 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new C–C bond formation model based on the quantum chemical topology of electron density†
Luis R. Domingo
*
Universidad de Valencia, Departamento de Química Orgánica, Dr Moliner 50, E-46100 Burjassot, Valencia, Spain. E-mail: domingo@utopia.uv.es; Web: www.luisrdomingo.com
First published on 8th July 2014
Abstract
ELF topological analyses of bonding changes in non-polar, polar and ionic organic reactions involving the participation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(X) double bonds make it possible to establish a unified model for C–C bond formation. This model is characterised by a C-to-C coupling of two pseudoradical centers generated at the most significant atoms of the reacting molecules. The global electron density transfer process that takes place along polar and ionic reactions favours the creation of these pseudoradical centers at the most nucleophilic/electrophilic centers of the reacting molecules, decreasing activation energies. The proposed reactivity model based on the topological analysis of the changes in electron density throughout a reaction makes it possible to reject the frontier molecular orbital reactivity model based on the analysis of molecular orbitals.
C(X) double bonds make it possible to establish a unified model for C–C bond formation. This model is characterised by a C-to-C coupling of two pseudoradical centers generated at the most significant atoms of the reacting molecules. The global electron density transfer process that takes place along polar and ionic reactions favours the creation of these pseudoradical centers at the most nucleophilic/electrophilic centers of the reacting molecules, decreasing activation energies. The proposed reactivity model based on the topological analysis of the changes in electron density throughout a reaction makes it possible to reject the frontier molecular orbital reactivity model based on the analysis of molecular orbitals.
 Luis R. Domingo | Luis R. Domingo obtained his PhD in Organic Chemistry in 1987. In 1990 he became an Associate Professor, and in 2010 obtained his current position of Full Professor at the Department of Organic Chemistry of the University of Valencia. In 1995 he came up with his first studies in the field of Theoretical Organic Chemistry. So far, he has published more than 220 theoretical studies in different fields of Organic Chemistry. His research interest comprises the study of molecular mechanisms of cycloaddition reactions, the study of organocatalysis, and the application of the DFT reactivity indices in Organic Chemistry. He says: while the distribution of the electron density is responsible for the molecular shape and physical properties, the capability for changes in electron density is responsible for the reactivity. |
Establishing organic molecular mechanisms based on quantum chemistry calculations and the transition state theory
From the advance of numerical computation at the end of the 20th century, computational quantum chemistry (CQC) has been greatly accepted by organic chemists due to its practicability in the study of reaction mechanisms involving actual molecules with 50–70 atoms. CQC enables the localisation and characterisation of the reagents, products, transition state structures (TS) and intermediates involved in organic reactions, thus making the study of molecular mechanisms of organic reactions possible. The comparison of computed activation parameters obtained from the transition state theory (TST)1–3 with those experimentally obtained by kinetic experiments allows performing an analysis of available computational models. Feasible competitive reaction channels can be theoretically studied, and thus, explain experimental outcomes.Two appealing data are first obtained through CQC in a straightforward manner: (i) total electronic energies; and (ii) molecular geometries. Although electronic energies associated with the stationary points involved in a chemical reaction are very dependent on the computational level, geometries are less dependent on it. Thus, while ab initio Hartee–Fock (HF) calculations4 widely used along the two last decades of the 20th century allowed the obtention of good TS geometries, that theoretical level yielded very high activation energies overestimating TS energies. Consequently, very time-consuming post-HF energy calculations4 were performed related to HF optimised geometries. The development of the density functional theory (DFT)5 at the end of the 20th century, whose calculations provided activation energies closer to experimental values, allowed the standardisation of DFT computations in the study of organic reactions. Thus, several DFT functionals such as B3LYP,6,7 MPWB1K8 and more recently M06-2X,9 which provide accurate energies, have been developed, allowing the study of organic reaction with a computational demand similar to HF calculations.
First, I would like to comment on some data obtained from CQC. Organic reactions involving the formation of new C–C single bonds are the most significant ones within the arsenal of the reactions in organic synthesis since they enable the construction of complex molecules. Among the diverse possibilities of creating a new C–C single bond, those involving the participation of CC(X) double bonds are the most significant methods. Diels–Alder (DA) reactions,10 which enable the creation of six-membered carbocyclic structures, are the most studied organic reactions due to their significance both from an experimental and theoretical point of view. Given the high potential of these reactions, the DA reaction between butadiene 1 and ethylene 2, presented in all textbooks as the prototype, but not experimentally performed in the laboratory, has been the most studied one (see Scheme 1).
 | ||
| Scheme 1 DA reaction between butadiene 1 and ethylene 2. | ||
In 1996, studying the DA reaction between butadiene 1 and ethylene 2, Houk performed an evaluation of the emerging B3LYP calculations developed within DFT by comparing them with different ab initio methods (see Table 1).11
| Method | ΔEact | ΔEreac. | d(C–C) |
|---|---|---|---|
| RHF/6-31G* | 47.4 | 36.0 | 2.201 |
| MP2/6-31G* | 20.0 | 45.9 | 2.286 |
| MP4/6-31G* | 49.1 | ||
| CASSCF/6-31G* | 47.4 | 2.223 | |
| B3LYP/6-31G* | 24.8 | 36.6 | 2.273 |
| Experimental | 27.5 | 38.4 |
As can be observed, while the reaction energies, ΔEreac., were found in the range of 36.0 to 49.1 kcal mol−1, the activation energies, ΔEact, ranged from 20.0 (MP2) to 47.4 (HF) kcal mol−1. Note that the predicted B3LYP activation energy, 24.8 kcal mol−1, was found to be closer to that experimentally estimated, 27.5 kcal mol−1.
Interestingly, in spite of these wide ranges of energies, TS geometries were found not to be dependent on computational methods. Thus, in all the methods studied, the distance between the two carbons involved in the formation of the two new single bonds in the synchronous TS1 were found to be ca. 2.2 Å (see Fig. 1). That is, while the energies of the stationary points involved in this DA reaction were found to be very dependent on the computational method, the geometry of TS1, which depends on its electronic structure, proved to be very similar.
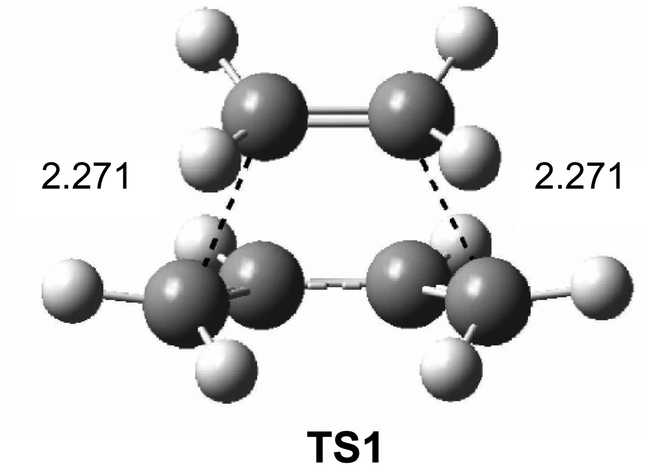 | ||
| Fig. 1 B3LYP/6-31G* transition structure TS1 associated with the DA reaction between butadiene 1 and ethylene 2. Distances are given in Angstroms. | ||
However, the TS geometries in different organic reactions are not invariable, being dependent on the nature of the bonds involved in the chemical process. In addition, for a given reaction, TS geometries can also be dependent on the substitution. Thus, among DA reactions both synchronous TSs, in which the two C–C single bonds are being formed at the same time, and highly asynchronous TSs, in which the two C–C single bonds are formed in two different stages of the reaction, can be found. It is noteworthy that the synchronous TS1 given in Fig. 1 is not representative of DA reactions, asynchronous TSs being the most common ones.
Therefore, as TS geometry depends on its electronic structure, the different TSs observed are a consequence of the different degree of the evolution of the bonding changes along the reaction. It is remarkable that the TS does not present any special characteristic in bonding changes along a reaction; it only corresponds to a structure of maximum energy along the intrinsic reaction coordinates12 (IRC).
Classification of organic reactions into non-polar, polar and ionic reactions
In order to establish the mechanism of DA reactions taking place experimentally under mild conditions, the DA reactions of cyclopentadiene (Cp, 4) with twelve ethylenes of different electron-withdrawing substitution were studied (see Scheme 2).13 | ||
| Scheme 2 DA reactions of Cp 4 with ethylenes of increased electrophilic character. | ||
While for the DA reaction with ethylene 2 a synchronous TS similar to TS1 was found, the asymmetric substitution on the ethylene yielded highly asynchronous TSs. Interestingly, while the B3LYP/6-31G* relative energies of the TSs of the studied DA reactions fluctuate from 22.0 kcal mol−1 for the most unfavourable reaction of Cp 4 towards methyl vinyl ether, an electron-rich ethylene, to −5.1 kcal mol−1 for the most favourable reaction towards an iminium cation, the strongest electron-deficient ethylene of the series, TS geometries showed in most cases a high asynchronicity in the C–C single bond formation, thus suggesting similar electronic structures. Note that in the DA reaction between Cp 4 and the iminium cation, formation of a pre-reaction complex with a relative energy of −9.6 kcal mol−1 makes the activation energy positive. The distance between the two carbons involved in the formation of the first C–C single bond at the asynchronous TSs were found in the narrow range from 2.17 to 1.96 Å. In order to analyse some significant characteristics of the C–C bond formation in DA reactions, three different reactions, i.e. the DA reactions of Cp 4 with styrene 5, with 1,1-dicyanoethylene (DCE) 6, and with iminium cation 7, have been selected (see Scheme 3). Relative energies and TS geometries are given in Fig. 2.
 | ||
| Scheme 3 DA reactions of Cp 4 with styrene 5, with DCE 6, and with iminium cation 7. | ||
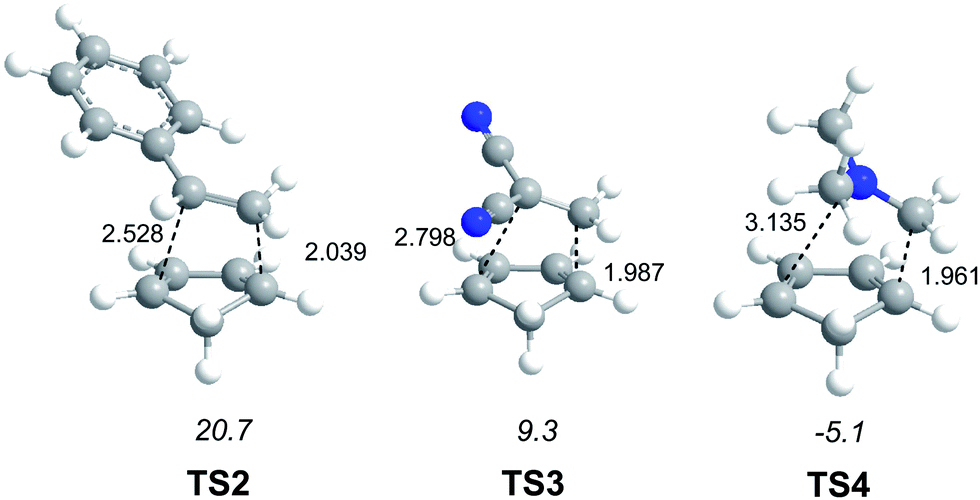 | ||
| Fig. 2 TSs involved in the DA reactions of Cp 4 with styrene 5, TS2, with dicyanoethylene 6, TS3, and with iminium cation 7, TS4. Relative energies are given in kcal mol−1, while distances are given in Angstroms. | ||
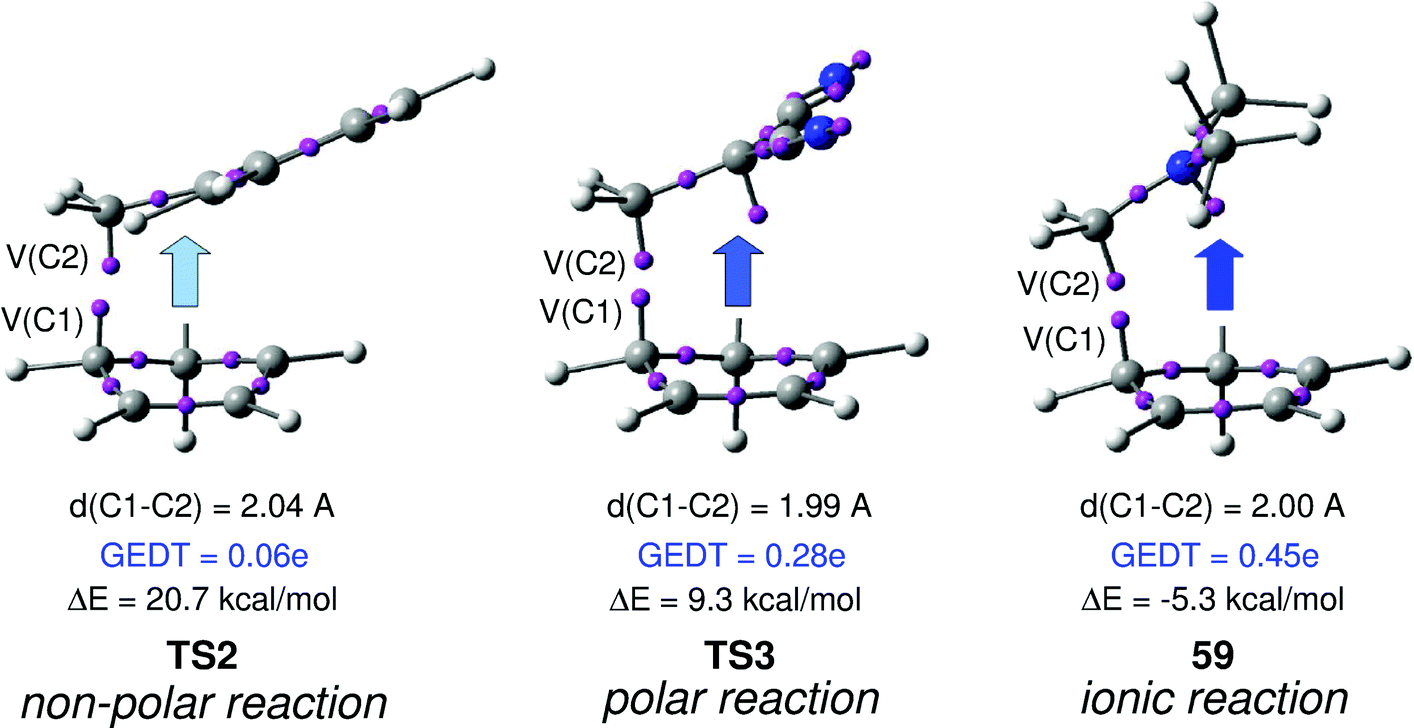
Interestingly, as can be seen in Fig. 2, while the relative energies associated with these DA reactions were found in a wide range from 21 to −5 kcal mol−1, the TS geometries were found to be very similar; the three TSs showed an asynchronous C–C single bond formation. If the most unfavourable TS2 is ruled out, TS3 and TS4 are found to be highly asynchronous. On the other hand, the three TSs showed a similar C–C single bond formation, the distances between the two interacting carbons at the TSs being in the narrow range from 2.04 to 1.96 Å. The C–C distances were only 0.2 Å shorter than the distances found at the very unfavourable synchronous TS1. Unfortunately, no information about the evolution of the bonding changes in the reactions can be obtained from these energy- and geometrical parameters; thus, with this information the nature of the molecular mechanism remains unresolved.
The results obtained from numerous theoretical studies devoted to DA reactions, pose an important question: what is the origin for the different activation energies found in TSs having similar geometries? The response was obtained after analysing a large number of theoretical studies devoted to cycloaddition reactions.
In 1999, in an earlier paper, the DA reactions of nitroethylene 11 with three ethylenes of increased electron-rich character were studied (see Scheme 4).14 A good correlation between the activation energies of the reactions and the nucleophilic/electrophilic behaviours of the reagents was established. The increase of the electron-rich character of the ethylene, i.e. its nucleophilic character, goes together with a decrease of the activation energy associated with the C–C bond formation.
 | ||
| Scheme 4 DA reactions of nitroethylene 11 with three ethylenes of increased nucleophilic character. | ||
This finding was quantitatively ascertained to analyse the electrophilicity ω index15 of the reagents involved in DA reactions.16 This first DFT study enabled the establishment of a unique scale of electrophilicity, in which both dienes and dienophiles were included,16 thus permitting the establishment of a good correlation between the difference of the electrophilicity ω indices of the reagents, Δω, and the feasibility of the DA reactions.16 The more electrophilic a reagent, i.e. when it is located at the top of the scale, and more nucleophilic the other reagent, i.e. when it is located at the bottom of the table, the more polar and faster the reaction.
Later, in 2003 the DA reactions between Cp 4 and the cyanoethylene series 14, experimentally studied by Sauer in 1964 (ref. 17) and given in most organic textbooks as an example of the effects of the electron-withdrawing substitution in DA reactions,18 were analysed (see Scheme 5).19
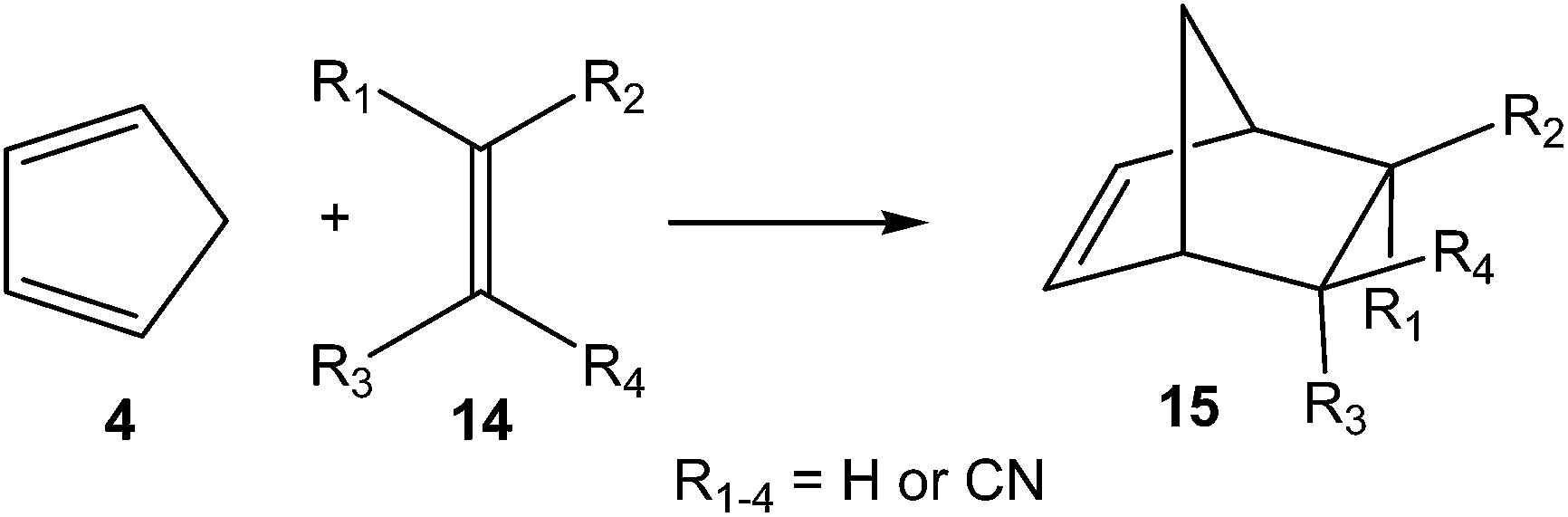 | ||
| Scheme 5 DA reactions between Cp 4 and cyanoethylene series 14. | ||
Some appealing conclusions were obtained from that DFT study: (i) both synchronous and asynchronous TSs were found in this series of DA reactions, depending on the symmetric substitution of the ethylene; i.e. while the symmetrically substituted 1,2-dicyanoethylenes maleonitrile (14a, R1 = R3 = CN, R2 = R4 = H) and fumaronitrile (14b, R1 = R4 = CN, R2 = R3 = H) yielded synchronous TSs, the asymmetric substituted DCE 6 (14c, R1 = R2 = CN, R3 = R4 = H) yielded a highly asynchronous TS;19 (ii) the synchronicity in the C–C single bond formation appears to be an unfavourable factor, i.e. the DA reaction with DCE 6 is ca. 1000 times faster than that with symmetric maleonitrile 14a and fumaronitrile 14b; and (iii) interestingly, a very good correlation between the global electron density transfer (GEDT) found at the TSs and the logarithm of the experimental rate constant was established, indicating that the GEDT could be one of the key factors in the activation energy (see Fig. 3).13 The GEDT at the TSs is computed by sharing the natural charges at the TSs obtained by natural bond orbital (NBO) analysis20,21 between the nucleophilic and the electrophilic frameworks.13 The GEDT concept comes from the observation that the electron density transfer that takes place along polar and ionic reactions is not a local process, but a global flux of electron density taking place from the nucleophile to the electrophile, and not being dependent on the approach mode of both reagents.22
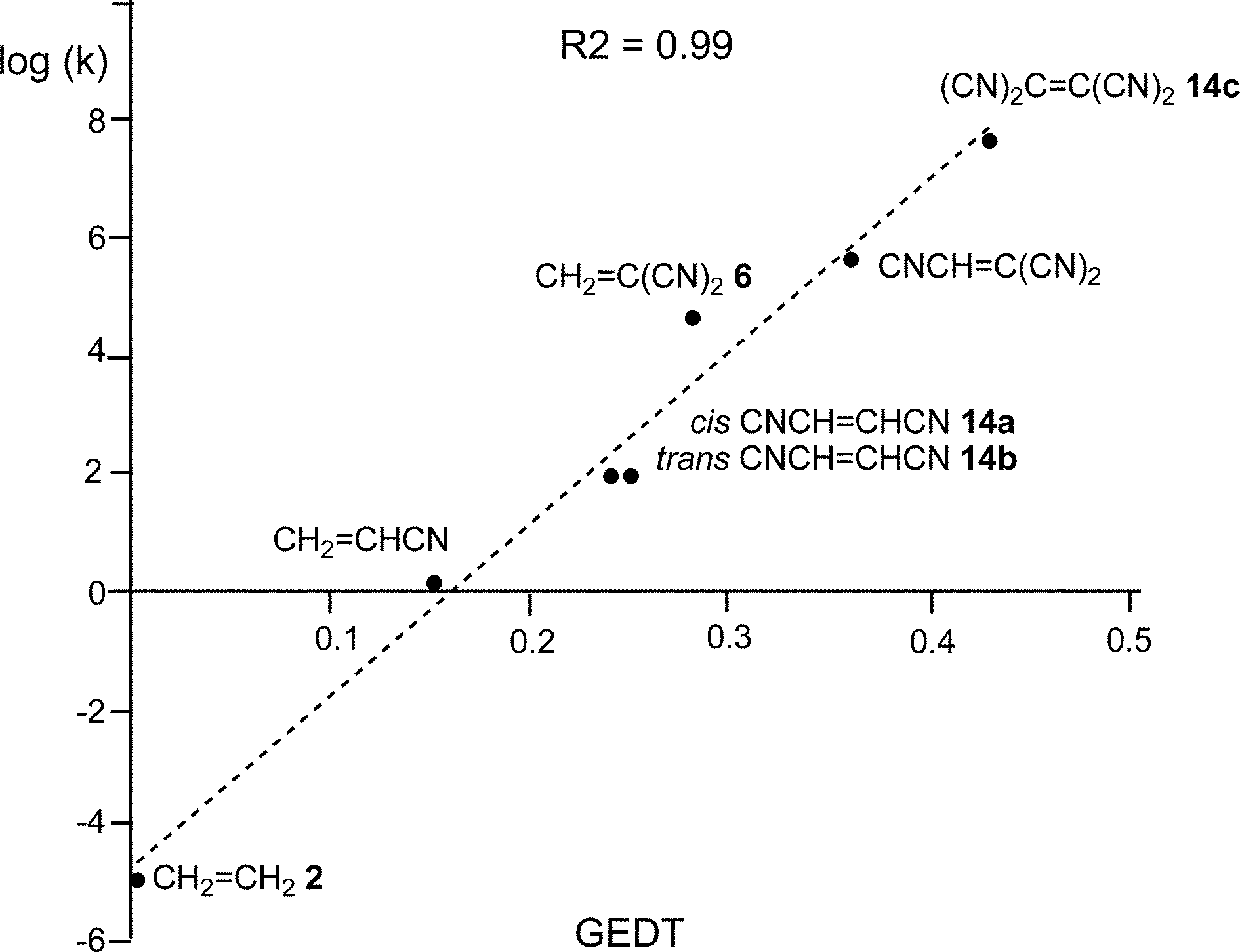 | ||
| Fig. 3 Plot of the logarithm of the experimental rate constant k vs. GEDT, in e, R2 = 0.99, for the DA reactions of Cp 4 with ethylene 2, and the cyanoethylene series. | ||
These results made it possible to establish the polar Diels–Alder (P-DA) mechanism,13 in which favourable electrophilic/nucleophilic interactions along the polar reaction are responsible for the GEDT found at the TSs, and consequently, for the feasibility of the reaction. The proposed polar mechanism for DA reactions has two very significant repercussions: (i) most DA reactions do not take place through a pericyclic mechanism23 as proposed in all textbooks,24–26 (ii) DA reactions are not a special type of organic reactions. Note that most of the DA reactions taking place through highly asynchronous TSs present a C–C bond formation similar to that found in most polar organic reactions.
From these findings, the different activation energies of the DA reactions given in Scheme 3 can be explained. The computed GEDT values at the TSs given in Fig. 2 are 0.06e at TS2, 0.28e at TS3 and 0.45e at TS4. Although only three reactions are dealt with herein, a very good correlation between the GEDT and the computed activation energy can be established (R2 = 0.98, see Fig. 4). Note that when the aforementioned twelve DA reactions are considered, this correlation remains at R2 = 0.89.13 The lineal correlation between polarity of the reaction, measured by the GEDT at the TS, and the corresponding activation energy, allowed the establishment of an appealing classification of DA reactions into non-polar Diels–Alder (N-DA) reactions, characterised by a GEDT below 0.2e, and P-DA reactions characterised by a GEDT above 0.2e.13 While N-DA reactions are of little synthetic interest as they demand harsh reaction conditions, the feasibility of a P-DA reaction increases with the polar character of the reaction; i.e. the electrophilic/nucleophilic character of the reagents. Interestingly, these behaviours can easily be anticipated by analysing the electrophilicity ω (ref. 15) and the nucleophilicity N indices,27,28 defined within the conceptual DFT,29,30 at the ground state of the reagents. A good correlation between the activation energies of the DA reactions of Cp 4 with ethylenes of different electron-withdrawing substitution shown in Scheme 2 and the global electrophilicity ω indices was found, R2 = 0.92.13 Similarly, a good correlation between the logarithm of the reaction rate constant of 5-substituted indoles with a series of benzhydryl cations, experimentally studied by Mayr,31 with the global nucleophilicity N indices has also been established, R2 = 0.98.28
 | ||
| Fig. 4 Plot of the activation energies ΔE, in kcal mol−1, vs. GEDT, in e, for the DA reactions given in Scheme 3. | ||
Of the twelve DA reactions studied, the fastest one was that between Cp 4, a nucleophilic neutral molecule, and the iminium cation 7, a very high electrophilic cationic species (see Scheme 3). To distinguish the P-DA reactions in which the polarity is only evidenced in the course of the reaction from those reactions in which all species are ionic, the latter reaction was classified as an ionic Diels–Alder (I-DA) reaction.13
What is the origin of the GEDT in polar and ionic organic reactions? An appealing concept defined within DFT is the electronic chemical potential μ (ref. 32) which correlates with the absolute electronegativity χ through the simple equation χ = −μ.33 The electronic chemical potential μ, defined as μ = (∂E/∂N)v, is associated with the feasibility to exchange electron density of a molecule with the environment at ground state.32 Sanderson suggested the electronegativity equalisation principle34 in chemistry, in which electronegativity tends to equalise. The correctness of Sanderson's principle immediately comes from the fact that the electronic chemical potential μ is a property of an equilibrium state. Consequently, when two molecules A and B, with μA < μB, approach one another, there is a flux of electron density from A, the less electronegative species, towards B, the more electronegative one, to equilibrate the electronic chemical potential μAB in the new interacting system. The larger the electronic chemical potential difference, ΔμA–B, the larger the GEDT. It is noteworthy that in P-DA reactions taking place along high asynchronous TSs, the maximum GEDT value is reached after passing the TS, and when the first C–C single bond is formed. In experimental stepwise P-DA reactions,35,36 this value is reached with the formation of the corresponding zwitterionic intermediate.
In spite of this appealing finding above-mentioned, some issues remained unresolved: how do the bonding changes in both N-DA and P-DA reactions take place? and, how can the GEDT modify the pattern in bonding changes along the reaction? That is, how can the polarity of the reaction modify the reaction mechanism? To answer these questions a complete analysis of the bonding changes along a reaction must be performed. This task demands a complete quantum topology analysis of the electron density obtained from the quantum wave function along the IRC of an elemental reaction.
Establishing organic reaction mechanisms based on the quantum chemical topology analysis of electron density
Since the introduction of the chemical bond concept by G. N. Lewis at the beginning of the 20th century37 many theoretical models have been developed to understand matter structure and chemical reactivity. Quantum chemical tools based on the valence bond (VB) theory,38–40 the molecular orbital (MO) theory,4 and most recently DFT5 have proven to be useful to chemists. However, in spite of the advances made in this field, the characterisation of chemical bonds, and more specifically the breaking/forming processes along a reaction, appear to be unresolved.41 Like many other chemical concepts, chemical bonds are defined in a rather ambiguous manner as they are not observable, but rather belong to a representation of the matter at a microscopic level which is not fully consistent with quantum mechanical principles.To harmonise the chemical description of matter with quantum chemical postulates, several mathematical models have been developed. Among them, the theory of dynamical systems,42 convincingly introduced by Bader through the theory of atoms in molecules (AIM),43 has become a powerful method of analysis. The AIM theory enables a partition of the electron density within the molecular space into basins associated with atoms. The development of the AIM theory was the origin of a significant contribution to conceptual chemistry in the definition of concepts such as the atom inside a molecule or bond critical points.44–46 For many years, Popelier has been studying the AIM quantum chemical topology (QCT) of the electron density for characterising chemical bonds, having performed a great deal of work in different fields such as the characterisation of heterocyclic rings,47 atomic properties of aminoacids,48 pKa predictions,49 and radicals.50
Another appealing procedure that provides a more straightforward connection between the electron density distribution and the chemical structure is the QCT analysis of the electron localisation function (ELF) of Becke and Edgecombe.51 In this sense, Silvi and Savin presented the ELF in a very chemical fashion, using their topological analysis as an appealing model of chemical bonding.52–55 After an analysis of the electron density, ELF divides the electron density of a molecule into basins, i.e. domains in which the probability of finding an electron pair is maximal. Basins are classified as core basins and valence basins. The latter are characterised by the synaptic order, i.e. the number of atomic valence shells in which they participate.56 Thus, there are monosynaptic, disynaptic, trisynaptic basins and so on. Monosynaptic basins, labelled V(A), correspond to lone pairs or non-bonding regions, while disynaptic basins, labelled V(A, B), connect the core of two nuclei A and B and, thus, correspond to a bonding region between A and B. This description recovers the Lewis bonding model, providing a very suggestive graphical representation of the molecular system. Analysis of the ELF valence basin populations N provides similar GEDT values of than those obtained by NBO analysis.22,57
On the other hand, the characterisation of the electron density reorganisation to evidence the bonding changes along a reaction path is the most attractive method to characterise a reaction mechanism.58–60 To quantitatively perform these analyses, the bonding evolution theory (BET), consisting of the joint-use of ELF topology and Thom's catastrophe theory61–63 (CT) was proposed by Krokidis et al.64 as a new tool for analysing the electronic changes in chemical processes, being applied to different elementary reactions.65–78 In this field, Andrés performed a systematic investigation characterising the mechanisms of significant organic reactions such as DA reactions,58,79 [3 + 2] cycloaddition (32CA) reactions,80 the Bergman cyclisation,81 the Cope rearrangement,82 and the Nazarov cyclisation.83
ELF topological model for the C–C bond formation in non-polar reactions
In 2003, the molecular mechanism of the DA reaction between butadiene 1 and ethylene 2, given in Scheme 1, was characterised using BET.58 Two appealing conclusions were obtained from this QCT analysis of the bonding changes along the one-step mechanism: (i) the reaction consists of seven differentiated phases characterised by 10 catastrophes, within of the classification given by Thom,84 belonging to the fold and cusp types. Each one of these phases is characterised by a bonding change in the Lewis structure with respect to the previous phase; (ii) the formation of the C–C single bond in this DA reaction takes place through a C-to-C coupling between two pseudoradical centers85,86 generated through the breaking of the CC double bonds, at a C–C distance of 2.04 Å (see Fig. 5).
 | ||
| Fig. 5 Most relevant ELF attractors in the structures of phase V, structure 16, and phase VI, structure 17, involved in the synchronous C–C single bond formation in the N-DA reaction between butadiene 1 and ethylene 2. The pseudoradical centers involved in the C–C bond formation are characterised by the four V(C) monosynaptic basins present in structure 16. | ||
The seven phases characterising this N-DA reaction have recently been categorised into four groups associated with significant chemical bonding changes (see Fig. 6):87 (i) in the first one, A (in red colour), the three CC double bonds present in butadiene 1 and ethylene 2 break, phases I to III; (ii) in the second group, B (in green colour), the formation of two pseudoradical structures takes place by gathering electron density at the end carbons of the two unsaturated reagents, phases IV and V (see the four V(C) monosynaptic basins, integrating ca. 0.5e each one, in structure 16 in Fig. 5). The electron density demanded for the creation of the pseudoradical centers comes from the depopulation of the CC double bonds of the butadiene and ethylene moieties; (iii) in the third group, C (in blue colour), which is constituted only by the most relevant phase VI, the formation of the two new C–C single bonds takes place through the C-to-C coupling between the pseudoradical centers generated in the previous group B (see the two disynaptic basins V(Cx, Cy), integrating ca. 1.0e each one, in structure 17 in Fig. 5) (iv) in the fourth group, D (in violet colour), while the formation of the two C–C single bonds is completed, the formation of the new CC double bonds takes place at the end of the IRC. This QCT analysis of the bonding changes along the one-step mechanism of the N-DA reaction between butadiene 1 and ethylene 2 shows that the bonding changes are non-concerted.87
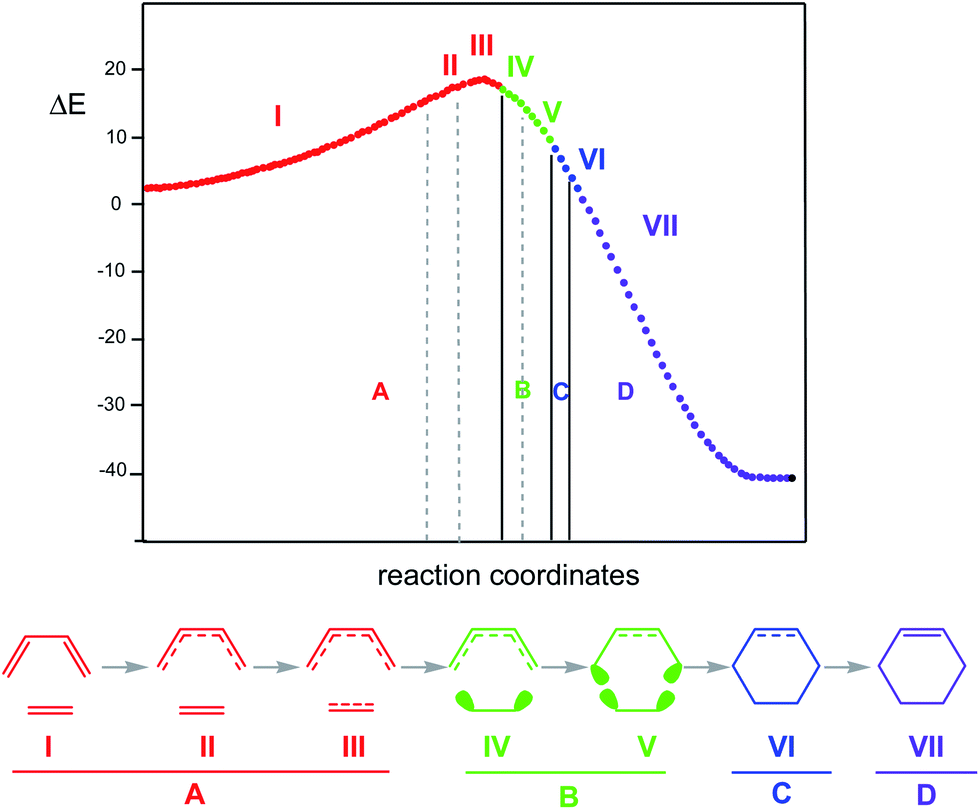 | ||
| Fig. 6 Reaction path calculated by means of the IRC method for the DA reaction between butadiene 1 and ethylene 2. ΔE are in kcal mol−1. A bonding between atoms in all phases is demonstrated by the standard Lewis representation; however, in the case of phases IV and V, ellipses reflect the non-bonding electron density concentrated in the C atoms. The four colours represent the four main groups in which the bonding changes can be categorised. | ||
Later, an ELF topological comparative study between the one-step and stepwise mechanisms of the N-DA of Cp 4 with ethylene 2 was performed in other to characterise these competitive molecular mechanisms (see Scheme 6).88 As expected, BET analysis of the one-step mechanism of the N-DA reaction of Cp 4 with ethylene 2 was found to be very similar to that found in the N-DA reaction of butadiene 1 with ethylene 2.58 Again, ELF topological analysis showed that the synchronous C–C single bond formation takes place at a C–C distance of 1.84 Å by the C-to-C coupling of two pseudoradical centers generated in Cp 4 and in ethylene 2. Interestingly, a similar pattern for the C–C single bond formation along the first step of the stepwise mechanism was found.88 In the stepwise mechanism, the formation of the first C–C single bond takes place also by a C-to-C coupling between two radical centers, yielding the formation of the diradical intermediate 18 (see Scheme 6). In this very unfavourable step, the C–C bond formation begins at a distance of 1.77 Å.
 | ||
| Scheme 6 One-step and stepwise pathways of the N-DA reaction between Cp 4 and ethylene 2. | ||
Some appealing conclusion was obtained from this comparative ELF topological analysis: (i) although the one-step mechanism and the stepwise mechanism appear to have a dissimilar electron reorganisation, note that the one-step mechanism is still classified as a pericyclic reaction,23 topologically, they present many similarities. The C–C bond formation along the one-step mechanism takes place by coupling of two pseudoradical centers (see pseudodiradical structure 20 in Fig. 7), while along the stepwise mechanism it takes place by coupling of two radical centers (see the diradical structure 21 in Fig. 7).88 Note that while the pseudodiradical structures are obtained by B3LYP restricted calculations to have a closed shell, the diradical structures such as 18 and 21 must be obtained by UB3LYP unrestricted calculations to have an open shell; (ii) ELF topological analysis of bonding changes along N-DA reactions of butadiene 1 and Cp 4 with ethylene 2, showed the non-concerted nature of breaking and forming bonds along these symmetric cycloaddition reactions, making it possible to reject the pericyclic mechanism for these reactions.87
 | ||
| Fig. 7 Semblance between the pseudodiradical structure 20 involved in the one-step mechanism, and the diradical structure 21 involved in the stepwise mechanism of the N-DA reaction between Cp 4 and ethylene 2. | ||
An analysis of bonding changes taking place along the energy profiles of N-DA reactions, allowed explaining the high activation energy required in non-polar processes. In all the studied reactions involving closed-shell molecules, the formation of the C–C single bonds takes place after passing the TS. The proposed model for the C–C bond formation in non-polar reactions suggests that it takes place by merging the electron density of two carbons having non-bonding electron density, which comes from the depopulation of electron density of the bonding region of the CC double bonds present in these reagents. ELF topological analysis of the corresponding pseudodiradical structures indicates that at this stage of the reaction the CC double bonds are already broken. Consequently, the high activation energy associated with non-polar reactions can be related to the unfavourable energy associated with the breaking of the CC double bond in these non-concerted processes.88
Several studies devoted to the characterisation of the molecular mechanism of non-polar reactions have supported this model for the C–C bond formation in non-polar processes. Thus, an ELF analysis of the electron reorganisation along the non-polar 32CA reaction of carbonyl ylide 22 with tetramethylethylene 23 (see Scheme 7) showed that the C–C bond formation in this 32CA reaction takes place at a C–C distance of 2.18 Å by a C-to-C coupling of two pseudoradical centers.89
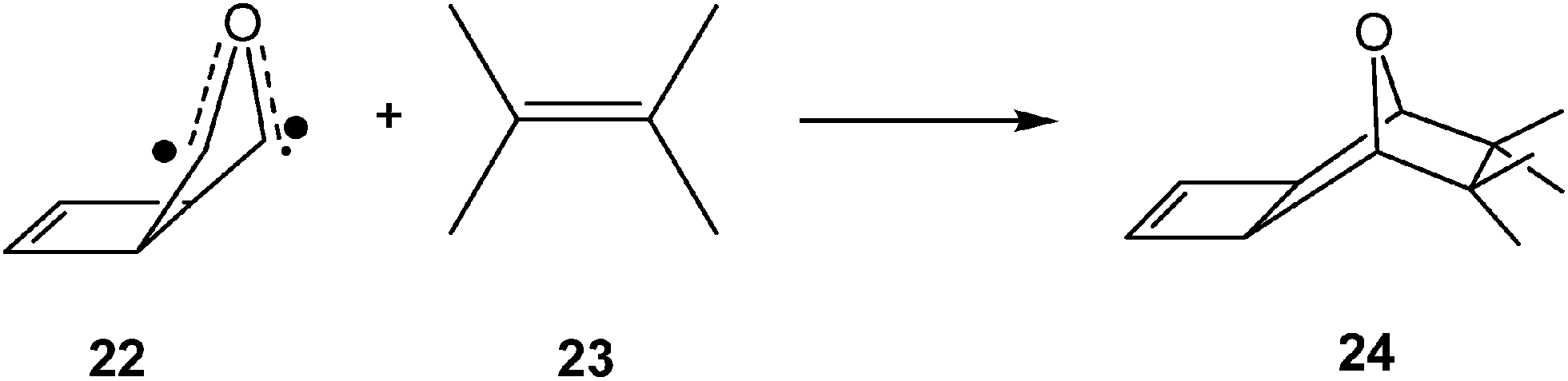 | ||
| Scheme 7 32CA reaction of carbonyl ylide 22 with tetramethylethylene 23. | ||
Interestingly, carbonyl ylide 22 already has a pseudodiradical character at its ground state (see structure 22 in Scheme 7). This behaviour accounts for the very low activation energy found in this 32CA reaction,89,90 and for the advanced character of the C–C bond formation. Note that organic reactions involving open-shell radical species are fast.
Recently, an ELF analysis for the C–C bond formation step in the N-heterocyclic carbene (NHC) catalysed hydroacylation of unactivated C–C double bonds (see Scheme 8) showed that in this non-polar reaction the C–C bond formation takes place also by a C-to-C coupling of a pseudodiradical structure 27 generated at the C1 and C2 carbons along the non-polar reaction (see Fig. 8).91
 | ||
| Scheme 8 The C–C bond formation step in the NHC catalysed hydroacylation of unactivated C–C double bonds. | ||
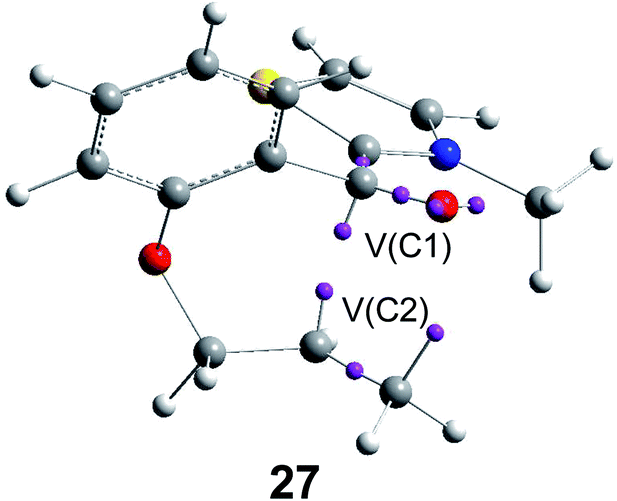 | ||
| Fig. 8 Pseudodiradical structure 27 generated along the C–C bond formation step in the N-heterocyclic carbene-catalysed hydroacylation of unactivated C–C double bonds. | ||
On the other hand, BET analysis along the cyclisation reactions of 1,3-butadiene 1,92 1,3,5-hexatriene 28,92 and 1,3,5,7-octatetraene 29 (ref. 93) (see Scheme 9) showed that the C–C bond formation in these electrocyclic reactions also takes place by a C-to-C coupling of two pseudoradical centers generated at the end of these conjugated systems.
 | ||
| Scheme 9 Electrocyclic reactions of the conjugated polyenic systems 1, 28 and 29. | ||
Very recently, Andrés studied the cycloheptatriene 33/norcaradiene 34 isomerisation (see Scheme 10).60 As in the isomerisation of hexatriene 28, this reaction is an electrocyclic reaction allowing the formation of a new C–C single bond between the end carbons of the hexatriene system of 33. The authors commented “once the TS has been reached, two new monosynaptic basins appear at the end carbons of the hexatriene system. This phase is very short, immediately these two monosynaptic basins merge into the disynaptic V(C1, C6) basin accounting for the closing of the cyclopropane ring”60 (see the merger of the V(C1) and V(C6) monosynaptic basins present in structure 35 into the V(C1, C6) disynaptic basin present in structure 36 in Scheme 10).
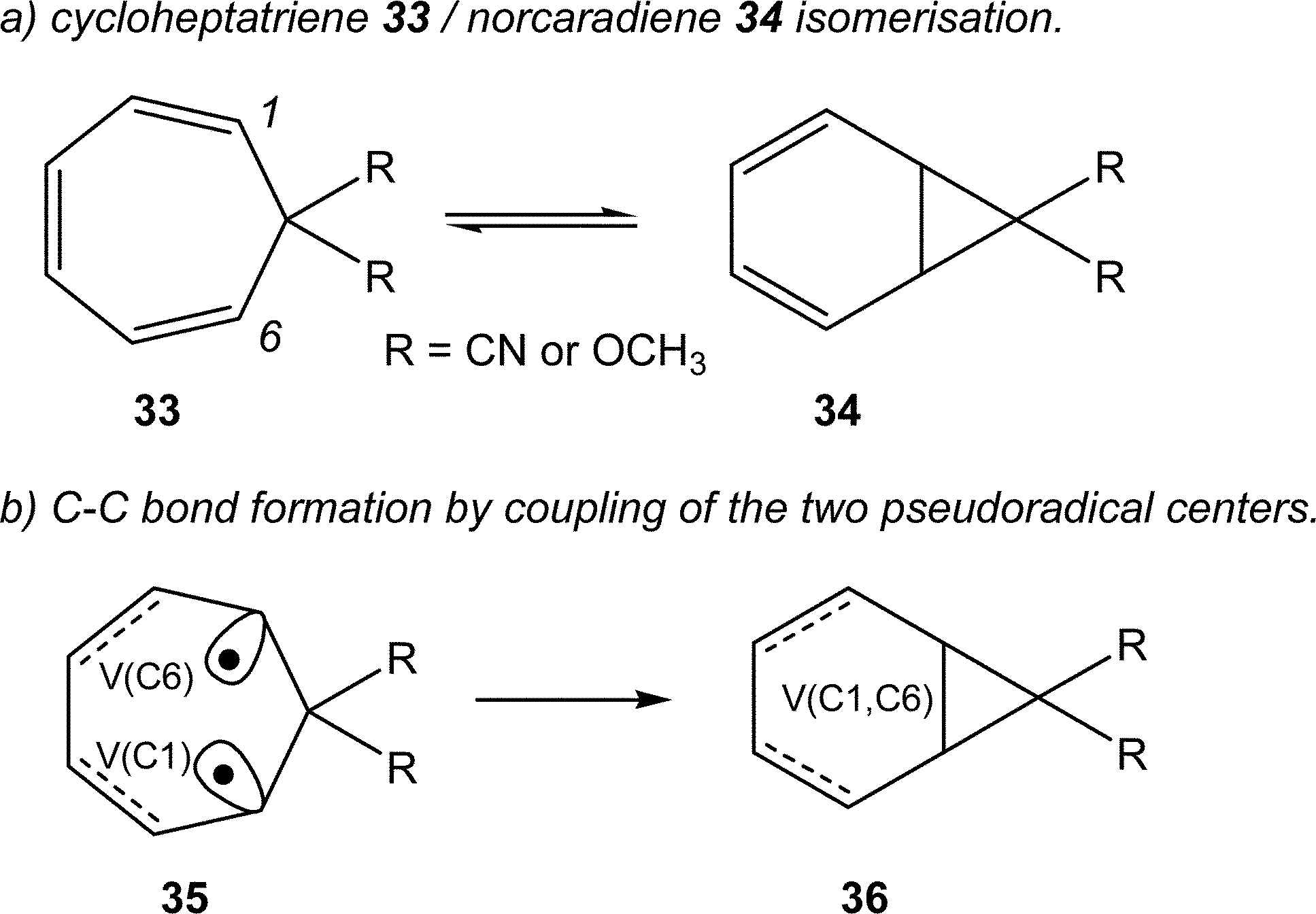 | ||
| Scheme 10 Cycloheptatriene 33/norcaradiene 34 isomerisation reaction. | ||
ELF topological model for the C–C bond formation in polar reactions
After performing a BET analysis of the N-DA of butadiene 1 with ethylene 2, the molecular mechanism of the P-DA reactions between butadiene 1 and acrolein 37, in the absence and the presence of a Lewis acid catalyst, BH3, were investigated in 2006 by using BET (see Scheme 11).79 These P-DA reactions take place by a one-step mechanism. BET analysis of the bonding changes along the two P-DA reactions indicated that they take place along eleven, in the non-catalysed reaction, and ten, in the LA-catalysed reaction, differentiated phases. In P-DA reactions, taking place along highly asynchronous TSs, the formation of the two C–C single bonds takes place in two differentiated phases of the IRC, thus characterising these reactions by a two-stage one-step mechanism.94 Interestingly, the formation of the first C–C single bond in these P-DA reactions showed a similar pattern to that found in the N-DA reaction between butadiene 1 and ethylene 2; i.e. the formation of the first C–C single bond takes place after passing the TSs, at a C–C distance of 1.92 Å, by a C-to-C coupling of two pseudoradical centers, in both non-catalysed and LA-catalysed reactions.79 | ||
| Scheme 11 P-DA reactions between butadiene 1 and acrolein 37, in the absence and the presence of a Lewis acid catalyst. | ||
Later, an ELF topological study on the origin of the synchronicity in bond formation in the P-DA reactions of Cp 4 with DCE 6, and with tetracyanoethylene (TCE) (14d, R1 = R3 = R2 = R4 = CN) established that, as the N-DA reaction between Cp 4 and styrene 5 or ethylene 1,88 the C–C single bond formation in these polar reactions takes place also by a C-to-C coupling of two pseudoradical centers generated along the reaction (see Fig. 9).95
 | ||
| Fig. 9 ELF attractors at selected points of the IRC of the P-DA reactions between Cp 4 and DCE 6, 39 and 40, and TCE 14d, 41 and 42. | ||
Interestingly, as in the P-DA reaction between butadiene 1 and acrolein 37, ELF analysis of the C–C bond formation at the P-DA reaction of Cp 4 with DCE 6 showed that the formation of the first C–C single bond in the two-stage one-step mechanism takes place at a C–C distance of 1.95 Å through the most favourable two-center interaction between the one of the most nucleophilic centers of Cp 4 and the most electrophilic center of DCE 6.95 This favourable interaction, which is responsible for the high regioselectivity experimentally observed in most P-DA reactions,22,96 is in agreement with the creation of the two pseudoradical centers at the most nucleophilic center of Cp 4 and the most electrophilic center of DCE 6 (see structure 39 in Fig. 9).
ELF topological studies of DA reactions have shown that while in N-DA reactions the electron density demanded for the formation of the pseudoradical centres is reached mainly by the depopulation of the CC double bonds present in the reagents,88 in P-DA reactions this electron density mainly comes from the GEDT that takes place along the polar process. Thus, the pseudoradical center created in the electrophilic species is formed mainly in the most electrophilic center of the molecule, which is the center with the highest spin density achieved through the GEDT process.97 These findings allowed proposing, within the conceptual DFT, first the nucleophilic Pk− and electrophilic Pk+ Parr functions,98,99 and later the radical Pk0 Parr functions100 in order to characterise the most relevant reactive centers within an organic molecule (see Fig. 10). Parr functions provide comparable information to that obtained using Fukui functions,101 but they are conceptually different; Fukui functions are approximated through the HOMO and LUMO frontier molecular orbitals (FMOs),102 while Parr functions are based on the changes of atomic spin density associated to GEDT processes.98
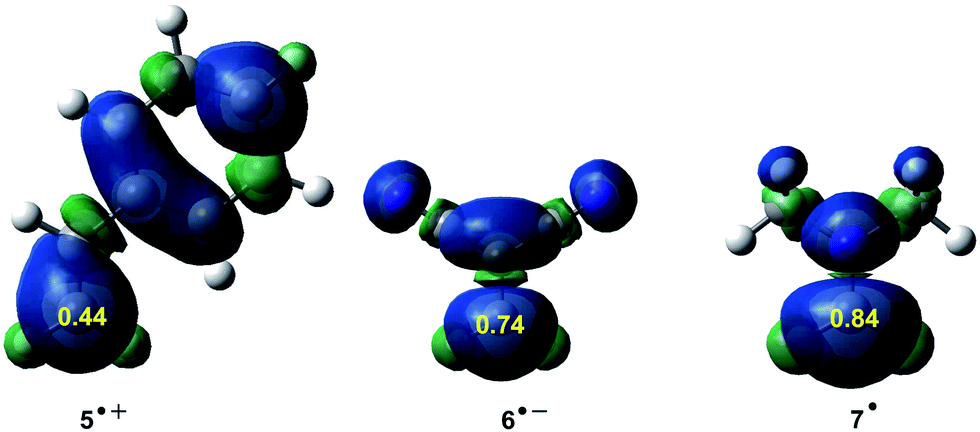 | ||
| Fig. 10 Nucleophilic Pk− function of styrene 5, electrophilic Pk+ Parr function of DCE 6, and radical Pk0 Parr functions of iminium cation 7. | ||
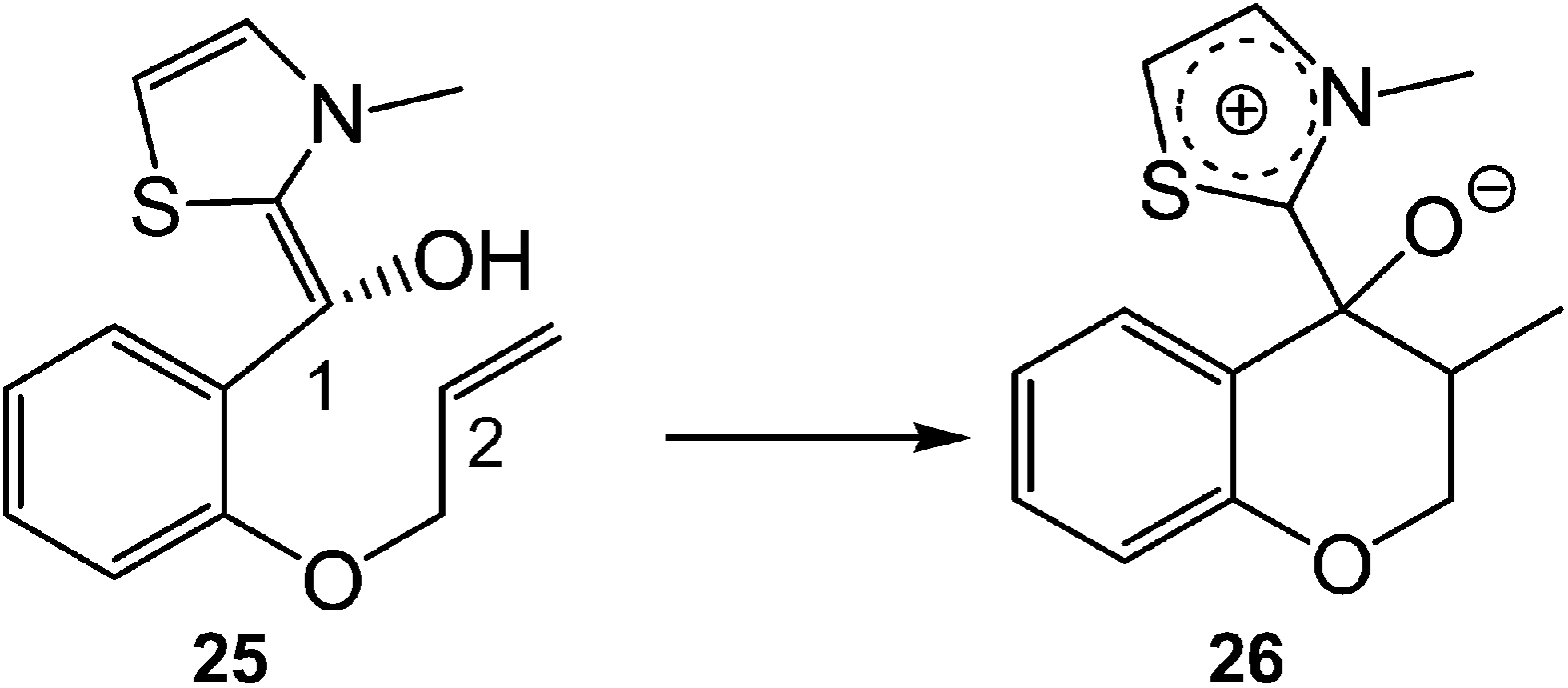
This model for the C–C bond formation in polar reactions has been supported by a number of ELF topological studies devoted to the characterisation of the molecular mechanism in diverse polar reactions. Thus, an ELF topological analysis for the C–C bond formation step in the intramolecular Stetter reaction103 (see Scheme 12) established that it takes place also by a C-to-C coupling of two pseudoradical centers generated at the most nucleophilic center of Breslow intermediate 43, the C1 carbon, and the most electrophilic center of the α,β-unsaturated ester framework of 43, the C2 carbon. Interestingly, TS5 already showed the presence of the pseudoradical centers at the C1 and the C2 carbons (see Fig. 11). Note that TS5, which was associated with a Michael-type addition, presents a similar topology to that found in the non-polar structure 27.
 | ||
| Scheme 12 The C–C bond formation step in the intramolecular Stetter reaction. | ||
 | ||
| Fig. 11 TS5 associated with the C–C bond formation in Breslow intermediate 43. | ||
A very recent ELF topological analysis for the C–C bond formation step in the NHC catalysed Michael addition of enols to α,β-unsaturated acyl-azoliums (see Scheme 13) showed that it takes place also at a C–C distance of 2.00 Å by a C-to-C coupling of two pseudoradical centers generated at the most nucleophilic center of enol 45, the C1 carbon, and the most electrophilic center of the α,β-unsaturated acyl-azoliums 46, the conjugated C2 carbon.104
 | ||
| Scheme 13 The C–C bond formation step in the NHC catalysed Michael addition of enols to α,β-unsaturated acyl-azoliums. | ||
Finally, to assert this model for the C–C bond formation in polar reactions, an ELF topological analysis of the Friedel–Crafts reaction between indole 48 and the electrophilically activated nitroethyelene 49 was performed (see Scheme 14).57 This hydrogen-bond catalysed Friedel–Crafts reaction presents a two-step mechanism. The first step is associated with the C–C bond formation between the most nucleophilic center of N-methyl indole 48, the C1 carbon, and the most electrophilic center of nitroethylene 49, the C2 carbon, yielding a zwitterionic intermediate 50.57
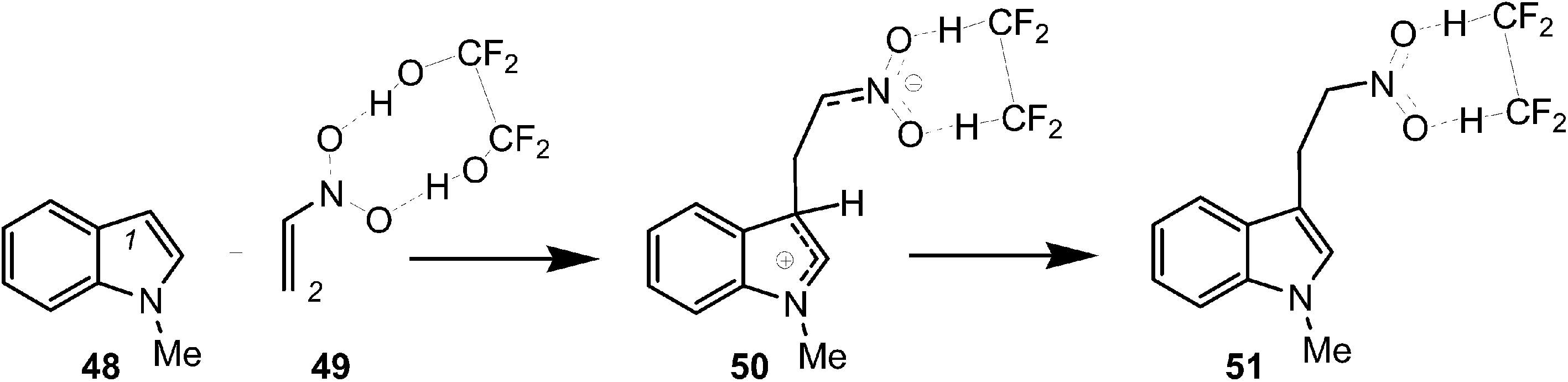 | ||
| Scheme 14 Friedel–Crafts reaction between indole 48 and the electrophilically activated nitroethyelene 49. | ||
ELF bonding analysis along the first step provided a complete characterisation of the changes of electron density along the C–C single bond formation, which begins at a C–C distance of 1.97 Å by a C-to-C coupling of two pseudoradical centers located at the most nucleophilic center of N-methyl indole 48, the C1 carbon, and the most electrophilic center of the nitroethylene complex 49, the C2 carbon (see the merger of the V(C1) and V(C2) monosynaptic basins present in structure 52 into the V(C1, C2) disynaptic basin present in structure 53 in Fig. 12).57 Interestingly, along this step of the reaction, a large amount of GEDT was found at the corresponding TS, 0.60e.
 | ||
| Fig. 12 Selected structures 52 and 53 involved the formation of the C–C single bond formation in the Friedel–Crafts reaction shown in Scheme 13. | ||
ELF topological model for the C–C bond formation in ionic reactions
After establishing a theoretical model for the C–C single bond formation in non-polar and polar reactions, there remained only one way to explain how the C–C single bond formation takes place in ionic processes. This question was very recently resolved in an ELF topological study for the mechanism of ionic [4+ + 2] I-DA reactions.105 In that study, the I-DA reactions of the oxonium cation 54, a cationic heterodiene, with cyclopentene 55 was selected as a computational model (see Scheme 15).105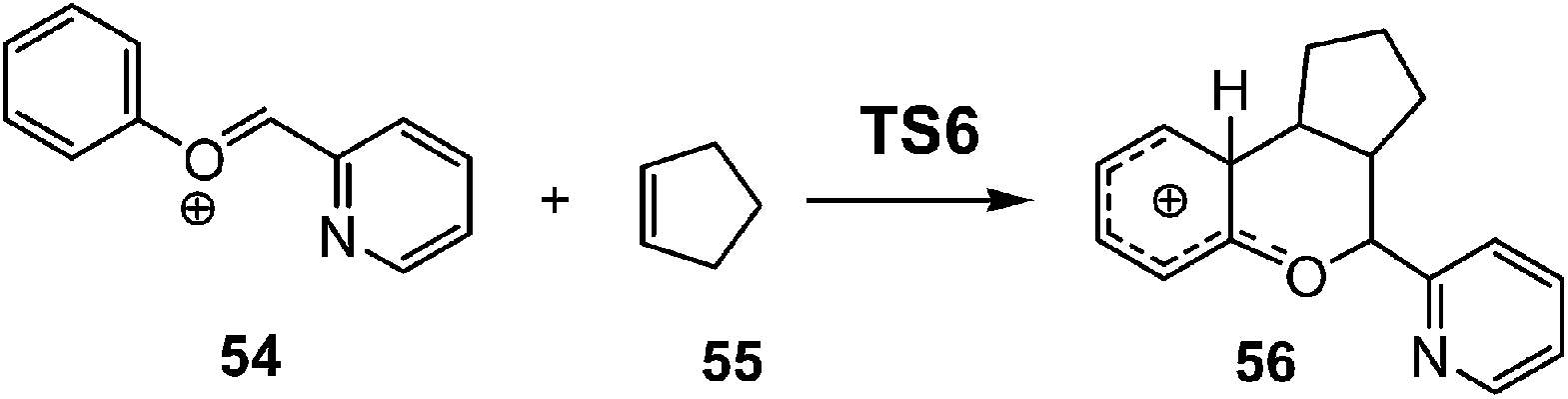 | ||
| Scheme 15 [4++2] I-DA reaction between oxonium cation 54 and cyclopentene 55. | ||
This I-DA reaction takes place through a two-stage one-step mechanism via a highly asynchronous TS6 (see Fig. 13). As expected, TS6 showed a large GEDT, 0.42e, as a consequence of the very high electrophilic character of oxonium cation 54. One of the most appealing findings of this study was the description provided by the ELF topology for the formation of the first C–C single bond in this I-DA reaction. Interestingly, just as in non-polar and polar reactions, the formation of the C–C single bond takes place also at a C–C distance of 1.90 Å by a C-to-C coupling of two pseudoradical centers generated at the most electrophilic center of oxonium cation 54 and the most nucleophilic center of cyclopentene 55 (see Fig. 14). In this way, the large GEDT found at the structure 57 with a C–C distance of 2.00 Å, 0.42e, is responsible for the formation of the pseudoradical centers in oxonium cation 53.105
 | ||
| Fig. 13 TS6 associated with the [4++2] I-DA reaction between the oxonium cation 54 and cyclopentene 55. | ||
 | ||
| Fig. 14 Selected structures 57 and 58 of the IRC associated with the formation of the first C–C single bond in I-DA reactions between oxonium cation 54 and cyclopentene 55. | ||
In order to highlight this appealing finding, and thus to generalise the model based on the C-to-C coupling of pseudoradical centers for the C–C formation in ionic reactions, an ELF topological analysis for the formation of the C–C single bond in the [4 + 2+] I-DA reaction between Cp 4 and N,N-dimethyliminium cation 7, a cationic heterodienophile, was very recently performed (see Scheme 3).106
Just as in the [4++2] I-DA reaction between oxonium cation 54 and cyclopentene 54, this [4 + 2+] I-DA reaction takes place through a two-stage one-step via a very highly asynchronous TS (see TS4 in Fig. 2).107 As aforementioned, a large amount of GEDT was observed at TS4, 0.45e, as a consequence of the high electrophilic character of iminium cation 7. As expected, the C–C bond formation in this [4 + 2+] I-DA reaction takes place also at a C–C distance of 1.96 Å via a C-to-C coupling between two pseudoradical centers generated at the most electrophilic center of the iminium cation, the carbon atom, and one of the two most nucleophilic centers of Cp 4. The non-bonding electron density of the pseudoradical center generated at the most electrophilic carbon of iminium cation, 0.32e, comes mainly from the GEDT which takes place along the reaction (see the V(C2) monosynaptic basin in structure 59 in Fig. 15).106
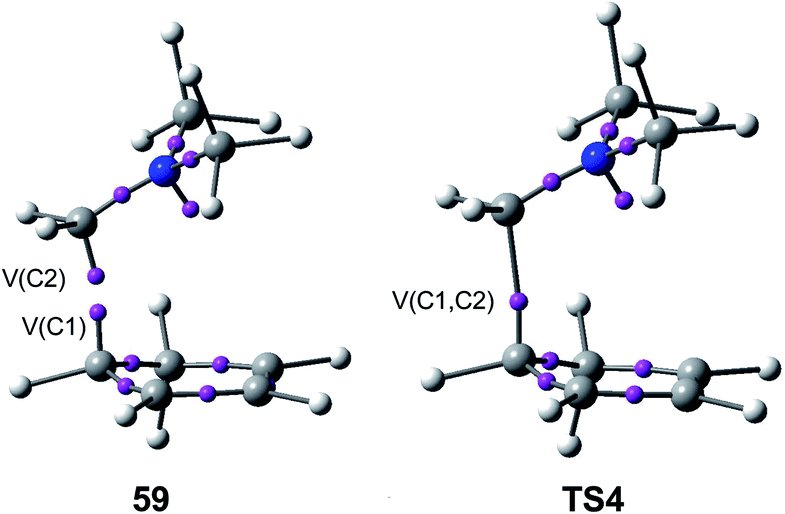 | ||
| Fig. 15 The most relevant ELF attractors in structure 59 and TS4 involved in the C–C single bond formation in the I-DA reaction between Cp 4 and iminium cation 7. | ||
A unified ELF topological model for the C–C bond formation in organic reactions
ELF topological analyses of bonding changes in the N-DA reaction of Cp 4 with styrene 5, in the P-DA reaction of Cp 4 with DCE 6, and in the I-DA reaction of Cp 4 with iminium cation 7 given in Scheme 3 make it possible to establish a unified model for the C–C bond formation in organic reactions involving the participation of CC(X) double bonds. This model is characterised by the C-to-C coupling of two pseudoradical centers generated at the most significant atoms of the reacting molecules. Fig. 16 shows the pseudodiradical structures involved in the C–C bond formation in these selected reactions. These three structures present a large similarity in their topology, the main difference being in the population of the corresponding basins as a consequence of the GEDT that takes place along these reactions.
 | ||
| Fig. 16 Most relevant data of pseudodiradical structures involved in the C–C bond formation in the N-DA reaction of Cp 4 with styrene 5, in the P-DA reaction of Cp 4 with DCE 6, and in the I-DA reaction of Cp 4 with iminium cation 7. The blue arrow indicates the direction of GEDT. | ||
ELF topological analyses along a large number of studies devoted to the characterisation of the molecular mechanism of different organic reactions involving the participation of CC(X) double bonds make it possible to establish some appealing conclusions about the C–C bond formation in these organic reactions:
(i) non-polar, polar and ionic reactions present a similar pattern for the formation of C–C single bonds;
(ii) this process is characterised by a C-to-C coupling of two pseudoradical centers generated along the reaction;
(iii) the formation of the C–C single bonds takes place commonly in a short region of the IRC with a C–C distance in the narrow range from 2.0 to 1.9 Å;
(iv) while along non-polar reactions the pseudoradical centers are generated by the homolytic breaking of the CC double bonds present in the two reagents, in polar and ionic reactions the formation of the pseudoradical centers are mainly generated by the GEDT that takes place from the nucleophile to the electrophile;
(v) for the three TSs associated with the reaction models, TS2, TS3 and TS4, which present a similar geometry, a good correlation between the computed activation energy and the GEDT at the TS is found; i.e. the polarity of the reaction appears to be an important factor determining the feasibility of the reaction;
(vi) the GEDT taking place in polar and ionic reactions causes significant changes in the electron density in both nucleophiles and electrophiles. In asymmetric molecules, an asymmetric electron density rearrangement takes place along the GEDT process. Thus, while in the nucleophilic species some atoms lose less electron density, in the electrophilic species some atoms gather more electron density. These relevant atoms correspond to the more nucleophilic and more electrophilic centres of the molecules.
(vii) in polar and ionic reactions, these relevant centers, which are suitably characterised by an analysis of the nucleophilic Pk− and electrophilic Pk+ Parr functions,98,99 in neutral species, and the radical Pk0 Parr functions,100 in ionic species, are the centers in which the pseudoradical centers will be formed as a consequence of the GEDT process taking place along the nucleophilic/electrophilic interaction. This behaviour is responsible for the chemo- and regioselectivity found in polar and ionic reactions involving asymmetric molecules since in these reactions the most favourable channels correspond to those with the most favourable nucleophilic/electrophilic two-center interaction.
This reaction model based on the quantum topological analysis of the changes in electron density along an organic reaction makes it possible to reject the reaction model based on the FMO theory, which made the fundamental assumption that a majority of chemical reactions should take place at the position and the direction of maximum overlapping of the HOMO and the LUMO frontier orbitals of the reacting molecules.108
MOs obtained as an approximation to Schrodinger's quantum wave function do not have any physical meaning; only the electron density is physically observable. On the other hand, if the occupied MOs do not have any physical meaning, virtual MOs coming from matrix calculations used in Schrodinger's equation resolution do physically not exist. In addition, recent DFT studies have emphasised that the HOMO–LUMO energy gaps used in FMO analyses are too high, above 3.0 eV (i.e. 70 kcal mol−1), to be reached in experimental reaction conditions.22,98 HOMO–LUMO energy gaps, usually above 100 kcal mol−1, can only be reached photochemically, and not thermodynamically which is the way most organic reactions are performed.
Consequently, in the molecular ground state, electrons can not reach MOs that do physically not exist; the reactions usually begin by an early electron density reorganisation in the interacting molecules, which can easily be predicted by analysis of the global and local electrophilicity ω and nucleophilicity N indices. The higher the GEDT, the easier the bonding changes and the faster the reaction.
In the proposed reactivity model, while in non-polar reactions involving closed-shell neutral molecules, the high energy demanded for the breaking of the CC double bonds is responsible for the high activation energies, the high GEDT taking place in ionic reactions makes bonding changes possible without any energetic effort.
References
- H. Eyring and M. Z. Polanyi, Z. Phys. Chem., Abt. B, 1931, 12, 279 CAS.
- H. Eyring, Chem. Rev., 1935, 17, 65 CrossRef CAS.
- K. J. Laidler and M. C. King, J. Phys. Chem., 1983, 87, 2657 CrossRef CAS.
- W. J. Hehre, L. Radom, P. v. R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- Y. Zhao and G. D. Truhlar, J. Phys. Chem. A, 2004, 108, 6908 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- O. Diels and K. Alder, Justus Liebigs Ann. Chem., 1928, 460, 98 CrossRef CAS.
- E. Goldstein, B. Beno and K. N. Houk, J. Am. Chem. Soc., 1996, 118, 6036 CrossRef CAS.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161 CrossRef CAS.
- L. R. Domingo and J. A. Sáez, Org. Biomol. Chem., 2009, 7, 3576 CAS.
- L. R. Domingo, M. Arnó and J. Andrés, J. Org. Chem., 1999, 64, 5867 CrossRef CAS.
- R. G. Parr, L. von Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922 CrossRef CAS.
- L. R. Domingo, M. J. Aurell, P. Pérez and R. Contreras, Tetrahedron, 2002, 58, 4417 CrossRef CAS.
- J. Sauer, H. Wiest and A. Mielert, Chem. Ber., 1964, 97, 3183 CrossRef CAS.
- W. Carruthers, Some Modern Methods of Organic Synthesis, Cambridge University Press, Cambridge, 1978 Search PubMed.
- L. R. Domingo, M. J. Aurell, P. Pérez and R. Contreras, J. Org. Chem., 2003, 68, 3884 CrossRef CAS PubMed.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- L. R. Domingo, P. Pérez and J. A. Sáez, Tetrahedron, 2013, 69, 107 CrossRef CAS PubMed.
- K. N. Houk, J. Gonzalez and Y. Li, Acc. Chem. Res., 1995, 28, 81 CrossRef CAS.
- M. B. Smith and J. March, March's Advanced Organic Chemistry, Reactions, Mechanisms and Structure, John Wiley & Sins, Inc., New York, 2001 Search PubMed.
- F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry. Paet A: Structure and Mechanisms, Springer, New York, 2007 Search PubMed.
- I. Fleming, Molecular Orbitals and Organic Chemical Reactions, Wiley, 2009 Search PubMed.
- L. R. Domingo, E. Chamorro and P. Pérez, J. Org. Chem., 2008, 73, 4615 CrossRef CAS PubMed.
- L. R. Domingo and P. Pérez, Org. Biomol. Chem., 2011, 9, 7168 CAS.
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793 CrossRef CAS PubMed.
- D. H. Ess, G. O. Jones and K. N. Houk, Adv. Synth. Catal., 2006, 348, 2337 CrossRef CAS.
- S. Lakhdar, M. Westermaier, F. Terrier, R. Goumont, T. Boubaker, A. R. Ofial and H. Mayr, J. Org. Chem., 2006, 71, 9088 CrossRef CAS PubMed.
- R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- R. G. Parr, R. A. Donnelly, M. Levy and W. E. Palke, J. Chem. Phys., 1978, 68, 3801 CrossRef CAS PubMed.
- R. T. Sanderson, Science, 1955, 121, 207 CAS.
- V. Y. Korotaev, A. Y. Barkov, P. A. Slepukhin, M. I. Kodess and V. Y. Sosnovskikh, Mendeleev Commun., 2011, 21, 112 CrossRef CAS PubMed.
- S. Lakhdar, F. Terrier, D. Vichard, G. Berionni, N. El Guesmi, R. Goumont and T. Boubaker, Chem.–Eur. J., 2010, 16, 5681 CrossRef CAS PubMed.
- G. N. Lewis, J. Am. Chem. Soc., 1916, 38, 762 CrossRef CAS.
- J. C. Slater, Phys. Rev. B: Solid State, 1931, 37, 481 CAS.
- J. C. Slater, Phys. Rev. B: Solid State, 1931, 38, 1109 CAS.
- L. Pauling, Nature, 1948, 161, 1019 CrossRef CAS.
- B. T. Sutcliffe, Int. J. Quantum Chem., 1996, 58, 645 CrossRef CAS.
- R. H. Abraham and C. D. Shaw, Dynamics: The Geometry of Behavior, Addison-Wesley, Redwood City, CA, 1992 Search PubMed.
- R. F. W. Bader, Atoms in Molecules. A Quantum Theory, Claredon Press, Oxford, UK, 1990 Search PubMed.
- R. F. W. Bader, S. G. Anderson and A. J. Duke, J. Am. Chem. Soc., 1979, 101, 1389 CrossRef CAS.
- R. F. W. Bader, T. T. Nguyendang and Y. Tal, J. Chem. Phys., 1979, 70, 4316 CrossRef CAS PubMed.
- R. F. W. Bader, Theor. Chem. Acc., 2001, 105, 276 CrossRef CAS.
- M. Griffiths and P. Popelier, J. Chem. Inf. Model., 2013, 53, 1714 CrossRef CAS PubMed.
- P. Popelier and F. Aicken, ChemPhysChem, 2003, 4, 824 CrossRef CAS PubMed.
- A. Harding, D. Wedge and P. Popelier, J. Chem. Inf. Model., 2009, 49, 1914 CrossRef CAS PubMed.
- N. Singh, P. Popelier and P. O'Malley, Chem. Phys. Lett., 2006, 426, 219 CrossRef CAS PubMed.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397 CrossRef CAS PubMed.
- A. Savin, A. D. Becke, J. Flad, R. Nesper, H. Preuss and H. G. von Schnering, Angew. Chem., Int. Ed., 1991, 30, 409 CrossRef.
- B. Silvi and A. Savin, Nature, 1994, 371, 683 CrossRef CAS.
- A. Savin, B. Silvi and F. Colonna, Can. J. Chem., 1996, 74, 1088 CrossRef CAS.
- A. Savin, R. Nesper, S. Wengert and T. F. Fassler, Angew. Chem., Int. Ed., 1997, 36, 1808 CrossRef CAS.
- B. Silvi, J. Mol. Struct., 2002, 614, 3 CrossRef CAS.
- L. R. Domingo, P. Pérez and J. A. Sáez, RSC Adv., 2013, 3, 7520 RSC.
- S. Berski, J. Andrés, B. Silvi and L. R. Domingo, J. Phys. Chem. A, 2003, 107, 6014 CrossRef CAS.
- V. Polo, J. Andrés, S. Berskit, L. R. Domingo and B. Silvi, J. Phys. Chem. A, 2008, 112, 7128 CrossRef CAS PubMed.
- J. Andrés, P. González-Navarrete and V. Safont, Int. J. Quantum Chem., 2014 DOI:10.1002/qua.24665.
- R. Thom, Structural Stability and Morphogenesis: An Outline of a General Theory of Models, Inc.:Reading, MA, 1976 Search PubMed.
- A. E. R. Woodcock and T. Poston, A Geometrical Study of Elementary Catastrophes, Spinger-Verlag, Berlin, 1974 Search PubMed.
- R. Gilmore, Catastrophe Theory for Scientists and Engineers, Dover, New York, 1981 Search PubMed.
- X. Krokidis, S. Noury and B. Silvi, J. Phys. Chem. A, 1997, 101, 7277 CrossRef CAS.
- S. Berski and Z. Latajka, Int. J. Quantum Chem., 2011, 111, 2378 CrossRef CAS.
- S. Berski, F. R. Sensato, V. Polo, J. Andrés and V. S. Safont, J. Phys. Chem. A, 2011, 115, 514 CrossRef CAS PubMed.
- E. Chamorro, J. C. Santos, B. Gomez, R. Contreras and P. Fuentealba, J. Chem. Phys., 2001, 114, 23 CrossRef CAS PubMed.
- E. Chamorro, J. C. Santos, B. Gomez, R. Contreras and P. Fuentealba, J. Phys. Chem. A, 2002, 106, 11533 CrossRef CAS.
- P. Chaquin and A. Scemama, Chem. Phys. Lett., 2004, 394, 244 CrossRef CAS PubMed.
- D. B. Chesnut and L. J. Bartolotti, Chem. Phys., 2000, 257, 175 CrossRef CAS.
- I. Fourre, B. Silvi, P. Chaquin and A. Sevin, J. Comput. Chem., 1999, 20, 897 CrossRef CAS.
- F. Fuster, A. Sevin and B. Silvi, J. Phys. Chem. A, 2000, 104, 852 CrossRef CAS.
- N. Gillet, R. Chaudret, J. Contreras-Garcia, W. T. Yang, B. Silvi and J. P. Piquemal, J. Chem. Theory Comput., 2012, 8, 3993 CrossRef CAS PubMed.
- X. Krokidis, V. Goncalves, A. Savin and B. Silvi, J. Phys. Chem. A, 1998, 102, 5065 CrossRef CAS.
- X. Krokidis, N. W. Moriarty, W. A. Lester and M. Frenklach, Chem. Phys. Lett., 1999, 314, 534 CrossRef CAS.
- I. M. Ndassa, B. Silvi and F. Volatron, J. Phys. Chem. A, 2010, 114, 12900 CrossRef PubMed.
- V. Polo, P. González-Navarrete, B. Silvi and J. Andrés, Theor. Chem. Acc., 2008, 120, 341 CrossRef CAS PubMed.
- J. P. Salinas-Olvera, R. M. Gómez and F. Cortés-Gúzman, J. Phys. Chem. A, 2008, 112, 2906 CrossRef CAS PubMed.
- S. Berski, J. Andrés, B. Silvi and L. R. Domingo, J. Phys. Chem. A, 2006, 110, 13939 CrossRef CAS PubMed.
- V. Polo, J. Andrés, R. Castillo, S. Berski and B. Silvi, Chem.–Eur. J., 2004, 10, 5165 CrossRef CAS PubMed.
- J. C. Santos, J. Andrés, A. Aizman, P. Fuentealba and V. Polo, J. Phys. Chem. A, 2005, 109, 3687 CrossRef CAS PubMed.
- V. Polo and J. Andrés, J. Comput. Chem., 2005, 26, 1427 CrossRef CAS PubMed.
- V. Polo and J. Andrés, J. Chem. Theory Comput., 2007, 3, 816 CrossRef CAS.
- R. Thom, Stabilité Structurelle et Morphogénèse, Paris, 1972 Search PubMed.
- L. A. Errede, J. M. Hoyt and R. S. Gregorian, J. Am. Chem. Soc., 1960, 53, 5224 CrossRef.
- L. R. Domingo, E. Chamorro and P. Pérez, Lett. Org. Chem., 2010, 432 CrossRef CAS.
- L. R. Domingo, Org. Chem. Curr. Res., 2013, 2, 120 Search PubMed.
- L. R. Domingo, E. Chamorro and P. Pérez, Org. Biomol. Chem., 2010, 8, 5495 CAS.
- L. R. Domingo and J. A. Sáez, J. Org. Chem., 2011, 76, 373 CrossRef CAS PubMed.
- L. R. Domingo and S. R. Emamian, Tetrahedron, 2014, 70, 1267 CrossRef CAS PubMed.
- L. R. Domingo, J. A. Saéz and M. Arnó, RSC Adv., 2012, 2, 7127 RSC.
- J. Andrés, S. Berski, L. R. Domingo, V. Polo and B. Silvi, Curr. Org. Chem., 2011, 15, 3566 CrossRef.
- J. Andrés, S. Berski, L. R. Domingo and P. González-Navarrete, J. Comput. Chem., 2012, 33, 748 CrossRef PubMed.
- L. R. Domingo, J. A. Saéz, R. J. Zaragozá and M. Arnó, J. Org. Chem., 2008, 73, 8791 CrossRef CAS PubMed.
- L. R. Domingo, P. Pérez and J. A. Sáez, Org. Biomol. Chem., 2012, 10, 3841 CAS.
- L. R. Domingo, P. Pérez, M. J. Aurell and J. A. Sáez, Curr. Org. Chem., 2012, 16, 2343 CrossRef CAS.
- L. R. Domingo, M. J. Aurell, P. Pérez and J. A. Sáez, RSC Adv., 2012, 2, 1334 RSC.
- L. R. Domingo, P. Pérez and J. A. Sáez, RSC Adv., 2013, 3, 1486 RSC.
- E. Chamorro, P. Pérez and L. R. Domingo, Chem. Phys. Lett., 2013, 582, 141 CrossRef CAS PubMed.
- L. R. Domingo and P. Pérez, Org. Biomol. Chem., 2013, 11, 4350 Search PubMed.
- R. G. Parr and W. Yang, J. Am. Chem. Soc., 1984, 106, 4049 CrossRef CAS.
- P. W. Ayers, C. Morell, F. De Proft and P. Geerlings, Chem.–Eur. J., 2007, 13, 8240 CrossRef CAS PubMed.
- L. R. Domingo, R. J. Zaragozá, J. A. Saéz and M. Arnó, Molecules, 2012, 17, 1335 CrossRef CAS PubMed.
- L. R. Domingo, J. A. Sáez and M. Arnó, Org. Biomol. Chem., 2014, 12, 895 CAS.
- L. R. Domingo, M. J. Aurell and P. Pérez, RSC Adv., 2014, 4, 16567 RSC.
- L. R. Domingo and P. Pérez, Phys. Chem. Chem. Phys., 2014, 16, 14108 Search PubMed.
- L. R. Domingo, J. Org. Chem., 2001, 66, 3211 CrossRef CAS.
- K. Fukui, Molecular Orbitals in Chemistry Physics and Biology, New York, 1964 Search PubMed.
Footnote |
| † Dedicated to Prof B. Silvi for his contribution to the development of the ELF topology analysis of electron density in the study of structure and reactivity. |
| This journal is © The Royal Society of Chemistry 2014 |