
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improved environmental stability of organic lead trihalide perovskite-based photoactive-layers in the presence of mesoporous TiO2†
Flannan T. F.
O'Mahony
a,
Yong Hui
Lee
b,
Cameron
Jellett
a,
Stoichko
Dmitrov
a,
Daniel T. J.
Bryant
a,
James R.
Durrant
a,
Brian C.
O'Regan
a,
Michael
Graetzel
b,
Mohammad K.
Nazeeruddin
b and
Saif A.
Haque
*a
aDepartment of Chemistry and Centre for Plastic Electronics, Imperial College London, South Kensington Campus, SW7 2AZ, UK. E-mail: s.a.haque@imperial.ac.uk; Tel: +44 (0)207 5941886
bLaboratory of Photonics and Interfaces, Swiss Federal Institute of Technology (EPFL), Station 6, Lausanne, CH 1015, Switzerland
First published on 13th March 2015
Abstract
Impressive hybrid photovoltaic device performances have been realised with the methylammonium lead triiodide (MAPbI3) perovskite absorber in a wide range of device architectures. However, the question as to which of these systems represents the most commercially viable long-term prospect is yet to be answered conclusively. Here, we report on the photoinduced charge transfer processes in MAPbI3 based films measured under inert and ambient conditions. When exposed to ambient conditions, the coated mesoporous Al2O3 and bilayer systems show a rapid and significant degradation in the yield of long-lived charge separation. This process, which does not affect sensitized-mesoporous TiO2 films, is only found to occur when both light and oxygen are present. These observations indicate that the presence of a mesostructured TiO2 electron acceptor to rapidly extract the photoexcited electron from the perovskite sensitizer may be crucial for fundamental photovoltaic stability and significantly increases innate tolerance to environmental conditions. This work highlights a significant advantage of retaining mesoscale morphological control in the design of perovskite photovoltaics.
Organic lead halide perovskites are currently generating enormous interest and excitement with respect to their application in low-cost, solution-processable photovoltaics. Whilst impressively high power conversion efficiencies have been achieved with both methylammonium lead triiodide and mixed halide absorbers, the architectural versatility of these materials has proven to be truly remarkable, with efficiencies exceeding 10% achieved in a wide range of configurations, whether as a sensitiser for mesoporous TiO2 (mp-TiO2),1 a structured absorber and electron transport material on a mesoporous Al2O3 scaffold (mp-Al2O3),2,3 or in a planar thin-film configuration.4,5 However, this versatility also encourages new questions to be posed; namely, what are the merits and shortcomings of the respective systems and which architecture is most promising in terms of long-term commercial viability? To consider these issues, and in the hope of improving understanding of the way in which the different systems work, we have used visible pump, visible-NIR probe transient absorption spectroscopy to compare and contrast the interfacial charge separation processes at the operational heart of device function for the main MAPbI3-based architectures. To date, few studies have directly addressed such questions, although recent work has explored the effect of UV light assisted degradation of mp-TiO2/MAPbI3 based devices.6 Whilst the problems associated with UV irradiation can be readily addressed by adding a UV blocking filter within the device structure, in the present study we focus on the effect of visible light irradiation – so crucial to effective photocurrent generation – on the environmental stability of the MAPbI3 based photoactive layers.
Fig. 1 shows the possible interfacial charge transfer processes occurring in the MAPbI3 based photoactive layers studied herein. When used as a sensitizer for mesoporous TiO2, electron injection has been reported to take place from the perovskite absorber to the metal oxide,7 in similar fashion to the dye-sensitised solar cell (DSSC).8,9 Subsequently, hole-transfer from the sensitiser to the hole transport material (HTM, typically spiro-OMeTAD) occurs, thus regenerating the absorber ground state. When TiO2 is replaced with an otherwise analogous Al2O3 mesostructure, electron injection is blocked by virtue of the high-energy metal oxide conduction band. In this case, interfacial charge separation is effected by hole transfer to the HTM and electrons are transported through the perovskite absorber.3,10 For the non-structured thin film assembly, as for the Al2O3-based system, no electron injection takes place, but photogenerated holes are likely to have further to travel before reaching the interface with spiro-OMeTAD and separating across the heterojunction.
