
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural determination of MCOFs: status, challenges and perspectives
A.
Boran
 ,
R.
González-Gómez
,
S.
Hennessey
and
P.
Farràs
*
,
R.
González-Gómez
,
S.
Hennessey
and
P.
Farràs
*
School of Biological and Chemical Sciences, Ryan Institute, University of Galway, H91 CF50 Galway, Ireland. E-mail: pau.farras@universityofgalway.ie
First published on 24th June 2025
Abstract
Porous materials have many promising characteristics, including tuneable chemical and optical properties, modifiable porosities and large surface areas. Long-range order frameworks have effective evaluation methods as a result of their crystallinities and in this context, nanoscale analysis, namely single-crystal X-ray diffraction, is a particularly useful approach for optimising the structure–property relationships. Metal–covalent organic frameworks (MCOFs), synthesised by incorporating a metal complex into a stable covalent organic framework (COF) backbone, have shown considerable promise for a variety of applications. Nonetheless, their wide-scale implementation remains hindered due to difficulties in structurally mapping them; their typically reduced crystallinities result in major challenges for their structural determination. By classifying MCOFs as metalated COFs (MeCOFs) and metalloligand COFs (MLCOFs), the characterisation of these lower crystallinity frameworks can be carried out according to their distinctive architecture using a combination of complementary structural analysis techniques. This perspective highlights examples of a synergistic approach to the structural elucidation of MLCOFs to overcome obstacles related to their crystalline nature, generating an atomic map through a combination of nano and macroscale characterisation procedures supported by theoretical modelling tools. The effective use of structural characterisation methods is considered in this perspective, which can reveal key information regarding the structure–activity relationships as they relate to MLCOFs.
1. Introduction
Porous materials have garnered considerable interest in recent years due to their attractive properties such as high porosity, large surface area, thermal and chemical stability which make them ideal materials for a wide range of applications, including gas storage, sensing, and catalysis. In this regard, metal–organic frameworks (MOFs) and covalent organic frameworks (COFs), both archetypal porous materials, have been the subject of much attention recently, owing to their tuneable structural features and relatively facile synthesis, making them versatile and multifunctional.1–4 Furthermore, their characteristics enable precise control and optimisation of their electronic properties towards more efficient and sustainable processes, including hydrogen evolution, carbon dioxide reduction and photocatalysis.1,2Despite their unique properties, limitations have reduced their efficiency and hindered their applicability. For instance, MOFs often degrade upon exposure to harsh chemical environments or high temperatures.5 Conversely, although COFs feature strong covalent bonds improving the systems' stability, the absence of metal reactive sites demotes their applicability. In terms of light-driven catalysis, MOFs have exhibited limitations in light absorption, in addition to rapid recombination of photo-generated electron–hole pairs and poor electronic conductivities.6 Whereas COFs have shown improved electron transport throughout their networks due to their well-known conjugation system and stacking arrangements, quenching processes have been described owing to non-radiative competing pathways, diminishing their usefulness.7 Despite the fact that transition metals have been integrated into porous networks to enhance the materials' catalytic activity, the desired efficiency and stability have not been achieved yet.8
Tailored novel frameworks that combine the properties of both MOFs and COFs, complementing each other's shortcomings while retaining their applicability, are highly desired. Metal–covalent organic frameworks (MCOFs) arise as breakthrough versatile porous materials, which incorporate the advantageous properties of MOFs and the structural stability provided by COFs into a unique network.9 These materials demonstrate enhanced optical properties compared to their individual counterparts, as well as improved electronic communication, advancing their potential in diverse areas, such as energy storage and catalysis.10–12 Nevertheless, alongside MOFs and COFs, their continuous development and multifunctionality are intrinsically related to the understanding of their structure–property relationship.
Different definitions have been given to the MCOFs, making them difficult to explore and evaluate,13 recently Dong et al., have simplified its description, defining an MCOF as a porous COF that incorporates metal ions into its network.14 MCOFs can be classified based on their synthesis procedure, which generally falls into three categories: direct synthesis, subcomponent self-assembly and post-synthetic metalation.
Nonetheless, details regarding their formation requirements and structural definition are not yet completely accounted for. It is relevant to notice that the presence of metallic species is not always imperative for generating the frameworks, such frameworks being categorised as metalated covalent organic frameworks (MeCOFs). Conversely, when a coordinated covalent bond is essential for the material's formation, it can be classified as metalloligand covalent organic frameworks (MLCOFs) (Fig. 1). MeCOFs can be formed either via pre- or post-synthetic metalation, without significant structural change regardless of the synthetic procedure. These materials resemble that of COFs, and under suitable conditions, their crystal structures can be obtained, facilitating their description at the nanoscale.15 Although it still remains a challenging task, Peng et al., have demonstrated the growth of large-sized 2D COF single crystals up to hundreds of micrometres.16 Metalated macrocycles and linkers with available chelating ligands are the building blocks usually used to form MeCOFs. On the other hand, MLCOFs can only be formed in the presence of metal-coordinating units.17 The structures of these materials are highly dependent on the metalloligand building block, and the complexity of coordination compounds in terms of geometry and flexibility, introduces an additional degree of freedom, creating further challenges in obtaining crystal structures.
 | ||
| Fig. 1 Classification of MCOFs according to the formation requirements and structural definition. | ||
Nanoscale structural characterisation has been pivotal for the design and development of porous materials. However, in contrast to MOFs, this has proven to be particularly challenging in the case of MCOFs due to their low crystallinity, resembling the COF systems which do not often present solvable crystal structures. The presence of covalent bonds within the framework characteristic of COFs and MCOFs leads to issues controlling the structural regularity of the material, resulting in the formation of small crystalline domains. In addition, dynamic bonds commonly present in both materials can undergo hydrolysis or isomerisation, making the growth of single crystals significantly more difficult.18 As a result, long-range disordered materials containing defects rather than highly ordered systems are produced, making it a demanding task to obtain solvable crystal structures.19
Being able to design better MCOF materials can be accomplished with a thorough understanding of their structure–property relationships at both macro and nanoscale dimensions, making it plausible to engineer more efficient materials, allowing their properties to be harnessed for the development of enhanced multifunctional systems. Nevertheless, due to their complex nature, obtaining a detailed structural atomic map is an ambitious task. Therefore, a combination of traditional characterisation techniques, in addition to advanced microscopy methodologies and computational tools should be used to obtain a comprehensive characterisation, especially in the case of MLCOFs.
In this perspective, we aim to establish an overview of characterisation techniques and computational methodologies utilised in synergy to produce beyond doubt, an accurate structural atomic map of the MCOFs, thus overcoming the obstacles for generating solvable crystal structures. We address the importance of using advanced and emerging characterisation methodologies to provide noteworthy insights for the structural determination of these materials. By recognising their similarities with MOFs and in particular, with COFs, we intend to point out the benefits of having a well-understood structural characterisation for improving their applicability and implementing this approach in the design of more functional MCOFs.
2. Structural determination of porous frameworks
Due to their inherent properties including porosity, large surface area, good electronic communication, thermal and chemical stability, MCOFs share many structural similarities with MOFs and COFs. When considering their development, a detailed atomic-scale characterisation has been fundamental. Therefore, a comprehensive understanding of the process of structural determination of MOFs and COFs is important for the future advancement of MCOFs towards more individualised and specialised applications.Although atomic mapping of MCOFs is challenging, it can be achieved through a mixture of cooperative characterisation methodologies. Yet single-crystal X-ray diffraction (SC-XRD) remains as the most preferred technique, as it provides precise information relating to the electronic environment, bonding and atomic arrangements of the material. However, structural disorder and low crystallinities found in MCOFs makes them unsuitable candidates for developing single crystals.
In the upcoming sections, we will discuss the characterisation of framework materials that exhibit long-range or short-range order, demonstrating the practicality of unveiling their structures to improve their properties and design more advanced materials.
2.1. Long-range ordered frameworks
Structural determination of long-range ordered porous materials is achievable due to their repetitive units' directional growth, enabling a single crystal of the material to be formed. MOFs, a prime example, consist of metal ion nodes successively linked via organic bridging ligands to produce dynamic coordinate covalent bonds successively arranged until becoming thermodynamically stable. These highly ordered networks possess large surface areas, significant porosity and structural diversity, along with unique characteristics and functionalities derived from both the metal nodes and the organic building blocks.20,21Redox properties, coordination preferences and overall framework geometry are influenced by the metal centres, which govern the surface properties and functionality of the MOFs. Likewise, the organic linker selection can greatly impact the functionality of MOFs by altering coordination preferences, leading to variations in π–π stacking and geometry of the network, thereby modifying their porosity and surface area. Alterations in the π-electron arrangement by integrating conjugated systems can facilitate charge transport through the network and enhance host–guest interactions. Altogether, MOFs are considered as highly tailored functional materials due to their controllable porosity and topology. In turn, this makes them desirable for applications in sensing, gas adsorption and separation, alongside a variety of catalytic processes, namely CO2 reduction (CO2RR), hydrogen evolution (HER) and photodegradation of organic pollutants.22–25
The highly ordered nature of MOFs makes crystal engineering plausible, allowing the synthesis of materials suitable for analysis through various XRD techniques. This offers an accurate atomic-scale mapping and a better understanding of the structure–property relationships of these frameworks.26
Ghobakhloo et al. demonstrated the usefulness of powder X-ray diffraction (PXRD) analysis in confirming the anchoring of a Schiff-base-Cu complex to the MOF and identifying, to a large degree, the primary region of attachment.27 In a similar vein, understanding the MOF structure made it possible to obtain higher catalytic efficiencies for the Knoevenagel condensation–Michael addition–cyclization of a Zr-based UiO-66-NH2 MOF modified with copper ions when compared to the non-functionalised MOF.27 Reports from Elcheikh Mahmoud et al. highlighted the importance of tackling structural characterisation challenges to perform SC-XRD, achieving structural modulation by incorporating Ru–polypyridine complex into a Zr-MOF system and improving its photocatalytic ability towards CO2 reduction.28 Furthermore, detailed atomic mapping regarding pore size, shape and sorption behaviour was achieved using XRD techniques by Zhang et al. to fine-tune a CeIV-based MOF, successfully augmenting the selectivity for CO2 adsorption (Fig. 2).29
 | ||
| Fig. 2 (a) One-dimensional inorganic subunit of the metal oxide chain in MIL-140-14F. (b) Illustration of the crystal structure and pore chemistry of MIL-140-4F. (c and d) The Hirshfeld surface with de (electrostatic potential) and binding sites in CeIV- (c) and ZrIV-MIL-140-4F (d) (red-to-blue color indicates the high-to-low transition of electron density). (e) Experimental and simulated PXRD patterns of CeIV-MIL-140-4F. (f) Experimental (black) and Pawley refined (red) PXRD patterns of ZrIV-MIL-140-4F. Reproduced from ref. 29 with permission from Wiley-VCH, copyright 2021. | ||
2.2. Low-crystallinity frameworks
In contrast to higher crystalline porous materials, less ordered frameworks, such as COFs, are significantly more demanding to characterise at the atomic level. Structural elucidation by SC-XRD is often not possible due to structural irregularities and the presence of defects in some instances.30 However, a combination of various characterisation techniques is regularly used to determine their structure at the nanoscale, making it feasible to tailor the frameworks to improve their properties.Nevertheless, in recent years, novel synthesis methodologies have enabled the growth of single crystals of COFs suitable for SC-XRD. Ma et al. produced a single crystal of an imine-based COF-300 along with its hydrated form, allowing for complete structural elucidation by SC-XRD. Notably, hydrated COF-300 experienced distortion, a feature which would have not been noted using only PXRD, underscoring the significance of the technique.34
Properties of COFs can be strengthened when a deeper understanding of their structure is obtained, as demonstrated in the work of Zhang et al. that utilised a variety of organic linkers to modulate the pore size of Schiff-base COFs (specifically COF-SDU1, COF-SDU2 and COF-SDU3), significantly improving their CO2 storage capabilities. At the nanoscale range, materials were characterised by combining insights gained from PXRD and computational simulations, as single crystals were not produced (Fig. 3).35 Furthermore, in the context of catalysis, the structural comprehension of the COFs' extended π-conjugation facilitated network modifications to augment their performance, as illustrated by Bi et al. The authors developed three variants of 2D COF with trans-disubstituted C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond linkages (g-C40N3-COF, g-C31N3-COF and g-C37N3-COF), showing superior properties of the most conjugated system, g-C40N3-COF, for photocatalytic water splitting. The systems' porous nature and atomic arrangements were unveiled by a combination of methods and techniques, including PXRD and high-resolution transmission electron microscopy (HR-TEM), as well as Fourier-transform infrared (FT-IR) spectroscopy, the latter of which was employed to investigate the bonding and extent of polymerisation within the frameworks.36
C bond linkages (g-C40N3-COF, g-C31N3-COF and g-C37N3-COF), showing superior properties of the most conjugated system, g-C40N3-COF, for photocatalytic water splitting. The systems' porous nature and atomic arrangements were unveiled by a combination of methods and techniques, including PXRD and high-resolution transmission electron microscopy (HR-TEM), as well as Fourier-transform infrared (FT-IR) spectroscopy, the latter of which was employed to investigate the bonding and extent of polymerisation within the frameworks.36
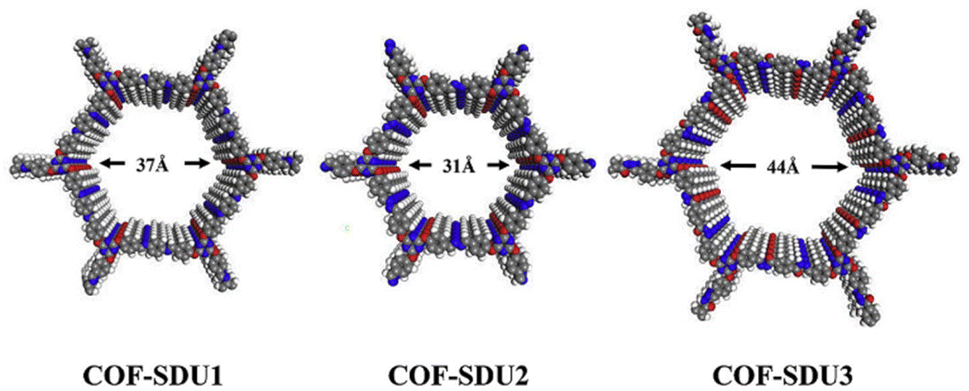 | ||
| Fig. 3 Schematic structures of COF-SDU with different building units. Reproduced from ref. 35 with permission from Elsevier, copyright 2019. | ||
 | ||
| Fig. 4 Design and synthesis of MeCOFs; pre- and post-synthetic approaches. | ||
Organic building blocks, such as porphyrins or phthalocyanines, can be metalated before the MeCOF assembly, yet limitations can arise during the framework formation attributed to the often harsh conditions applied to synthesise the materials, causing metals to leach from the networks.38 Alternatively, post-synthetic metalation is commonly preferred as a procedure to construct MeCOFs, as it allows for a broader range of synthesis conditions when forming the COF backbone.17 Typically, COF metalation is carried out in the presence of chelating species, like macrocycles and nitrogen-base pincer ligands, through either ligand exchange or direct coordination of the metal ions into the framework.
Chen et al. applied a pre-synthetic metalation approach to their porphyrin-based COFs (MPor-DETH-COF; M = H2, Co, Ni, and Zn).39 Their structural analysis and photophysical measurements showed that the photo-redox properties could be altered through the incorporation of different metal ions, tuning the structure–property relationships towards optimised light-driven HER. Consequently, the ZnPor-DETH-COF was identified as an ideal material for the photocatalytic reaction due to its improved electron movement and suitable band gap. The structural determination at the macroscale was initially done by FT-IR and solid-state NMR (nuclear magnetic resonance) spectroscopy (ss-NMR), confirming the formation of the hydrazone bond, meanwhile, ultraviolet-visible spectroscopy (UV-Vis) and X-ray photoelectron spectroscopy (XPS) results illustrated the formation of extended conjugated networks within the MeCOFs.
The post-synthetic metalation approach was employed by Xiang et al. to produce a highly π-conjugated sp2c-COFdpy-based (dpy = 2 2′-bipyridine) MeCOF capable of photocatalytic reduction of CO2.40 A comparative study was conducted by embedding a variety of metals (Fe, Co, Ni, and Cu) into the COF structure, showing an enhancement in catalytic activity, particularly when introducing copper. The formation of the frameworks was confirmed using PXRD, revealing the retention of a high degree of crystallinity after metalation and in addition, the structural analysis was further supported by 13C-NMR and FT-IR measurements. Morphological effects on the materials were investigated through HR-TEM, energy-dispersive X-ray spectroscopy (EDX) and scanning electron microscopy (SEM), displaying homogeneous distributions of the catalytically active metal sites throughout the framework (Fig. 5).
 | ||
| Fig. 5 (a) Synthesis of sp2c-COFdpy and (b) sp2c-COFdpy-Co. (c) Schematic CO2 photoreduction on the as-prepared sp2c-COFdpy-Co. Reproduced from ref. 40 with permission from Elsevier, copyright 2020. | ||
Whereas metalation of COFs has proven to be a convenient method for improving their properties, these materials still experience limitations including poor stabilities and control difficulties when polymerising, hindering their large-scale implementation.41 Exploring the structural properties of COFs through an effective use of characterisation techniques has provided valuable insights for designing upgraded materials, enabling future advancements in the field. Nevertheless, increased complexity arises when lower crystalline MCOFs are explored, such as MLCOFs. Consequently, it is necessary to employ a synergistic combination of experimental methods and theoretical tools beyond the traditional ones to obtain a precise atomic map and gain deeper knowledge of the properties exhibited by these materials.
3. Metalloligand covalent organic frameworks (MLCOFs)
MLCOF networks are formed by assembling a functionalised metal complex (metalloligand) with organic linking molecules (Fig. 6). Unlike MeCOFs, which can be produced either pre- or post-synthetically, the structural integrity of the MLCOFs relies on the presence of the metalloligands and the absence of metal entities results in the decomposition of the network.13 | ||
| Fig. 6 Design and synthesis of MLCOFs. | ||
The electron withdrawing or electron-donating nature of the metalloligand is utilised to adjust the structural and electronic characteristics of the MLCOFs, and in addition, varying the metal centre is often responsible for altering the geometry of the framework. In a similar vein, the choice of organic building blocks should be considered carefully, as it directly affects the π-conjugation of the framework, modifying the movement of electrons within the framework, the interlayer π–π relationships in the z-direction and changing the electronic and photophysical properties of the system.35
The nature and spatial arrangement of the building blocks make MLCOFs low crystalline materials, and in various cases, the obtention of single crystals is unattainable. In such instances, a wide range of complementary characterisation methods should be employed to represent their structure precisely at the nanoscale level. Overcoming the challenges associated with uncovering their atomic disposition provides deeper knowledge for advancing the understanding of MLCOFs structure–property relationships and expanding their application scope.42,43
3.1. Resolving the structure of MLCOFs
To effectively modulate the properties of MLCOFs or to design new structures, their atomic arrangement should be resolved. In this context, we aim to highlight various characterisation methods, as well as denote potential future advancements for MLCOFs across three sections: macroscale techniques, nanoscale tools and theoretical modelling methodologies. The synergistic use of these characterisation methods will subsequently be explored to obtain a thorough understanding of the MLCOFs' properties and how the structural insights gained could be harnessed to improve their efficiency and applicability.Qualitative investigations assessing the network formation are often initially deduced using FT-IR spectroscopy, where vibrations of newly formed functional groups are registered. Distinctive linkage, such as imine bonds characteristic of Schiff-base MLCOFs, can be recognised, along with information regarding the polymerisation of the framework.44,45 Alternatively, ss-NMR techniques are commonly used to support FT-IR findings, recognising changes in the nuclei chemical environment of the building blocks, confirming the network formation due to the presence of new bonds.
Liu et al. demonstrated the convenience of utilising FT-IR and ss-NMR to validate the imine bond formation in their Schiff-base copper-MLCOF (COF-505), supporting the findings from SC-XRD.46 FT-IR analysis showed new vibrational peaks attributed to the CN functional group. Additionally, 13C-ss-NMR indicated the absence of aldehyde signals corresponding to the starting material, nonetheless, the cross-polarization magic angle spinning (CPMAS) technique was unsuccessful in differentiating the imine group signal from the phenanthroline building unit. Cross-polarisation and polarisation inversion (CPPI) methodology was performed to isolate the CHN signal and corroborate the presence of the imine group (Fig. 7).
 | ||
| Fig. 7 (a) FT-IR spectrum of activated COF-505, (b) solid-state 13C-CP/MAS NMR spectrum of COF-505 and its molecular analogue and (c) schematic representation of COF-505. Reproduced from ref. 46 with permission from AAAS, copyright 2016. | ||
Chemical composition and photophysical and electronic properties of the MLCOFs can be evaluated using UV-Vis spectroscopy, as has been observed with MOFs. Particularly in cases where SC-XRD is optional, optical spectroscopies can be helpful for data validation and photoelectronic investigations. In this context, photoabsorption analyses were conducted by Nguyen et al. on a titanium-MOF (MOF-901), synthesised through the imine condensation of benzene-1,4-dialdehyde and titanium oxo cluster.47 UV-Vis diffuse reflectance spectroscopy (UV-Vis-DRS) argued the presence of the framework's building blocks by studying its absorption properties in the visible region, moreover, a Tauc plot was generated, and its band gap was calculated to be 2.65 eV. In parallel analyses, structural characterisation techniques, namely PXRD, FT-IR and ss-NMR, were also utilised to corroborate the formation of the framework and the presence of the building blocks.
Likewise, fluorescence spectroscopy methodologies can be applied to examine the excitation and emission profiles of MLCOFs. Photophysical effects associated with the incorporation of new metal sites or fluorophores, whether through new metalloligands or organic linkers, can be investigated. In addition, fluorescence techniques such as fluorescence lifetime imaging (FLIM) can be used to assess the purity and stability of MLCOFs as demonstrated by Martins et al., revealing the effects of metal incorporation on the electron transfer processes within the photo-MOF.48
Thermal decomposition processes of MLCOFs are frequently studied to recognise the coexistence of the frameworks' units and to evaluate their thermal stability. TGA is often used for this purpose, as shown by Han et al., who presented a Ru/Re-MCOF stable over 320 °C suitable for photocatalytic CO2 reduction, demonstrating the presence of its monomers and its workability across a wide temperature range.49
Oxidation state and confirmation of the presence of redox-active metal entities within the backbone of MLCOFs can be distinguished using CV. Furthermore, insights into the electrochemical stabilities and recyclability of the frameworks can also be assessed. As demonstrated in the electrochemical characterisation of Ag/Ag2O doped Co-MCOF conducted by Wang et al., who distinguished the metallic species in the network and described its efficiency for the oxygen reduction reaction (ORR) by carrying out testing in N2 against O2-saturated solutions. The peak densities of the CV curves were evaluated, exhibiting better efficiency of Ag/Ag2O doped Co-MCOF catalyst compared to others.50
Technological advances have made in situ characterisation techniques both feasible and increasingly important for studying porous materials. Unlike standard methods like ss-NMR or FT-IR, which offer only structural information pertaining to the final product, in situ NMR, IR, and XRD techniques provide real-time insights into dynamic processes, including framework formation and crystallisation processes. Additionally, host–guest interactions can be described by in situ methods, allowing to see structural changes not otherwise observable.
L. Jones et al. have emphasised the value of in situ methodologies by monitoring the formation MOF networks with in situ liquid-state NMR, which effectively showcased its use to analyse the formation and crystallisation process of a nickel-based MOF (MFM-500(Ni)).51 Two nuclei, 1H and 31P NMR were recorded in situ to monitor the reaction progress at fixed temperatures ranging from 60 to 100 °C (Fig. 8). A comparison of resulting spectra revealed key insights relating to the aggregation of the 1,3,5-benzene-tri-p-phenylphosphonic acid (BTPPA) linker, which was attributed to being promoted by a thermal process. Furthermore, in situ31P NMR offered a deeper understanding of the reaction progress by showing the gradual decrease of the phosphonic acid-based linker signals in the solution phase as the insoluble product, the MOF, formed. In addition, 1H in situ NMR was also able to identify the timescale for metal coordination by observing downfield shifts of the aromatic protons over the course of the reaction.
 | ||
| Fig. 8 Intensity contour plots of the 1H NMR spectra recorded as a function of time in the in situ NMR study of MFM-500(Ni) synthesis, and individual spectra selected at specific times (indicated by horizontal dashed lines in the contour plots), at (a) 60 °C, (b) 70 °C, (c) 80 °C, (d) 90 °C and (e) 100 °C. Assignments of the three peaks due to aromatic 1H environments (denoted Ha, Hb and Hc) in the BTPPA linker are shown in (f). The spectra are shown without normalization. Reproduced from ref. 51 with permission from RSC, copyright 2021. | ||
In situ NMR has also been used to monitor framework materials under working conditions by assessing host–guest interactions to identify structural alterations. The research conducted by Roztocki et al. illustrates a combination of in situ solid-state 13C NMR, FT-IR and PXRD on a zinc-based JUK-8 MOF to infer a structural map of the framework indirectly by using CO2 as a guest molecule.52 Shifts in CO2 characteristic NMR signals indicated that the adsorbed molecules existed in two distinct chemical environments within the framework's pores due to a breathing and swelling phenomenon that occurred in the polymer, later on corroborated by FT-IR and PXRD. The authors identified the acylhydrazone pockets as regions for CO2 adsorption, outlining the selectivity of the network when studying the MOF's specificity in a CO2/CH4 mixture.
Although in situ NMR is a powerful tool for real-time monitoring of the framework formation, it faces significant challenges, limiting its widespread use for the analysis of MLCOFs. One major issue is the complexity of the technique, which requires reaction temperature windows appropriate for the NMR probe and suitable deuterated solvents. Another limiting factor relates to restrictions regarding the sensitivity of the instrument when monitoring reaction intermediates and product formation, therefore, small-scale reactions should be conducted to prevent oversaturation of the solvent.
Alternatively, Raman spectroscopy is an effective tool for uncovering structural information concerning key functional groups. Additionally, in situ applications of this technique can enable the frameworks' host–guest interactions with other molecules or reaction processes to be observed. As evidenced by Embrechts et al. who utilised Raman spectroscopy to uncover structural information about functional groups and to monitor the reaction kinetics in the formation of a MOF (MIL-53).53 By using in situ Raman spectroscopy, the reaction progress was tracked over a temperature range of 60 to 100 °C, identifying three new bands assigned to the νC–C, νC–H, and ring breathing of terephthalates in the MOF, proving the polymerisation of the framework. Applying in situ methodologies to address some of the current limitations that hinder deeper insights into the formation of MLCOFs and their structural transformations when implemented in real scenarios is highly desired to improve their feasibility.
While macroscale analysis methods provide detailed insights into the structural bonding and optical, thermal and electrochemical properties of MLCOFs, further detailed characterisation is still needed to show the atomic spatial arrangement of these materials. In this context, nanoscale analyses and their combination with previously described techniques are essential for obtaining a precise structural elucidation of MLCOFs.
 | ||
| Fig. 9 PXRD patterns of (a) JNM-3-AA and (b) JNM-3-ABC with the experimental profiles in black, difference curve in light blue, and calculated profiles of AA (orange) and ABC (purple) packing modes. (Herein, the preferred orientation along with the (110) plane was considered in the calculated ABC model). Top (c) and side (e) views of the corresponding refined 2D crystal structure of JNM-3-AA. Top (d) and side (f) views of the corresponding refined 2D crystal structure of JNM-3-ABC. (g) The pore-size distribution profiles of JNM-3-AA calculated by nonlocal DFT modelling based on N2 adsorption data, showing a uniform pore size of 3.70 nm. (h) The N2 adsorption (filled) and desorption (open) isotherm profiles of JNM-3-AA and JNM-3-ABC at 77 K. Reproduced from ref. 55 with permission from RSC, copyright 2021. | ||
Although PXRD and BET are advantageous tools for characterising low-crystallinity MLCOFs, in most cases, a diverse variety of microscopy techniques should also be utilised in parallel to accurately determine their structure at an atomic level. Techniques such as TEM, HR-TEM, SEM, field emission SEM, scanning tunnelling microscopy, Raman microscopy, and atomic force microscopy are commonly combined for assessing the structural properties and nanoscale morphological details of MLCOFs.46,55–57
The usefulness of applying different microscopy techniques was highlighted by Wei et al.58 In their work on Cu-MCOFs, SEM was used to visually detect the synthesis conditions' effects on the crystallinity and morphology of the frameworks, showing that the produced films consist of either closely packed lamellae with smooth surfaces or ball-shaped microcrystals, depending on the MLCOF system (Fig. 10). As well, the formation of micrometre-sized crystals based on synthesis conditions was also observed by SEM, providing insights into the crystal growth process of Cu-MCOFs. After sample exfoliation, HR-TEM was carried out, suggesting that the 2D material exhibited long-range order with a hexagonal honeycomb lattice morphology in the [001] direction. Moreover, the porosity of the nanosheets was indicated by periodic bright spots in the crystal lattice, with a pore size estimated to be approximately 2.6 nm. The significance of modifying the morphology of MLCOFs was stressed through catalytic testing of the systems in hydroboration reactions of alkynes. Exfoliated nanosheets showed enhanced catalytic activity attributed to the superior efficiency of contact between the catalytic centres and the substrate, alongside the increased exposure of the Cu(I) catalytic centres.58
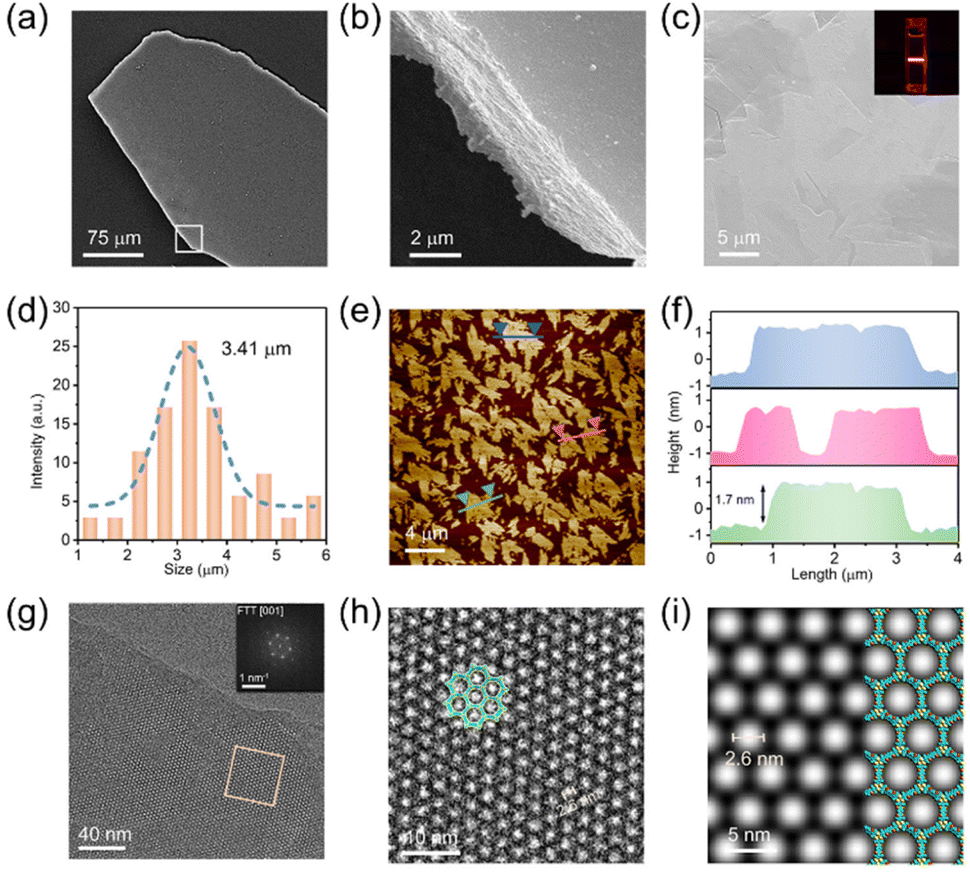 | ||
| Fig. 10 (a) SEM image of the film-like morphology of 2D-JNM-4. (b) Enlarged image of area in the white box in part a. (c) SEM image of JNM-4-Ns. Inset, Tyndall effect of JNM-4-Ns dispersion in EtOH. (d) Lateral size distribution histogram and Gaussian fit curve of JNM-4-Ns. (e) AFM image of JNM-4-Ns. (f) Corresponding height curves for the selective areas in part (e). (g) HR-TEM image of JNM-4-Ns. Inset, fast Fourier transform (FFT) image. (h) Enlarged image of selected area in part (g). (i) Simulated TEM image of JNM-4-Ns along the [100] direction. Reproduced with permission from ref. 58 with permission from ACS, copyright 2022. | ||
The importance of utilising nanoscale characterisation methods was also demonstrated by Zhou et al. through their effective application of microscopy techniques to support the PXRD findings and modelling results for both AA and ABC stackings of their aforementioned CTU-based Cu-MCOFs.55 TEM micrographies provided insights into the stacking arrangements of the MLCOFs, revealing a hexagonal structure associated with the eclipsed AA stacking model. In contrast, a highly ordered striped structure was observed in the other investigated Cu-MLCOF, suggesting the presence of a [110] reflection plane attributed to the staggered ABC stacking structure; both observations aligned with PXRD analyses. Additionally, SEM showed nanotube morphologies or layered stacking structures, depending on the MLCOF studied. Furthermore, EDX confirmed the uniform distribution of copper throughout the network in each system.
Real-time information regarding the development and evolution of MLCOFs' crystallites can be obtained by monitoring the framework formation through in situ XRD analysis. Despite these methodologies not yet being adopted for the characterisation of MLCOFs, they have shown potential due to their successful results with MOFs. Nevertheless, the technique can only be applied to crystalline systems, which restrict its application for MLCOFs. The promising potential of in situ XRD methodologies were illustrated by the work of Lyu et al., which investigated the phase transitions of a zirconium-based MOF.59 Their study involved variable temperature PXRD (VT-PXRD) to monitor the phase transitions that the framework underwent, resulting in two distinct topologies (Fig. 11). The microporous NU-906 was transformed into the mesoporous NU-1008 with a cubic square (csq-net) and a simple cubic (scu-net) topology, respectively, over time as different temperatures were applied. During the characterisation, at lower temperatures, the diffraction peak at 5° experienced a decrease in relative intensity, which was assigned to a reduction in the crystallites of NU-906; meanwhile, at 70 °C, a new signal at low angle (2.5°) was detected, suggesting the formation of the NU-1008 MOF. Additionally, using in situ variable temperature liquid cell TEM, the phase transition from the NU-906 MOF, characterised by oval morphologies, to the NU-1008 MOF, which displayed a road-like appearance, was identified. The images also showed a non-negligible change in the lattice spacing of the MOF, expanding from 1.7 nm to 3.5 nm, thereby confirming the phase transition in the framework.
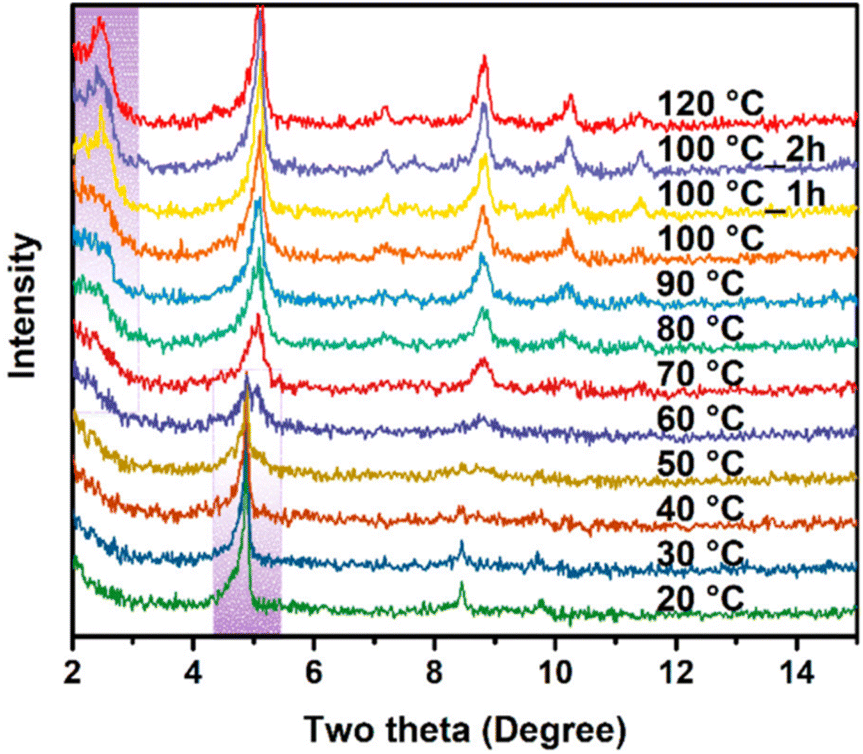 | ||
Fig. 11
In situ VT-PXRD patterns from NU-906 to NU-1008 within DMF/formic acid (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) taken with a Cu Kα radiation source. Reproduced from ref. 59 with permission from ASC, copyright 2020. :1) taken with a Cu Kα radiation source. Reproduced from ref. 59 with permission from ASC, copyright 2020. | ||
Fast Fourier transform (FFT) analysis, when combined with TEM, has proven to be an effective method for unveiling structural features of framework materials, including MOFs and COFs; it is envisaged to be also applied to MLCOFs in the coming years. Castano et al. obtained detailed information about nanoscale features, including point defects, orientations and shapes of their 2D COF crystalline domains using this combined strategy.60 An automated postprocessing Fourier-mapping approach was applied to TEM images to determine the arrangement of 2D COFs on a graphene substrate in various orientations, where they presented as small crystallites with irregularly shaped domains, resulting due to uncontrolled growth and nucleation processes. Moreover, FFT-TEM mapping of the COFs has also shown the overlap of 2D COF sheets and provided insights into the tilt grain boundaries by analysing variations in the FFTs across the mapped area. Nevertheless, this approach can also be applied when the domains in the FFT pattern are tilted, reducing its practical application to other frameworks.
Coupled techniques, such as atomic force microscopy-infrared (AFM-IR), are powerful tools which leverage the combination of absorption spectroscopy and the spatial resolution of AFM.61 This method has been utilised with MOFs, to study the growth of HKUST-1 films,62 however, it is anticipated to be used to characterise MLCOFs in the near future.
Non-destructive characterisation techniques are often targeted due to difficulties encountered in the scaling-up process of certain frameworks. X-ray Absorption Spectroscopy (XAS) has emerged as a useful methodology for the analysis of both crystalline and amorphous materials, holding significant potential for the characterisation of MLCOFs. XAS techniques, including extended X-ray absorption fine structure (EXAFS) and X-ray absorption near edge structure (XANES), have been utilised for the examination of electronic configurations and structural determination of MOFs and low-crystalline COFs. Romero-Muñiz et al. demonstrated the utility of XAS in describing the coordination environment of palladium ions embedded within a TAPB-BTCA COF, (1,3,5-tris-(4- aminophenyl)benzene (TAPB); 1,3,5 benzene-tricarbaldehyde (BTCA)).63 EXAFS analyses indicated that the bonding between metallic species and COF (Pd–N) remained unchanged after exposure to Suzuki–Miyaura catalysis, highlighting the interaction strength. In contrast, other ligands, namely halogens, were found to be replaced during the reaction process. Then, XANES methodology showed a shift to lower energy values during the reaction process, revealing the reduction of palladium ions to Pd(0) during the catalytic cycle. Additional findings showed the presence of Pd–Pd bonding, suggesting the nucleation and growth of Pd nanoparticles throughout the reaction. With the advancement of modern synchrotron radiation sources, in situ XAS monitoring is envisaged to be developed in the following years, potentially making it a cutting-edge technique for understanding structural modifications and working mechanisms of MLCOF materials.
Although a comprehensive structural analysis of MLCOFs can be achieved through macro and nanoscale characterisations, its combination has proven to be effective when mapping the material, however, theoretical simulations can also play an important role, corroborating experimental findings with precise models.
Gao et al. effectively conducted an atomic analysis of MLCOFs using DFT; information obtained was employed to explain the properties of a calcium-intercalated COF. H2 storage capacity was evaluated and accurately predicted for different organic building units and covalent linkers. Interestingly, their findings revealed that although calcium displayed strong interactions with C5H5, it does not necessarily correlate with strong H2 binding interactions, revealing the requirement for high Ca–H2 affinity (Fig. 12).64 Based on the validated initial DFT model, an optimised 3D CaCOF was constructed, identifying the most promising framework for H2 storage. This research underscores the value of DFT methods in developing new porous materials and predicting their properties.
 | ||
| Fig. 12 (a) Side and (b) top view of charge density difference after Ca binding onto benzene. Isosurfaces with values of ±0.005 e Å−3 are shown. Red and blue clouds correspond to electron depletion and accumulation, respectively. (c) Same as (b) but for both two Ca and 10H2 binding to benzene. (d) and (e) Same as (a) and (b) but for Ca binding on five-membered C5H5. (f) The H2 binding energy as a function of distance (z) between Ca and H2 on C5H5. Reproduced from ref. 64 with permission from Nature Portfolio, copyright 2013. | ||
DFT was also utilised by Ke et al. to investigate the structure–activity relationships in both the node and linker of Ni-MLCOFs with lithium depositions. The model was corroborated by macro and nanoscale structural characterisation that were in agreement with the simulated IR and XRD, as well as with its pore size. DFT was further used to predict Li-deposition energies, exploring the differences between the metal complex sites and benzaldehyde as well as the amine-based linking units. Modelling showed that the primary Li-deposition sites were located in the Ni-complex units, while the linking units behaved as secondary deposition sites, highlighting the importance of obtaining a detailed atomic map of the frameworks.65
Theoretical modelling tools are highly useful to support experimental findings and predict properties related to the structure of the materials. Through integrating these methods synergistically alongside macro and nanoscale characterisation analyses, a precise and trustworthy atomic map can be generated, guiding in this way the design and optimisation of MLCOFs systems to a superior level.
3.2 MLCOF atomic mapping: synergistic characterisation techniques
Relationships between the structural characteristics of MLCOFs and their attributed properties can be better understood by using a combination of macro and nanoscale characterisation techniques and theoretical tools in synergy (Fig. 13). Their application in parallel or in conjunction has facilitated the design and development of new and improved MLCOF materials. | ||
| Fig. 13 Structure elucidation of MLCOFs, a synergetic approach. | ||
Han et al. illustrated the effectiveness of this approach in characterising three 2,2′-bipyridine-based Ru-MLCOFs by a combination of experimental analyses supported by computational modelling to precisely elucidate the structure of these materials. Initially, the polymerization of the network was investigated using FT-IR alongside 13C-ss-NMR, displaying vibrational peaks and signals typically associated with imine bonding. Following this, PXRD analysis showed a series of new diffraction peaks not observed in the precursors, confirming the formation of new materials. Pawley and Rietveld refinements provided additional information on the unit cell parameters of the materials, which was used alongside microscopy analyses to generate reliable structural models. Geometrical energy minimization was next utilised, indicating that the stp topology with a P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group represented the most stable configuration for all three of the frameworks. The simulations were in good agreement with experimental PXRD values and ordered lattice fringes observed in HR-TEM analyses (Fig. 14).8 The performance of the materials was evaluated through HER photocatalytic testing, revealing that the Ru-MCOF that incorporated (aminolphenyl)benzene-based building blocks exhibited superior catalytic activity among tested MLCOFs. Based on a precise characterisation of the materials and the development of an atomic map, it was concluded that an enhanced activity towards HER is related to the availability and degree of freedom of the Ru(bpy)2+ (bpy = 2,2′-bipyridine) photosensitizing units within the materials, resulting in a more stable and efficient photocatalyst.
space group represented the most stable configuration for all three of the frameworks. The simulations were in good agreement with experimental PXRD values and ordered lattice fringes observed in HR-TEM analyses (Fig. 14).8 The performance of the materials was evaluated through HER photocatalytic testing, revealing that the Ru-MCOF that incorporated (aminolphenyl)benzene-based building blocks exhibited superior catalytic activity among tested MLCOFs. Based on a precise characterisation of the materials and the development of an atomic map, it was concluded that an enhanced activity towards HER is related to the availability and degree of freedom of the Ru(bpy)2+ (bpy = 2,2′-bipyridine) photosensitizing units within the materials, resulting in a more stable and efficient photocatalyst.
 | ||
| Fig. 14 The SEM (left), TEM (right) and HAADF-STEM with corresponding elemental mapping (bottom) images of (a) RuCOF-ETTA, (b) RuCOF-TPB and (c) RuCOF-ETTBA. The PXRD patterns of (d) RuCOF-ETTA, (e) RuCOF-TPB and (f) RuCOF-ETTBA: the experimental (red), Pawley refined(black), simulated (blue), and difference between experimental and refined (green). Insets: the structures, powders and unit cell parameters of corresponding RuCOFs. Reproduced from ref. 8 with permission from Wiley-VCH, copyright 2022. | ||
A similar synergetic methodology was undertaken by Sun et al. to study the photocatalytic activity towards HER of Schiff-base Ni-MLCOFs, noting differences based on their morphology, either rod-like or spherical-like. These insights were derived from the elaboration of detailed atomic maps of the materials. Structure elucidation began by proving the formation of an imine bond; FT-IR showed a significant decrease in the characteristic vibrational bands of the starting materials (amine and aldehyde), while new imine signals were identified. The results, which were corroborated by 13C-ss-NMR, confirmed the presence of the imine. Diffraction patterns corresponding to a preferential AA stacking were obtained by PXRD, a finding further validated by theoretical models. Lattice stripes were also identified through TEM, suggesting a long-range ordered material. Nevertheless, the complete map at different scales was obtained by SEM. The surface analysis revealed that the choice of organic building blocks, pyrene- or benzene-based molecules, led to the formation of either rod-like or spherical-like morphologies, which influenced their catalytic behavior.66 Comparisons drawn by using this approach gave important information regarding the structure–activity relationships of Ni-MLCOFs, enabling, in this way, its future development.
In a fundamental study, Hennessey et al. reported the complete atomic mapping of a Ru-based MCOF composed of pyrene units covalently linked via an imine bond to a [Ru(tpy)2]2+ (tpy: 2,2′,6,2′′-terpyridine) metalloligand by strategically combining experimental and computational methodologies.67 The synergetic approach not only displayed the structural composition of the framework but also provided information about the stacking arrangement of the material. Initially, the identification of the imine linkage was conducted using FT-IR and 13C-NMR. This was followed by photophysical investigations that demonstrated the presence of both photoactive building units and a broadening effect on the light-active window by integrating light-active units into the framework, highlighting its potential for photocatalytic applications. PXRD data exhibited the low-crystalline nature of the MLCOF, which was further validated through HR-TEM measurements. Interestingly, scanning tunneling microscopy (STM) analysis showed an ordered material at the nanoscale; however, the measured distances between building units could not be fitted within a monolayer model. Subsequently, optimised DFT models were built to find stable configurations of the material wherein a multilayer model was plausible, revealing that the MCOF formed a shifted-stacking AB bilayer, results that were corroborated by microscopy analyses, including AFM, HR-TEM and STM (Fig. 15). The authors evidenced the advantages of carefully combining macro and nanoscale experimental analyses alongside theoretical calculations to generate a comprehensive atomic map of low-crystalline MLCOFs, revealing important structural features for improved material design.
 | ||
| Fig. 15 Scanning probe microscopies characterisation of the Ru-pyrene MCOF. (a) High-resolution STM image showing the Ru-pyrene MCOF imaged at the heptanoic acid/HOPG interface. The measured distances and angle between repeating units obtained from calibrated STM images are: a = 1.7 ± 0.1 nm; b = 1.8 ± 0.1 nm; γ = 80 ± 3°. Imaging parameters: Vbias = −0.4 V, Iset = 150 pA. (b) Molecular models of the Ru-pyrene MCOF and Ru-metalloligand 3 (inset). Relevant intramolecular and intralayer distances between Ru atoms are given in nm. The STM pattern is overlaid in grey. (c) AFM image showing the layered morphology of the Ru-pyrene MCOF. (d) Line profile across one of the islands in (c) showing the step height of ca. 0.6 nm. Reproduced from ref. 67 with permission from Wiley-VCH, copyright 2025. | ||
3.3 Cutting-edge advanced characterisation technique
Pair distribution funtion (PDF) has emerged as an effective tool for analysing low crystalline porous materials using synchrotron X-ray or neutron radiation. This method allows the obtention of key information, including atomic distances, coordination number, and particle size, among others, offering data about materials' structures and crystallisation processes. The method calculates atom–atom distances based on Bragg peaks and diffuse scattering signal from X-ray radiation, followed by fitting the information using computational methods to generate radial distribution plots.68Although it has not yet been widely applied to MLCOFs, the applications of PDF analysis have been demonstrated by the characterisation of MOFs and COFs, as highlighted by C. Ashworth,69 who achieved the structural determination of an amorphous MIL-100 MOF and compared it to its crystalline analogue. The atomic map of the short-range ordered framework, Fe-BTC MOFs (1,3,5-benzenetricarboxylate (BTC)) was described firstly with XAS, identifying trinuclear building blocks; however, the data was limited to the first coordination sphere of the framework. To overcome this, the authors employed advanced electron microscopy techniques followed by PDF analysis using total scattering experiments with synchrotron X-ray radiation. The results allowed a thorough examination of the long-range structure of both the amorphous and the crystalline MOFs, showing that, in contrast to the amorphous material, 39% to 64% of the trimers form tetrahedra structures but with low long-range order being the reason for the material crystallinity.
Crystallinity defects formed during the synthesis of palladium-loaded COF systems were demonstrated by Romero-Muñiz et al. through PDF analyses.70 Initially, the short-range order of the frameworks and their local chemical environment were characterised using EXAFS studies. Nonetheless, to assess the longer-range order of the modified COFs, a PDF methodology was conducted. Notably, the effects of the loaded Pd on the atom–atom distances within the COF could be assessed by subtracting the PDF data obtained from the non-metallated COF from that of the loaded material. The results showed the evolution of the framework when metals were embedded, finding two atom–atom distances assigned to the coordination of Pd with chlorine and nitrogen atoms. Moreover, the value of PDF was also shown after catalysis (Suzuki–Miyaura coupling), where face-centred cubic (fcc) Pd(0) diffraction peaks, corresponding to the formation of nanoparticles, were detected. This information was then confirmed by TEM analysis. A similar PDF analysis was performed on Pd-COFs synthesised using different methods, showing that in situ metallation led to the stabilisation of Pd nanoparticles within the pores of the COF rather than on the surface of the framework.
The PDF methodology in combination with theoretical simulations was successfully utilised by Han et al. for the structural determination of a TiCOF-spn 3D MLCOF, which consists of Ti(IV) complexes connected by TAPT (1,3,5-tris(4-aminophenyl) triazine) linkers (Fig. 16).71 As with other frameworks, initial investigations aimed to detect the formation of new imine bonds, employing both FT-IR and 13C (CP/MAS) NMR techniques. A deeper characterisation was then performed using XPS to confirm the presence of both the Ti–O and C–O bonds within the framework, followed by studies on the crystalline structure through a combination of PXRD and PDF analyses. Theoretical calculations and structural models based on PXRD measurements suggested that the TiCOF-spn adopted a non-interpenetrated spn topology, which was further verified through PDF analysis using synchrotron X-ray radiation to calculate the atom–atom distances. The results showed diffraction peaks related to Ti–O and Ti–Ti bonding, which aligned with a non-interpenetrated spn topology, as predicted by theoretical modelling. This example illustrates the advantages of PDF analysis for the structural determination of low-crystalline frameworks, emphasizing its usefulness in characterising the long-range order of the frameworks, as well as obtaining information relating to their bonding.
 | ||
| Fig. 16 (a) Representation of the (3,6)-connected eea and spn topologies, which were deconstructed into trigonal antiprismatic and planar triangle linkers with their corresponding chemical equivalents Na2Ti(2,3-DHTA)3 and TAPT. (b) The experimental (black) and Pawley refined (red) PXRD patterns of TiCOF-spn, the difference between the experimental and refined PXRD patterns (green), and the simulated PXRD patterns based on eea net (purple) and spn net (blue). (c) The 2D scattering image of TiCOF-spn was reduced to 1D data. (d) The PDF data and calculated PDF pattern with spn net for TiCOF-spn. Reproduced from ref. 71 with permission from Elsevier, copyright 2022. | ||
Although the PDF methodology has significant potential for characterising a wide variety of low-crystalline materials, access to high-energy radiation sources is imperative, which limits its widespread adoption. X-ray synchrotron sources are often preferred, as the frameworks need to be deuterated before using alternative radiation sources such as neutrons. Despite the current complexity of the technique, emerging technologies which simplify the analysis are becoming more popular, including differential PDF analysis (dPDF), which highlights differences between PDF profiles by subtracting them.
3D electron diffraction (3DED) has emerged as another promising method for determining the structure of materials with low crystallinities, especially when used alongside PXRD and ab initio calculations.72 The technique has been effective in comparing and confirming PXRD findings of MOFs and COFs that typically form nano- and submicron-sized crystals. Additionally, 3DED can be utilised to monitor host–guest interactions within framework materials by locating the positions of the guest molecules.72 Nevertheless, challenges that limit its application still remain. High guest molecule occupancy and cryogenic conditions are generally required when using 3DED. In addition, the technique is unsuitable for materials sensitive to electron beams, such as MLCOFs.60 Moreover there is a need for more advanced analytical software for 3DED, as the existing programs were originally developed for SC-XRD.
The development of MeCOFs and MLCOFs is still an emerging field; synthetic challenges, such as scalability, optimisation and control over the crystallinity hinder their applicability. Consequentially, a more comprehensive understanding of the structural impacts of synthesis conditions is crucial when striving for a high degree of morphological control over their properties, including porosity, crystallinity and surface areas. Therefore, improvements in their structural characterisation and computational modelling are necessary for the development of more accurate atomic maps of the frameworks, which will enable better control over their properties. As described, the combination of characterisation techniques, at the macro and nanoscale, along with theoretical modelling is the most appropriate approach to resolve their structures, requiring the collection of a high amount of data. However, it is necessary to better understand the structure–property relationships of the MeCOFs and MLCOFs, providing a starting point for predicting their electronic and chemical properties, resulting in the design of more effective materials for a wide range of applications.
4 Summary and outlook
Advancements in porous material research have been progressing rapidly, with applications in fields including gas adsorption, catalysis and electronics. Despite the challenges faced for the future development of MLCOFs, the repeating metalloligand units, as well as the ability to alternate the metal centres, creates a high degree of compositional variety to the frameworks. The presence of both coordinate and covalent bonds can add an element of enhanced stability in contrast with other MOF or COF alternatives. The support of well-established structural mapping techniques, specifically XRD analysis, has been hugely beneficial for the establishment of structure–property relationships and optimisation of such materials. The recent development of porous materials with long-range order can be partly attributed to the ease by which their crystallinities enable their accurate atomic scale analysis, thereby offering key structural insights which enable further advancements in their applications.However, in some instances the use of SC-XRD or PXRD in conjunction with structure refinement can lead to structures that do not fully represent the nature of the material. Regarding single crystals, they may only account for 1–2% of a sample, whereas for PXRD, hidden structures in low quantities are difficult to identify and isolate. One such example has been elucidated by using 3DED, where MOF structures of different atomic arrangements were found on a sample, which was considered pure using the traditional characterisation techniques.73 However, access to such facilities is still restricted and therefore a combination of techniques is required to have a full picture of the nanomaterials obtained when synthesising porous materials.
We investigated the emergence of MCOFs, classifying them as MeCOFs and MLCOFs to describe a synergistic approach to their structural evaluation. Specifically, we have highlighted the challenges faced when carrying out structural determination on MLCOFs which typically result in low-crystallinity frameworks.
The slow but steady rise in the development of MLCOFs is drawing attention towards the need for a definitive method for the atomic mapping of such materials. Structural determination plays a key role in establishing a fundamental understanding of the properties of framework materials. As many examples have shown herein, the usual characterisation techniques can be misleading when hypothesising atomic mapping of porous materials, and the exact disposition of the monomers is important when optimising structure–activity relationships.
Devising a more straightforward approach to structurally mapping MLCOFs could progress the development and structural optimisation of these materials greatly going forward. By highlighting the characterisation of MLCOFs from a macro and nanoscale perspective, supported by theoretical modelling tools, we illustrate how a combination of techniques can be used to atomically map such materials using previous work on MLCOFs as guiding examples. The discussed approaches could bridge the gap in devising a synergistic approach to the structural elucidation of this rapidly developing area of materials chemistry.
Although challenges remain to be addressed, particularly regarding the synthesis of increasingly crystalline materials, we have highlighted a pathway to overcome these obstacles. It is evident that the priority should be to find synthesis methods which result in materials with better crystallinity, however many applications have demonstrated that amorphous or low-crystalline materials may have even better properties in certain applications. With the range of techniques at our disposal, there should be no excuse not to attempt to provide a better description of the materials synthesised, even if it means using advanced techniques or a combination of thereof.
Emphasis was placed on carefully selecting a combination of analysis techniques which work in synergy with one another, providing a strategy for the atomic mapping of framework materials. Advanced techniques are required for some of the challenging materials, as illustrated by the examples shown for MLCOFs, where the higher degree of flexibility of some of the building units compared to the more established MOF and COF facilities add the extra degree of difficulty to obtain the complete structure of the material at the atomic level. As seen throughout the perspective, theoretical tools provide further insights by combination with experiments. However, theoretical models are usually hindered by the lengthy calculations, which lead to models that do not fully represent the experimental materials. It is very important to validate any theoretical model by supporting it with experimental data that represents the bulk of the material. Overall, a better understanding of the structure–property relationships will prompt significant progress for the future advancement of MCOFs and their applications in gas storage, catalysis and sensing.
Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Author contributions
All authors contributed to the writing and revision of the manuscript and have approved the final version of this perspective article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. B., R. G.-G., S. H. and P. F. acknowledge funding support from the European Union's Horizon 2020 research and innovation programme (project NEFERTITI) under Grant Agreement No. 101022202.Notes and references
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- R. Chen, J. Zhang, J. Chelora, Y. Xiong, S. V Kershaw, K. F. Li, P.-K. Lo, K. W. Cheah, A. L. Rogach, J. A. Zapien and C.-S. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 5699–5708 CrossRef CAS PubMed.
- Y. Qian, F. Zhang and H. Pang, Adv. Funct. Mater., 2021, 31, 2104231 CrossRef CAS.
- Y. Li, M. Karimi, Y.-N. Gong, N. Dai, V. Safarifard and H.-L. Jiang, Matter, 2021, 4, 2230–2265 CrossRef CAS.
- L. Feng, K.-Y. Wang, G. S. Day, M. R. Ryder and H.-C. Zhou, Chem. Rev., 2020, 120, 13087–13133 CrossRef CAS PubMed.
- P. Zhou, J. Lv, X. Huang, Y. Lu and G. Wang, Coord. Chem. Rev., 2023, 478, 214969 CrossRef CAS.
- T. Skorjanc, D. Shetty and M. Valant, ACS Sens., 2021, 6, 1461–1481 CrossRef CAS PubMed.
- W.-K. Han, Y. Liu, X. Yan, Y. Jiang, J. Zhang and Z.-G. Gu, Angew. Chem., Int. Ed., 2022, 61, e202208791 CrossRef CAS PubMed.
- C. Lin, H. Ma, J.-R. He, Q. Xu, M. Song, C.-X. Cui, Y. Chen, C.-X. Li, M. Jiao and L. Zhai, Small, 2024, 2403775 CrossRef CAS PubMed.
- H. Peng, J. Raya, F. Richard, W. Baaziz, O. Ersen, A. Ciesielski and P. Samorì, Angew. Chem., Int. Ed., 2020, 59, 19602–19609 CrossRef CAS PubMed.
- T. Skorjanc, D. Shetty, M. E. Mahmoud, F. Gándara, J. I. Martinez, A. K. Mohammed, S. Boutros, A. Merhi, E. O. Shehayeb, C. A. Sharabati, P. Damacet, J. Raya, S. Gardonio, M. Hmadeh, B. R. Kaafarani and A. Trabolsi, ACS Appl. Mater. Interfaces, 2022, 14, 2015–2022 CrossRef CAS PubMed.
- Y. Meng, Y. Luo, J.-L. Shi, H. Ding, X. Lang, W. Chen, A. Zheng, J. Sun and C. Wang, Angew. Chem., Int. Ed., 2020, 59, 3624–3629 CrossRef CAS PubMed.
- R.-J. Wei, X. Luo, G.-H. Ning and D. Li, Acc. Chem. Res., 2025, 58, 746–761 CrossRef CAS PubMed.
- J. Dong, X. Han, Y. Liu, H. Li and Y. Cui, Angew. Chem., Int. Ed., 2020, 59, 13722–13733 CrossRef CAS PubMed.
- C. Kang, K. Yang, Z. Zhang, A. K. Usadi, D. C. Calabro, L. S. Baugh, Y. Wang, J. Jiang, X. Zou, Z. Huang and D. Zhao, Nat. Commun., 2022, 13, 1370 CrossRef CAS PubMed.
- L. Peng, Q. Guo, C. Song, S. Ghosh, H. Xu, L. Wang, D. Hu, L. Shi, L. Zhao, Q. Li, T. Sakurai, H. Yan, S. Seki, Y. Liu and D. Wei, Nat. Commun., 2021, 12, 5077 CrossRef CAS PubMed.
- J. L. Segura, S. Royuela and M. Mar Ramos, Chem. Soc. Rev., 2019, 48, 3903–3945 RSC.
- Y. Qin, X. Zhu and R. Huang, Biomater. Sci., 2023, 11, 6942–6976 RSC.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS PubMed.
- B. J. Burnett, P. M. Barron and W. Choe, CrystEngComm, 2012, 14, 3839–3846 RSC.
- L. Jiao, Y. Wang, H.-L. Jiang and Q. Xu, Adv. Mater., 2018, 30, 1703663 CrossRef PubMed.
- D. A. Anito, T.-X. Wang, H.-P. Liang, X. Ding and B.-H. Han, Polym. Chem., 2021, 12, 4557–4564 RSC.
- G. Lan, Y.-Y. Zhu, S. S. Veroneau, Z. Xu, D. Micheroni and W. Lin, J. Am. Chem. Soc., 2018, 140, 5326–5329 CrossRef CAS PubMed.
- M. Y. Masoomi, A. Morsali, A. Dhakshinamoorthy and H. Garcia, Angew. Chem., Int. Ed., 2019, 58, 15188–15205 CrossRef CAS PubMed.
- C. Jiang, X. Wang, Y. Ouyang, K. Lu, W. Jiang, H. Xu, X. Wei, Z. Wang, F. Dai and D. Sun, Nanoscale Adv., 2022, 4, 2077–2089 RSC.
- Y.-H. Yu, S.-L. Huang and G.-Y. Yang, CrystEngComm, 2022, 24, 3160–3164 RSC.
- F. Ghobakhloo, D. Azarifar, M. Mohammadi, H. Keypour and H. Zeynali, Inorg. Chem., 2022, 61, 4825–4841 CrossRef CAS PubMed.
- M. Elcheikh Mahmoud, H. Audi, A. Assoud, T. H. Ghaddar and M. Hmadeh, J. Am. Chem. Soc., 2019, 141, 7115–7121 CrossRef CAS PubMed.
- Z. Zhang, S. B. Peh, R. Krishna, C. Kang, K. Chai, Y. Wang, D. Shi and D. Zhao, Angew. Chem., Int. Ed., 2021, 60, 17198–17204 CrossRef CAS PubMed.
- E. Lin, Z. Wang and Z. Zhang, J. Mater. Chem. A Mater, 2024, 12, 21704–21715 RSC.
- Y. He, X. Chen, C. Huang, L. Li, C. Yang and Y. Yu, Chin. J. Catal., 2021, 42, 123–130 CrossRef CAS.
- L. Liu, X.-X. Wang, X. Wang, G.-J. Xu, Y.-F. Zhao, M.-L. Wang, J.-M. Lin, R.-S. Zhao and Y. Wu, J. Hazard. Mater., 2021, 403, 123917 CrossRef CAS PubMed.
- H. R. Abuzeid, A. F. M. EL-Mahdy and S.-W. Kuo, Giant, 2021, 6, 100054 CrossRef CAS.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS PubMed.
- M. Zhang, R. Zheng, Y. Ma, R. Chen, X. Sun and X. Sun, Microporous Mesoporous Mater., 2019, 285, 70–79 CrossRef CAS.
- S. Bi, C. Yang, W. Zhang, J. Xu, L. Liu, D. Wu, X. Wang, Y. Han, Q. Liang and F. Zhang, Nat. Commun., 2019, 10, 2467 CrossRef PubMed.
- Q. Guan, L.-L. Zhou and Y.-B. Dong, Chem. Soc. Rev., 2022, 51, 6307–6416 RSC.
- Q. Guan, L.-L. Zhou and Y.-B. Dong, Chem. Soc. Rev., 2022, 51, 6307–6416 RSC.
- R. Chen, Y. Wang, Y. Ma, A. Mal, X.-Y. Gao, L. Gao, L. Qiao, X.-B. Li, L.-Z. Wu and C. Wang, Nat. Commun., 2021, 12, 1354 CrossRef CAS PubMed.
- Y. Xiang, W. Dong, P. Wang, S. Wang, X. Ding, F. Ichihara, Z. Wang, Y. Wada, S. Jin, Y. Weng, H. Chen and J. Ye, Appl. Catal., B, 2020, 274, 119096 CrossRef CAS.
- V. Hasija, S. Patial, P. Raizada, A. Aslam Parwaz Khan, A. M. Asiri, Q. Van Le, V.-H. Nguyen and P. Singh, Coord. Chem. Rev., 2022, 452, 214298 CrossRef CAS.
- N.-Y. Huang, J.-Q. Shen, X.-W. Zhang, P.-Q. Liao, J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2022, 144, 8676–8682 CrossRef CAS PubMed.
- H.-S. Lu, W.-K. Han, X. Yan, C.-J. Chen, T. Niu and Z.-G. Gu, Angew. Chem., Int. Ed., 2021, 60, 17881–17886 CrossRef CAS PubMed.
- L.-H. Li, X.-L. Feng, X.-H. Cui, Y.-X. Ma, S.-Y. Ding and W. Wang, J. Am. Chem. Soc., 2017, 139, 6042–6045 CrossRef CAS PubMed.
- C. Xiong, Z. Shao, J. Hong, K. Bi, Q. Huang and C. Liu, Int. J. Miner., Metall. Mater., 2023, 30, 2297–2309 CrossRef CAS.
- Y. Liu, Y. Ma, Y. Zhao, X. Sun, F. Gándara, H. Furukawa, Z. Liu, H. Zhu, C. Zhu, K. Suenaga, P. Oleynikov, A. S. Alshammari, X. Zhang, O. Terasaki and O. M. Yaghi, Science, 2016, 351, 365–369 CrossRef CAS PubMed.
- H. L. Nguyen, F. Gándara, H. Furukawa, T. L. H. Doan, K. E. Cordova and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 4330–4333 CrossRef CAS PubMed.
- L. Martins, L. K. Macreadie, D. Sensharma, S. Vaesen, X. Zhang, J. J. Gough, M. O'Doherty, N.-Y. Zhu, M. Rüther, J. E. O'Brien, A. L. Bradley and W. Schmitt, Chem. Commun., 2019, 55, 5013–5016 RSC.
- W.-K. Han, J. Li, R.-M. Zhu, M. Wei, S.-K. Xia, J.-X. Fu, J. Zhang, H. Pang, M.-D. Li and Z.-G. Gu, Chem. Sci., 2024, 15, 8422–8429 RSC.
- M. Wang, C. Wang, J. Liu, F. Rong, L. He, Y. Lou, Z. Zhang and M. Du, ACS Sustain. Chem. Eng., 2021, 9, 5872–5883 CrossRef CAS.
- C. L. Jones, C. E. Hughes, H. H.-M. Yeung, A. Paul, K. D. M. Harris and T. L. Easun, Chem. Sci., 2021, 12, 1486–1494 RSC.
- K. Roztocki, M. Rauche, V. Bon, S. Kaskel, E. Brunner and D. Matoga, ACS Appl. Mater. Interfaces, 2021, 13, 28503–28513 CrossRef CAS PubMed.
- H. Embrechts, M. Kriesten, K. Hoffmann, W. Peukert, M. Hartmann and M. Distaso, J. Phys. Chem. C, 2018, 122, 12267–12278 CrossRef CAS.
- N. Keller and T. Bein, Chem. Soc. Rev., 2021, 50, 1813–1845 RSC.
- H.-G. Zhou, R.-Q. Xia, J. Zheng, D. Yuan, G.-H. Ning and D. Li, Chem. Sci., 2021, 12, 6280–6286 RSC.
- J. Fu, S. Das, G. Xing, T. Ben, V. Valtchev and S. Qiu, J. Am. Chem. Soc., 2016, 138, 7673–7680 CrossRef CAS PubMed.
- J. Du, S. Liang, M. Wang, Y. Wang, T. Yi, Z. Zhang, F. Pan and Z. Jiang, J. Memb. Sci., 2023, 683, 121840 CrossRef CAS.
- R.-J. Wei, P.-Y. You, H. Duan, M. Xie, R.-Q. Xia, X. Chen, X. Zhao, G.-H. Ning, A. I. Cooper and D. Li, J. Am. Chem. Soc., 2022, 144, 17487–17495 CrossRef CAS.
- J. Lyu, X. Gong, S.-J. Lee, K. Gnanasekaran, X. Zhang, M. C. Wasson, X. Wang, P. Bai, X. Guo, N. C. Gianneschi and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 4609–4615 CrossRef CAS PubMed.
- I. Castano, A. M. Evans, R. dos Reis, V. P. Dravid, N. C. Gianneschi and W. R. Dichtel, Chem. Mater., 2021, 33, 1341–1352 CrossRef CAS PubMed.
- J. J. Schwartz, D. S. Jakob and A. Centrone, Chem. Soc. Rev., 2022, 51, 5248–5267 RSC.
- G. Delen, Z. Ristanović, L. D. B. Mandemaker and B. M. Weckhuysen, Chem. Eur. J., 2018, 24, 187–195 CrossRef CAS PubMed.
- I. Romero-Muñiz, A. Mavrandonakis, P. Albacete, A. Vega, V. Briois, F. Zamora and A. E. Platero-Prats, Angew. Chem., Int. Ed., 2020, 59, 13013–13020 CrossRef PubMed.
- F. Gao, Z. Ding and S. Meng, Sci. Rep., 2013, 3, 1882 CrossRef PubMed.
- S.-W. Ke, Y. Wang, J. Su, K. Liao, S. Lv, X. Song, T. Ma, S. Yuan, Z. Jin and J.-L. Zuo, J. Am. Chem. Soc., 2022, 144, 8267–8277 CrossRef CAS PubMed.
- L. Sun, M. Lu, Z. Yang, Z. Yu, X. Su, Y.-Q. Lan and L. Chen, Angew. Chem., Int. Ed., 2022, 61, e202204326 CrossRef CAS PubMed.
- S. Hennessey, R. González-Gómez, N. Arisnabarreta, A. Ciotti, J. Hou, N. V Tarakina, A. Bezrukov, K. S. Mali, M. Zaworotko, S. De Feyter, M. García-Melchor and P. Farràs, Adv. Mater., 2025, 37(13), 2502155 CrossRef CAS PubMed.
- I. Romero-Muñiz, P. Albacete, A. E. Platero-Prats and F. Zamora, ACS Appl. Mater. Interfaces, 2021, 13, 54106–54112 CrossRef.
- C. Ashworth, Nat. Rev. Chem., 2021, 5, 298 CrossRef.
- I. Romero-Muñiz, A. Mavrandonakis, P. Albacete, A. Vega, V. Briois, F. Zamora and A. E. Platero-Prats, Angew. Chem., Int. Ed., 2020, 59, 13013–13020 CrossRef PubMed.
- W.-K. Han, H.-S. Lu, J.-X. Fu, X. Liu, X. Zhu, X. Yan, J. Zhang, Y. Jiang, H. Dong and Z.-G. Gu, Chem. Eng. J., 2022, 449, 137802 CrossRef CAS.
- Z. Huang, E. S. Grape, J. Li, A. K. Inge and X. Zou, Coord. Chem. Rev., 2021, 427, 213583 CrossRef CAS.
- T. Yang, T. Willhammar, H. Xu, X. Zou and Z. Huang, Nat. Protoc., 2022, 17, 2389–2413 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |