (HER), CO2 reduction reaction (CO2RR), and nitrogen/nitrate reduction reaction (N2RR/NOxRR). The integration of MPcs with carbon supports such as graphene and carbon nanotubes significantly enhances their catalytic performance. From a mechanistic point of view, these carbon substrates provide a high surface area and electrical conductivity, facilitating efficient electron transfer during catalysis. Additionally, strong π–π interactions between MPcs and the carbon matrix promote uniform dispersion of active sites and prevent aggregation, thereby increasing the number of accessible catalytic centers. Moreover, the carbon-based supports also induce charge redistribution and structural modulation at the interface, which optimizes the adsorption of reaction intermediates and improves reaction kinetics. In general, these synergistic effects contribute to enhanced activity, selectivity, and stability of MPc-based catalysts in various electrochemical reactions. Advances in computational methods, such as grand-canonical DFT (GC-DFT) and ab initio molecular dynamics (AIMD), have elucidated the mechanistic details of MPc-catalyzed processes, revealing potential-dependent pathways, solvent effects, and interfacial dynamics. Machine learning (ML) further accelerates catalyst discovery by bridging computational efficiency with predictive accuracy, enabling rapid screening of substituents, metal centers, and substrate interactions to tailor catalytic selectivity and activity.Looking ahead, the field faces challenges in translating theoretical insights into practical applications. Future research should mainly focus on enhancing the durability and scalability of MPc-based catalysts under operational conditions. Up to now, the most recently reported dimethylamino group-decorated NiPc catalysts for CO production reached nearly 100% CO2-to-CO reduction selectivity, exhibiting long-term stability by preserving more than 98% CO selectivity for over 40 hours at a current density of 100 mA cm−2.143 However, both the current density and the duration of stable operation remain below the thresholds required for practical commercial application. Besides, MPc catalysts need to address competitive side reactions like the HER through advanced electronic and geometric engineering. Moreover, integrating multi-scale computational frameworks—combining DFT, AIMD, and ML—with experimental validation will refine predictive models and uncover novel descriptors for catalytic performance. The AI-driven modulation and screening based on the descriptors provides efficient investigations with low computational cost, offering the chance to unravel unknown mechanisms in a more complicated situation. Apart from the DFT + ML hybrid approach, the AIMD + ML hybrid approach, considering the potential or free energy change, is also considered to be a promising strategy to supply accurate, scalable, and cost-effective simulations in the future, expanding the scope of molecular and materials modelling to more complex systems and longer time scales. Such integrations are able to alleviate the gap between theoretical calculations and practical experiments, which offer more significant insights into the reaction processes and accelerates the advances of current catalyst designs.144 The future DFT and AIMD techniques intersected by ML in mechanism studies and environment simulations will enable the application of MPcs in practical applications.
Data availability
No primary research results, software, or code have been included, and no new data were generated or analysed as part of this review.Author contributions
Z. L. performed the literature search, analyzed the published results, and wrote the manuscript. Z. Z., M. S., T. W., Q. L., L. L., B. C., C. H. C., and H. H. W. assisted with the writing of the manuscript. B. Huang provided key advice and supervised the preparation of the review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the support from the National Key R&D Program of China (2021YFA1501101), Research Grant Council of Hong Kong (15304023, 15304724, and C1003-23Y), National Natural Science Foundation of China/Research Grant Council of Hong Kong Joint Research Scheme (N_PolyU502/21), National Natural Science Foundation of China/Research Grants Council of Hong Kong Collaborative Research Scheme (CRS_PolyU504/22), Shenzhen Fundamental Research Scheme-General Program (JCYJ20220531090807017), and Natural Science Foundation of Guangdong Province (2023A1515012219).References
- M. B. Ross, P. De Luna, Y. Li, C.-T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- C. Hu and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 11736–11758 CrossRef CAS.
- Z. Liao, Y. Wang, Q. Wang, Y. Cheng and Z. Xiang, Appl. Catal., B, 2019, 243, 204–211 CrossRef CAS.
