
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preliminary toxicity and ecotoxicity assessment of methyltrioxorhenium and its derivatives†
S.
Stolte
*abc,
H. T. T.
Bui
ab,
S.
Steudte
ab,
V.
Korinth
d,
J.
Arning
ab,
A.
Białk-Bielińska
abc,
U.
Bottin-Weber
ab,
M.
Cokoja
d,
A.
Hahlbrock
e,
V.
Fetz
e,
R.
Stauber
e,
B.
Jastorff
ab,
C.
Hartmann
f,
R. W.
Fischer
g and
F. E.
Kühn
*d
aUniversity of Bremen, Sustainable Chemistry, Bremen, Germany. E-mail: stefan.stolte@uni-bremen.de; Fax: ++49 4212189863370; Tel: ++49 42121863370
bUniversity of Bremen, Centre for Environmental Research and Sustainable Technology, Leobener Strasse, Bremen, Germany
cDepartment of Environmental Analytics, Faculty of Chemistry, University of Gdańsk, Wita Stwosza 63, 80-308 Gdańsk, Poland
dChair of Inorganic Chemistry/Molecular Catalysis, Catalysis Research Center, Technische Universität München, Ernst-Otto-Fischer-Straße 1, 85747 Garching bei München, Germany. E-mail: fritz.kuehn@ch.tum.de
eMolecular and Cellular Oncology, Mainz Screening Centre, University Medical Centre of Mainz, Langenbeckstr. 1, 55131 Mainz, Germany
fAtlantiChem GmbH, Industrie Center Obernburg, 63784 Obernburg, Germany
gMuniCat, Technische Universität München, Catalysis Research Center, Ernst-Otto-Fischer-Straße 1, 85747 Garching bei München, Germany
First published on 10th November 2014
Abstract
Methyltrioxorhenium (MTO) is one of the best examined catalysts for the activation of H2O2 in oxidation reactions, particularly olefin epoxidation reactions. The broad applicability and high activity of MTO and MTO Lewis base adducts combined with their selectivity for a variety of processes have also generated industrial interest. In compliance with the basic principles of green chemistry, however, the optimization of the technological properties of chemicals should be investigated in parallel with the minimization of their (eco)toxicological hazard potential. The toxicity of MTO and its complexes towards human health and the environment has not been investigated so far. Performed in close cooperation between industry and academia, the present study aims to fill this knowledge gap by systematically assessing the (eco)toxicity of MTO and its derivatives at different levels of biological organisation. Instrumental analysis was used to investigate the stability and solubility of the target compounds in biological media. The initial evaluation of the hazard potential of MTO shows that it causes strong short-term (eco)toxicological effects and offers preliminary indications of its genotoxic potential. In this light it appears important to prevent the release of MTO into the environment and the exposure of operators; the necessary safety precautions should be taken. The fast decomposition of MTO in aqueous media, on the other hand, leads to perrhenates, which exhibit a much lower (eco)toxicological hazard potential.
Introduction
A synthesis procedure for methyltrioxorhenium(VII) (MTO) was first reported in 1979.1 The breakthrough regarding the exploration of both its reaction chemistry and its applications came after an improved synthesis method had been developed almost 10 years later.2 It was soon discovered that MTO is an efficient catalyst, particularly for olefin epoxidation reactions,3,4 aldehyde olefination, olefin metathesis,5–8 dehydration of alcohols,9 deoxydehydration of diols to olefins and many other organic reactions.10–13 Recently it has even been found to act as a catalyst for the cleavage of C–O bonds of lignin model compounds.14 The broad applicability and high activity of MTO combined with its selectivity in a variety of processes generated both academic and industrial interest, e.g. in the production of food additives.15,16 Nevertheless, the synthesis of MTO remained expensive (price of rhenium and intrinsic restriction to 50% yield) and involved highly toxic organotin reagents until a new synthetic route was developed starting from perrhenates.17,18 Based on this optimized procedure, a pilot plant synthesis was implemented by Süd-Chemie AG (now Clariant AG), making MTO available with improved operational safety, in larger amounts and at a lower cost.In the context of olefin epoxidation it turned out that the addition to MTO of either an excess of complex-forming aromatic Lewis bases19,20 or stoichiometric amounts of Schiff bases21,22 leads to a significant increase in both epoxide yield and selectivity.
The use of MTO and derivatives in catalytic reactions may comply with the 12 Principles of Green Chemistry of Paul Anastas and John C. Warner23 in terms of, for example, the atomic and energy efficiency of reactions and the reduced work-up of products. However, in accordance with the 4th principle, chemicals should also be designed to preserve efficacy of function while reducing toxicity. The toxicity of MTO and its complexes towards human health and the environment has not yet been investigated. Apart from a few studies focusing on the toxicology of the radioactive isotopes 186/188Re24 and Re(I) complexes25,26 as radio- and chemotherapeutic agents27 in cancer therapy, rhenium compounds have very infrequently been investigated. This is primarily due to Re being one of the rarest elements in the earth's crust and because it is not considered an essential element for animals or plants. Thus its organometallic chemistry was of little practical interest until its broad applicability in catalysis was discovered.
An interdisciplinary industry–academia collaborative team, including groups from organic synthesis, toxicology and ecotoxicology as well as the manufacturer of MTO, aims to fill this knowledge gap by systematically assessing the influence of MTO and its derivatives. For the comparative hazard assessment presented here, not only MTO but also the (4-tert-butylpyridine) methyltrioxorhenium (MTO-TBP) complex, ammonium perrhenate (NH4ReO4) and 4-tert-butylpyridine (TBP) (Fig. 1, test set 1) were examined through comprehensive (eco)toxicity testing. Additionally a cytotoxicity screening for a series of Re-based catalysts with various Lewis bases was conducted (Fig. 2, test set 2). These sets of compounds enabled us to explore the (eco)toxicity of the catalytically active Re-centre and to find out whether the observed effects could be modulated by aromatic Lewis bases.
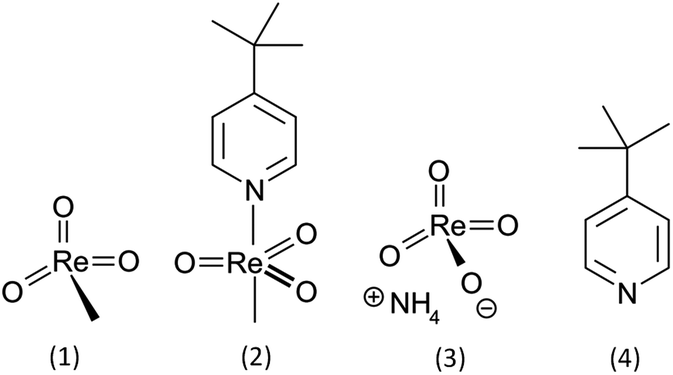 | ||
| Fig. 1 Structures of the compounds investigated in detail (test set 1): (1) methyltrioxorhenium (MTO), (2) (4-tert-butylpyridine)-methyltrioxorhenium (MTO-TBP), (3) ammonium perrhenate (NH4ReO4), (4) 4-tert-butylpyridine (TBP). | ||
 | ||
| Fig. 2 Structures of the various MTO-based complexes and selected Lewis bases used for cytotoxicity screening (test set 2): (5) (4,4-dimethyl-2,2-bipyridine)-methyltrioxorhenium (MTO-BiPy), (6) 4,4-dimethyl-2,2-bipyridine (BiPy), (7) (((4-chlorophenyl)imino)methyl)phenol-methyltrioxorhenium (MTO-ClPh), (8) (((4-chlorophenyl)imino)methyl)phenol (ClPh), (9) (1,2-cyclohexanediylbis[(E)-nitrilomethylidyne]bis[4-bromophenol])-methyltrioxorhenium (MTO-BrPh), (10) 1,2-cyclohexanediylbis[(E)-nitrilomethylidyne]bis[4-bromophenol] (BrPh), (11) (pyrazole)-methyltrioxorhenium (MTO-Pz), (12) (4-cyanopyridine)-methyltrioxorhenium (MTO-CNPy), (13) (1,10-phenanthroline)-methyltrioxorhenium (MTO-Phen). | ||
Since the (eco)toxicity testing was performed in aqueous solution and MTO is known to hydrolyse rapidly in alkaline and slowly in acidic media forming methane and perrhenate,28 the ReO4− anion was also taken into consideration. For the same reason TBP, a degradation product of MTO-TBP, was examined.29
For a preliminary toxicity evaluation we investigated the inhibition of isolated electric eel acetylcholinesterase (AChE) and applied 48 h in vitro cytotoxicity assays with IPC-81 leukaemic rat cells and HepG2 human liver hepatocellular carcinoma cells to measure general cell viability as a toxicological endpoint. Moreover, the genotoxic potential of MTO and NH4ReO4 was analysed in the HeLa human epithelial cervical cancer cell line. The ecotoxicity of the compounds (Fig. 1, test set 1) was assessed in acute tests with the marine bacteria Vibrio fischeri, the limnic green algae Raphidocelis subcapitata, the higher aquatic plant Lemna minor (duckweed) and the water flea Daphnia magna. These test systems represent different levels of organization or trophic levels that are well established and have been proven useful for determining the hazard potential of various industrial chemicals.30–33 The (eco)toxicological tests were supported by instrumental analysis with respect to the solubility and stability of the target compounds using High Performance Liquid Chromatography with UV detection (HPLC-UV), Ion Chromatography (IC) and Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES). These data are essential for assessing the speciation and bioavailability of the compounds tested in different media.
Current chemical legislation, such as the REACH (Registration, Evaluation, Authorization and Restriction of Chemicals) directive, requires (eco)toxicological data of chemicals if they are produced or imported in amounts >1 t per annum. In this context, registration will probably never apply to MTO or any other organometallic catalysts, since they are used in comparatively small quantities. Nevertheless, specific toxic effects of very high concern to humans and the environment, e.g. genotoxicity, can render substances subject to authorization and/or restriction processes under REACH, regardless of their annual production volume. Hence, such effects need to be included in a holistic hazard assessment of chemicals. In view of this, our investigations are guided by the ideas of green chemistry and responsible care to proactively assess the potential safety and environmental and health problems of MTO and its derivatives that may occur when working with them, and to deal with traces of catalysts remaining in synthetic food additives and other products or when released into the environment via process effluents, for example.
Material and methods
Solubility studies
The solubility of MTO was determined using an inductively coupled plasma optical emission spectrometer (Optima 7300 DV, PerkinElmer, Waltham, Massachusetts, USA) equipped with an autosampler (CETAC). The following parameters were used for the analysis: wavelength: 197.248 nm; plasma gas flow rate: 15 L min−1; auxiliary gas flow rate: 0.2 L min−1; nebulizer gas flow rate: 0.80 L min−1; sample flow rate: 1.50 mL min−1; plasma view: axial. In order to obtain the calibration curve, five concentrations (in the range between 0.1 and 60 mg L−1 Re) of a Re-Standard (1000 ppm, Sigma-Aldrich) dissolved in different test media containing 1% HNO3 (p.a.) were prepared. The test samples of MTO were prepared in a similar way: the stock solution as used in the different toxicity tests was prepared in media containing 1% HNO3 (p.a.) and diluted to achieve a Re-concentration within the calibration range.The solubility of NH4ReO4 was determined using an ion chromatograph (Metrohm 881 Compact IC) with an online eluent degasser, 20 μL injection loop, a Metrosep A supp ion exchange column (4.0 × 50 mm, 5 μm particle size) coupled with a Metrosep A Supp 4/5 Guard and a Metrosep RP Guard, a self-regenerating Suppressor Module (MSM) and a CO2-suppressor (MCS); a conductometric detector and Metrohm software (MagICNet version 1.1 compact), all purchased from Metrohm, Herisau, Switzerland. The eluent contained 3.2 mM Na2CO3, 1.0 mM NaHCO3 and 15% acetonitrile; the flow rate was adjusted to 0.7 mL min−1. The calibration curve was obtained by injecting five concentrations of 25–500 μM NH4 ReO4 in the specific media. The stock solutions as used in the toxicity tests were diluted to the calibrated concentration range, measured and the concentration recalculated.
For determining the difference between the nominal and real concentrations of TBP a VWR Hitachi HPLC-UV system (containing the L-2130 HTA-pump, L-2130 degasser, L-2200 autosampler, L-2300 column oven, L-2450 diode array-detector and the EZChrom Elite software, VWR, Darmstadt, Germany) was used. Separation was achieved with a CN-column (LiChrospher 100 CN, 125 × 4 mm, 5 μm particle size, Merck KGaA, Darmstadt, Germany) using 10% acetonitrile and 90% phosphate buffer (3 mM KH2PO4, 2 mM K2HPO4, pH 7.0) and a flow rate of 1 mL min−1. A detection wavelength of 254 nm was used for the quantification. The calibration curve was obtained by injecting five concentrations of 5–500 μM TBP into the specific media. The stock solutions as used in the toxicity tests were diluted as needed, measured and the concentration recalculated.
