
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Furosemide's one little hydrogen atom: NMR crystallography structure verification of powdered molecular organics†
Cory M.
Widdifield
,
Harry
Robson
and
Paul
Hodgkinson
*
Department of Chemistry, University of Durham, Durham DH1 3LE, UK. E-mail: paul.hodgkinson@durham.ac.uk
First published on 26th April 2016
Abstract
The potential of NMR crystallography to verify molecular crystal structures deposited in structural databases is evaluated, with two structures of the pharmaceutical furosemide serving as examples. While the structures differ in the placement of one H atom, using this approach, we verify one of the structures in the Cambridge Structural Database using quantitative tools, while establishing that the other structure does not meet the verification criteria.
Significant progress has been made in solid-state nuclear magnetic resonance (SSNMR). With the development of high applied magnetic fields1 and fast magic-angle spinning (MAS) probes,2,3 studies of chemical systems that would have been intractable using SSNMR as recently as 10 years ago are becoming routine. An area of consistent recent development is ‘NMR crystallography’.4–6 As the name suggests, it offers insights into the structures of crystalline materials, but is distinct from diffraction methods as it uses SSNMR experiments to collect system data. Alongside the SSNMR advances, developments in density functional theory (DFT) software operating under periodic boundary conditions that use pseudopotentials to describe core electron states and plane waves to describe the valence electrons have enabled the efficient computation of NMR parameters for periodic systems.4,7,8 When relativistic effects may be ignored, the accuracy of these quantum chemical tools is such that, when paired with SSNMR data, this combined approach has been able to refine and determine molecular crystal structures in lieu of diffraction,9–11 although this approach is not yet a general method for crystal structure determinations.
Diffraction-based crystal structure determination methods have been crucial in the advancement of chemistry.12 Crystal structure databases, such as the Cambridge Structural Database (CSD), are important repositories of crystal structures determined using diffraction methods. While metrics are available to the crystallographic community to quantify the quality of a crystal structure refinement (e.g., R-factors), systematic studies of the CSD have shown that a significant portion of deposited structures possess unrealistic structural features.13–15 Independent verification of crystal structures determined using diffraction methods could be of significant general value. Indeed, recent work by van de Streek and Neumann has shown that dispersion-corrected DFT (dc-DFT) offers the potential to verify crystal structures by observing the changes in the non-H atomic positions under a full structural relaxation.16 However, due to the difficulties in locating hydrogen atom positions by X-ray diffraction (XRD), prior studies have not included them in their verification procedures.
In principle, new single-crystal XRD (scXRD) experiments may be performed to confirm a crystal structure, although this can be a significant challenge for systems that do not readily form sufficiently large single crystals. Secondly, the use of XRD methods to verify XRD structures, e.g. by comparison against established distributions of interatomic distances, is obviously cyclical in nature. NMR crystallography methods can address both these aspects. As NMR experiments are sensitive local probes of structure and dynamics, they probe matter in a different fashion than XRD, and do not require single crystal samples. Based upon the successes of powder NMR crystallography for structure determinations,2,9–11 refinements17 and distinctions,18,19 NMR may have an important role in the verification of crystal structures deposited in chemical structure databases.
As a test case of NMR crystallography methods to verify crystal structures, we consider furosemide, which is an important pharmaceutical listed in the World Health Organization's List of Essential Medicines.20 Furosemide has therapeutic applications related to relieving fluid accumulation in the heart, liver and kidney due to the partial or total failure of these organs,21 and in treating hypertension.22 Due to its poor bioavailability, recent studies have focused on the synthesis of furosemide co-crystals,23–26 but prior SSNMR data are known for pure furosemide (form 1).24,27 There are several crystal structure determinations in the CSD for furosemide (ca. 10), which provides the opportunity to compare and contrast selected structures using NMR crystallography methods.