- K. Chen, K. Liu, P. An, H. Li, Y. Lin, J. Hu, C. Jia, J. Fu, H. Li, H. Liu, Z. Lin, W. Li, J. Li, Y.-R. Lu, T.-S. Chan, N. Zhang and M. Liu, Nat. Commun., 2020, 11, 4173 CrossRef CAS PubMed.
- H. Zhong, M. Wang, M. Ghorbani-Asl, J. Zhang, K. H. Ly, Z. Liao, G. Chen, Y. Wei, B. P. Biswal, E. Zschech, I. M. Weidinger, A. V. Krasheninnikov, R. Dong and X. Feng, J. Am. Chem. Soc., 2021, 143, 19992–20000 CrossRef CAS PubMed.
- X. Yu, S. Lai, S. Xin, S. Chen, X. Zhang, X. She, T. Zhan, X. Zhao and D. Yang, Appl. Catal., B, 2021, 280, 119437 CrossRef CAS.
- S. Xu, Y. Ding, J. Du, Y. Zhu, G. Liu, Z. Wen, X. Liu, Y. Shi, H. Gao, L. Sun and F. Li, ACS Catal., 2022, 12, 5502–5509 CrossRef CAS.
- J. Mukherjee, S. Paul, A. Adalder, S. Kapse, R. Thapa, S. Mandal, B. Ghorai, S. Sarkar and U. K. Ghorai, Adv. Funct. Mater., 2022, 32, 2200882 CrossRef CAS.
- S. Paul, S. Sarkar, A. Adalder, A. Banerjee and U. K. Ghorai, J. Mater. Chem. A, 2023, 11, 13249–13254 RSC.
- W. Zhu, S. Liu, K. Zhao, Y. Su, Y. Yang, K. Huang and Z. He, Adv. Funct. Mater., 2024, 34, 2402537 CrossRef CAS.
- W. Duan, Y. Sun, Z. Li, P. Sun, Y. Zhuang and X. Zhi, ACS Appl. Nano Mater., 2024, 7, 24833–24846 CrossRef CAS.
- S. Sarkar, A. Adalder, S. Paul, S. Kapse, R. Thapa and U. K. Ghorai, Appl. Catal., B, 2024, 343, 123580 CrossRef CAS.
- B. Chen, H. Zou, L. Gong, H. Zhang, N. Li, H. Pan, K. Wang, T. Yang, Y. Liu, L. Duan, J. Liu and J. Jiang, Appl. Catal., B, 2024, 344, 123650 CrossRef CAS.
- A. Fauzi, X. Chen, H. Zhao, S. Cao, L. Kong, S. Huang, S. Zhang and X. Ma, Next Energy, 2023, 1, 100045 CrossRef.
- A. Kumar, P. K. Prajapati, M. S. Aathira, A. Bansiwal, R. Boukherroub and S. L. Jain, J. Colloid Interface Sci., 2019, 543, 201–213 CrossRef CAS PubMed.
- Q. Li, H.-g. Wang, Y. Li, Y. Li and Q. Duan, Dyes Pigm., 2018, 149, 261–267 CrossRef CAS.
- H. Zhong, K. H. Ly, M. Wang, Y. Krupskaya, X. Han, J. Zhang, J. Zhang, V. Kataev, B. Büchner, I. M. Weidinger, S. Kaskel, P. Liu, M. Chen, R. Dong and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 10677–10682 CrossRef CAS PubMed.
- M.-D. Zhang, D.-H. Si, J.-D. Yi, Q. Yin, Y.-B. Huang and R. Cao, Sci. China:Chem., 2021, 64, 1332–1339 CrossRef CAS.
- B. Han, X. Ding, B. Yu, H. Wu, W. Zhou, W. Liu, C. Wei, B. Chen, D. Qi, H. Wang, K. Wang, Y. Chen, B. Chen and J. Jiang, J. Am. Chem. Soc., 2021, 143, 7104–7113 CrossRef CAS PubMed.
- S. Huang, K. Chen and T.-T. Li, Coord. Chem. Rev., 2022, 464, 214563 CrossRef CAS.