 | ||
| Fig. 1 Schematic to illustrate charge photogeneration in various device architectures incorporating MAPbI3 as an absorber. | ||
The requirement that hole transfer takes places from MAPbI3 to spiro-OMeTAD is ultimately common to all three systems. As such, the transient existence of photooxidised spiro-OMeTAD, which is known to absorb strongly in the near-infrared,3,11 is a valuable indicator of the generation of potential useful charges. Fig. 2 shows the transient absorption spectra of an mp-TiO2/MAPbI3/spiro-OMeTAD film (Fig. 2a) and the equivalent mp-Al2O3-based system (Fig. 2b), recorded 10 μs after a 567 nm laser excitation. Both spectra reflect the characteristic absorption profile of oxidised spiro-OMeTAD, as shown by the similarity of shape with the absorption spectrum of a chemically oxidised solution of spiro-OMeTAD (blue dashed line in Fig. 2a). Additionally, strong quenching of the MAPbI3 emission is observed when the HTM is introduced on both mp-TiO2 and mp-Al2O3 substrates (Fig. S1 – ESI†). These observations are consistent with the theory that photoinduced hole-transfer from the perovskite absorber to the hole transport material (HTM) can occur independently of electron injection and importantly, that the transient products of the reaction absorb strongly at 1600 nm.10 We note that when no spiro-OMeTAD is present and the pristine metal oxide/MAPbI3 films are measured, no transient absorption signal is recorded at 1600 nm further indicating that the signal at this probe wavelength is due to the transient existence of photooxidised spiro-OMeTAD. Therefore, the intensity of transient absorption at 1600 nm for MAPbI3/spiro-OMeTAD-based assemblies provides a measure of the relative yield of charge separation.
 | ||
| Fig. 2 Transient absorption spectra of (a) TiO2/MAPbI3/spiro-OMeTAD and (b) Al2O3/MAPbI3/spiro-OMeTAD films. The blue trace in (a) represents the absorption spectrum of a solution of spiro-OMeTAD that has been chemically oxidised with N(PhBr)3SbCl6. Laser excitation energy density = 25 μJ cm−2 at 567 nm. | ||
In Fig. 3a, the yield of long-lived photogenerated spiro-OMeTAD cations ([spiro-OMeTAD]+) is monitored as a function of time after pulsed laser excitation by measuring the change in absorption (ΔOD) at 1600 nm for mp-TiO2/MAPbI3/spiro-OMeTAD (black), mp-Al2O3/MAPbI3/spiro-OMeTAD (red) and bilayer MAPbI3/spiro-OMeTAD (blue). The decay in signal represents the recombination of the charge-separated states. In accordance with the work of Marchioro et al.,11 a longer charge separation lifetime is seen for the mp-TiO2-based assembly (τ1/2 (1 μs) = 1 ms) than for the mp-Al2O3 analogue (τ1/2 (1 μs) = 100 μs). This is consistent with holes transferred to spiro-OMeTAD recombining more quickly with electrons in the MAPbI3 (as on the Al2O3 scaffold) than with those in transferred to TiO2. Charge separation at the pristine TiO2/MAPbI3 interface is supported by the quenching of MAPbI3 photoluminescence relative to the analogous Al2O3-based system (Fig. S1 – ESI†). It is pertinent to note that the lifetime of spiro-OMeTAD holes in the planar-junction type bilayer assembly appears almost identical to those in the mp-Al2O3 system. However, the differing dynamics of recombination (TiO2versus Al2O3 and planar geometry based samples) may be a result of increased spatial separation between electrons and holes when the photogenerated electrons on the MAPbI3 are transferred to the TiO2 (as opposed to remaining in the MAPbI3), which would be expected to retard the back electron transfer process. In essence, the longer charge separated state lifetime in the mp-TiO2/MAPbI3/spiro-OMeTAD (as compared to mp-Al2O3/MAPbI3/spiro-OMeTAD) is indicative of electron injection at the mp-TiO2/MAPbI3 heterojunction.
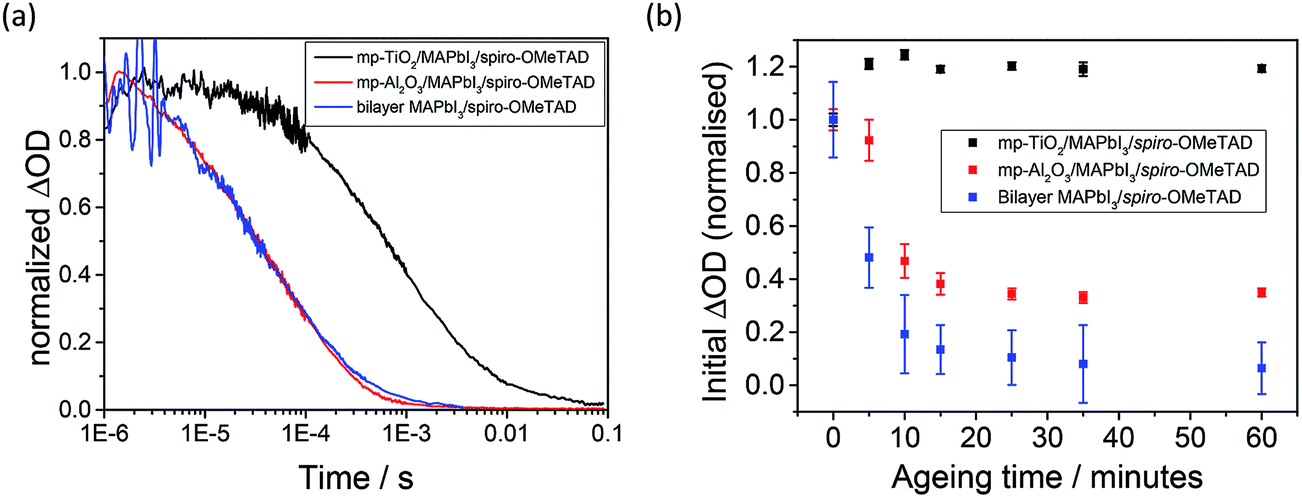 | ||
| Fig. 3 (a) Time-resolved transient absorption at 1600 nm for pristine mp-TiO2/MAPbI3/spiro-OMeTAD (black), mp-Al2O3/MAPbI3/spiro-OMeTAD (red) and bilayer MAPbI3/spiro-OMeTAD (blue) films. All traces normalized to ΔOD = 1 at 1 μs to highlight differences in recombination kinetics. (b) Relative yield of long-lived charge separation as a function of ageing time in atmospheric conditions under ambient light. Data points determined from the mean amplitude of ΔOD at 1600 nm between 2 and 5 μs for each measurement. All films were excited at 567 nm with a 25 μJ cm−2 laser pulse. | ||
We now consider the relative yield of charge separation (i.e. long-lived [spiro-OMeTAD]+) in samples exposed to ambient air. In these studies, the transient absorption signal at 1600 nm was obtained at regular time intervals over the course of 1 hour, with the films exposed to ambient light and air (but not the laser pulse or probe light) in the interim periods. The transient absorption measurements themselves were conducted with the sample sealed under flowing nitrogen. Fig. 3b shows the initial amplitude of ΔOD (averaged between 2 and 5 μs) as a function of ageing time. Raw data (ΔOD versus time as function of ageing) are presented in the ESI (Fig. S3†). On TiO2 (Fig. 3b, black markers) the transient absorption signal remains relatively consistent over the ageing process, barring a small increase in intensity after a short period of exposure to atmospheric conditions. For the bilayer and mp-Al2O3-based films, the yield of long-lived spiro-OMeTAD holes decreases dramatically with ageing time, (red and blue data sets in Fig. 3b for Al2O3 and bilayer systems, respectively). This suggests that hole transfer is rapidly and critically inhibited by the exposure of these systems to environmental conditions. Importantly, this process is not accompanied by any noticeable reduction in steady-state absorption in the near IR region where the perovskite sensitizer exhibits a strong absorption (Fig. S3 – ESI†), which would be expected if the water-soluble perovskite sensitiser were simply interacting with moisture in the air.
In order to try and identify what causes the decay in the yield of long-lived charge separation for Al2O3-based and bilayer systems, the effects of film ageing in different environmental conditions are investigated. Fig. 4 shows the time-resolved transient absorption signals for an Al2O3/MAPbI3/spiro-OMeTAD at 1600 nm before (black) and after (pink) 90 minutes of ageing in controlled conditions (where (a) is exposed to ambient light and sealed under flowing N2, (b) is in light and ambient air, (c) is in light and dry air (H2O < 2 vpm) and (d) is in the dark and ambient air). The data presented in Fig. 4 shows that the degradation of hole-transfer also takes place in dry air with light, but not in the dark. Similarly, the yield of long-lived charge separation is not affected when the samples are stored in nitrogen, whether it be in the dark or under illumination. It can thus be concluded that the degradation process is light-induced and as it is independent of moisture, is likely to be due to reaction of the perovskite sensitizer with oxygen in the air. It is noteworthy that for the Al2O3-based system, the observed degradation process is also accompanied by an increase in transient lifetime. It is unclear as to whether this long-lived component reflects an increase in the lifetime of the spiro-OMeTAD holes, or represents the contribution of a product of the parasitic reaction. The possible origins of these effects are discussed below.
 | ||
| Fig. 4 Effect of exposure to varying environmental conditions for two hours on the transient absorption signal at 1600 nm for a mesoporous Al2O3/MAPbI3/spiro-OMeTAD film: (a) ambient light, sealed under flowing N2, (b) ambient light, non-sealed, (c) ambient light, sealed under flowing dry air (H2O < 2 vpm), (d) dark, non-sealed. Laser excitation energy density = 25 μJ cm−2 at 567 nm. | ||
To further investigate the impact of air and light exposure on Al2O3 samples, femtosecond transient absorption spectroscopy of an mp-Al2O3/MAPbI3/spiro-OMeTAD film was carried out both in inert conditions and in the presence of ambient air and light (Fig. 5). Femtosecond transient absorption spectroscopy has previously been used to study the charge dynamics in various perovskite films with and without hole and electron blocking layers.7,11–14 In the present study, similar transient spectra in the visible to near infrared were obtained for the pristine sample to those reported previously, including in particular a strong negative signal peaking at 780 nm. This feature has recently assigned to ground-state bleaching caused by phase space filling, as well as a possible contribution from stimulated emission,10,12,13 and is therefore an indicator as to the occupation of the initial perovskite excited state. Fig. 5 presents the decay kinetics of this transient absorption signal at 780 nm. It is apparent that the decay dynamics of this feature and therefore the carrier lifetimes in the pristine mp-Al2O3/MAPbI3/spiro-OMeTAD film are significantly longer than those observed once the film has been exposed to air and ambient light. The shortened decay dynamics in the presence of air and light therefore suggest the existence of an alternative quenching mechanism that can compete with the hole transfer process and which may be associated with the decomposition process. Further studies into the exact mechanism of decomposition of the Al2O3 based films in the presence of light and air are currently underway and will be reported in due course.