We highlight the structures of two determinations of furosemide (Fig. 1), referred to here using their CSD reference codes, FURSEM0128 and FURSEM17.29 The diffraction measurement temperatures were similar between both structures (ambient), and both have relatively low R-factors (6.8% and 5.7%, respectively). This eliminates complicating factors, such as temperature-dependent polymorphism and obviously poor structures (i.e., R-factor > 10%).30
 | ||
| Fig. 1 An overlay of the asymmetric units of FURSEM01 (C atoms in grey) and FURSEM17 (C atoms in green). H atoms are generally omitted, but a critical difference in the carboxyl H atom positioning is shown. Arrow head points toward the H position of FURSEM17. | ||
A review of furosemide structures in the CSD by Karami et al.31 used simulated powder XRD (pXRD) patterns to show that several deposited structures were essentially the same polymorphic form (form 1), and were most correctly described by FURSEM01. In contrast to the two other known polymorphic forms of furosemide (forms 2 and 3),32 the structure of FURSEM17 is very similar to FURSEM01, but contains a chemically non-intuitive hydrogen bonding motif where one of the COOH hydrogen atoms is directed away from forming an O–H⋯O dimer. The refined structures possess very similar unit cell dimensions and non-H atomic positions (non-H atomic root-mean-squared difference (RMSD) value of 0.015 Å for 16 overlaid molecules), with Z′ = 2. As the major difference between these crystal structures lies in the placement of a single hydrogen atom, (Fig. 1) the calculated pXRD patterns are nearly identical (ESI†), and thus pXRD cannot distinguish between these two structures. In contrast, NMR experiments are expected to be sensitive to this structural difference, given their local nature. However, it is unknown if the sensitivity to this single local difference is sufficient to distinguish between FURSEM01 and FURSEM17 over the total crystal structure.
We obtained powders of furosemide from two sources. The first was used as-is from Sigma-Aldrich (S-A), with phase purity verified by pXRD (ESI†). A second sample was obtained by following the re-crystallisation procedure advocated by the authors who obtained the structure of FURSEM17 (ReCryst). This yielded a sufficiently large single crystal to solve its crystal structure, which is referred to as FURSEM-NEW.‡
Fast 1H MAS, 13C and 15N cross-polarization (CP)/MAS, and 1H–13C refocused frequency-switched Lee-Goldburg dipolar heteronuclear correlation (FSLG-HETCOR) SSNMR experiments were used to achieve a reasonably complete assignment of the NMR peaks to sites in the furosemide molecules (Fig. 2 and ESI†). The present assignment is largely consistent with that of Longhi et al.,27 however the peak at ca. 138 ppm, assigned here to C–Cl groups, was apparently not observed in the prior account, leading to a misassignment. By using variable contact time FSLG-HETCOR experiments (ESI†), and analysing systematic trends in computed magnetic shielding values from gauge-including projector augmented-wave density functional theory (GIPAW DFT) calculations (ESI†),33 we could distinguish between nearly all of the H and C chemical sites in the two furosemide molecules of the asymmetric unit. Further 13C–13C correlation experiments could provide data for a complete assignment, but as the 1H spin–lattice relaxation value (T1) is over 40 s at room temperature (ESI†), this experiment is not feasible without 13C isotopic enrichment. Signal enhancement methods such as dynamic nuclear polarization (DNP),34,35 or the addition of paramagnetic dopants may also prove fruitful.
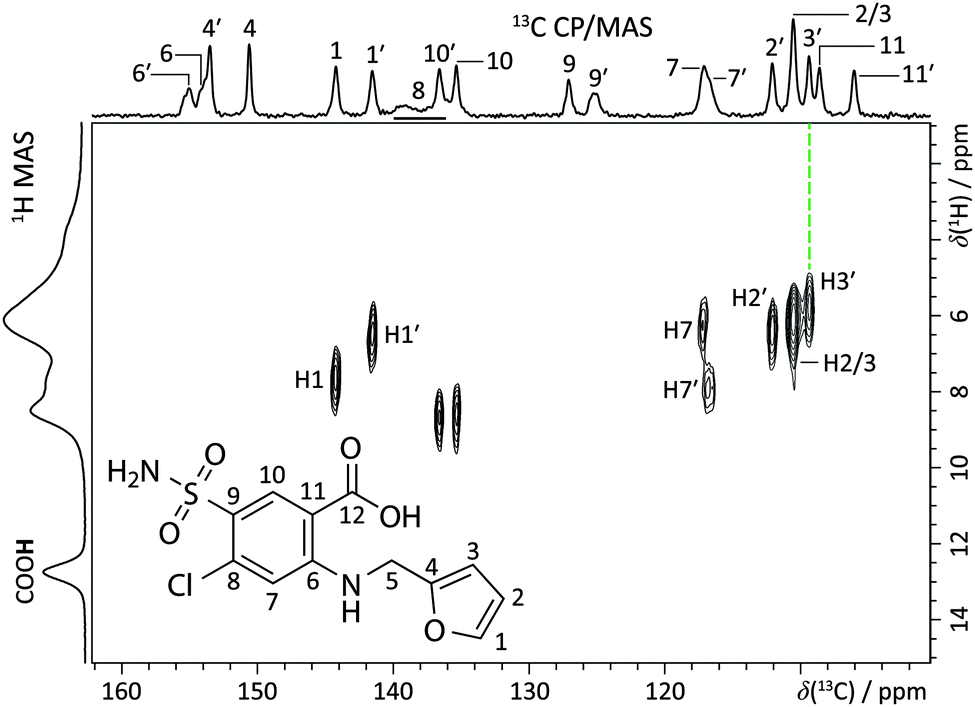 | ||
| Fig. 2 Partial 1H–13C refocused dipolar FSLG-HETCOR NMR spectrum (B0 = 11.7 T, T = 293(1) K; νMAS = 13.24 kHz, contact time = 100 μs) with site assignments for the furosemide molecule (inset, S-A sample). The top horizontal axis shows the 13C CP/MAS NMR spectrum, while the left vertical axis corresponds to the 60 kHz 1H MAS NMR spectrum of the same material. See ESI† for a detailed account of the assignment. | ||
The assignment facilitates the comparison between experimentally measured isotropic chemical shift values (δiso) with those calculated using GIPAW DFT, quantified through an RMSD. This contrasts with earlier work where differences in H atom positioning could be distinguished by simple qualitative comparisons of either 1H fast MAS spectra18 or 13C NMR spectra.19 The quantitative ensemble approach presented here has been used in NMR crystallography structure determinations of organics,9,10 but has not previously been used to verify hydrogen positioning. 1H RMSD values of 0.33 ± 0.16 ppm, and 13C RMSD values of 1.9 ± 0.4 ppm are ranges typical for valid molecular organic structures.§ Accordingly, the crystal structures of FURSEM01, FURSEM17, and FURSEM-NEW were H-optimised, followed by GIPAW DFT calculations using CASTEP.36–38 The calculated δiso values for 1H and 13C were compared against those measured experimentally to yield δiso RMSD values, Fig. 3(a and b). In Fig. 3c we compare the calculated 1H NMR spectra (see ESI,† for calculated vs. experimental correlation plots) for FURSEM01 and FURSEM17 with the experimental spectrum of S-A.
 | ||
| Fig. 3 RMSD values for δiso(1H) (a) and δiso(13C) (b) between experimentally measured δiso of S-A and ReCryst with those computed using GIPAW DFT on the H-optimised structures indicated. Grey bands correspond to RMSD ranges established using a series of benchmark organics.§ The 1H RMSD is highly sensitive to the COOH hydrogen atom position, as shown by RMSD values which include (blue) and do not include (red) this site. (c) compares calculated (FURSEM01/FURSEM17) and experimental (S-A) 1H NMR spectra. Red traces in (c) correspond to the carboxyl H atom from each crystallographically unique furosemide molecule. | ||
Although there is some disagreement when considering calculated and experimental δiso(1H) values for the OH sites in FURSEM01, this is not sufficient to invalidate its structure; as discussed in the ESI,† structure changes of a few pm can significantly reduce the 1H RMSD value. Importantly, the overall agreement between the calculated and experimental data is clearly much better for the structures of FURSEM01 and FURSEM-NEW, while the calculated values using the FURSEM17 structure are in poor agreement with experimental values. Further, the differences in the computed 1H and 13C RMSD values for the FURSEM01 and FURSEM17 structures allow them to be distinguished using NMR crystallography, with a significance of ca. 2σ in both 1H and 13C. For FURSEM01 and FURSEM-NEW however, the differences between both the atomic positions (non-H atomic RMSD over the unit cell = 0.08 Å), and the computed δiso values are minor, especially when noting that FURSEM-NEW was determined at 120 K, while FURSEM01 was determined at room temperature. As such these two structures cannot be distinguished by NMR crystallography, although the slightly lower RMSD values for FURSEM-NEW may reflect the lower temperature at which the data were acquired.39 Despite following the crystallisation procedure of the authors of FURSEM17, we do not arrive at the same crystal structure.
Due to the good agreement between calculation and experiment for FURSEM01 (RMSD(1H) = 0.46 ppm and RMSD(13C) = 2.01 ppm), we state that its structure (and that of FURSEM-NEW) is verified using NMR crystallography; however, we are unable to verify the structure of FURSEM17, as both associated RMSD values are quite large (RMSD(1H) = 0.77 ppm and RMSD(13C) = 2.78 ppm, Fig. 3(a and b)). The largest contributions to the 1H RMSD value of FURSEM17 (ESI†) are due to the 1H shift value for the COOH hydrogen from one of the two furosemide molecules (see the variation in the computed values in the spectra provided in Fig. 3c). This is consistent with the previously highlighted structural difference between FURSEM01 and FURSEM17. Omitting this datum from the 1H RMSD calculations significantly reduces the calculated 1H RMSD value for FURSEM17 (Fig. 3a, red columns).
Being unable to verify a crystal structure using NMR crystallography does not infer a structure is incorrect. Similarly, the approach we outline should complement existing verification methods, and does not serve as a replacement. We consider additional computational information to assess the likelihood that FURSEM17 is an isolable polymorph of furosemide. From the dc-DFT structure verification method of van de Streek and Neumann,16FURSEM01 is validated, as its non-H atomic positions, when subjected to a dc-DFT calculation that optimises the unit cell and all atoms, do not change significantly (non-H atomic RMSD for one unit cell = 0.093 Å, where deviations greater than 0.25 Å are considered significant). From a dc-DFT geometry optimisation, FURSEM17 is found to be close to a local energy minimum and hence a theoretically viable furosemide polymorph. However, the non-H atomic RMSD after optimisation is 0.327 Å over the unit cell, and so FURSEM17 should be flagged for closer inspection. NMR crystallography provides an obvious way to perform such an inspection according to the above dc-DFT protocol.
Finally, the DFT-calculated enthalpies for the fully-optimised structures of FURSEM01 and FURSEM17 are distinct, with the optimised structure of FURSEM17 being ca. 25 kJ mol−1 molecule−1 above FURSEM01 (ESI†). Based upon recent findings of the crystal structure prediction (CSP) community,40,41 such a high energy structure would be very unlikely to occur under typical conditions. Indeed, this would establish a new record for an energy difference between two non-conformational polymorphic forms by a margin of 7 kJ mol−1 molecule−1.
In conclusion, we have demonstrated the potential value of NMR crystallography for the verification of crystal structures. We verify the structure of FURSEM01via NMR crystallography, and show that this conclusion is consistent with other methods, such as energy calculations using dispersion-corrected DFT. However, for the crystal structure of FURSEM17, the DFT-computed chemical shift data is inconsistent with NMR experiments, and hence its structure is not verified. This conclusion is consistent with current protocols from the CSP community, and also when using the method of van de Streek and Neumann.16 We are also able to distinguish between the FURSEM01 and FURSEM17 structures with ca. 2σ confidence, which is noteworthy as the structures are essentially identical save for the position of one hydrogen atom. While the diffraction community has many available structure verification tools, the ability to ensure the quality of crystal structures in databases in a way which is independent of the chosen experimental method is potentially important, as these structures are often starting points when screening for new pharmaceuticals, testing computational methods, and designing new materials. We are refining and extending this protocol as part of a systematic study on how to verify crystal structures using NMR crystallography tools.
We gratefully acknowledge Dr Dmitry Yufit (Durham University crystallography service) for acquiring the scXRD data, and solving the crystal structure for FURSEM-NEW. Mr Gary Oswald (Durham) is acknowledged for acquiring pXRD data for S-A. This work benefitted from discussions within the context of the EPSRC-funded collaborative computational project for NMR crystallography (CCP-NC). We acknowledge Dr Jason C. Cole (Cambridge Crystallographic Data Centre) and Prof. Judith A. K. Howard (Durham) for helpful discussions and critical readings of the manuscript. This work is supported under EPSRC grant number EP/L012243/1.
Notes and references
- K. Hashi, S. Ohki, S. Matsumoto, G. Nishijima, A. Goto, K. Deguchi, K. Yamada, T. Noguchi, S. Sakai, M. Takahashi, Y. Yanagisawa, S. Iguchi, T. Yamazaki, H. Maeda, R. Tanaka, T. Nemoto, H. Suematsu, T. Miki, K. Saito and T. Shimizu, J. Magn. Reson., 2015, 256, 30–33 CrossRef CAS PubMed.
- V. Agarwal, S. Penzel, K. Szekely, R. Cadalbert, E. Testori, A. Oss, J. Past, A. Samoson, M. Ernst, A. Böckmann and B. H. Meier, Angew. Chem., Int. Ed., 2014, 53, 12253–12256 CrossRef CAS PubMed.
- R. Zhang, M. K. Pandey, Y. Nishiyama and A. Ramamoorthy, Sci. Rep., 2015, 5, 11810 CrossRef PubMed.
- NMR Crystallography, ed. R. K. Harris, R. E. Wasylishen and M. J. Duer, John Wiley & Sons Ltd, Chichester, West Sussex, United Kingdom, 2009 Search PubMed.
- C. Martineau, J. Senker and F. Taulelle, Annu. Rep. NMR Spectrosc., 2014, 82, 1–57 CrossRef.
- C. Martineau, Solid State Nucl. Magn. Reson., 2014, 63–64, 1–12 CrossRef CAS PubMed.
- C. Bonhomme, C. Gervais, F. Babonneau, C. Coelho, F. Pourpoint, T. Azaïs, S. E. Ashbrook, J. M. Griffin, J. R. Yates, F. Mauri and C. J. Pickard, Chem. Rev., 2012, 112, 5733–5779 CrossRef CAS PubMed.
- T. Charpentier, Solid State Nucl. Magn. Reson., 2011, 40, 1–20 CrossRef CAS PubMed.
- M. Baias, C. M. Widdifield, J.-N. Dumez, H. P. G. Thompson, T. G. Cooper, E. Salager, S. Bassil, R. S. Stein, A. Lesage, G. M. Day and L. Emsley, Phys. Chem. Chem. Phys., 2013, 15, 8069–8080 RSC.
- M. Baias, J.-N. Dumez, P. H. Svensson, S. Schantz, G. M. Day and L. Emsley, J. Am. Chem. Soc., 2013, 135, 17501–17507 CrossRef CAS PubMed.
- E. Salager, G. M. Day, R. S. Stein, C. J. Pickard, B. Elena and L. Emsley, J. Am. Chem. Soc., 2010, 132, 2564–2566 CrossRef CAS PubMed.
- T. Sumner, Science, 2014, 343, 1092–1093 CrossRef PubMed.
- B. H. Toby, Powder Diffr., 2006, 21, 67–70 CrossRef CAS.
- Y. L. Slovokhotov, Cryst. Growth Des., 2014, 14, 6205–6216 CAS.
- J. van de Streek, Acta Crystallogr., Sect. B: Struct. Sci., 2006, B62, 567–579 CAS.
- J. van de Streek and M. A. Neumann, Acta Crystallogr., Sect. B: Struct. Sci., 2010, B66, 544–558 Search PubMed.
- T. Pawlak and M. J. Potrzebowski, J. Phys. Chem. B, 2014, 118, 3298–3309 CrossRef CAS PubMed.
- M. Kibalchenko, D. Lee, L. Shao, M. C. Payne, J. J. Titman and J. R. Yates, Chem. Phys. Lett., 2010, 498, 270–276 CrossRef CAS.
- R. K. Harris, P. Hodgkinson, V. Zorin, J.-N. Dumez, B. Elena-Herrmann, L. Emsley, E. Salager and R. S. Stein, Magn. Reson. Chem., 2010, 48, S103–S112 CrossRef CAS PubMed.
- http://www.who.int/medicines/publications/essentialmedicines .
- K. M. Ho and B. M. Power, Anaesthesia, 2010, 65, 283–293 CrossRef CAS PubMed.
- F. Andreasen, O. L. Pedersen and E. Mikkelsen, Eur. J. Clin. Pharmacol., 1978, 14, 237–244 CrossRef CAS PubMed.
- H. E. Kerr, L. K. Softley, K. Suresh, A. Nangia, P. Hodgkinson and I. R. Evans, CrystEngComm, 2015, 17, 6707–6715 RSC.
- N. R. Goud, S. Gangavaram, K. Suresh, S. Pal, S. G. Manjunatha, S. Nambiar and A. Nangia, J. Pharm. Sci., 2012, 101, 664–680 CrossRef CAS PubMed.
- B. I. Harriss, L. Vella-Zarb, C. Wilson and I. R. Evans, Cryst. Growth Des., 2014, 14, 783–791 CAS.
- T. Ueto, N. Takata, N. Muroyama, A. Nedu, A. Sasaki, S. Tanida and K. Terada, Cryst. Growth Des., 2012, 12, 485–494 CAS.
- C. Garnero, A. K. Chattah and M. Longhi, Carbohydr. Polym., 2013, 94, 292–300 CrossRef CAS PubMed.
- J. Lamotte, H. Campsteyn, L. Dupont and M. Vermeire, Acta Crystallogr., 1978, B34, 1657–1661 CrossRef CAS.
- O. Bolukbasi and A. Yilmaz, Vib. Spectrosc., 2012, 62, 42–49 CrossRef CAS.
- J. van de Streek and S. Motherwell, Acta Crystallogr., Sect. B: Struct. Sci., 2005, B61, 504–510 CAS.
- S. Karami, Y. Li, D. S. Hughes, M. B. Hursthouse, A. E. Russell, T. L. Threlfall, M. Claybourn and R. Roberts, Acta Crystallogr., Sect. B: Struct. Sci., 2006, B62, 689–691 CrossRef CAS PubMed.
- N. J. Babu, S. Cherukuvada, R. Thakuria and A. Nangia, Cryst. Growth Des., 2010, 10, 1979–1989 CAS.
- X. Filip and C. Filip, Solid State Nucl. Magn. Reson., 2015, 65, 21–28 CrossRef CAS PubMed.
- Q. Z. Ni, E. Daviso, T. V. Can, E. Markhasin, S. K. Jawla, T. M. Swager, R. J. Temkin, J. Herzfeld and R. G. Griffin, Acc. Chem. Res., 2013, 46, 1933–1941 CrossRef CAS PubMed.
- A. J. Rossini, A. Zagdoun, M. Lelli, A. Lesage, C. Copéret and L. Emsley, Acc. Chem. Res., 2013, 46, 1942–1951 CrossRef CAS PubMed.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CAS.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245101 CrossRef.
- J. R. Yates, C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 024401 CrossRef.
- M. Dračínský and P. Hodgkinson, CrystEngComm, 2013, 15, 8705–8712 RSC.
- H. P. G. Thompson and G. M. Day, Chem. Sci., 2014, 5, 3173–3182 RSC.
- J. Nyman and G. M. Day, CrystEngComm, 2015, 17, 5154–5165 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Molecular label specifications, full experimental disclosure, pXRD data for S-A, crystal structure data for FURSEM-NEW, tables of chemical shifts, 15N CP/MAS NMR spectrum, additional 1H–13C HETCOR spectra, details of computational results (shielding values and energies), shift/shielding correlation plots, computed 13C NMR spectrum, sample RMSD calculation. CCDC 1458460. Original research data can be accessed through data DOI: 10.15128/s1784k724. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc02171a |
| ‡ CCDC 1458460. |
| § These values were established by benchmarking ca. 30 molecular systems of known correct structure using the GIPAW DFT method in CASTEP. The grey banded areas correspond to regions equal to ±1σ about the mean values (dashed lines). Details for these systems are in preparation for submission. |
| This journal is © The Royal Society of Chemistry 2016 |