- Y. Fu, D. Xu, Y. Wang, X. Li, Z. Chen, K. Li, Z. Li, L. Zheng and X. Zuo, ACS Sustain. Chem. Eng., 2020, 8, 8338–8347 CrossRef CAS.
- Q. Liu, J. Wang, J. Zhang, Y. Yan, X. Qiu, S. Wei and Y. Tang, Catal. Sci. Technol., 2020, 10, 450–457 RSC.
- G. Liao, L. Zhang, C. Li, S.-Y. Liu, B. Fang and H. Yang, Matter, 2022, 5, 3341–3374 CrossRef CAS.
- X. Wang, S. Sun, J. Yao, H. Wan, R. Ma and W. Ma, Energy Environ. Mater., 2024, 7, e12709 CrossRef CAS.
- Y.-C. Feng, X. Wang and D. Wang, Mater. Chem. Front., 2024, 8, 228–247 RSC.
- A. Kumar, J. Kaur, M. Ubaidullah and R. K. Gupta, Mol. Catal., 2024, 561, 114161 CAS.
- J. H. Wu, J. W. Wang, B. M. Aramburu-Troselj, F. J. Niu, L. J. Guo and G. Ouyang, Nanoscale, 2024, 16, 11496–11512 RSC.
- S. Li, S. Li, Z. Wu, L. Qin, J. Liu, W. Zhou, H. Kang, L. Gong and L. Zhang, Fuel, 2024, 357, 129760 CrossRef CAS.
- L. Xie, Y. Wang, Q. Kong and R. Cao, ChemCatChem, 2024, 16, e202400956 CrossRef CAS.
- X. Shang, X. Yang, G. Liu, T. Zhang and X. Su, Chem. Sci., 2024, 15, 4631–4708 RSC.
- Z. Li, S. Wang and H. Xin, Nat. Catal., 2018, 1, 641–642 CrossRef.
- B. Ryu, L. Wang, H. Pu, M. K. Y. Chan and J. Chen, Chem. Soc. Rev., 2022, 51, 1899–1925 RSC.
- Z. Yao, Y. Lum, A. Johnston, L. M. Mejia-Mendoza, X. Zhou, Y. Wen, A. Aspuru-Guzik, E. H. Sargent and Z. W. Seh, Nat. Rev. Mater., 2023, 8, 202–215 CrossRef.
- R. Batra, L. Song and R. Ramprasad, Nat. Rev. Mater., 2021, 6, 655–678 CrossRef.
- J. Wang, H. Kong, J. Zhang, Y. Hao, Z. Shao and F. Ciucci, Prog. Mater. Sci., 2021, 116, 100717 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. L. Stephens and J. Rossmeisl, Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef PubMed.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957–3971 CrossRef CAS.
- Y. Jiang, Y. Lu, X. Lv, D. Han, Q. Zhang, L. Niu and W. Chen, ACS Catal., 2013, 3, 1263–1271 CrossRef CAS.
- Y. Wang, H. Yuan, Y. Li and Z. Chen, Nanoscale, 2015, 7, 11633–11641 RSC.
- X. Wang, B. Wang, J. Zhong, F. Zhao, N. Han, W. Huang, M. Zeng, J. Fan and Y. Li, Nano Res., 2016, 9, 1497–1506 CrossRef CAS.
- J. Yang, F. Toshimitsu, Z. Yang, T. Fujigaya and N. Nakashima, J. Mater. Chem. A, 2017, 5, 1184–1191 RSC.
- M. Ladouceur, G. Lalande, D. Guay, J. P. Dodelet, L. Dignard-Bailey, M. L. Trudeau and R. Schulz, J. Electrochem. Soc., 1993, 140, 1974 CrossRef CAS.
- C. Li, T. Huang, Z. Huang, J. Sun, C. Zong, J. Yang, W. Deng and F. Dai, Dalton Trans., 2019, 48, 17258–17265 RSC.