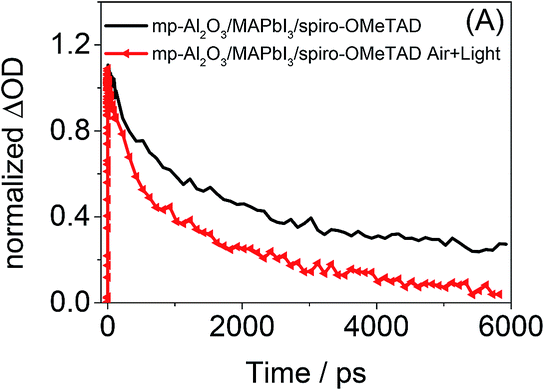 | ||
| Fig. 5 Time-resolved transient absorption at 780 nm for pristine mp-Al2O3/MAPbI3/spiro-OMeTAD (black) and exposed to air and ambient light MAPbI3/spiro-OMeTAD (red) films. Traces normalized to ΔOD = 1 at 1 ps time-delay to highlight differences in recombination kinetics. Laser excitation energy density = 25 μJ cm−2 at 567 nm. | ||
The next question that arises relates to why degradation occurs on Al2O3 and in a bilayer, but not on mesoporous TiO2. At present, however, we can only speculate on the origins of these observations. The most fundamental difference between using a mesoporous TiO2 electrode and an insulating scaffold is that in the former case, the TiO2 is able to accept photoinduced electrons from the sensitizer.7 Conversely, when no electron acceptor is present, photogenerated electrons remain within the sensitizer layer until they either recombine, or in a full device, reach a contact and are extracted. For DSSC's, from which the present perovskite systems have evolved, it has long been established that extending the lifetime of the photoexcited (or photoinduced) electrons within the sensitizer material increases the probability of destructive side-reactions taking place and can pose serious stability problems.15 Considering one example, Kohle et al., have shown that the N3 dye is fundamentally unstable under extended illumination in ethanolic solution, as the photoexcited electron reacts with oxygen and causes system to degrade.16 However, when anchored to an electron accepting TiO2 surface, rapid deactivation of the dye-excited state via interfacial electron injection precludes the parasitic side reaction and the assembly displays remarkable photostability. It is therefore reasonable to suppose that the increased lifetime of the photoexcited electron within the perovskite experienced in the Al2O3-based and bilayer systems could increase the probability of reaction with external species, such as molecular oxygen. Moreover, it is possible that a reaction between the photogenerated electron on the perovskite absorber and molecular oxygen could take place resulting in photodegradation of the MAPbI3 pervoskite absorber leading to products that hinder interfacial charge transfer. For example, electron reduction of molecular oxygen generating superoxide has been widely reported to result in photo-degradation of molecular materials. As such, one possibility is that the photo-degradation of MAPbI3 (as a result of exposure to light and oxygen) leads to the formation of lead oxide or lead iodide. Support for this hypothesis comes from consideration of the absorption spectra for Al2O3 and planar-junction based perovskite systems before and after ageing in air. It is evident from the data presented in Fig. S3† that the ageing of both the Al2O3 and planar junction systems is accompanied by an increase in the absorption between 450–550 nm. This increase in absorption could be due to the presence lead iodide as this material is known to have a strong absorption in this region.17 Furthermore, if the lead iodide were to form at the interface between the spiro-MeOTAD and MAPbI3 (as a result of a photodegradation reaction) it could function as a blocking layer reducing the yield of charge separation as observed here. Further, more detailed investigations, addressing the mechanism of the degradation processes are currently underway and will be reported in due course. Additionally, at present, it is unclear as to whether the presence of the TiO2 electron acceptor completely eliminates this degradation pathway or simply delays it; this question is currently the focus of further studies.
An important consequence of the sensitivity of the non-electron accepting architectures to ambient conditions appears to be a significantly reduced tolerance to processing environment. All of the films considered thus far in the present work have been prepared by spin-coating and annealing the MAPbI3 layer in a nitrogen glove box. When the Al2O3/MAPbI3/spiro-OMeTAD and bilayer films are prepared in ambient conditions, transient absorption spectroscopy of the as-new films already shows significant signs of decay (Fig. S4 – ESI†). In contrast, the equivalent TiO2-based films are more tolerant to processing conditions and even show signs of improvement when prepared in air (Fig. S4 – ESI†).
In order to test the practical implications of these effects, preliminary devices studies were undertaken. The results obtained from these studies along with the relevant experimental details are presented in Fig. S5.† Briefly, the devices were fabricated in a glove box (inert atmosphere). After fabrication, the devices were removed from the glove box and aged in dry air and ambient light prior to solar cell efficiency measurements. We note that the solar cell efficiency measurements were performed on unsealed devices. Taken together, our solar cell results support the assertion that mp-TiO2/MAPI3 based cells are more stable to light and air exposure than those based on mp-Al2O3/MAPI3 cell architectures. It is apparent from the data presented in Fig. S5† that exposure of the mp-Al2O3/MAPI3/spiro-MeOTAD devices to dry air and light for just 72 hours results in a significant drop in the device power conversion efficiency (∼50%). In contrast, the analogous mp-TiO2 devices exhibit a smaller drop in efficiency (∼25%) when exposed to dry air and light for the same length of time. We note that the drop in performance of the mesoporous TiO2 devices is most likely due to a fraction of the perovskite film behaving in a similar manner to the perovskite in the Al2O3 devices and not injecting electrons in the mesoporous TiO2. Moreover, further evidence for an enhanced environmental stability of mp-TiO2 based devices comes from our recently published work (ref. 18). Specifically, we observed that sealed cells with mp-TiO2/MAPI3/spiro-MeOTAD photoactive layers can survive 60 hours under 40 suns irradiation with only ≤10% decrease in Jsc.18
In summary, transient absorption spectroscopy has been used to probe yields and lifetimes of photoinduced, long-lived charge separation in the three major architectural variations of MAPbI3-sensitised solar cells. It is confirmed that hole transfer to spiro-OMeTAD takes place in all cases and can therefore occur independently of electron extraction. When exposed to ambient conditions, the coated mp-Al2O3 and bilayer systems show a rapid and significant degradation in the yield of long-lived charge separation. This process, which does not affect sensitized mesoporous TiO2 films, is only found to occur when both light and oxygen are present. It is therefore thought to be a result of parasitic reactions taking place between the photogenerated electron on the perovskite absorber and molecular oxygen. Investigations of functioning photovoltaic devices indicate that the mesoporous TiO2 based devices are more stable than the analogous Al2O3 based devices. Moreover, for the non mp-TiO2 based systems, a profound sensitivity to ambient conditions can be expected to levy a costly toll on the economy of large-scale device production, whilst also imposing the need for more stringent and expensive standards of encapsulation. This work indicates a potential benefit of retaining mesoscale morphological control in hybrid photovoltaics, even when it is not required for the achievement of initially high intrinsic yields of charge separation.
Methods
Sample preparation
Mesoporous TiO2 films were deposited on glass microscope slides from Ti-nanoxide T37 paste (37 nm particles, Solaronix) by spin coating and sintering at 450 °C. Resultant films were measured to be ∼400 nm in thickness by profilometry. Mesoporous Al2O3 films of same thickness were prepared in a similar fashion. Methylammonium lead trihalide (MAPbI3) perovskite-based films for transient absorption spectroscopy studies reported herein were prepared following a previously published procedure (ref. 9). All samples were prepared in a glove box (controlled moisture and O2 environment) apart from the studies presented in ESI Fig. S3.† Where relevant, spiro-OMeTAD was spin coated from a chlorobenzene solution with no lithium salt or tert-butyl pyridine (TBP) additives.Optical characterization
Steady-state absorption spectra were obtained using a Perkin Elmer Lambda 25 UV-vis spectrometer (absorbance measurements). Transmission and reflectance spectra (to calculate %absorption) were measured on a Perkin-Elmer Lambda 750 UV-vis-n-IR absorption spectrometer fitted with an integrating sphere. Photoluminescence emission spectra were measured at room temperature using a Horiba Jobin Yvon Fluorolog-3 spectrofluorimeter.Transient absorption spectroscopy
For pump-probe micro to millisecond transient absorption spectroscopy, films were excited by a dye laser (Photon Technology International GL-301, sub-nanosecond pulse width) pumped by a pulsed nitrogen laser (Photon Technology International GL-3300). A quartz halogen lamp (Bentham IL1) was passed through a monochromator and used to probe changes in the absorption characteristics of the film as a function of time after the laser excitation. The probe light was detected using home-built silicon (≤1000 nm) or InxGa1−xAs (>1000 nm) photodiodes and an oscilloscope. Unless otherwise stated, films were kept under flowing N2 during the measurements. All micro to millisecond transient absorption spectroscopy measurements were conducted employing 567 nm laser pulses (25 μJ cm−2). Femtosecond transient absorption spectroscopy: mesoporous Al2O3/MAPbI3/spiro-OMeTAD films were characterised using commercially available broadband transient absorption spectrometer HELIOS (Ultrafast systems), seeded with a 1 kHz, 800 nm, 90 femtosecond train of pulses generated by a Ti:sapphire regenerative amplifier Solstice (Newport Ltd). Films were excited with 567 nm, 25 μJ cm−2 pulses, the output of an optical parametric amplifier TOPAS (light conversion).Acknowledgements
S. A. H acknowledges financial support from the Engineering and Physical Science Research Council (EPSRC) through the UK-India (EP/H040218/2). S. A. H, M. K. N & M. G. acknowledge financial support acknowledge financial support from the European Community's Seventh Framework Programme (NanoMatcell, grant agreement no. 308997).References
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Gratzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- D. Bi, S.-J. Moon, L. Haggman, G. Boschloo, L. Yang, E. M. J. Johansson, M. K. Nazeeruddin, M. Graetzel and A. Hagfeldt, RSC Adv., 2013, 3, 18762–18766 RSC.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395–398 CrossRef CAS PubMed.
- O. Malinkiewicz, A. Yella, Y. H. Lee, G. Minguez Espallargas, M. Graetzel, M. K. Nazeeruddin and H. J. Bolink, Nat. Photonics, 2014, 8, 128–132 CrossRef CAS.
- T. Leijtens, G. E. Eperon, S. Pathak, A. Abate, M. M. Lee and H. J. Snaith, Nat. Commun., 2013, 4, 2885 Search PubMed.
- C. S. Ponseca Jr, T. J. Savenije, M. Abdellah, K. Zheng, A. Yartsev, T. Pascher, T. Harlang, P. Chabera, T. Pullerits, A. Stepanov, J.-P. Wolf and V. Sundstrom, J. Am. Chem. Soc., 2014, 136, 5189–5192 CrossRef PubMed.
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef.
- A. Listorti, B. O'Regan and J. R. Durrant, Chem. Mater., 2011, 23, 3381–3399 CrossRef CAS.
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Graetzel and N.-G. Park, Sci. Rep., 2012, 2, 591 Search PubMed.
- A. Marchioro, J. Teuscher, D. Friedrich, M. Kunst, R. van de Krol, T. Moehl, M. Graetzel and J.-E. Moser, Nat. Photonics, 2014, 8, 250–255 CrossRef CAS.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Graetzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS PubMed.
- F. Deschler, M. Price, S. Pathak, L. E. Klintberg, D.-D. Jarausch, R. Higler, S. Hüttner, T. Leijtens, S. D. Stranks, H. J. Snaith, M. Atatüre, R. T. Phillips and R. H. Friend, J. Phys. Chem. Lett., 2014, 5, 1421–1426 CrossRef CAS.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- M. Gratzel, C. R. Chim., 2006, 9, 578–583 CrossRef PubMed.
- O. Kohle, M. Gratzel, A. F. Meyer and T. B. Meyer, Adv. Mater., 1997, 9, 904–906 CrossRef CAS.
- Y. Wu, A. Islam, X. Yang, C. Qin, J. Liu, K. Zhang, W. Peng and L. Han, Energy Environ. Sci., 2014, 7, 2934–2938 CAS.
- C. Law, L. Miseikis, S. Dimitrov, P. Shakya-Tuladhar, X. Li, P. R. F. Barnes, J. Durrant and B. C. O'Regan, Adv. Mater., 2014, 26, 6268–6273 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ta01221j |
| This journal is © The Royal Society of Chemistry 2015 |