 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of pyrene derivatives on Au(111): substituent effects on intermolecular interactions†
Tuan Anh
Pham
*a,
Fei
Song
a,
Manh-Thuong
Nguyen
b and
Meike
Stöhr
*a
aZernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: t.a.pham@rug.nl; m.a.stohr@rug.nl
bThe Abdus Salam International Centre for Theoretical Physics, Strada Costiera 11, I-34151 Trieste, Italy
First published on 30th May 2014
Abstract
The adsorption behaviour as well as the influence of bromine substituents on the formation of highly-ordered two-dimensional structures of pyrene derivatives on Au(111) are studied by a combination of scanning tunnelling microscopy (STM) and density functional theory (DFT) calculations.
Halogen bonding (X-bonding) is nowadays considered as a highly important tool in the field of crystal engineering and supramolecular chemistry.1–4 In comparison to hydrogen bonding (H-bonding), X-bonding offers a tuneable interaction strength by properly choosing the halogen atoms as well as a higher directionality.5–8 These unique properties make X-bonding an interesting alternative to the often employed and well-established H-bonding for achieving desired molecular architectures, which is considered to open up new pathways for the fabrication of molecular devices by the bottom-up approach. However, despite these exciting advantages, the use of X-bonding for controllably constructing on-surface supramolecular assemblies has only recently been recognised. For example, Lackinger et al. demonstrated the formation of two-dimensional (2D) supramolecular structures from tris-bromophenyl derivatives mediated by X-bonding at the solid–vacuum interface,9a while Perepichka et al. reported the formation of 2D chiral structures from brominated tetrathienoanthracene derivatives at the solid–liquid interface.9b So far, however, only a few more studies investigated the influence of the chemical nature of the halogen substituents on the surface-supported self-assembly process.10–13 It has been recognised that for the construction of such 2D structures, halogen substituents may play the determining role in the structural formation by balancing the delicate interplay between molecule–molecule and molecule–substrate interactions. Therefore, an in-depth understanding of the effects of halogen substituents on the resulting intermolecular interactions is of utmost importance for the usage of X-bonds for the construction of molecular devices.
Herein, we investigated the influence of the position and number of bromine substituents on the formation of highly-ordered self-assembled structures as well as the resulting intermolecular interactions of pyrene derivatives on Au(111) under UHV conditions. For this purpose, two different pyrene derivatives, 1,3,6,8-tetrabromopyrene (Br4Py) and 2,7-dibromopyrene (Br2Py), were chosen, which possess four and two functional bromine groups, respectively, at different substituent positions. In order to minimize the effect of the substrate on the structural formation, we employed Au(111) as a substrate since it is generally considered less reactive than other noble metals.14
Scheme 1 shows the chemical structure and the corresponding calculated electrostatic potential distribution of these molecules. Due to an anisotropic charge distribution around the halogen atom in a halogen–carbon bond, the bromine substituents in these molecules can act as both electrophiles and nucleophiles, allowing two different binding motifs at the same time.1,5,8,15
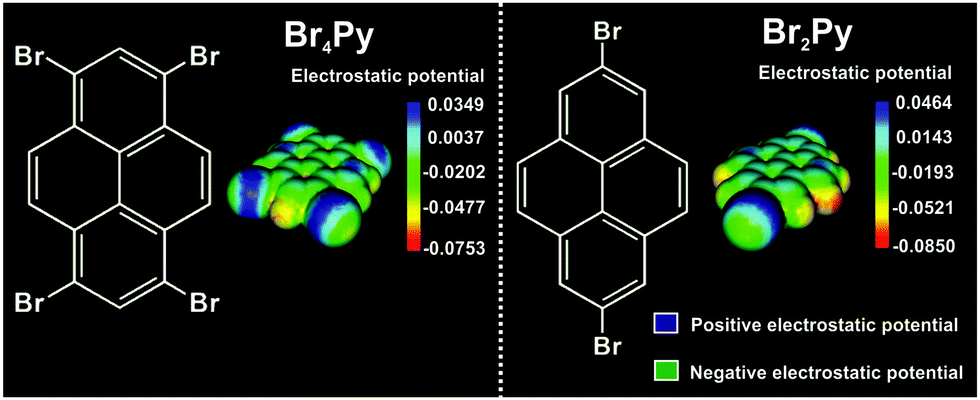 | ||
| Scheme 1 Chemical structure and corresponding electrostatic potential distributions of 1,3,6,8-tetrabromopyrene (Br4Py) (left) and 2,7-dibromopyrene (Br2Py) (right), respectively, showing the positive potential in blue and the negative potential in green/yellow at isodensity surfaces. | ||
Upon deposition of Br4Py on Au(111) held at room temperature, well-ordered 2D patterns were observed by STM under UHV conditions. Close to monolayer coverage, two different molecular arrangements coexist: a parallel arrangement (Fig. 1a, top, and Fig. 1b) and a square arrangement (Fig. 1a, bottom, and Fig. 1d), labelled phase I and II in the following, respectively. It should be noted that also at room temperature and for submonolayer coverage, Br4Py forms islands exhibiting phase I. Phase I was found to be the dominant phase (occurrence of around 90%). In overview STM images for Br4Py self-assembled into phase I (Fig. S1, ESI†), the characteristic herringbone reconstruction of Au(111) is visible through the molecular adlayer. This shows that the Au reconstruction is neither modified nor lifted upon adsorption of the molecules, indicating a weak molecule–substrate interaction. One axis of the molecular unit cell is rotated by approximately (7 ± 1)° with respect to a principal Au direction. Interestingly, the molecules aligned in this unit cell direction undergo a small lateral shift with respect to each other at the positions at or close to the soliton walls of the herringbone reconstruction (Fig. S1c, ESI†).20 The co-existence of three rotational domains having an angle of 120° to each other was observed, reflecting the threefold symmetry of the underlying substrate (Fig. S2, ESI†). The molecular orientation in each of these domains is the same with respect to the underlying substrate. Taking these findings together, it turns out that the substrate has a weak but not negligible influence on the self-assembly process. Because the molecular lattice direction does not run parallel to a principal Au direction, it is expected that for each of the rotational domains, a mirror domain exists. Indeed, in STM images we observed rotational and mirror domains at the same time (Fig. S3, ESI† shows the existence of domains Am and Bm, which are the mirror counterparts of domain A and B, respectively). The unit cell directions of “normal” and mirror domains enclose an angle of ±(7 ± 1)° with a principal Au direction which is the mirror plane direction.
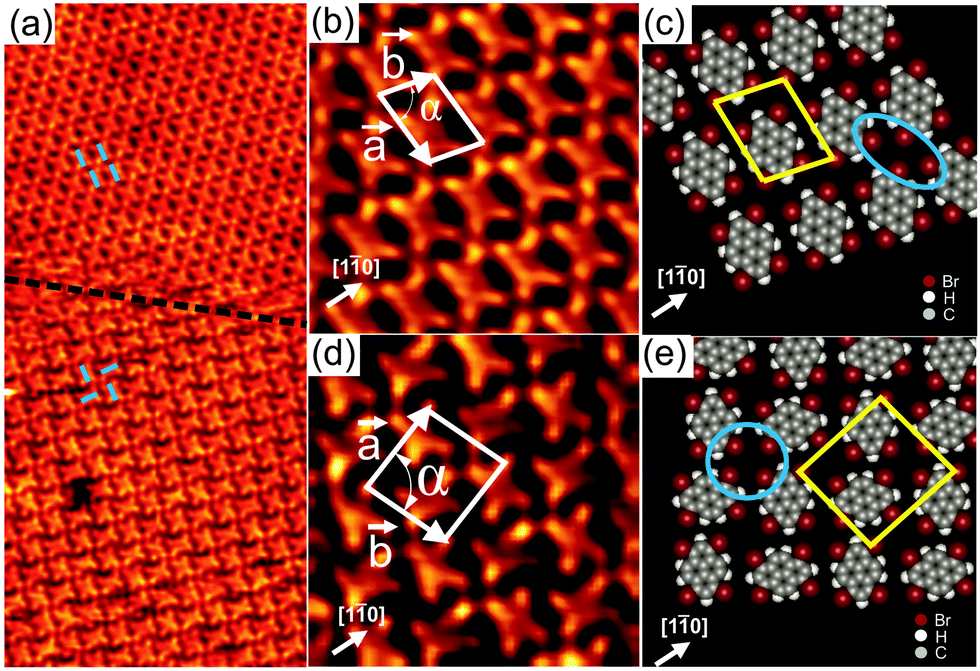 | ||
| Fig. 1 (a) Overview STM image (30 × 14 nm2) for 1 ML Br4Py on Au(111), showing the co-existence of two different molecular arrangements, labelled phase I (top) and phase II (bottom). (b, c) and (d, e) Close-up STM images (5 × 5 nm2) and corresponding proposed molecular models for phase I and II, respectively. The STM images were taken at room temperature with U = −1.2 V, I = 60 pA. | ||
In the high-resolution STM image in Fig. 1b, each Br4Py molecule in phase I can be clearly discerned. Each molecule exhibits four bright protrusions corresponding to the four bromine substituents which is in good agreement with its chemical structure (Scheme 1). To precisely determine the unit cell parameters, also with respect to the underlying Au(111) substrate, low energy electron diffraction (LEED) measurements were performed (Fig. S4, ESI†). An incommensurate superstructure was found for the molecules arranged into phase I. The unit cell contains one molecule and its lattice dimensions are found to be 11.7 × 8.8 Å2 with an internal angle of 78°. It should be noted that in LEED we did not obtain any spots arising from phase II. This further supports the fact that phase I, indeed, is the dominant phase upon adsorption of Br4Py on Au(111) at monolayer coverage. Fig. 1c shows the proposed molecular model for phase I. The molecules align parallel with respect to each other. Four Br substituents of four neighbouring molecules point toward each other to form a fourfold node (marked by a blue ellipse in Fig. 1c). Each molecule connects to four such fourfold nodes. Because of the non-spherical charge distribution, each Br substituent can attractively interact with a negatively charged Br or a positively charged H atom of adjacent molecules, forming so-called triangular binding motifs consisting of Br–Br and Br–H bonds, which are responsible for the network formation.9b
In Fig. 1d individual Br4Py molecules arranged into phase II are visible. The unit cell contains two molecules and its dimensions are found to be a = (15.6 ± 0.2) Å, b = (14.4 ± 0.2) Å and α = (87 ± 1)°, as determined from STM measurements. The packing density of this phase is ∼0.9 molecules nm−2, whereas that of phase I is higher, ∼1 molecule nm−2. Within this phase, each molecule is surrounded by four neighbouring molecules rotated by 90° with respect to the central molecule. In the proposed molecular model (Fig. 1e), four molecules meet in a fourfold node formed by four Br substituents (marked by a blue circle in Fig. 1e), similar to what was observed for phase I. However, each molecule only participates in two such fourfold nodes what is in contrast to phase I. As observed for phase I, the molecular network is stabilised by triangular binding motifs based upon Br–Br and Br–H bonds. The basic unit of this molecular network is a chiral pinwheel-like tetramer resulting in organizational chirality for the assembly. Both left- and right-handed homochiral domains were observed (Fig. S6, ESI†).
In order to examine the influence of the position and number of the Br substituents on the resulting intermolecular interactions responsible for the network formation, Br2Py was deposited onto Au(111) held at room temperature. For monolayer coverage, the molecules arrange in a 2D brick wall-like pattern which is labelled phase III (Fig. 2). Again, the characteristic herringbone reconstruction of Au(111) is observed through the molecular adlayer which is indicative of a weak molecule–substrate interaction. Each molecule exhibits two bright terminal protrusions corresponding to the two Br substituents (Fig. 2b). The unit cell contains one molecule and its lattice parameters are determined to be a = (11 ± 0.2) Å, b = (12.7 ± 0.2) Å and α = (52 ± 2)°. The packing density of this phase is found to be approximately 0.87 molecules nm−2 which is lower than that of phase I and II. As can be seen in the proposed molecular model (Fig. 1c), the molecules align in parallel rows with their Br substituents in an antiparallel fashion, forming twofold nodes (marked by a blue circle in Fig. 2c). Importantly, this molecular arrangement does not form Br–Br bonds. Instead, the supramolecular network is stabilized by triangular binding motifs based on Br–H bonds enabled by the opposite charge regions of Br and H atoms within the twofold nodes.
 | ||
| Fig. 2 (a) Overview STM image (24 × 15 nm2) for 1 ML of Br2Py on Au(111). (b) Close-up STM image (6.5 × 6.5 nm2) and (c) corresponding proposed molecular model. The STM images were taken at 77 K with U = −1.8 V, I = 30 pA. | ||
The absence of Br–Br bonds in phase III could be explained by taking thermodynamic considerations into account. It can be assumed that the interplay of entropy and enthalpy plays an important role in the network formation.16,17 The decrease in entropy during self-assembly must be compensated by a gain in enthalpy via the formation of favourable intermolecular interactions. Since Br2Py has two terminal Br substituents, two adjacent molecules will preferably bind via Br–H bonds to maximize attractive interactions and to minimize repulsive Br–Br interactions. In this way a minimum in the Gibbs free energy is obtained corresponding to a stable state of the overall system.
In order to obtain a more detailed understanding of the intermolecular interactions, we carried out DFT calculations with van der Waals corrections included. The optimized intermolecular bonds are shown in Fig. 3 and the corresponding computed results are summarised in Fig. S7 (ESI†). It should be noted that previously two different types of X-bonds were classified based on the bonding angle of the C–X groups. Type-I X-bonds are repulsive while type-II X-bonds are based on electrostatic attractive interactions.18 Combining this classification with our calculated results leads to an intuitive explanation for the presence of intermolecular interactions in the three networks. Both in phases I and II, the molecular networks are mainly stabilized by H1⋯Br4⋯Br1 and H3⋯Br2⋯Br3 triangular binding motifs formed by Br–Br bonds (type-II) and Br–H bonds in each fourfold node. The main difference between these phases lies in the type-I repulsive interactions. The computed Br1 to Br3 distance shows that this repulsive interaction does not occur in phase II, but it may contribute to the sum of the driving forces for the network formation in phase I. More explanation for these intermolecular bonds can be found in the ESI† (Fig. S7). For phase III, the molecular network is mainly driven by H1⋯Br1⋯H5 and H3⋯Br2⋯H6 triangular binding motifs formed by only Br–H bonds in each twofold node. The computed distances of Br1⋯H4, Br2⋯H2 and Br1⋯Br2 imply that the role of intermolecular interactions between parallel molecular rows running along the shorter unit cell axis can be neglected for the network formation.19
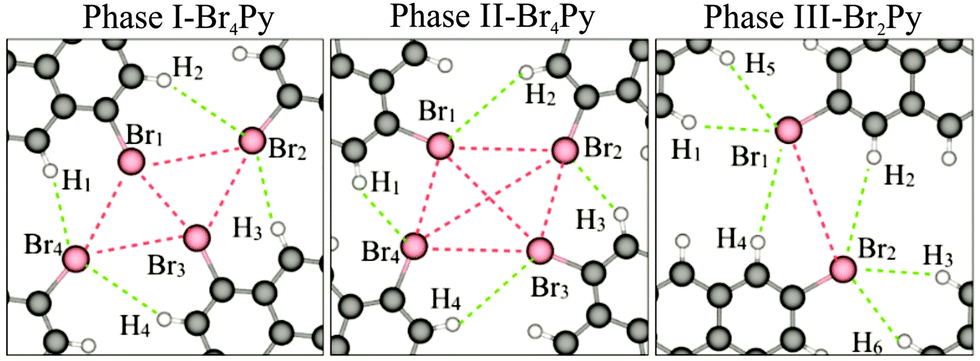 | ||
| Fig. 3 DFT optimized intermolecular interactions for phase I and II formed by Br4Py and for phase III formed by Br2Py molecules. The intermolecular Br–Br and Br–H bonds are marked by red and green dashed lines, respectively. | ||
The calculated unit cells are entirely consistent with the experimental results (Fig. S8, ESI†). The binding energies per molecule obtained from DFT calculations are 0.3 eV and 0.28 eV for phase I and phase II, respectively, whereas that of phase III is lower (0.21 eV). These results demonstrate that there are less intermolecular bonds per molecule in the case of phase III compared to that of phases I and II due to the differences in the position and number of Br substituents. However, the DFT calculations do not fully explain why for monolayer coverage almost exclusively phase I is observed. The reason for this could be substrate effects (presence of the herringbone reconstruction) which were not taken into account in our calculations.
In conclusion, we compared the self-assembly of pyrene derivatives on Au(111) under UHV conditions by combining STM experiments and DFT calculations. The Br4Py molecules arrange into two different 2D patterns which are stabilized by X-bonds and Br–H bonds at the same time. On the other side, the 2D self-assembled pattern of Br2Py is exclusively driven by Br–H bonds. These results successfully demonstrate that the positions and number of Br substituents determine the final supramolecular networks of pyrene derivatives on Au(111).
The authors acknowledge financial support from the Netherlands Organization for Scientific Research (NWO, Chemical Sciences, VIDI-grant No. 700.10.424) and the European Research Council (ERC-2012-StG 307760-SURFPRO).
Notes and references
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114 CrossRef CAS PubMed.
- L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2013, 5, 42 CrossRef CAS PubMed.
- R. W. Troff, T. Makela, F. Topic, A. Valkonen, K. Raatikainen and K. Rissannen, Eur. J. Org. Chem., 2013, 1617 CrossRef CAS.
- H. M. Titi, R. Patra and I. Goldberg, Chem. – Eur. J., 2013, 19, 14941 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748 RSC.
- C. B. Aakeroy, M. Baldrighi, J. Desper, P. Metrangolo and G. Resnati, Chem. – Eur. J., 2013, 19, 16240 CrossRef PubMed.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686 CrossRef CAS PubMed.
- S. M. Huber, J. D. Scanlon, E. J. Izal, J. M. Ugalde and I. Infante, Phys. Chem. Chem. Phys., 2013, 15, 10350 RSC.
- (a) H. Walch, R. Gutzler, T. Sirtl, G. Eder and M. Lackinger, J. Phys. Chem. C, 2010, 114, 12604 CrossRef CAS; (b) R. Gutzler, O. Ivasenko, C. Fu, J. L. Brusso, F. Rosei and D. F. Perepichka, Chem. Commun., 2011, 47, 9455 RSC.
- (a) K. H. Chung, J. Park, K. Y. Kim, J. K. Yoon, H. Kim, S. Han and S. J. Kahng, Chem. Commun., 2011, 47, 11492 RSC; (b) K. H. Chung, H. Kim, W. J. Jang, J. K. Yoon, S. J. Kahng, J. Lee and S. Han, J. Phys. Chem. C, 2013, 117, 302 CrossRef CAS.
- (a) J. K. Yoon, W. J. Son, H. Kim, K. H. Chung, S. Han and S. J. Kahng, Nanotechnology, 2011, 22, 275705 CrossRef PubMed; (b) Y. Makoudi, B. Baris, J. Jaeannoutot, F. Palmin, B. Grandidier and F. Cherioux, ChemPhysChem, 2013, 14, 900 CrossRef CAS PubMed.
- B. Cui, H. J. Yan, D. Wang and L. J. Wan, J. Electroanal. Chem., 2013, 668, 237 CrossRef PubMed.
- N. M. Jenny, H. Wang, M. Neuburger, H. Fuchs, L. Chi and M. Mayor, Eur. J. Org. Chem., 2012, 2738 CrossRef CAS.
- F. S. Tautz, Prog. Surf. Sci., 2007, 82, 479 CrossRef CAS PubMed.
- K. E. Riley and P. Hobza, Phys. Chem. Chem. Phys., 2013, 15, 17742 RSC.
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312 CAS.
- N. Wintjes, J. Hornung, J. L. Checa, T. Voigt, T. Samuely, C. Thilgen, M. Stöhr, F. Diederich and T. A. Jung, Chem. – Eur. J., 2008, 14, 5794 CrossRef CAS PubMed.
- T. T. T. Bui, S. Dahaoui, C. Lecomte, G. R. Desiraju and E. Espinosa, Angew. Chem., Int. Ed., 2009, 48, 3838 CrossRef CAS PubMed.
- H. F. Lieberman, R. J. Davey and D. M. T. Newsham, Chem. Mater., 2000, 12, 490 CrossRef CAS.
- I. Chizhov, G. Scoles and A. Kahn, Langmuir, 2000, 16, 4358 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, additional STM images, DFT calculations, and LEED measurements for Br4Py. See DOI: 10.1039/c4cc02753a |
| This journal is © The Royal Society of Chemistry 2014 |