- S. Aralekallu, V. A. Sajjan, M. Palanna, K. Prabhu C P, M. Hojamberdiev and L. K. Sannegowda, J. Power Sources, 2020, 449, 227516 CrossRef CAS.
- X. Wan, H. Niu, Y. Yin, X. Wang, C. Shao, Z. Zhang and Y. Guo, Catal. Sci. Technol., 2020, 10, 8339–8346 RSC.
- X. Zhang, X. Li, W. Wang and Z. Yang, Surf. Sci., 2022, 717, 122000 CrossRef CAS.
- I. S. Kwon, I. H. Kwak, J. Y. Kim, H. G. Abbas, T. T. Debela, J. Seo, M. K. Cho, J. P. Ahn, J. Park and H. S. Kang, Nanoscale, 2019, 11, 14266–14275 RSC.
- Y. Song, X. Zhang, K. Xie, G. Wang and X. Bao, Adv. Mater., 2019, 31, 1902033 CrossRef CAS PubMed.
- H. S. Shafaat and J. Y. Yang, Nat. Catal., 2021, 4, 928–933 CrossRef.
- J. Ran, M. Jaroniec and S.-Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- E. Gong, S. Ali, C. B. Hiragond, H. S. Kim, N. S. Powar, D. Kim and H. Kim, Energy Environ. Sci., 2022, 15, 880–937 RSC.
- Ž. Kovačič, B. Likozar and M. Huš, ACS Catal., 2020, 10, 14984–15007 CrossRef.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- M. Dunwell, W. Luc, Y. Yan, F. Jiao and B. Xu, ACS Catal., 2018, 8, 8121–8129 CrossRef CAS.
- N. J. Firet and W. A. Smith, ACS Catal., 2017, 7, 606–612 CrossRef CAS.
- S. Meshitsuka, M. Ichikawa and K. Tamaru, J. Chem. Soc., Chem. Commun., 1974, 158–159 RSC.
- X. Zhang, Y. Wang, M. Gu, M. Wang, Z. Zhang, W. Pan, Z. Jiang, H. Zheng, M. Lucero, H. Wang, G. E. Sterbinsky, Q. Ma, Y.-G. Wang, Z. Feng, J. Li, H. Dai and Y. Liang, Nat. Energy, 2020, 5, 684–692 CrossRef CAS.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- J. Shen, M. J. Kolb, A. J. Göttle and M. T. M. Koper, J. Phys. Chem. C, 2016, 120, 15714–15721 CrossRef CAS.
- A. J. Göttle and M. T. M. Koper, Chem. Sci., 2017, 8, 458–465 RSC.
- N. Han, Y. Wang, L. Ma, J. Wen, J. Li, H. Zheng, K. Nie, X. Wang, F. Zhao, Y. Li, J. Fan, J. Zhong, T. Wu, D. J. Miller, J. Lu, S.-T. Lee and Y. Li, Chem, 2017, 3, 652–664 CAS.
- F. Franco, C. Rettenmaier, H. S. Jeon and B. Roldan Cuenya, Chem. Soc. Rev., 2020, 49, 6884–6946 RSC.
- N. Han, P. Ding, L. He, Y. Li and Y. Li, Adv. Energy Mater., 2020, 10, 1902338 CrossRef CAS.
- F. Lv, N. Han, Y. Qiu, X. Liu, J. Luo and Y. Li, Coord. Chem. Rev., 2020, 422, 213435 CrossRef CAS.
- H. Zhang, X. Wang, C. Chen, X. Yang, C. Dong, Y. Huang, X. Zhao and D. Yang, Chin. J. Struct. Chem., 2023, 42, 100089 Search PubMed.
- E. Boutin, M. Wang, J. C. Lin, M. Mesnage, D. Mendoza, B. Lassalle-Kaiser, C. Hahn, T. F. Jaramillo and M. Robert, Angew. Chem., Int. Ed., 2019, 58, 16172–16176 CrossRef CAS.
- L. Yao, J. Ding, X. Cai, L. Liu, N. Singh, C. C. L. McCrory and B. Liu, ACS Nano, 2024, 18, 21623–21632 CrossRef CAS.
