Rational design of new bifunctional inhibitors of type II dehydroquinase†
Miguel D. Toscanoa, Kirsty A. Stewartb, John R. Cogginsc, Adrian J. Lapthornb and Chris Abell*a
aUniversity Chemical Laboratory, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: ca26@cam.ac.uk; Fax: 44 1223 336362; Tel: 44 1223 336405
bDept. of Chemistry, Joseph Black Building, University of Glasgow, Glasgow, UK G12 8QQ
cInstitute of Biomedical and Life Sciences, Division of Biochemistry and Molecular Biology, University of Glasgow, Glasgow, UK G12 8QQ
First published on 1st August 2005
Abstract
Selective inhibitors of type II dehydroquinase were rationally designed to explore a second binding-pocket in the active-site. The molecular modelling, synthesis, inhibition studies and crystal structure determination are described.
The shikimate pathway is involved in the biosynthesis of aromatic amino acids and other key metabolites, and has been found in plants, bacteria, fungi and some parasites, but not in mammals.1 It is a recognised target in the development of novel herbicides and antimicrobial agents. The third step in this pathway is the dehydration of 3-dehydroquinate (1) to 3-dehydroshikimate (2) (Scheme 1), catalysed by dehydroquinase (EC 4.2.1.10, 3-dehydroquinate dehydratase).2 This step is also part of the quinate pathway in fungi.1 There are two types of dehydroquinase, which are structurally distinct and catalyse the same transformation by different mechanisms.3,4 Type I dehydroquinases are dimeric proteins with a 26–28 kDa subunit and catalyse the syn dehydration of 1 through the formation of a Schiff base.2,5 Type II dehydroquinases are dodecamers of subunit 12–18 kDa and catalyse the anti elimination of water via an enolate intermediate.6 Since only type II dehydroquinases are present in important pathogens such as Mycobacterium tuberculosis7 and Helicobacter pylori,8 specific inhibitors of these enzymes have therapeutic potential.
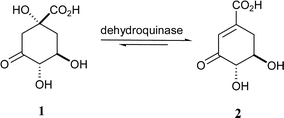 | ||
| Scheme 1 The reaction catalysed by dehydroquinase. | ||
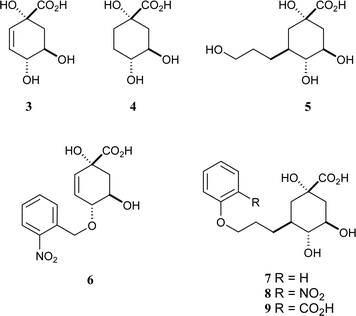
The first generation of mechanism-based inhibitors was based on mimicking the flattened enolate intermediate.9 The anhydro compound 3 had a KI of 30 µM against Streptomyces coelicolor type II dehydroquinase and was significantly more potent than the reduced analogue 4
(KI
= 600 µM). A crystal structure of S. coelicolor type II dehydroquinase with 3 bound in the active site revealed a second binding-pocket, which was occupied by a glycerol molecule (PDB code: 1GU1)
(Fig. 1a).10 Subsequently, bifunctional compounds with a quinate core and side-chains from C-3 to reach into this second binding site were designed. The primary alcohol 5 was the most potent of these compounds but had a modest KI of 180 µM against S. coelicolor type II dehydroquinase.11 In order to derive more benefit from this second binding site, a range of substituted benzyl groups were attached to the C-1 and C-4 hydroxyl groups of 3.12 Molecular docking suggested that the substituted benzyl groups would bind at the “glycerol” pocket, and the most potent compound (6, KI
= 8 µM) included a 2-nitrobenzyl substituent. We have now combined these strategies and designed compounds 7–9 to have pendant aromatic groups attached to side chains attached to C-3 of the quinate core.
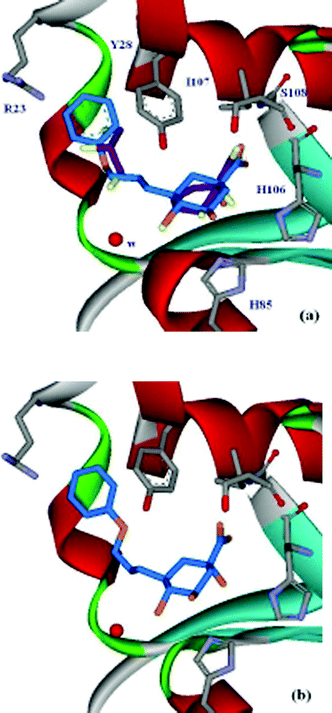 | ||
| Fig. 1 15 (a) Active-site view of the S. coelicolor type II dehydroquinase crystal structure with 3 and glycerol (purple) bound (PDB code: 1GU110). Comparison between the position of 3 and the docking result of 7 (blue); (b) active-site view of the S. coelicolor type II dehydroquinase crystal structure with 7 (blue) bound (PDB code: 2BT4). | ||
Molecular docking with the program GOLD (version 2.1)13 was used to predict the binding modes of 7–9. These ligands were docked to the type II dehydroquinase structure (1GU1), which was prepared using Sybyl6.5.14 The result from 7 is shown in Fig. 1a, compared with the position of 3 in the crystal structure. Both 8 and 9 docked in the same orientation (see ESI†). The quinate ring docks in a similar position to 3, and the side-chain reaches to the second binding-pocket, which appears to be large enough to accommodate the phenyl ring. The modelling suggests ring-stacking between the ligands' phenyl group and the side-chain of Tyr-28, and a possible interaction of any ortho-substituent with Arg-23.
The target compounds (7–9) were synthesised from quinic acid (10) according to the method outlined in Scheme 2. The intermediate 11 was prepared in three steps using known methods16 and the free hydroxyl was acetylated with acetic anhydride in pyridine in 96% yield to give 12. The anti-Markovnikov bromination of the allyl side-chain to form 13 was achieved in high yield (97%) by bubbling HBr through a solution of 12 in carbon tetrachloride in the presence of catalytic AIBN. This terminal bromide was the key branch-point in the synthetic strategy. It was reacted with phenol, 2-nitrophenol and methyl 2-hydroxybenzoate, in the presence of sodium hydride and potassium iodide (catalytic), and heated to reflux in acetonitrile to give 14 (69% yield), 15 (11%) and 16 (23%), respectively. The yields reflect the nucleophilicity of the phenols (PhOH > 2-MeO2CPhOH > 2-O2NPhOH). This strategy potentially allows the attachment of a wide range of nucleophiles, such as other alcohols, amines or thiols, to the propyl side-chain of these quinate analogues. Full deprotection of all compounds (14–16) was achieved by treatment with sodium hydroxide in a MeCN–H2O (1 : 1) solution at room-temperature and the products 7–9 were obtained as a 1 : 1 mixture with benzoate. The pure ammonium salts of the target compounds were obtained after HPLC purification through a C-18 reverse-phase column eluting with a gradient of H2O–MeCN and treatment with ammonium bicarbonate.
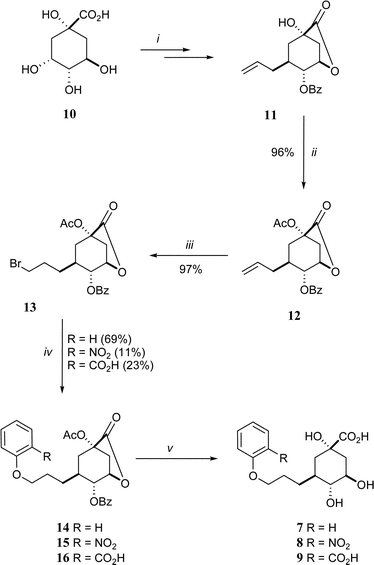 | ||
| Scheme 2 Reagents and conditions: (i) ref. 16; (ii) Ac2O, pyridine, RT; (iii) HBr, CCl4, RT; (iv) 2-RPhOH, NaH, NaI, MeCN, Δ; (v) 1. NaOH, H2O–MeCN (1 : 1), 2. HPLC purification (reverse phase, H2O–MeCN gradient). | ||
The analogues 7–9 were tested for inhibition against type I (Salmonella typhi) and type II (S. coelicolor) dehydroquinases by monitoring the formation of product at 232 nm.17 The results are shown in Table 1. None of the compounds showed any inhibition against the type I enzyme within the sensitivity of the assay, which is consistent with all the results obtained on C-3 substituted quinate analogues. However all three compounds showed competitive reversible inhibition against type II dehydroquinase. Compound 7 is the best inhibitor of the S. coelicolor type II enzyme, with a KI of 33 µM. This inhibitor has a similar KI to 3 and is 20 times more potent than the reduced analogue 4, and 6 times more potent than 5, which emphasises the importance of the benzyl ring. The analogues with a 2-substituted phenyl ring, 8 and 9, had KI values of 84 and 220 µM respectively, which is in contrast with the positive effect of the 2-nitro group on 6. These results show a trend where increase of hydrophilicity at the 2-position in the ring is unfavourable, suggesting that there is no positive interaction with the Arg-23 side-chain.
| Compounds | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|
| a KM values of 16 µM (S. typhi type I dehydroquinase) and 250 µM (S. coelicolor type II dehydroquinase) were obtained, using the standard assay.17b Ref. 9.c Ref. 11.d Ref. 12. | |||||||
| Type I (S. typhi)a | 3000 ± 1000b | 4500 ± 500b | >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000c 000c | — | >20000 | >20000 | >20000 |
| Type II (S. coelicolor)a | 30 ± 10b | 600 ± 200b | 180 ± 20c | 8 ± 2d | 33 ± 5.4 | 84 ± 13 | 220 ± 50 |
The best inhibitor of this set, 7, was crystallised with S. coelicolor type II dehydroquinase and the crystal structure shows the compound binding at the active-site (Fig. 1b). The quinate ring is in a similar position as 3 in the main binding-pocket with the exception of C-2, which in the absence of a double bond is moved by 0.85 Å, the quinate ring adopting a full chair conformation. The carboxyl interactions with the backbond amides of Ile-107 and Ser-108 are essentially the same, while the H-bond from the hydroxyl group at C-1 with the side chain of His-106 is longer (0.1 Å). The quinate ring of 7 is moved away from the protein by ∼0.3 Å and as a result H-bonding distances between the hydroxyl groups at C-4 and C-5 with His-85 and Arg-117 (not shown) are slightly changed (by +0.1 and −0.2 Å, respectively). This movement is accompanied by a 0.5 Å movement in the water conserved in all S. coelicolor type II dehydroquinase structures and implicated in the mechanism.10 This water movement is caused by the side-chain at C-3 extending into the “glycerol” binding-pocket, where there is ring-stacking between the benzyl ring and Tyr-28. In addition, the benzene ring displaces two water molecules and effects the position of a third buried water.
Despite some disruption to the structure, the side-chain at C-3, which extends into the “glycerol” binding-pocket must account for the increased potency of 7. The position of 7 in the active site agrees well with the binding position predicted by molecular docking (Fig. 1a vs. 1b), validating the strategy and methods used in this project. Further optimisation of 7, to reduce the unfavourable interactions identified from the crystal structure should lead to significantly more potent specific inhibitors of type II dehydroquinase than are currently available.
In summary, we have rationally designed a set of compounds to explore specific interactions at a second binding-pocket in the active site of type II dehydroquinase, achieving an increase in potency, with 7 in particular, and high selectivity. Structural information showed 7 in a position consistent with molecular docking experiments, and identified key interactions to optimise in future compounds.
Acknowledgements
We would like to thank Fundação para a Ciência e a Tecnologia for financial support.References
- C. Abell, in Enzymology and Molecular Biology of the Shikimate Pathway; Comprehensive Natural Products Chemistry, ed. U. Sankawa, Pergamon, Elsevier Science Ltd., Oxford, 1999, pp. 573–607 Search PubMed.
- C. Kleanthous, R. Deka, K. Davis, S. M. Kelly, A. Cooper, S. E. Harding, N. C. Price, A. R. Hawkins and J. R. Coggins, Biochem. J., 1992, 282, 687–695 CAS.
- V. B. Patel, M. Schweizer, C. C. Dyskstra, S. R. Kushner and N. H. Giles, Proc. Natl. Acad. Sci. USA, 1981, 78, 5783–5787 CAS.
- D. G. Gourley, A. K. Shrive, I. Polikarpov, T. Krell, J. R. Coggins, A. R. Hawkins, N. W. Isaacs and L. Sawyer, Nat. Struct. Biol., 1999, 6, 521–525 CrossRef CAS.
- (a) C. Kleanthous, M. Reilly, A. Cooper, S. Kelly, N. C. Price and J. R. Coggins, J. Biol. Chem., 1991, 266, 10893–10898 CAS; (b) R. K. Deka, C. Kleanthous and J. R. Coggins, J. Biol. Chem., 1992, 267, 22237–22242 CAS; (c) A. P. Leech, R. James, J. R. Coggins and C. Kleanthous, J. Biol. Chem., 1995, 270, 25827–25836 CrossRef CAS.
- (a) J. Harris, C. Kleanthous, J. R. Coggins, A. R. Hawkins and C. Abell, J. Chem. Soc., Chem. Commun., 1993, 270, 1080–1081 RSC; (b) A. Shneier, J. Harris, C. Kleanthous, J. R. Coggins, A. R. Hawkins and C. Abell, Bioorg. Med. Chem. Lett., 1993, 3, 1399–1402 CrossRef CAS; (c) J. M. Harris, C. Gonzalez-Bello, C. Kleanthous, A. R. Hawkins, J. R. Coggins and C. Abell, Biochem. J., 1996, 319, 333–336 CAS.
- T. Garbe, S. Servos, A. Hawkins, G. Dimitriadis, D. Young, G. Dougan and I. Charles, Mol. Gen. Genet., 1991, 228, 385–392 CrossRef CAS.
- J. R. Bottomley, C. L. Clayton, P. A. Chalk and C. Kleanthous, Biochem. J., 1996, 319, 559–565 CAS.
- M. Frederickson, E. J. Parker, A. R. Hawkins, J. R. Coggins and C. Abell, J. Org. Chem., 1999, 64, 2612–2613 CrossRef CAS.
- A. W. Roszak, D. A. Robinson, T. Krell, I. S. Hunter, M. Fredrickson, C. Abell, J. R. Coggins and A. Lapthorn, Structure, 2002, 10, 493–503 CrossRef.
- M. D. Toscano, M. Frederickson, D. P. Evans, J. R. Coggins, C. Abell and C. González-Bello, Org. Biomol. Chem., 2003, 1, 2075–2083 RSC.
- C. González-Bello, E. Lence, M. D. Toscano, L. Castedo, J. R. Coggins and C. Abell, J. Med. Chem., 2003, 46, 5735–5744 CrossRef CAS.
- (a) G. Jones, P. Willet and R. C. Glen, J. Mol. Biol., 1995, 245, 43–53 CrossRef CAS; (b) G. Jones, P. Willet, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS.
- SYBYL. 6.5. 1699 South Hanley Road, St. Louis, Missouri, 63144, USA: Tripos Inc.
- WebLab ViewerPro4.0. 9685 Scranton Road, San Diego, CA 92121, USA: Accelrys Inc.
- T. Widlanski, S. L. Bender and J. R. Knowles, Biochemistry, 1989, 28, 7572–7582 CrossRef CAS.
- UV assay monitored the formation of 3-dehydroshikimate at 234 nm using black-walled quartz cuvettes. All assays were done in 1 ml reaction volumes, at 25 °C and pH 7.0 (50 mM potassium phosphate buffer for type I dehydroquinase; 50 mM Tris-HCl buffer for type II dehydroquinase).
Footnote |
| † Electronic supplementary information (ESI) available: experimental details. See http://dx.doi.org/10.1039/b507156a |
| This journal is © The Royal Society of Chemistry 2005 |