Hydrolysis studies
The hydrolysis of MTO-TBP was determined by the release of the free base TBP via HPLC-UV. The method and system used are described in detail in the solubility studies section. Approximately 10 mg of the adduct was dissolved in 100 mL water or methanol and injected immediately after dissolving. The samples were injected at regular intervals (ca. 15 min).The hydrolysis of MTO yielded the hydrolysis product ReO4−via IC (for the detailed method description, see the solubility studies section). A solution of 500 μM MTO in the specific buffer was measured immediately, illuminated and heated as required and re-measured at regular intervals, depending on the duration of the toxicity test (30, 60 and 90 min for acetylcholinesterase and Vibrio fischeri, 1, 2, 6, 24 and 48 h for cells and Daphnia magna, 2, 6, 24, 48 and 72 h for Raphidocelis subcapitata, daily for Lemna minor). The increasing ReO4− concentration was the measure of MTO degradation and was plotted against the time assuming first order kinetics (eqn (1)):
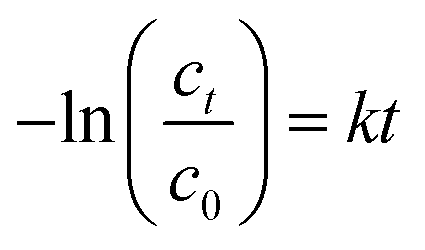 | (1) |
Acetylcholinesterase inhibition assay
The inhibition of AChE was measured using a colorimetric assay based on the reduction of the dye 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB) by the enzymatically formed thiocholine moiety from the AChE substrate acetylthiocholine iodide. The assay is described in detail in Stock et al.34 Briefly, a dilution series of the test substances in phosphate buffer (0.02 M, pH 8.0) containing max. 1% methanol was prepared directly in the wells of a 96-well microtitre plate. DTNB (2 mM, 0.185 mg mL−1 NaHCO3 in phosphate buffer pH 8.0) and the enzyme (0.2 U mL−1, 0.25 mg mL−1 bovine serum albumin in phosphate buffer pH 8.0) were added to each well. The reaction was started by the addition of acetylthiocholine iodide (2 mM in phosphate buffer). The final test concentrations were 0.5 mM of DTNB and acetylthiocholine iodide and 0.05 U mL−1 AChE. Each plate contained blanks (no enzyme) and controls (no toxicant). In each experiment each substance was tested with three replicates of nine different concentrations. Two independent experiments with different stock solutions of each substance were performed. Enzyme kinetics were measured at 405 nm at 30 second intervals in a microplate-reader (MRX Dynatech) for 5 minutes. The enzyme activity was expressed as the slope of optical density (in OD min−1) from a linear regression.Cell viability assay with IPC-81 cells and HepG2
Briefly, promyelocytic rat cells from the IPC-81 cell line were incubated with the test substances for 48 h. Subsequently, cells were incubated for 4 h in 96-well plates with 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulphophenyl)-2H-tetrazolium monosodium salt (WST-1) reagent. Each plate contained blanks (no cells) and controls (no toxicant).The HepG2 cell line was obtained from the University of Oldenburg (Germany) and cultured in 10 mL flasks in a culture medium consisting of 90% RPMI 16/40 medium and 10% foetal calf serum supplemented with 2 mg mL−1 NaHCO3, 2 mM L-glutamine, 100 U per mL penicillin and 100 μg per mL streptomycin. The cultures were maintained at 37 °C in a humidified atmosphere containing 5% CO2, and the culture medium was replaced once a week. The cells were trypsinated and seeded to new flasks when reaching 70%–80% confluence. To measure the cell viability, cells were incubated with the test substances for 48 h. Subsequently, cells were incubated for 4 h in 96-well plates with 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulphophenyl)-2H-tetrazolium monosodium salt (WST-1) reagent. Each plate contained blanks (no cells) and controls (no toxicant).
The cell viability assays with IPC-81 and HepG2 were generally carried out for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dilution series. Each dose response curve was recorded for 9 parallel dilution series on three different 96-well plates. Two independent experiments with different stock solutions of each compound were performed. Positive controls with Carbendazim were checked at regular intervals.
:1 dilution series. Each dose response curve was recorded for 9 parallel dilution series on three different 96-well plates. Two independent experiments with different stock solutions of each compound were performed. Positive controls with Carbendazim were checked at regular intervals.
Luminescence inhibition assay with marine bacteria Vibrio fischeri
This test with the marine bacterium Vibrio fischeri was performed according to DIN EN ISO 11348-2.23. The freeze-dried bacteria were purchased from Dr Lange GmbH (Düsseldorf, Germany). The tests were carried out twice for each substance. First a range-finding was undertaken with two replicates per dilution series. Those results were validated within a second test using three replicates for each concentration. Within each test at least 4 controls (2% NaCl solution, phosphate buffered) were used. To exclude pH-effects all the substances were prepared as phosphate-buffered solutions (0.02 M, pH 7.0, including 2% sodium chloride). The tests were performed at 15 °C using thermostats (LUMIStherm, Dr Lange GmbH, Düsseldorf, Germany). The luminescence was measured with a luminometer (LUMIStox 300, Dr Lange GmbH, Düsseldorf, Germany). The liquid-dried bacteria were rehydrated according to the test protocol, then 500 μL aliquots of the bacterial solution were pre-incubated for 15 min at 15 °C. After measuring the initial luminescence 500 μL of the diluted samples were added to the pre-aliquoted bacteria. The bioluminescence was measured again after an incubation time of 30 min. The relative toxicity of the samples was expressed as percentage inhibition compared to the controls. Luminescent bacteria assays were done without any co-solvents.Reproduction inhibition assay with limnic green algae Raphidocelis subcapitata
For this assay the unicellular limnic green algae Raphidocelis subcapitata (Culture Collection of Algae, Universität Göttingen, Göttingen), formerly known as Selenastrum capricornutum or Pseudokirchneriella subcapitata, was used and toxicity tests were done using a synchronized culture. The stock culture was grown under photoautotrophic conditions at 28 °C (±0.5 °C) in an inorganic, sterilized medium (50.0 mg L−1 NaHCO3, 15.0 mg L−1 NH4Cl, 12.0 mg L−1 MgCl2·6H2O, 18.0 mg L−1 CaCl2·2H2O, 15.0 mg L−1 MgSO4·7H2O, 1.60 mg L−1 KH2PO4, 0.064 mg L−1 FeCl3·6H2O, 0.100 mg L−1 Na2EDTA·2H2O, 0.185 mg L−1 H3BO3, 0.415 mg L−1 MnCl2·4H2O, 0.003 mg L−1 ZnCl2, 0.0015 mg L−1 CoCl2·6H2O, 0.007 mg L−1 NaMoO4·2H2O, 0.00001 mg L−1 CuCl2·2H2O, pH 8.1) with saturating white light (intensity of 22 to 33 kilolux) (Lumilux Daylight L 36 W-11 and Lumilux Interna L 36 W-41, Osram, Berlin, Germany). Cells were aerated with 1.5 vol% CO2 and synchronized using a 14 h light and 10 h darkness cycle. The stock culture was diluted every day to a cell density of 5 × 105 cells mL−1.The toxicity tests started with autospores (young algal cells at the beginning of the growth cycle). Algae were exposed to the test substances for 24 h. The endpoint of this assay was the inhibition of algal reproduction measured as inhibition of population growth. All cell numbers (stock culture and test) were determined with the Coulter Counter Z2 (Beckmann, Nürnberg, Germany). The tests were performed in sterilized glass tubes (20 mL Pyrex tubes sealed with caps containing a gas tight Teflon membrane), algae were stirred over the whole test period of 24 h and the test conditions were the same as for the stock culture except for the CO2 source. Here the NaHCO3 present in the medium is sufficient to cover the CO2 demand. The methods for stock culturing and testing are described in detail in ref. 32. Laboratory facilities allowed parallel testing of up to 60 tubes. All substances were tested twice: first a range of findings was taken (4 concentrations, two replicates) and in a second test the results were verified with 6 concentrations per substance in two replicates. The growth inhibition was calculated using the cell counts of the treated samples in relation to the untreated controls (pure medium). For each assay at least 6 controls were used.
Acute immobilization assay with Daphnia magna
The 48 h acute immobilization test with the crustacean Daphnia magna was assessed using the commercially available Daphtoxkit F (MicroBioTest Incorporation, Gent, Belgium) in accordance with the ISO standard (ISO 6341). The tests with neonates less than 24 h old, obtained by the hatching of ephippia, were performed at 20 °C in the dark. 5 Pre-fed animals were incubated with the test substances in 10 mL of mineral medium (67.75 mg L−1 NaHCO3, 284 mg L−1 CaCl2·2H2O, 123.25 mg L−1 MgSO4·7H2O, 5.75 mg L−1 KCl). The number of immobilized or dead organisms was checked after 24 and 48 h. The relative toxicity of the samples was expressed as a percentage of the unaffected organisms compared to the controls. All the substances were tested in three independent experiments.Growth inhibition assay with duckweed Lemna minor
The growth inhibition assay with Lemna minor was performed according to a modified version of the test protocol described by Drost et al.35 Stock cultures of the plants were grown in open Erlenmeyer flasks in sterilized Steinberg medium (pH 5.5 ± 0.2) in a climate chamber with a constant temperature of 21 ± 2 °C in accordance with OECD guideline 221. The chamber was illuminated continuously with a maximum of 6000 lx. The test was performed in six-well cell culture plates (Greiner Bio-One GmbH, Frickenhausen, Germany). For the test 1 plant with 3–4 fronds was exposed to the test substance for 7 days (culture conditions were the same as for the stock culture). The growth was determined on the basis of the frond area (mm2) with a Scanalyzer from Lemnatec GmbH (Würselen, Germany). The relative toxicity of the samples was expressed as a percentage of growth compared to the controls. To exclude the effects on plant growth caused by the acidity of the compounds the pH values were checked at the beginning of the test and at the end. All substances were tested three times with a minimum of 6 controls (pure Steinberg medium) for each test.Genotoxicity assay
The human epithelial cervical cancer cell line, HeLa, was maintained in DMEM medium (Gibco) with L-glutamine supplemented with 10% FCS and Pen/Strep (100 U/100 μg mL−1) (Gibco). For cytotoxicity studies cells were seeded into 96-well plates and further cultivated under standard conditions. Cells were exposed to different concentrations of MTO or ammonium perrhenate in DMEM medium. The impact of MTO and ReO4− exposure on cell vitality was assessed using the alamarBlue® assay (Invitrogen) as described.36 Immunoblotting was carried out as described in Knauer et al.37 Briefly, proteins were separated on a 10% SDS-polyacrylamide gel and transferred onto a polyvinylidene difluoride (PVDF) membrane. Membranes were blocked with 5% non-fat dry milk in PBS and the following antibodies were used: α-GAPDH (Santa Cruz); α-γH2AX (Bethyl Laboratories) and appropriate HRP-conjugated secondary antibodies (Santa Cruz). Automated high content microscopy (HCM) was employed to quantify the DNA-repair foci using the automated ArrayScan®VTI imaging platform (Thermo Fisher) as described in Bier et al.38 Briefly, HeLa cells were seeded into black-walled 96-well thin bottom μclear® plates (Greiner) and further cultivated for 24 h. Cells were exposed to MTO or ammonium perrhenate in DMEM medium for 8 h. Subsequently, cells were fixed for 15 min with 4% paraformaldehyde (PFA). DNA-repair foci were immunofluorescently labelled with α-γH2AX (Bethyl Laboratories) and appropriate FITC-labelled secondary antibody (Abcam). Cell nuclei were stained with Hoechst 33342 as outlined.39 To quantify the γ-H2AX foci per cell, images were analysed using a Spot Detector V4® assay. For every cell, a binary image mask was created from the Hoechst 33342 staining signal, marking the nucleus. In the second channel (green fluorescence) spots were counted within this nuclear region. Scans were performed with settings to give sub-saturating fluorescence intensity. A minimum of 1000 cells per well was recorded.Results and discussion
Nominal vs. real concentrations
The data derived for test set 1 from instrumental analysis indicate mainly a low deviation between nominal and measured concentrations of stock solutions used in ecotoxicity testing (Table S1; ESI†). The variation range was generally ±10%. However, especially for MTO, deviations of up to −25% could be determined, which has to be considered when assessing the hazard potential of this chemical.Stability
The stability of MTO and MTO-TBP was investigated over time to find out whether the observed effects could be attributed to the parent compound or whether they were due to the hydrolysis products formed under the test conditions. Therefore, the compounds were dissolved in six different biological test media and incubated under the given test conditions. The degradation of MTO was tracked by ion chromatographic quantification of the degradation product ReO4−. A linear correlation was not observed for all the media when the logarithm of the fraction of original reactant remaining [ln(c/c0)] was plotted against time (ESI†). This indicates that the hydrolysis of MTO does not necessarily follow pseudo-1st-order kinetics. In all media, however, there was significant hydrolysis of MTO, ranging from 20% degradation in the short-term test with acetylcholinesterase (30 min) to 92% decomposition in a 72 h test with algae (Table 1).| pH | Temperature [°C] | Illumination | Ionic strengtha [mM] | Initial MTO concentrationb [μM] | Estimated test durationc | Degraded MTO at end of the experimentd (in %) | |
|---|---|---|---|---|---|---|---|
| a Calculated via PHREEQCi (v 2.18). b Equal to the highest tested concentration in the respective test. c Including sample preparation. d Based on the released ReO4−. | |||||||
| AChE | 8.0 | 20 | No | 51 | 600 | 30 min | 22 |
| V. fischeri | 7.0 | 15 | No | 378 | 550 | 1 h | 30 |
| HepG2/IPC-81 | 6.8 | 37 | No | 144 | 500 | 48 h | 32 |
| D. magna | 7.5 | 20 | No | 8.3 | 500 | 48 h | 53 |
| R. subcapitata | 8.1 | 21 | Yes | 1.7 | 520 | 72 h | 92 |
| L. minor | 5.5 | 21 | Yes | 9.3 | 510 | 7 d | 80 |
Generally, the conditions (temperature, MTO concentration, ionic strength, illumination) varied widely from test to test, so that could have been responsible for the different hydrolysis rates. The pH in particular appears to be important, since MTO is known to hydrolyse faster in aqueous alkaline than in acidic solution.28
The stability of MTP-TBP was tested by dissolving the compound in particular media or distilled water and injected virtually simultaneously into HPLC. No signal due to MTO-TBP manifested itself in the HPLC-UV chromatograms, but a signal from the free Lewis base in a concentration corresponding to the initial MTO-TBP concentration was visible (data not shown). The results suggest spontaneous hydrolysis of MTO-TBP to MTO and TBP, but one cannot rule out the possibility that the hydrolysis took place during the separation process (9 min) in the stationary phase in contact with the mobile phase (10% acetonitrile, 90% 5 mM KH2PO4/K2HPO4 buffer; pH 7, temperature 30 °C).
(Eco)Toxicity
The half maximum effect or inhibitory concentrations (EC50 and IC50 in μM) and confidence intervals of MTO, MTO-TBP, TBP and NH4ReO4 are presented in Table 2. The results obtained from cytotoxicity screening of test set 2 are given in Table 3.| MTO | MTO-TBP | TBP | NH4 ReO4 | |
|---|---|---|---|---|
| a In the case of AChE. | ||||
| Test system | IC50a and EC50 values [μM] (confidence interval) |
|||
| AChE | 92.3 (84.6–100.7) | 227 (208–248) | 5600 (5400–6200) | >2360 |
| IPC-81 | 100 (94–108) | 44.9 (38.1–53.9) | 5000 (4600–5400) | >5150 |
| HepG2 | 45.3 (46.0–48.3) | 47.3 (42.6–52.7) | >6200 | >5150 |
| V. fischeri | 0.275 (0.252–0.300) | 0.217 (0.203–0.231) | 2.34 (1.87–2.91) | >11000 |
| R. subcapitata | 19.0 (13.1–27.8) | 23.8 (15.9–35.8) | 115 (87–135) | >1500 |
| L. minor | 14.60 (11.3–18.8) | 16.4 | 15.84 (4.21–19.10) | 420 (350–502) |
| D. magna | 1.58 (1.53–2.12) | 2.34 | 402 (357–473) | >2500 |
| Substance | EC50 values [μM] (confidence interval) |
|---|---|
| (5) MTO-BiPy | 99 (86–118) |
| (6) BiPy | >1000 |
| (7) MTO-ClPh | 76.8 (58.8–99.8) |
| (8) ClPh | >1000 |
| (9) MTO-BrPh | 98 (93–115) |
| (10) BrPh | 501 (455–583) |
| (11) MTO-Pz | 116 (91–146) |
| (12) MTO-CNPy | 108 (87–125) |
| (13) MTO-Phen | 9.12 (7.96–10.58) |
Comparison of test compounds
A comparison of MTO and MTO-TBP shows that the effects of concentrations for a particular test system lay within a similar range (Table 2). This finding supports the assumption that MTO-TBP is decomposed spontaneously into MTO and TBP in aqueous solution. The MTO released from the complex appears to be the biologically active component that probably causes the observed effects, whereas a lower toxicity was determined for TBP with EC50 values that were mostly at least one order of magnitude higher than those obtained for MTO and MTO-TBP. Hence, the overall contribution to the toxicity of the Lewis base liberated from MTO-TBP appears comparatively small.The same observation was made for a series of MTO- based complexes (Table 3). The EC50s of MTO-BiPy, MTO-ClPh, MTO-BrPh, MTO-Pz, MTO-CNPy towards IPC-81 cells were similar to the value found for MTO (100 μM), whereas the free Lewis bases BiPy, ClPh and BrPh caused significantly lower cytotoxic effects. Only in the case of MTO-Phen the EC50 value was much lower (9 μM), indicating the clear contribution of 1,10-phenanthroline, which is a known cytotoxic agent.40
NH4ReO4 was selected to investigate the influence of the perrhenate anion on (eco)toxicity. Since NH4Cl showed no effect within the tested concentration ranges in any of the test systems (data not shown), it can be assumed that NH4+ has no influence on the toxicity of the tested NH4ReO4; hence, the observed toxic activities are attributable to the anion alone. However, NH4ReO4 usually shows an EC50 of 1 mM or higher, which is an indication of its low toxicity (inhibition potential). Even the strongest effect observed for this compound with a recorded EC50 of 420 μM (Lemna minor) is still one to two orders of magnitude higher than the values determined for the other test compounds.
Comparison of test systems and effect levels
The EC50 and IC50 values generally span up to five orders of magnitude ranging between <1 μM and >10 mM for test set 1, depending on the test system and tested compounds. The ecotoxicological tests were generally more sensitive than the toxicological screening assays with enzymes and cell lines.The enzyme inhibition test with AChE is an important biological marker in toxicology for evaluating the influence of chemicals on the central nervous system of organisms. For NH4ReO4 no inhibition was observed and was not to be expected, considering the negative surface potential of AChE at the entrance to the catalytic centre. TBP analogues such as 4-tert-butyl bipyridinium are known to be very effective AChE inhibitors, since the pyridinium moiety forms strong π-cationic interactions and the tert-butyl residue increases lipophilicity, both supporting docking to the catalytic active centre.41 Under test conditions (pH 8) TBP is mainly deprotonated (pKa of the corresponding acid is about 5.5) and does not interact strongly with the catalytic site of AChE (IC50 = 5600 μM). MTO and MTO-TBP have respective IC50s of 92 and 227 μM, indicating a certain inhibition potential, but which is much lower than that of the effective AChE inhibitor Aldicarb (IC50 = 4.9 μM31). To date it is not known whether MTO and MTO-TBP inhibit the enzyme via direct binding to the catalytic triad or to the regulatory site such as the peripheral anionic site (PAS). Moreover, reduced enzyme activity may be due to non-regio-specific binding of the strong Lewis-acidic Re-centre to the Lewis-basic amino acids of the enzyme.
We used in vitro testing with the IPC-81 leukaemia rat cell line and human hepatocellular carcinoma to screen for the effects on basal cell functions and cell structures. Comparison of the EC50s from the experiments with IPC-81 and HepG2 shows that the values are quite similar for the same compounds. High EC50s were determined for TBP and NH4ReO4, but MTO and MTO-TBP display a greater toxicity with effective concentrations that are at least two orders of magnitude smaller.
EC50 values of 45 to 100 μM are comparable to the value (EC50 = 130 μM) found for N-methylisothiazol-3-one42 (a known cytotoxic agent used as a preservative in cosmetics), indicating considerable cytotoxicity.
The tests with Vibrio fischeri (ISO 11348-3), Raphidocelis subcapitata (OECD 201), Daphnia magna (ISO 6341) and Lemna minor (OECD 221) are widely used, highly standardized bio-tests to assess the short-term toxicity of chemicals. In all tests MTO and MTO-TBP exhibit distinct toxic effects, indicating their high ecotoxicological hazard potential. In particular, the marine bacteria Vibrio fischeri respond strongly to these compounds with EC50s of around 0.25 μM, corresponding to a higher toxicity also found for highly reactive biocides and the antifouling agent 4,5-dichloro-N-octylisothiazol-3-one (EC50 = 0.43 μM).42 Also, the effect of concentrations determined for Daphnia magna (1.5 and 2.3 μM) are very similar to the EC50s recorded for potassium dichromate ranging between 3 and 9 μM, which in this assay was used as the positive control. Moreover, the test reveals that the effects (immobilization) occur immediately (from seconds to minutes) after exposure of the daphnids to MTO and MTO-TBP. On the basis of this preliminary (eco)toxicity assessment, the mode of toxic action cannot be stated with certainty, but the results do indicate that the chemical reactivity of the MTO fragment might be the source of the observed toxicity. The strong Lewis-acidic Re-centre might bind to Lewis-basic side-chains of amino acid residues in peptides, proteins and DNA. The potential formation of DNA adducts may take place along with damage to DNA and the genetic information and is therefore relevant in terms of genotoxicity.
We therefore analysed whether MTO and NH4ReO4 induce an increase of γ-H2AX in human cancer cells following DNA damage (Fig. 3). The histone variant H2AX becomes immediately phosphorylated (γ-H2AX) and accumulates at damaged positions, and this is followed by the accumulation of proteins involved in the DNA repair process.43–45 A concentration-dependent increase in γ-H2AX was detected after exposure of HeLa cells to 0.01–1 mM MTO, but not for 0.01–1 mM NH4ReO4. Consistently, high content microscope-dependent analysis of immunofluorescently labelled γ-H2AX foci revealed an increase upon MTO treatment. This supports our hypothesis that MTO effects might be based on reactivity and interactions of biomolecules such as DNA. Their genotoxicity and mutagenicity need to be further investigated. Moreover, high concentrations of MTO (100 μM) significantly affected the cell viability of epithelial cell models (Fig. 3D). Whether this effect is predominantly mediated via MTO's genotoxicity and/or by additional molecular mechanisms remains to be investigated.
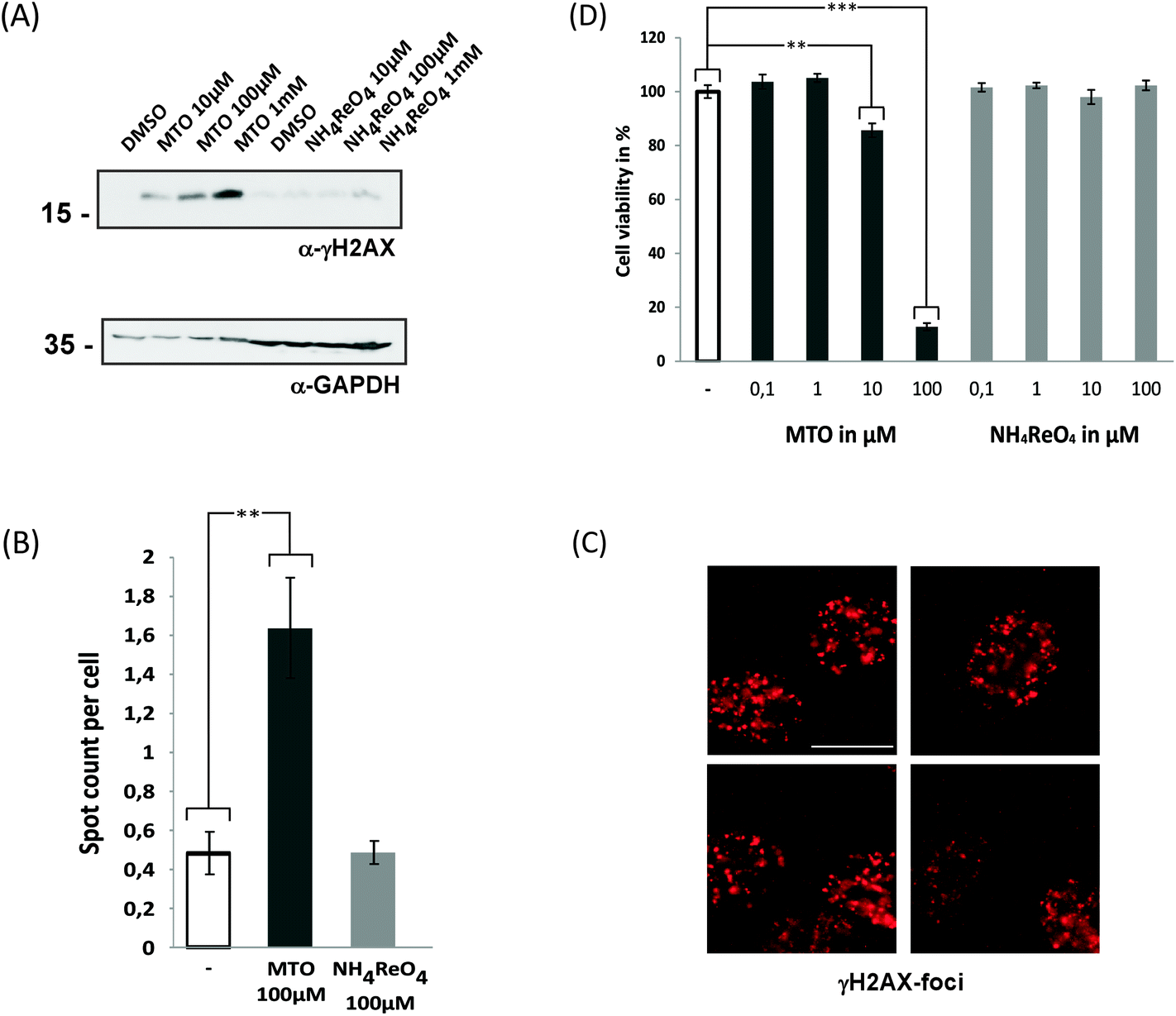 | ||
| Fig. 3 MTO treatment but not ammonium perrhenate is genotoxic and affects cell viability. (A/B/C) MTO but not ammonium perrhenate treatment results in enhanced phosphorylation of histone H2AX, indicative of DNA-damage. (A) Whole cell extracts of HeLa cells were analysed by western blotting using an anti-γ-H2AX- or GAPDH-specific antibody. Cells were treated with MTO or ammonium perrhenate for 48 h. GAPDH served as a load control. Molecular weight is indicated. (B) The number of γ-H2AX-foci per cell was analysed using high content microscopy. HeLa cells were treated with MTO or ammonium perrhenate for 8 h. γ-H2AX-foci were visualized using immunofluorescent staining. (C) Representative micrographs of γ-H2AX-foci are shown. Bar, 10 μm. (D) The impact of MTO-exposure on cell vitality was assessed using the alamarBlue® assay. * = P-value < 0.05; ** = P-value < 0.01; *** = P-value < 0.005. MTO treatment is genotoxic and affects cell viability. | ||
In all the test systems ReO4− exhibited only slight toxic effects, if any. Vibrio fischeri – the most sensitive test system towards MTO and MTO-TBP – was especially insensitive to perrhenates with EC50 > 10 mM. The perrhenate anion is generally known to be highly mobile in water and soil and to be efficiently taken up by plants (brown algae,46 white clover,47 radish48); however, toxic effects have not been reported. Re compounds are often used as surrogates for investigating the behaviour of radioactive 99Tc, owing to their similar chemistries.49 TcO4− can also be taken up by plants,50 but it has also been shown to be intracellularly reduced and to form Tc-cysteine, Tc-glutathione and Tc-protein adducts. Such adducts of ReO4− have not yet been investigated, but perrhenates generally appear to be more recalcitrant than Tc compounds to direct chemical (Sn(II)) and biogeochemical reduction.51 The reduction and binding of Re-species to biomolecules, however, could cause a time-shifted toxicity that might lead to chronic effects; these still need to be investigated.
Conclusions
The initial evaluation of the hazard potential of methyltrioxorhenium(VII) has shown that it causes strong, short-term (eco)toxicological effects at different trophic levels and has yielded the first indications of its genotoxicological potential.In this light it appears important to prevent the release of MTO into the environment and the exposure of operators and that suitable safety precautions are taken. The same conclusion can be drawn for the investigated 4-tert-butylpyridine and other Lewis-base adducts of MTO, which apparently spontaneously release MTO in aqueous solution. The acute toxicity of MTO in solution, however, seems to be limited with time, since its hydrolysis rapidly produces ReO4−, which did not exhibit any acute toxic effects in the test systems used. Perrhenate is also a typical decomposition product of MTO in catalytic reactions. Nevertheless, the chronic toxicity of perrhenates cannot be ruled out and still needs to be investigated. Additionally, the acute ecotoxicity of MTO is of relevance where a probably small but ongoing exposure of the aquatic environment is expected, e.g. by process waste water effluents. The aim for the future should be to develop MTO complexes that retain their catalytic activity but have reduced toxicity. Catalyst immobilization on carrier materials such as membranes or on the surface of nanomaterials might be worth examining.
Acknowledgements
We would like to thank the entire UFT Team, especially Carla Sernow and Julia Kolwek for their experimental support. The authors would like to acknowledge the financial support provided by the German Federal Foundation for the Environment (Deutsche Bundesstiftung Umwelt (DBU), Osnabrück/Germany), the ChemBioMed Zeiss-Stiftung and the German Federal Ministry for Education and Research (BMBF 03X0152). Ha Bui Thi Thu thanks the Vietnamese ministry of education and training for providing financial support.References
- I. R. Beattie and P. J. Jones, Inorg. Chem., 1979, 18, 2318–2319 CrossRef CAS.
- W. A. Herrmann, J. G. Kuchler, J. K. Felixberger, E. Herdtweck and W. Wagner, Angew. Chem., Int. Ed. Engl., 1988, 27, 394–396 CrossRef.
- S. A. Hauser, M. Cokoja and F. E. Kühn, Catal. Sci. Technol., 2013, 3, 552 CAS.
- F. E. Kühn, A. Scherbaum and W. A. Herrmann, J. Organomet. Chem., 2004, 689, 4149–4164 CrossRef PubMed.
- W. A. Herrmann, R. W. Fischer and D. W. Marz, Angew. Chem., Int. Ed. Engl., 1991, 30, 1638–1641 CrossRef.
- W. A. Herrmann, R. W. Fischer, D. W. Marz, W. Wagner, U. N. Flessner, U. Volkhardt and H. Komber, Angew. Chem., Int. Ed. Engl., 1991, 30, 1638–1641 CrossRef.
- W. A. Herrmann and M. Wang, Angew. Chem., Int. Ed. Engl., 1991, 30, 1641–1643 CrossRef.
- C. C. Romão, F. E. Kühn and W. A. Herrmann, Chem. Rev., 1997, 97, 3197–3246 CrossRef PubMed.
- T. J. Korstanje, J. T. B. H. Jastrzebski and R. J. M. K. Gebbink, ChemSusChem, 2010, 3, 695–697 CrossRef CAS PubMed.
- J. Yi, S. Liu and M. M. Abu-Omar, ChemSusChem, 2012, 5, 1401–1404 CrossRef CAS PubMed.
- M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2012, 51, 8082–8086 CrossRef CAS PubMed.
- M. Crucianelli, R. Saladino and A. De Francesco, ChemSusChem, 2010, 3, 524–540 CrossRef CAS PubMed.
- J. E. Ziegler, M. J. Zdilla, A. J. Evans and M. M. Abu-Omar, Inorg. Chem., 2009, 48, 9998–10000 CrossRef CAS PubMed.
- R. G. Harms, I. I. E. Markovits, M. Drees, H. C. M. W. A. Herrmann, M. Cokoja and F. E. Kühn, ChemSusChem, 2014, 7, 429–434 CrossRef CAS PubMed.
- M. M. Carril, P. Altmann, M. Drees, W. Bonrath, T. Netscher, J. Schuetz, F. E. Kühn and J. Schütz, J. Catal., 2011, 283, 55–67 CrossRef CAS PubMed.
- M. M. Carril, P. Altmann, W. Bonrath, T. Netscher, J. Schütz, F. E. Kühn and J. Schuetz, Catal. Sci. Technol., 2012, 2, 722–724 CAS.
- W. A. Herrmann, R. M. Kratzer and R. W. Fischer, Angew. Chem., Int. Ed. Engl., 1997, 36, 652–2654 CrossRef.
- W. A. Herrmann, A. M. J. Rost, J. K. M. Mitterpleininger, N. Szesni, S. Sturm, R. W. Fischer and F. E. Kühn, Angew. Chem., Int. Ed., 2007, 46, 7301–7303 CrossRef CAS PubMed.
- J. Rudolph, K. L. Reddy, J. P. Chiang and K. B. Sharpless, J. Am. Chem. Soc., 1997, 119, 6189–6190 CrossRef CAS.
- P. Ferreira, W. Xue and É. Bencze, Inorg. Chem., 2001, 5834–5841 CrossRef CAS PubMed.
- M.-D. Zhou, J. Zhao, J. Li, S. Yue, C.-N. Bao, J. Mink, S.-L. Zang and F. E. Kühn, Chem. – Eur. J., 2007, 13, 158–166 CrossRef CAS PubMed.
- Z. Xu, M.-D. Zhou, M. Drees, H. Chaffey-Millar, E. Herdtweck, W. A. Herrmann and F. E. Kühn, Inorg. Chem., 2009, 48, 6812–6822 CrossRef CAS PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, USA, 2000 Search PubMed.
- J. R. Dilworth and S. J. Parrott, Chem. Soc. Rev., 1998, 27, 43 RSC.
- J. Ho, W. Y. Lee, K. J. T. Koh, P. P. F. Lee and Y.-K. Yan, J. Inorg. Biochem., 2013, 119, 10–20 CrossRef CAS PubMed.
- J. Zhang, J. J. Vittal, W. Henderson, J. R. Wheaton, I. H. Hall, T. S. A. Hor and Y. K. Yan, J. Organomet. Chem., 2002, 650, 123–132 CrossRef CAS.
- S. Jürgens, W. A. Herrmann and F. E. Kühn, J. Organomet. Chem., 2014, 751, 83–89 CrossRef PubMed.
- G. Laurenczy, F. Lukács, R. Roulet, W. A. Herrmann and R. W. Fischer, Organometallics, 1996, 15, 848–851 CrossRef CAS.
- S. M. Nabavizadeh, A. Akbari and M. Rashidi, Dalton Trans., 2005, 2423–2427 RSC.
- B. Jastorff, K. Mölter, P. Behrend, U. Bottin-Weber, J. Filser, A. Heimers, B. Ondruschka, J. Ranke, M. Schaefer, H. Schroder, A. Stark, P. Stepnowski, F. Stock, R. Stormann, S. Stolte, U. Welz-Biermann, S. Ziegert, J. Thoming, H. Schröder, R. Störmann and J. Thöming, Green Chem., 2005, 7, 362–372 RSC.
- J. Arning, S. Stolte, A. Böschen, F. Stock, W.-R. Pitner, U. Welz-Biermann, B. Jastorff and J. Ranke, Green Chem., 2008, 10, 47 RSC.
- M. Matzke, S. Stolte, J. Arning, U. Uebers, J. Filser and U. Arning, Green Chem., 2008, 10, 584 RSC.
- J. Filser, D. Arndt, J. Baumann, M. Geppert, S. Hackmann, E. M. Luther, C. Pade, K. Prenzel, H. Wigger, J. Arning, M. C. Hohnholt, J. Köser, A. Kück, E. Lesnikov, J. Neumann, S. Schütrumpf, J. Warrelmann, M. Bäumer, R. Dringen, A. von Gleich, P. Swiderek and J. Thöming, Nanoscale, 2013, 5, 1034–1046 RSC.
- J. Ranke, K. Mölter, F. Stock, U. Bottin-Weber, J. Poczobutt, J. Hoffmann, B. Ondruschka, J. Filser and B. Jastorff, Ecotoxicol. Environ. Saf., 2004, 58, 396–404 CrossRef CAS.
- W. Drost, M. Matzke and T. Backhaus, Chemosphere, 2007, 67, 36–43 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef CAS PubMed.
- S. K. Knauer, B. Unruhe, S. Karczewski, R. Hecht, V. Fetz, C. Bier, S. Friedl, B. Wollenberg, R. Pries, N. Habtemichael, U.-R. Heinrich and R. H. Stauber, Hum. Mutat., 2013, 34, 395–404 CrossRef CAS PubMed.
- C. Bier, S. K. Knauer, D. Wünsch, L. Kunst, S. Scheiding, M. Kaiser, C. Ottmann, O. H. Krämer and R. H. Stauber, FASEB J., 2012, 26, 3421–3429 CrossRef CAS PubMed.
- V. Fetz, S. K. Knauer, C. Bier, J. P. von Kries and R. H. Stauber, Sensors, 2009, 9, 5423–5445 CrossRef CAS PubMed.
- P. P. Silva, W. Guerra, G. C. Dos Santos, N. G. Fernandes, J. N. Silveira, A. M. da Costa Ferreira, T. Bortolotto, H. Terenzi, A. J. Bortoluzzi, A. Neves and E. C. Pereira-Maia, J. Inorg. Biochem., 2014, 132, 67–76 CrossRef CAS PubMed.
- K. Musilek, J. Roder, M. Komloova, O. Holas, M. Hrabinova, M. Pohanka, V. Dohnal, V. Opletalova, K. Kuca and Y.-S. Jung, Bioorg. Med. Chem. Lett., 2011, 21, 150–154 CrossRef CAS PubMed.
- J. J. Arning, M. Matzke, S. Stolte, F. Nehen, U. Bottin-Weber, A. Böschen, S. Abdulkarim, B. Jastorff and J. Ranke, Chem. Res. Toxicol., 2009, 22, 1954–1961 CrossRef CAS PubMed.
- E. P. Rogakou, C. Boon, C. Redon and W. M. Bonner, J. Cell Biol., 1999, 146, 905–916 CrossRef CAS.
- T. T. Paull, E. P. Rogakou, V. Yamazaki, C. U. Kirchgessner, M. Gellert and W. M. Bonner, Curr. Biol., 2000, 10, 886–895 CrossRef CAS.
- L. B. Schultz, N. H. Chehab, A. Malikzay and T. D. Halazonetis, J. Cell Biol., 2000, 151, 1381–1390 CrossRef CAS.
- Y. Xiong, J. Xu, W. Shan, Z. Lou, D. Fang, S. Zang and G. Han, Bioresour. Technol., 2013, 127, 464–472 CrossRef CAS PubMed.
- C. Tzvetkova and O. Bozhkov, in Recent Advances in Environment, Ecosystems and Develpment, World Scientific and Engineering Academy and Society, 2009, pp. 123–126 Search PubMed.
- K. Tagami and S. Uchida, Appl. Radiat. Isot., 2004, 61, 1203–1210 CrossRef CAS PubMed.
- K. Tagami and S. Uchida, in Encyclopedia of Inorganic Chemistry, John Wiley & Sons, Ltd, 2006 Search PubMed.
- J. Hattink, A. V. Harms and J. J. M. de Goeij, Sci. Total Environ., 2003, 312, 59–65 CrossRef CAS.
- E. R. Maset, S. H. Sidhu, A. Fisher, A. Heydon, P. J. Worsfold, A. J. Cartwright and M. J. Keith-Roach, Environ. Sci. Technol., 2006, 40, 5472–5477 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4gc01919a |
| This journal is © The Royal Society of Chemistry 2015 |