- J. Ding, Z. Wei, F. Li, J. Zhang, Q. Zhang, J. Zhou, W. Wang, Y. Liu, Z. Zhang, X. Su, R. Yang, W. Liu, C. Su, H. B. Yang, Y. Huang, Y. Zhai and B. Liu, Nat. Commun., 2023, 14, 6550 CrossRef CAS PubMed.
- J. Li, B. Shang, Y. Gao, S. Cheon, C. L. Rooney and H. Wang, Nat. Synth., 2023, 2, 1194–1201 CrossRef CAS.
- C. L. Rooney, M. Lyons, Y. Wu, G. Hu, M. Wang, C. Choi, Y. Gao, C.-W. Chang, G. W. Brudvig, Z. Feng and H. Wang, Angew. Chem., Int. Ed., 2024, 63, e202310623 CrossRef CAS PubMed.
- L. Yao, K. E. Rivera-Cruz, P. M. Zimmerman, N. Singh and C. C. L. McCrory, ACS Catal., 2023, 14, 366–372 CrossRef.
- X. Ren, J. Zhao, X. Li, J. Shao, B. Pan, A. Salame, E. Boutin, T. Groizard, S. Wang, J. Ding, X. Zhang, W. Y. Huang, W. J. Zeng, C. Liu, Y. Li, S. F. Hung, Y. Huang, M. Robert and B. Liu, Nat. Commun., 2023, 14, 3401 CrossRef CAS PubMed.
- Y. Song, P. Guo, T. Ma, J. Su, L. Huang, W. Guo, Y. Liu, G. Li, Y. Xin, Q. Zhang, S. Zhang, H. Shen, X. Feng, D. Yang, J. Tian, S. K. Ravi, B. Z. Tang and R. Ye, Adv. Mater., 2024, 36, 2310037 CrossRef CAS.
- J. Su, C. B. Musgrave, Y. Song, L. Huang, Y. Liu, G. Li, Y. Xin, P. Xiong, M. M.-J. Li, H. Wu, M. Zhu, H. M. Chen, J. Zhang, H. Shen, B. Z. Tang, M. Robert, W. A. Goddard and R. Ye, Nat. Catal., 2023, 6, 818–828 CrossRef CAS.
- N. Furuya and H. Yoshiba, J. Electroanal. Chem. Interfacial Electrochem., 1989, 272, 263–266 CrossRef CAS.
- C. He, Z.-Y. Wu, L. Zhao, M. Ming, Y. Zhang, Y. Yi and J.-S. Hu, ACS Catal., 2019, 9, 7311–7317 CrossRef CAS.
- S. Bhardwaj, S. K. Das, A. Biswas, S. Kapse, R. Thapa and R. S. Dey, Chem. Sci., 2023, 14, 8936–8945 RSC.
- U. K. Ghorai, S. Paul, B. Ghorai, A. Adalder, S. Kapse, R. Thapa, A. Nagendra and A. Gain, ACS Nano, 2021, 15, 5230–5239 CrossRef CAS PubMed.
- S. Murmu, S. Paul, S. Kapse, R. Thapa, S. Chattopadhyay, A. N, S. N. Jha, D. Bhattacharyya and U. K. Ghorai, J. Mater. Chem. A, 2021, 9, 14477–14484 RSC.
- A. Adalder, S. R. Waghela, S. A. Shelukar, N. Mukherjee, S. Das and U. K. Ghorai, Eng. Rep., 2024, 6, e12705 CrossRef CAS.
- N. Chebotareva and T. Nyokong, J. Appl. Electrochem., 1997, 27, 975–981 CrossRef CAS.
- A. Adalder, S. Paul, N. Barman, A. Bera, S. Sarkar, N. Mukherjee, R. Thapa and U. K. Ghorai, ACS Catal., 2023, 13, 13516–13527 CrossRef CAS.
- N. J. Harmon, J. Li, B. T. Wang, Y. Gao and H. Wang, ACS Catal., 2024, 14, 3575–3581 CrossRef CAS.
- Y. J. Sa, H. Jung, D. Shin, H. Y. Jeong, S. Ringe, H. Kim, Y. J. Hwang and S. H. Joo, ACS Catal., 2020, 10, 10920–10931 CrossRef CAS.
- Q. Chang, Y. Liu, J. H. Lee, D. Ologunagba, S. Hwang, Z. Xie, S. Kattel, J. H. Lee and J. G. Chen, J. Am. Chem. Soc., 2022, 144, 16131–16138 CrossRef CAS PubMed.
- J. Yuan, S. Chen, Y. Zhang, R. Li, J. Zhang and T. Peng, Adv. Mater., 2022, 34, 2203139 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- J.-H. Liu, L.-M. Yang and E. Ganz, ACS Sustain. Chem. Eng., 2018, 6, 15494–15502 CrossRef CAS.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- X. Zhao and Y. Liu, J. Am. Chem. Soc., 2020, 142, 5773–5777 CrossRef CAS PubMed.
- K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2015, 6, 2663–2668 CrossRef CAS PubMed.
- K. Chan and J. K. Nørskov, J. Phys. Chem. Lett., 2016, 7, 1686–1690 CrossRef CAS PubMed.
- C. Choi, G. H. Gu, J. Noh, H. S. Park and Y. Jung, Nat. Commun., 2021, 12, 4353 CrossRef CAS PubMed.
- K. Letchworth-Weaver and T. A. Arias, Phys. Rev. B:Condens. Matter Mater. Phys., 2012, 86, 075140 CrossRef.
- R. Sundararaman, W. A. Goddard 3rd and T. A. Arias, J. Chem. Phys., 2017, 146, 114104 CrossRef PubMed.
- Y. A. Alsunni, A. W. Alherz and C. B. Musgrave, J. Phys. Chem. C, 2021, 125, 23773–23783 CrossRef CAS.
- Y. Ji, Y. Li, H. Dong, L. Ding and Y. Li, J. Mater. Chem. A, 2020, 8, 20402–20407 RSC.
- P. Brimley, H. Almajed, Y. Alsunni, A. W. Alherz, Z. J. L. Bare, W. A. Smith and C. B. Musgrave, ACS Catal., 2022, 12, 10161–10171 CrossRef CAS.
- T. Wang, L. Zhou, S. Xia and L. Yu, J. Phys. Chem. C, 2023, 127, 2963–2973 CrossRef CAS.
- Z. Xu, C. Ma, B. Tang, J. Dong and Q. Zhang, J. Phys. Chem. Lett., 2025, 16, 294–307 CrossRef CAS PubMed.
- R. Car and M. Parrinello, Phys. Rev. Lett., 1985, 55, 2471–2474 CrossRef CAS PubMed.
- K. Leung, S. B. Rempe and O. A. von Lilienfeld, J. Chem. Phys., 2009, 130, 204507 CrossRef PubMed.
- S. Nosé, Mol. Phys., 1984, 52, 255–268 CrossRef.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef PubMed.
- C. Jarzynski, Phys. Rev. Lett., 1997, 78, 2690–2693 CrossRef CAS.
- G. Ciccotti, R. Kapral and E. Vanden-Eijnden, ChemPhysChem, 2005, 6, 1809–1814 CrossRef CAS PubMed.
- R. Elber, A. P. Ruymgaart and B. Hess, Eur. Phys. J.: Spec. Top., 2011, 200, 211–223 CAS.
- G. M. Torrie and J. P. Valleau, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- A. Laio and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12562–12566 CrossRef CAS PubMed.
- K. Leung, I. M. B. Nielsen, N. Sai, C. Medforth and J. A. Shelnutt, J. Phys. Chem. A, 2010, 114, 10174–10184 CrossRef CAS PubMed.
- T. Liu, Y. Wang and Y. Li, JACS Au, 2023, 3, 943–952 CrossRef CAS PubMed.
- T. Cheng, H. Xiao and W. A. Goddard III, J. Am. Chem. Soc., 2016, 138, 13802–13805 CrossRef CAS PubMed.
- H. Li, Y. Shao, Z. Zhang, M. N. Tahir, T. Hou, L. Gan, F. Ding and J. Li, ACS Catal., 2024, 15, 139–147 CrossRef.
- Y.-L. Li, X.-L. Jiang, H. Cao, H.-Y. Zhao, J. Li and Y.-G. Wang, ACS Catal., 2024, 14, 9575–9585 CrossRef CAS.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Nature, 2018, 559, 547–555 CrossRef CAS PubMed.
- Z. W. Ulissi, A. J. Medford, T. Bligaard and J. K. Nørskov, Nat. Commun., 2017, 8, 14621 CrossRef.
- S. Back, K. Tran and Z. W. Ulissi, ACS Catal., 2019, 9, 7651–7659 CrossRef CAS.
- M. Sun, A. W. Dougherty, B. Huang, Y. Li and C.-H. Yan, Adv. Energy Mater., 2020, 10, 1903949 CrossRef CAS.
- N. J. O'Connor, A. S. M. Jonayat, M. J. Janik and T. P. Senftle, Nat. Catal., 2018, 1, 531–539 CrossRef.
- K. Tran and Z. W. Ulissi, Nat. Catal., 2018, 1, 696–703 CrossRef CAS.
- X. Wan, Z. Zhang, W. Yu and Y. Guo, Mater. Rep.Energy, 2021, 1, 100046 CAS.
- J. H. Friedman, Ann. Stat., 2001, 29, 1189–1232 Search PubMed.
- J. H. Friedman, Comput. Stat. Data Anal., 2002, 38, 367–378 CrossRef.
- A. Liaw and M. Wiener, R News, 2002, 2, 18–22 Search PubMed.
- P. Geurts, D. Ernst and L. Wehenkel, Mach. Learn., 2006, 63, 3–42 CrossRef.
- Y. Zhang, J. Duchi and M. Wainwright, J. Mach. Learn. Res., 2015, 16, 3299–3340 Search PubMed.
- H. Zou, J. Am. Stat. Assoc., 2006, 101, 1418–1429 CrossRef CAS.
- H. Zou and T. Hastie, J. R. Stat. Soc. Ser. B-Stat. Methodol., 2005, 67, 301–320 CrossRef.
- T. Cover and P. Hart, IEEE Trans. Inf. Theory, 1967, 13, 21–27 Search PubMed.
- A. J. Smola and B. Schölkopf, Stat. Comput., 2004, 14, 199–222 CrossRef.
- C. Williams and C. Rasmussen, Adv. Neural Inf. Process. Syst., 1995, 8, 514–520 Search PubMed.
- T. D. Sanger, Neural Networks, 1989, 2, 459–473 CrossRef.
- X. Wan, Z. Zhang, H. Niu, Y. Yin, C. Kuai, J. Wang, C. Shao and Y. Guo, J. Phys. Chem. Lett., 2021, 12, 6111–6118 CrossRef CAS PubMed.
- Z. Zhang and Y.-G. Wang, J. Phys. Chem. C, 2021, 125, 13836–13849 CrossRef CAS.
- Y. Wang, X. Huang, H. Fu and J. Shang, J. Mater. Chem. A, 2022, 10, 24362–24372 RSC.
- W. Xia, Z. Hou, J. Tang, J. Li, W. Chaikittisilp, Y. Kim, K. Muraoka, H. Zhang, J. He, B. Han and Y. Yamauchi, Nano Energy, 2022, 94, 106868 CrossRef CAS.
- L. Sun, T. Su, A. C. Fisher, J. Shan, W. Chen, H. Zhang and X. Wang, Sci. Adv., 2025, 11, eadu6915 CrossRef CAS PubMed.
- R. Martin-Barrios, E. Navas-Conyedo, X. Zhang, Y. Chen and J. Gulín-González, Int. J. Quantum Chem., 2024, 124, e27389 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |