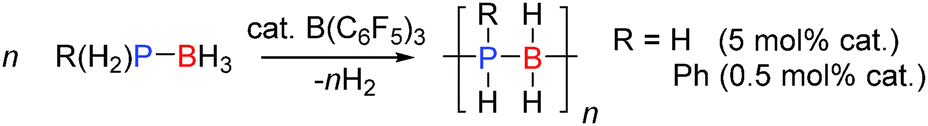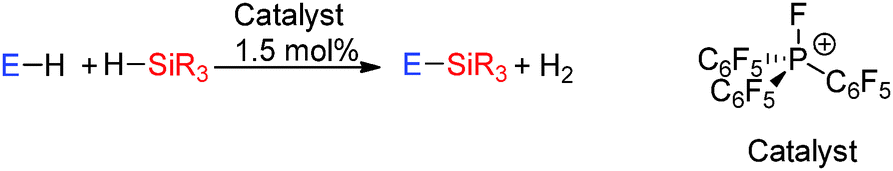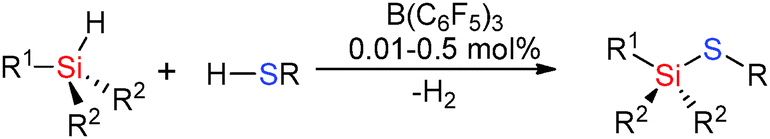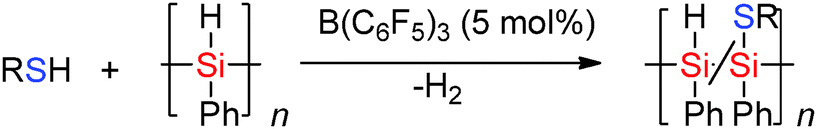DOI:
10.1039/C5CS00521C
(Tutorial Review)
Chem. Soc. Rev., 2016,
45, 775-788
Dehydrocoupling routes to element–element bonds catalysed by main group compounds†
Received
2nd July 2015
First published on 10th August 2015
Abstract
Dehydrocoupling reactions, i.e. reactions involving elimination of H2 between two E–H bonds, provide a clean route to E–E bonds within the main group. The products afforded from these reactions have applications in organic synthesis and materials chemistry, and in addition the H2 released during these reactions can also be useful as an energy source. Previous methods for dehydrocoupling involve both thermal and transition metal catalysed routes but recent developments have shown that main group compounds can be used as catalysts in these reactions. This tutorial review will focus on the development of main group catalysed dehydrocoupling reactions as a route to heteronuclear element–element bonds.

Rebecca L. Melen
| Rebecca Melen studied for her MSc and PhD degrees at the University of Cambridge, completing her PhD in 2012 with Prof. Dominic Wright. Her research focused on dehydrocoupling reactions using main group compounds. Following Postdoctoral studies with Prof. Douglas Stephan in Toronto and with Prof. Lutz Gade in Heidelberg, she took up a position at Cardiff University in 2014. Her research interests include diverse aspects of main group reactivity and catalysis. |
Key learning points
(1) Introduction to main group element–element bonds and their applications
(2) Dehydrocoupling reactions as a route to element–element bonds
(3) Main group compounds in dehydrocoupling reactions
(4) Comparison of reactivity pathways and mechanistic aspects of different main group dehydrocoupling catalysts
(5) Future prospects of main group catalysed dehydrocoupling
|
1. Introduction to dehydrocoupling
Dehydrocoupling involves the formation of element–element (E–E) bonds with the concurrent loss of hydrogen gas. Dehydrocoupling reactions between two of the same elements (homo-dehydrocoupling) or two different elements (hetero-dehydrocoupling) provide a convenient and clean method to synthesise E–E and E–E′ bonds with the release of H2 as the sole by-product (eqn (1)).| |
| E–E + H–E′ → E–E′ + H2 | (1) |
Both E–E and E–E′ bond-forming reactions are central to the development of many areas of main group chemistry including inorganic rings, chains and polymers. These reactions offer significant advantages over previous methods of main-group E–E′ bond formation which were often difficult. Classical syntheses involved element-specific reductive coupling, high temperature condensation reactions or salt metathesis reactions.1 Dehydrogenative coupling reactions have a wide range of important applications in terms of both the main group element products formed and the H2 gas released. The E–E′ bonded products have found applications in both organic synthesis and materials chemistry, whereas the H2 released during these reactions could be used as an energy source for a future Green Economy.1 In this Tutorial Review ‘classical’ thermal dehydrocoupling reactions as a method for E–E and E–E′ bond formation will be introduced, followed by a review of recent developments in main-group catalysed dehydrocoupling reactions specifically in hetero-dehydrocoupling reactions. In particular a comparison of the similarities and differences in catalytic activity between main group elements will be addressed, focusing on important factors which appear necessary for efficient main group catalysts. The development of E–E and E–E′ bond-forming reactions is as important to main group chemists as C–C bond forming reactions are to organic chemists.
One of the classical approaches to form E–E bonds is the thermolysis of binary element hydrides, EHn. In these cases the entropically favourable elimination of H2 appears a significant driving force in E–E bond formation. As early as 1880 the complete decomposition of silane (SiH4) at elevated temperatures (400 °C) was found to afford elemental Si and dihydrogen.2 Indeed, the thermal decomposition of SiH4 is an industrially important process and SiH4 is now used as a source of ultra-high purity silicon via chemical vapor deposition.3 However, the outcome of such dehydrocoupling reactions is often poorly defined. Whilst the ultimate product may be elemental E and dihydrogen, intermediate hydrides of formula ExHy can also be isolated under milder thermolysis conditions. In many cases a mixture of products can be observed in which the product distribution is sensitive to the temperature. For example, thermal decomposition of B2H6 is very complicated and leads to higher order boranes.4 The pyrolysis above 100 °C is a stepwise process (eqn (2)) initially affording B3H7 and subsequently higher boranes such as B4H10, B5H9, B5H11, B6H10, B6H12 and eventually yielding the more stable borane B10H14 along with polymeric BH boranes and higher order boranes. In these cases careful control of the reaction temperature, time and pressure is required to isolate the intermediate boranes in reasonable yields.5
The E–H bond cleavage occurring during pyrolysis broadly follows the strength of the E–H bond broken (
Table 1).
1 Thus, whilst methane has to be heated in the range 800–1000 °C to generate carbon black and polycyclic hydrocarbons,
7 silane completely decomposes
2 at 400 °C and the thermal decomposition of germane to form elemental germanium and dihydrogen commences at just 280 °C.
8 The weakening of the E–H bond on descending group 14 is reflected in other groups of the periodic table due to poorer E–H covalency on descending the group through orbital size/energy mismatch.
1 Conversely there is a general increase in E–H bond energy from left to right across a period due to the increasing ionic contribution to bonding associated with the greater bond polarity. Both the strength and polarity of the E–H bond are important factors when considering E–H dehydrocoupling processes.
1 Similar pyrolytic decomposition reactions have been used in E–E′ bond formation and is well-illustrated by the formation of boron nitride from thermal decomposition of NH
3BH
3.
9 These thermal methods are energy intensive and, in some cases, may lead to a distribution of products rather than a well-defined single product. As a consequence approaches to generate single products or a single major product under mild conditions are highly desirable.
9 While metal-catalysed carbon–carbon bond forming reactions are an extremely important part of modern synthetic organic chemistry, the catalytic formation of main group element–element bonds represents a comparatively new and exciting area.
10 Catalytic E–E bond formation was first reported by Sneddon in 1984 in the formation of homonuclear B–B bonds when they successfully linked two polyhedral boron cages using PtBr
2 as a catalyst.
10 Subsequently catalytic E–E bond formation has become an area of specific interest with many reports of efficient transition metal catalysts with long lifetimes and good turnover frequencies in the formation of a wide range of homo- and hetero-nuclear E–E bonds under mild conditions.
1,9–11 Most catalytic reactions are currently considered the domain of transition metals however, in recent years there has been an evolution of main group catalysis. An alternative strategy to E–E and E–E′ bond formation is to use main-group catalysts and these are the focus of this review.
Table 1 Approximate diatomic E–H bond enthalpies (kJ mol−1)6
| Group 13 |
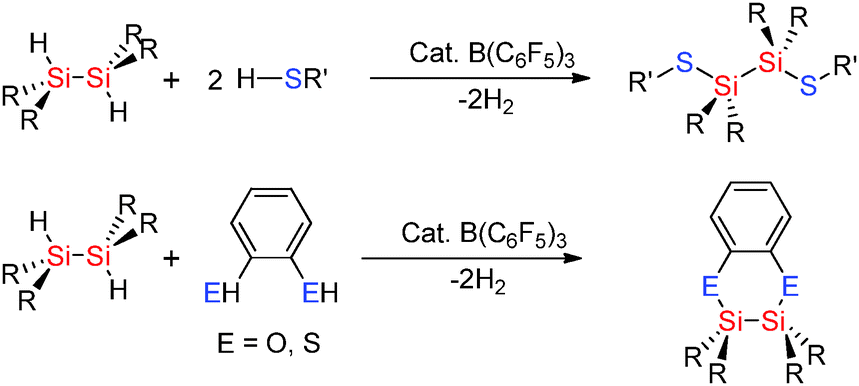
Group 14 |
Group 15 |
Group 16 |
Group 17 |
| B–H |
C–H |
N–H |
O–H |
F–H |
| 389 |
411 |
386 |
456 |
565 |
| Al–H |
Si–H |
P–H |
S–H |
Cl–H |
| 285 |
318 |
321 |
364 |
432 |
| Ga–H |
Ge–H |
As–H |
Se–H |
Br–H |
| <274 |
285 |
297 |
313 |
366 |
| In–H |
Sn–H |
Sb–H |
Te–H |
I–H |
| 243 |
264 |
257 |
270 |
298 |
2. Catalytic heteronuclear-dehydrocoupling
2.1 B–N bond formation
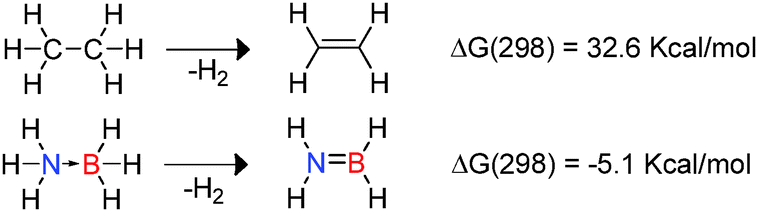
The use of hydrogen as a renewable, non-toxic and clean energy source in addition to its high energy density has attracted attention in recent years.9 In this context amine-borane compounds may be effective hydrogen storage materials since they offer high hydrogen content by mass (ca. 20% by mass hydrogen for NH3BH3). Indeed there is now a substantial body of research describing efficient approaches to the catalytic hetero-dehydrocoupling reactions of amine-boranes as hydrogen sources.1,9,10 These studies have also been driven by the interest in boron-nitrides such as hexagonal boron-nitride which has a range of applications in high temperature ceramics and as a lubricant inter alia.1 As a consequence one of the most well-studied areas of dehydrocoupling is the dehydrogenation of amine-borane compounds. The differing electronegativities of B and N with respect to H within the donor–acceptor adduct H3N → BH3 results in polarisation of both B–H and N–H bonds.9 This polarity enables facile release of hydrogen with the dehydrogenation of NH3BH3 to generate H2N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) BH2 being mildly exothermic and favourable on both entropic and enthalpic terms. This is in stark contrast to isoelectronic ethane for which dehydrogenation to form ethene is endothermic (Scheme 1).9 As a consequence, although dehydrocoupling of NH3BH3 is thermodynamically favoured at 298 K, much higher temperature are required for the dehydrogenation of ethane.12
BH2 being mildly exothermic and favourable on both entropic and enthalpic terms. This is in stark contrast to isoelectronic ethane for which dehydrogenation to form ethene is endothermic (Scheme 1).9 As a consequence, although dehydrocoupling of NH3BH3 is thermodynamically favoured at 298 K, much higher temperature are required for the dehydrogenation of ethane.12
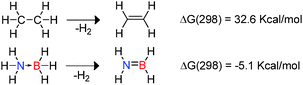 |
| | Scheme 1 Thermodynamics of dehydrogenation of ethane versus NH3BH3. | |
Owing to the mild conditions and increased selectivity in catalysed dehydrocoupling reactions, a wide range of transition metal catalysts have been developed and employed in the dehydrogenation/dehydrocoupling of a diverse spectrum of primary (RNH2BH3) and secondary (R2NHBH3) amine-boranes to afford monomeric or dimeric products, borazines or polymeric B–N compounds depending on the substrate.9 The synthesis of poly-amino-boranes has been explored to generate boron-nitrogen polymers which are analogues of polyolefins [R2NBR2′]n which may have potential applications in the synthesis of carbon free ceramics.1,9,10 There are currently many catalysts based upon transition metals which have been applied to amine-borane dehydrogenation reactions.9 Of these, the platinum group metals appear to offer some of the best catalytic reactions to date and have been widely studied. However, complexes of first row transition metals (e.g. Fe, Cu) and early transition metals (e.g. Ti, Zr) have also been explored.9 An important aspect of transition-metal catalysed dehydrocoupling appears to be associated with the Lewis acidic (or Lewis basic) character of the metal. For example a range of d0 metals e.g. Ti, Zr have high Lewis acidity and have been shown to be effective in the dehydrocoupling of amine-boranes.13 The diverse range of main group metals and oxidation states permit the Lewis acidity and basicity of as well as hard/soft character of the main group metal to be fine tuned. This opportunity to tune both Lewis acidity and basicity is at the heart of much of the renaissance in main group chemistry and has led to a range of both s- and p-block catalysts whose activities appear related to their Lewis acidity and/or the presence of strongly basic anions which are capable of deprotonating the amine-borane substrate.13
2.1.1 B–N bond formation using alkaline earth elements.
An important discovery in B–N dehydrocoupling was the observation that the closed shell group 2 ions can catalyse dehydrocoupling reactions. The alkaline earth (Ae) systems employed have been based upon magnesium, calcium and strontium bearing alkyl, amido ligands or β-diketiminate ligands (Fig. 1).
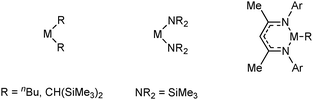 |
| | Fig. 1 Alkaline earth systems employed in dehydrocoupling reactions. | |
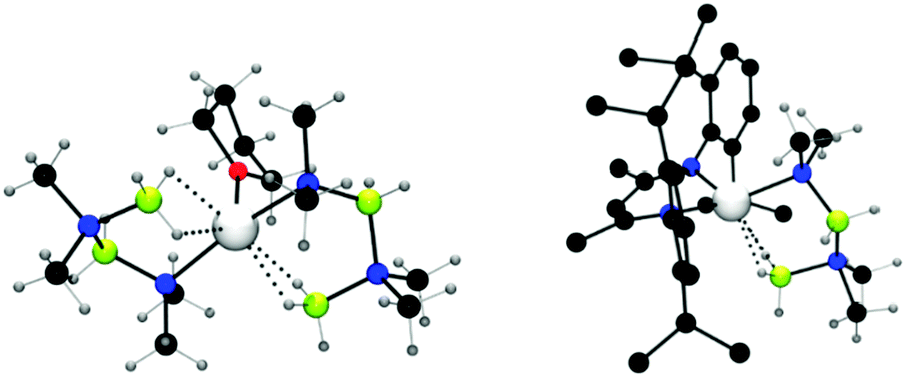
When Me2NHBH3 was reacted with MgBu2 or Mg[CH(SiMe3)2]2(thf)2 in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, a stoichiometric dehydrocoupling reaction occurred to liberate H2 and generate Mg(NMe2BH2NMe2BH3)2(thf) which was identified by multinuclear NMR and X-ray diffraction (Fig. 2, left).14 The crystal structure reveals the B2N2 chain coordinates to the magnesium centre through an Mg–N bond and also via an agostic Mg⋯HB interaction. Heating this species to 60 °C resulted in the elimination of [Me2NBH2]2 by an intramolecular σ-bond metathesis (δ-hydride elimination) presumably giving the magnesium hydride as a side-product.14 The liberation of the dehydrocoupled dimer [Me2NBH2]2 from the magnesium at elevated temperatures suggests the potential for a catalytic process. Whilst no direct evidence for Mg–H could be observed in this case, magnesium hydrides have been clearly identified elsewhere in this chemistry (vide infra).
:1 ratio, a stoichiometric dehydrocoupling reaction occurred to liberate H2 and generate Mg(NMe2BH2NMe2BH3)2(thf) which was identified by multinuclear NMR and X-ray diffraction (Fig. 2, left).14 The crystal structure reveals the B2N2 chain coordinates to the magnesium centre through an Mg–N bond and also via an agostic Mg⋯HB interaction. Heating this species to 60 °C resulted in the elimination of [Me2NBH2]2 by an intramolecular σ-bond metathesis (δ-hydride elimination) presumably giving the magnesium hydride as a side-product.14 The liberation of the dehydrocoupled dimer [Me2NBH2]2 from the magnesium at elevated temperatures suggests the potential for a catalytic process. Whilst no direct evidence for Mg–H could be observed in this case, magnesium hydrides have been clearly identified elsewhere in this chemistry (vide infra).
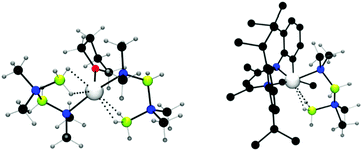 |
| | Fig. 2 X-ray structure of Mg(NMe2BH2NMe2BH3)2(thf) (left) and (Dipp-nacnac)Mg(NMe2BH2NMe2BH3) (right) showing Mg⋯HB interactions. | |
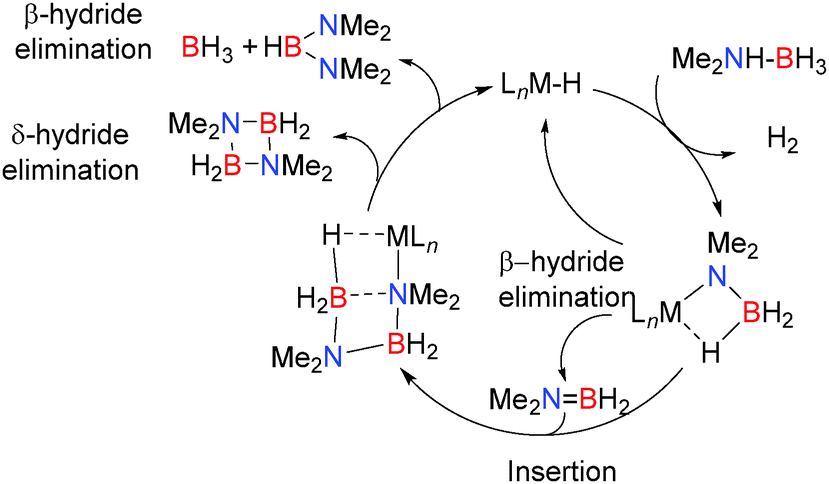
In order to test the catalytic potential of these reactions 5 mol% Mg[CH(SiMe3)2]2(thf)2 was employed in the dehydrocoupling of Me2NHBH3 resulting in formation of the dimer [Me2NBH2]2 after 72 h at 60 °C along with smaller quantities of (Me2N)2BH.14 The proposed mechanism shown in Scheme 2 is supported by NMR data and the identification of intermediates including both Ae–NMe2BH3 and Ae–NMe2BH2NMe2BH3 complexes in this and closely-related reactions.
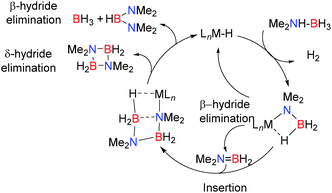 |
| | Scheme 2 Proposed mechanism for the catalytic dehydrocoupling of Me2NHBH3 group 2 metals. | |
Evidence for the proposed mechanism involving β-hydride elimination and insertion steps was provided by the stoichiometric reactions of alkyl–strontium complexes with secondary amine-boranes to give (Me3Si)2HC-Sr-NR2BH3 (R = Me, (CH2)2) which could undergo stoichiometric β-hydride elimination and Sr–C insertion reactions to give (Me3Si)2HC-BH-NR2.15 Similarly the stoichiometric reactions of M[N(SiMe3)2]2 (M = Mg, Ca, Sr, Ba) with Me2NHBH3 or iPr2NHBH3 were investigated.16 With the magnesium silylamide starting material the amino-boranes Me2NBH2 or iPr2NBH2 are formed in high yields via δ-hydride elimination, whereas with calcium (depending upon the stoichiometry) the yields of the amino-boranes are much lower and with strontium and barium only the metal amido-borane complexes (M–NMe2BH3 or M–NiPr2BH3) are formed with no evidence for β-hydride elimination.16 Thus it would appear that the reactivity for the elimination is in the order Mg > Ca > Sr > Ba. Such behaviour might be usefully compared to the tendency of d-block alkyls to undergo β-hydride elimination reactions in which the migration appears favourable for more electropositive (‘hard’) metal ions and proceeds via an agostic M⋯H interaction.17 Indeed, the dehydrogenation of iPr2NHBH3 using 5 mol% Mg[N(SiMe3)2]2 resulted in complete dehydrogenation to iPr2NBH2 after 1 h. Thus the most active catalysts appear to be those based on Mg.16
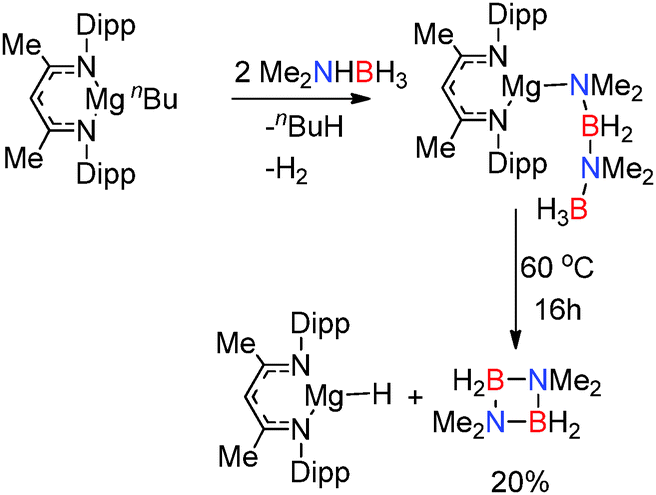
Replacement of the alkyl and amido ligands by the more strongly coordinating chelate nacnac type ligand affords more stable “heteroleptic” complexes,18 though the presence of at least one strongly basic M–H, M–R or M–NR2 group is necessary to assist the initial activation of the amine-borane substrate. The reaction of (Dipp-nacnac)MgnBu with two equivalents of Me2NHBH3 yielded the Mg-chain species (Dipp-nacnac)Mg(NMe2BH2NMe2BH3) which was structurally characterised and shows agostic Mg⋯HB interactions in the solid state (Fig. 2, right) similar to that for Mg(NMe2BH2NMe2BH3)2(thf) described above.14 This compound was found to be more thermodynamically stable than Mg(NMe2BH2NMe2BH3)2(thf) and only underwent 20% conversion to [Me2NBH2]2 at 60 °C after 16 h (Scheme 3). Significantly elimination of [Me2NBH2]2 resulted in the formation of the dimeric magnesium hydride [(Dipp-nacnac)MgH(thf)2]2 which, in this case, could be observed by 1H NMR spectroscopy, thus providing evidence of the formation of metal hydrides in these reactions.14
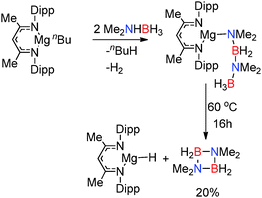 |
| | Scheme 3 Dehydrocoupling of Me2NHBH3 using [(Dipp-nacnac)MgnBu]2. | |
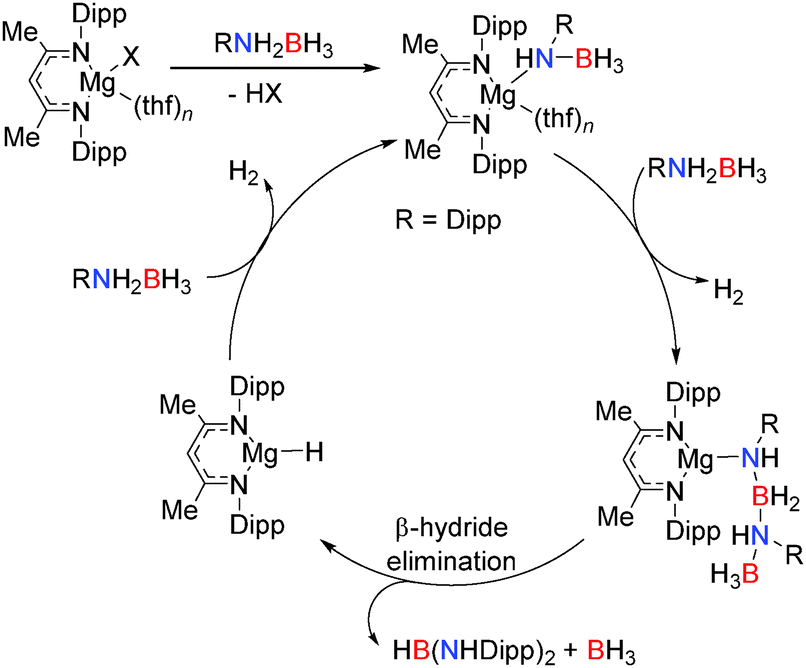
In the presence of more sterically demanding substrates, the outcome of the reaction appears somewhat modified. For example, the closely related bis(trimethylsilyl)amido derivative [(Dipp-nacnac)MgN(SiMe3)2]2 reacts with the sterically encumbered (Dipp)NH2BH3 with the rapid evolution of hydrogen and formation of the di-amino-borane HB[NH(Dipp)]2 rather than dimeric [DippNHBH2]2 (Scheme 4).19 This reaction was found to be catalytic with a mechanism essentially the same as that described in Scheme 2: the reaction proceeds via a magnesium amido-borane intermediate Mg-NH(Dipp)BH3 which reacts with the amine-borane (Dipp)NH2BH3 with release of H2 to generate the chain species Mg-NH(Dipp)BH2NH(Dipp)BH3. This then undergoes a β-hydride elimination (rather than δ-hydride elimination) to give the symmetrical di-amino-borane product, HB[NH(Dipp)]2 and BH3, generating a magnesium hydride which can then react with the amine-borane releasing H2 thereby regenerating the magnesium amido-borane and completing the catalytic cycle.19 Thus a common catalytic cycle seems prevalent within this series of compounds; initial deprotonation of the N–H group of the RNH2BH3 substrate occurs via the strongly basic Mg–R or Mg–NR2 group affording an Ae–NHRBH3 complex. Addition of a second equivalent of substrate leads to elimination of H2 and formation of Ae–N(R)HBH2NH(R)BH3 which then undergoes a δ-hydride elimination to generate the hydride Ae–H and release dimeric [RNHBH2]2 although β-hydride elimination can be a competing factor generating HB(NHR)2. In the case of the bulky amine-borane DippNH2BH3 β-hydride elimination is preferable over δ-hydride elimination generating HB[NH(Dipp)]2 as the product in this reaction. In both cases (β- or δ-hydride elimination) a magnesium hydride species forms which appears a sufficiently potent base to deprotonate another equivalent of amine-borane RNH2BH3 regenerating further Ae–NHRBH3 thus allowing a catalytic reaction to take place.
 |
| | Scheme 4 Synthesis of the diaminoborane HB[NH(Dipp)]2 using the pre-catalyst [(Dipp-nacnac)MgN(SiMe3)2]2. | |
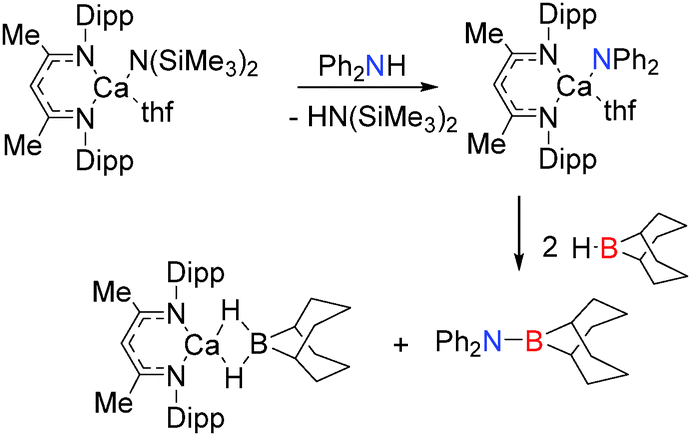
One of the first indications that the related calcium nacnac complexes may be used in B–N coupling was the report of the stoichiometric σ-bond metathesis reaction between a β-diketiminato calcium amide complex with 9-BBN to give the B–N product and a calcium borohydride complex (Scheme 5).20 Here again the Ae–NR2 group is used to deprotonate the secondary amine in the first step of the reaction. Similar stoichiometric reactions of [(Dipp-nacnac)CaH(thf)]2 with NH3BH3, MeNH2BH3 or iPrNH2BH3 have led to the isolation of calcium amido-borane intermediates which comprise an NBNB2− chain which is formed via a B–N coupling process with release of hydrogen (Scheme 6).18,21 For the bulky substrate DippNH2BH3 formation of the B2N22− chain is suppressed and the calcium complex, Ae–N(R)BH2 is formed with release of H2.21 The isolation of this complex suggest that, for Ca, the kinetics of reaction with a further equivalent of RNH2BH3 is slow in relation to elimination of H2 to form the bimetallic complex, whereas for Mg reaction with a further equivalent of substrate appears more favoured. Although [(Dipp-nacnac)Ca-NH2BH3(thf)] was found to be thermally unstable,18 [(Dipp-nacnac)Ca-NMe2BH3(thf)] demonstrated higher stability and at 80 °C underwent some decomposition to generate the monomeric amino-borane Me2NBH2 and (Me2N)2BH and the calcium hydride complex [(Dipp-nacnac)CaH(thf)2]2.14 These studies suggest that a β-hydride elimination/σ-bond metathesis reaction pathway is more prevalent for Ca than Mg which tended to undergo δ-hydride elimination.
 |
| | Scheme 5 Dehydrocoupling of 9-BBN and Ph2NH using a β-diketiminato calcium amide. | |
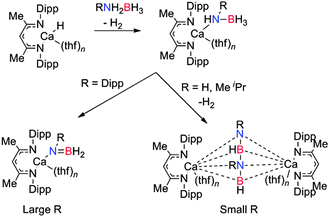 |
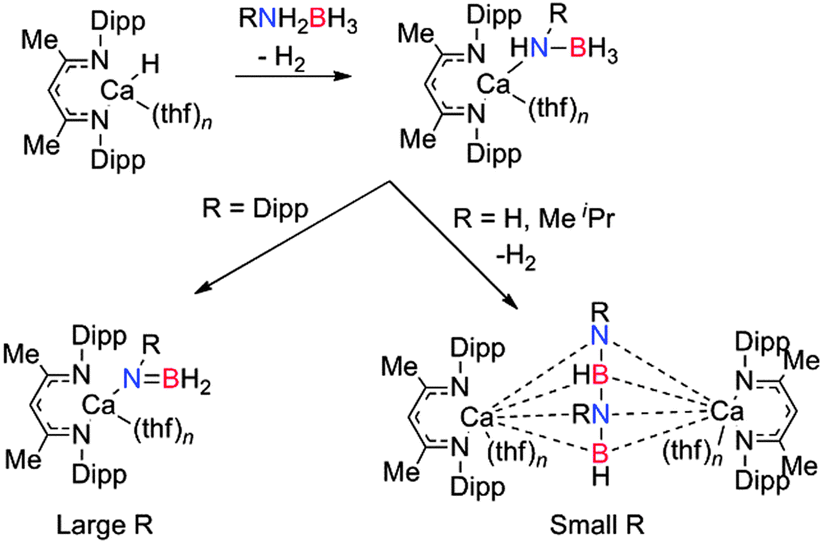
| | Scheme 6 Dehydrogenation of primary amine-boranes using a (Dipp-nacnac)CaH species. | |
In summary, NMR and crystallographic studies of the catalytic and stoichiometric dehydrocoupling of Me2NHBH3, together with the evidence of metal hydride intermediates in these systems enabled a general mechanism for the dehydrocoupling mechanism to be proposed (Scheme 2) which combines the aspects described for the mechanisms in Schemes 3–6. The amino-borane Me2NBH2 is thought to be formed from the β-hydride elimination/σ-bond metathesis of the M–NMe2BH3 complex. The alkene-analogue Me2NBH2 can then insert into the metal-bonded [Me2NBH3]− anion giving the metal-bonded [NMe2BH2NMe2BH3]− species. Subsequent δ-hydride elimination affords [Me2NBH2]2 whereas β-hydride elimination generates [(Me2N)2BH] (Scheme 2). The dehydrocoupling of the secondary amine-borane, pyrrolidine amine-borane (CH2)4NHBH3 using both Ca and Mg-based catalysts proves instructive in this regard. When (Dipp-nacnac)MgN(SiMe3)2 was reacted with one or two equivalents of (CH2)4NHBH3 then the compounds (Dipp-nacnac)MgN(CH2)4BH3 and (Dipp-nacnac)MgN(CH2)4BH2N(CH2)4BH3 were formed respectively, consistent with the rapid addition of a second equivalent of amine-borane over elimination of dihydrogen.16 Heating the later compound to 60 °C resulted in the formation of the cyclic dimer product [(CH2)4NBH2]2 as shown by the presence of a triplet in the 11B NMR spectrum at δ = 6.5 ppm, and in the 1H NMR spectrum a shift due to the potential magnesium hydride was observed suggesting a δ-hydride elimination, identical to that described in the catalytic cycle above for Me2NHBH3.16 With the heavier calcium analogue [(Dipp-nacnac)CaN(SiMe3)2(thf)] dehydrocoupling of one equivalent of (CH2)4NHBH3 gave the calcium amido-borane [(Dipp-nacnac)Ca-N(CH2)4-BH3] however subsequent reaction with a second equivalent of amine-borane did not occur.16 This is consistent with the observation that the calcium is less active in these processes than magnesium as [Ca]-NMe2BH3 also does not undergo β-hydride elimination or B–N coupling to form the Ca–NBNB chain.
The alkaline earth bis(trimethylsilyl)amides M[N(SiMe3)2]2 (M = Mg, Ca) were later found to be active in the synthesis of unsymmetrical di-amino-boranes from the dehydrocoupling of amine-boranes (R2NHBH3) and amines (R′NH2) to give (R2N)BH(NR2′).22 These reactions are proposed to proceed via metal hydride intermediates which can react with the amine-borane to form the previously observed metal bonded amido-borane which then undergoes β-hydride elimination yielding the amino-borane R2NBH2.22 In addition to reaction of the metal hydride with the amine-borane, the metal hydride may also react with the amine resulting in deprotonation of the amine, releasing hydrogen and generating a metal amido species. This metal amido intermediate can then react with the amino-borane R2NBH2 to give a metal bound di-amido-borate M–NR2′BH2NR2. Subsequent β-hydride elimination from this species releases the neutral unsymmetrical di-amino-boranes (R2N)BH(NR2′) and regenerates the metal hydride (Scheme 7).22 This methodology has significant advantages over previous synthetic approaches to prepare di-amino-boranes in which only symmetrical-di-amino-boranes (R2N)BH(NR2′) (RR′) could be synthesised from redistribution reactions of tris(dialkylamino)boranes [(R2N)3B] with B2H6.23
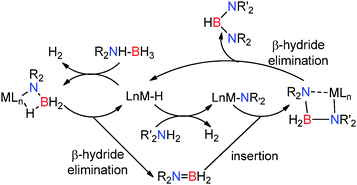 |
| | Scheme 7 Proposed mechanism for the group 2 catalysed dehydrocoupling of amines with amine-boranes. | |
In summary, the catalytic dehydrocoupling of a range of amines and boranes has been achieved using alkaline earth alkyl/amido or β-diketiminate complexes with the catalytic activity in the order Mg > Ca > Sc, consistent with the stronger Lewis acidity/hardness of the smaller group 2 metals. Importantly, in common with transition metal catalytic systems, the β-hydride elimination metal hydrides are proposed as key intermediates in the reactions. Significantly this work showed that higher catalytic activity appears to result from increased charge density of the metal and therefore the use of the more positively charged Al3+ and Ga3+ complexes are anticipated to be more active in the dehydrogenic coupling of amine-boranes.
2.1.2 B–N bond formation using group 3 metals.
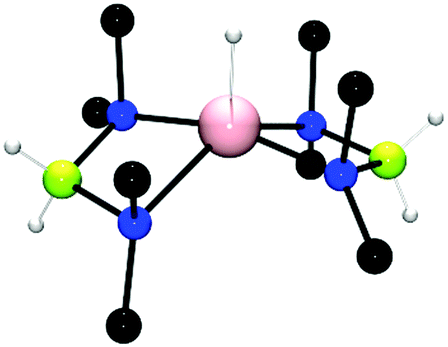
The catalytic dehydrocoupling using group 13 elements containing basic E–NR2 bonds has also been investigated and shows several similarities to the group 2 catalysts described above. In this regard, the dehydrocoupling of Me2NHBH3 with Al(NMe2)3 was explored.24 The reaction of Me2NHBH3 with 8 mol% Al(NMe2)3 was followed by in situ11B NMR studies and showed the rapid formation of (Me2NBH2)2 under ambient conditions in addition to small quantities of (Me2N)2BH and a triplet at (δ = 1.87 ppm, 1JBH = 107 Hz) which was subsequently assigned to the hypervalent 5-coordinate AlIII hydride [{(Me2N)2BH2}2AlH] (Fig. 3) which was isolated from the 1:1 stoichiometric reaction of Al(NMe2)3 with Me2NHBH3. The AlIII hydride was found to be a thermally stable catalyst for this reaction.24
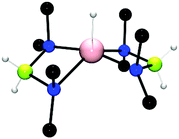 |
| | Fig. 3 The crystal structure of the AlIII hydride [{(Me2N)2BH2}2AlH]. | |
Although the activity of the pre-catalyst Al(NMe2)3 is relatively low at room temperature (the 8 mol% loading in toluene gives a ca. 80% conversion after 5 days), the reaction is significantly faster at elevated temperatures. For example a 5 mol% loading of Al(NMe2)3 at 50 °C in toluene gives complete conversion after 48 h.24 The reactivity at higher temperature is at the lower end of that observed for a range of transition metal catalysed systems.9 DFT studies revealed that the complex [{(Me2N)2BH2}2AlH] readily undergoes a β-hydride transfer from B to Al with a low activation energy (ΔG‡ ∼ +78.2 kJ mol−1 at 298 K) with simultaneous elimination of Me2NBHNMe2 (identified in the 11B NMR spectrum) which was energetically favourable (ΔG = −59.4 kJ mol−1) to form the four-coordinate aluminium dihydride (Me2NBH2NMe2)AlH2 which is the proposed active catalyst in this reaction.25 Notably the aluminium dihydride is structurally related to other aluminium dihydrides such as [iPr2NAlH2]2 which have also been shown to be active in dehydrocoupling reactions (see below).25 The reaction was observed to be first-order in the starting material. A mechanism involving dehydrogenation of Me2NHBH3 to give Me2NBH2 followed by rapid dimerisation to give (Me2NBH2)2 can be discounted since Me2NBH2 is relatively stable in solution and was not detected in the 11B NMR.25 A general mechanism for the aluminium hydride catalysed dehydrocoupling can be envisaged which proceeds via the Al–H acting as a base to deprotonate the substrate (analogous to the alkaline earth chemistry), leading to the formation of a metal-bound Me2NBH3− species which can undergo dehydrocoupling with a second equivalent of substrate to form an aluminium coordinated [Me2NBH2NMe2BH3]− intermediate reminiscent of that seen for group 2 catalysts. Subsequent δ-hydride elimination regenerates the dihydride catalyst and releases the product (Me2NBH2)2.25
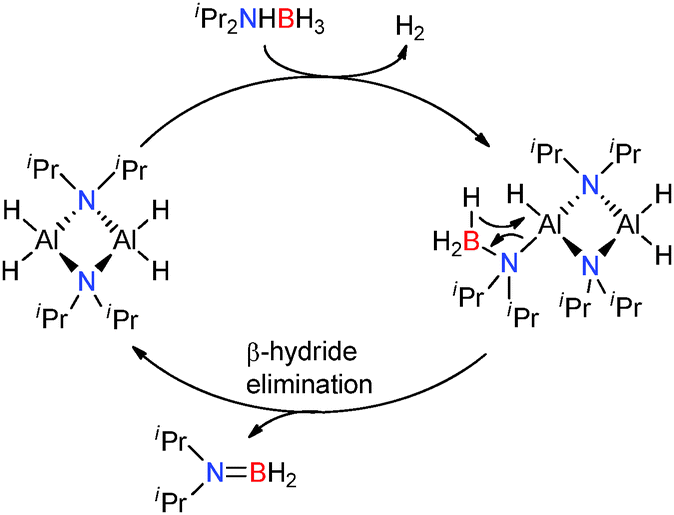
The 2:1 stoichiometric reaction of the related amine-borane iPr2NHBH3 with Al(NiPr2)3 afforded the centrosymmetric dimeric aluminium dihydride [H2Al(μ-NiPr2)]2. This catalyst once again exhibits the presence of a metal hydride which appears critical in these main-group catalysed dehydrocoupling reactions and notably [iPr2NAlH2]2 exhibits the same core structure as (Me2NBH2NMe2)AlH2 discussed above (by replacement of Al by B). This [H2Al(μ-NiPr2)]2 complex was found to be a very active catalyst for the dehydrogenation of iPr2NHBH3 to afford iPr2NBH2, e.g. a 0.5 mol% loading of crystalline [H2Al(μ-NiPr2)]2 resulted in the dehydrogenation of iPr2NHBH3 to iPr2NBH2 at 20 °C.25 Although low concentrations of highly reactive monomeric H2AlNiPr2 cannot be discounted, a mechanism based on a dimetallic aluminium catalyst [H2Al(μ-NiPr2)]2 as the active catalyst is proposed (Scheme 8). Initial deprotonation of the protic N–H group by the basic Al–H hydride, releasing H2 and generates the [Al]–NiPr2–BH3 species. A subsequent β-hydride shift from boron to aluminium affords iPr2NBH2 and regenerates the catalyst.25 This deprotonation/β-hydride elimination sequence is identical to that observed for group 2 metal catalysts in the dehydrocoupling of amine-boranes (Section 2.1.1). Since the [H2Al(μ-NiPr2)]2 catalyst is prepared in situ from iPr2NHBH3 and Al(NiPr2)3 then Al(NiPr2)3 can be used as a pre-catalyst. Thus a 10 mol% loading of Al(NiPr2)3 resulted in complete conversion of iPr2NHBH3 to iPr2NBH2 in 2 h at 60 °C, albeit with a relatively long induction period prior to release of H2 gas. Similar to the dehydrocoupling of Me2NHBH3 the reaction was shown to be first order in substrate.25 These are similar to the transition metal catalysts [Rh(1,5-cod)(μ-Cl)]2 and Cp2Ti which catalyse the same dehydrogenation reaction in 49% yield at 25 °C in 24 h using a 1 mol% catalyst or give quantitative yields in 1 h at 20 °C using a 2 mol% loading respectively.13,25
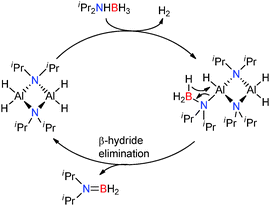 |
| | Scheme 8 Catalytic dehydrogenation of iPr2NHBH3 using [H2Al(μ-NiPr2)]2. | |
Further evidence for aluminium hydrides/dihydrides as the active catalyst is reflected in the observation that (i) LiAlH4 catalyses the dehydrocoupling of Me2NHBH3, although the mechanism is much more complicated than that observed for Al(NMe2)3 and with substantially lower catalytic activity; (ii) a series of tBuO-substituted aluminium hydrides also catalyse the dehydrocoupling of amine-boranes at elevated temperatures with the dihydride [tBuOAlH2]2 showing the highest activity.26
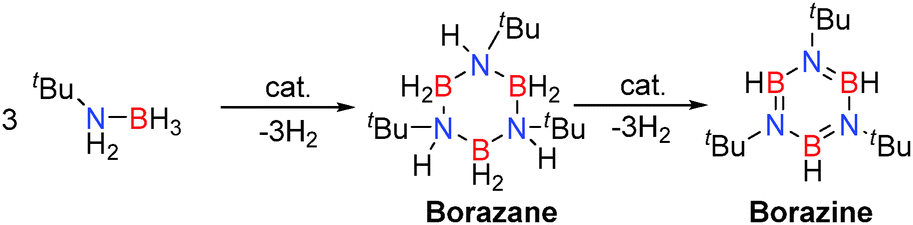
Replacement of AlIII by GaIII was interesting given their identical formal charges and similarity of their radii (due to the d-block contraction) thereby offering potentially similar Lewis acidity. Their reactivity is reflected through a comparison of the ability of E(NMe2)3 (E = Al, Ga) to effect the dehydrocoupling of the primary amine-borane, tBuNH2BH3. In these reactions up to two equivalents of hydrogen can be released per equivalent of starting material, offering the potential to form ring compounds such as borazanes (tBuNHBH2)3 and borazines (tBuNBH)3 by elimination of either one or two equivalents of H2 respectively, or formation of polymeric compounds such as [–(R)NB(H)–]n. Preparative scale reactions at reduced temperatures allowed the intermediate borazane (isoelectronic with cyclohexane derivatives) [tBuNHBH2]3 to be isolated and structurally characterised.25 Under ambient conditions (3 mol% Al(NMe2)3 or 5 mol% Ga(NMe2)3) further dehydrocoupling afforded the borazine (tBuNBH)3 in addition to an unidentified BN containing polymeric product.25 This is similar to transition metal processes, such as with the catalyst [Rh(1,5-cod)(μ-Cl)]2, in which borazanes are intermediates en route to borazines in the dehydrocoupling of primary amine-boranes (Scheme 9).13,27
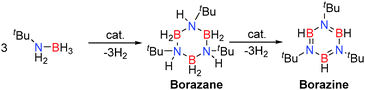 |
| | Scheme 9 Dehydrocoupling of primary amine-boranes to give borazines via borazane intermediates. | |
Notably gallium appeared to be more active than the corresponding aluminium complexes however precipitation of gallium metal during the reaction reflected a poorer catalyst lifetime. This appears to be due to reduced redox stability which resulted in precipitation of gallium metal during the reaction. Such redox instability was also observed in the reduction of tin(II) and arsenic(III) to either the metal or a Zintl phase during the homo-dehydrocoupling of phosphines.13,25,28
2.1.3 B–N bond formation using group 4 metals.
Tin(II) compounds have also been examined in the catalytic dehydrogenation of amine-boranes. Cp*2SnCl2 and Ph2SnCl2 were examined as pre-catalysts for the dehydrocoupling of Me2NHBH3 but were only found to be only slightly active giving 69% and 47% conversion in benzene at 65 °C using 10 mol% of the tin compound. SnCl2 was also found to be a poor catalyst for the reaction giving only 23% conversion under the same conditions. The precipitation of metallic tin from these reactions accounts for the reduced activity of the tin catalyst relative to the aluminium analogues above. The tin catalysts were also found to be less selective than the aluminium compounds giving a variety of different products from the reaction including the dimer [Me2NBH2]2, Me2NHBH2NMe2, H2BNMe2BH3, as well as other unidentified species (Table 2).29
Table 2 Dehydrocoupling of Me2NHBH3 using tin catalysts
| Catalyst |
[Me2NBH2]2 (%) |
Me2NHBH2NMe2 (%) |
H2BNMe2BH3 (%) |
Other (%) |
| Cp*2SnCl2 |
67 |
25 |
7 |
<5 |
| Ph2SnCl2 |
18 |
32 |
16 |
34 |
| SnCl2 |
39 |
12 |
2 |
21 |
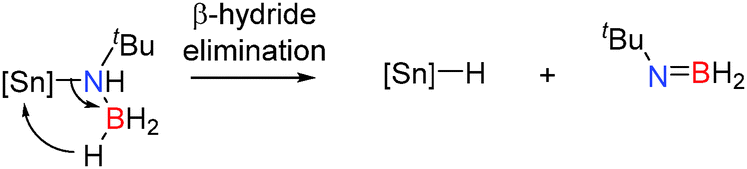
Similar dehydrocoupling studies using tBuNH2BH3 as a substrate with Cp*2SnCl2 (10 mol%), Ph2SnCl2 (10 mol%) and SnCl2 (5 mol%) were examined. After 4–5 days at 65 °C a range of dehydrocoupled products were obtained with 95%, 93% and 84% conversion respectively (Table 3).29 Whilst these reactions showed significantly higher conversions than the corresponding dehydrocoupling reactions with secondary amine-boranes (Table 2), they were also markedly less selective than the aluminium catalysts affording a wide range of product distribution and surprisingly little borazine product. Notably the lack of borazine product may offer distinct advantages in the context of hydrogen storage since borazines are known to poison fuel cells.9 It is interesting to note that whilst the SnIV catalysts gave quite a lot of the dehydrogenated tBuN(H)BH2 product, SnCl2 only yielded <5% of this product, consistent with the harder more Lewis acidic SnIV centre favouring β-hydride elimination. It is less clear what the active catalyst(s) is/are during these reactions, though the intermediacy of tin-hydrides is likely based on studies on group 2 and 3 metals. For example, dehydrogenation of tBuNH2BH3 most likely occurs via a β-hydride elimination from a tin amido-borane complex, affording the amino-borane tBuN(H)BH2 and generating a tin hydride (Scheme 10),29 in a similar manner to that observed for the dehydrocoupling of iPr2NHBH3 with Al(NiPr2)3 and the dehydrocoupling of amine-boranes with group 2 catalysts.
Table 3 Dehydrocoupling of tBuNH2BH3 using tin catalysts
| Catalyst |
Polymers (%) |
(tBuNBH)3 (%) |
t
BuNHBH2 (%) |
t
BuNHB2H5 (%) |
Other (%) |
| Cp*2SnCl2 |
20 |
0 |
16 |
26 |
30 |
| Ph2SnCl2 |
41 |
8 |
23 |
23 |
30 |
| SnCl2 |
13 |
6 |
<5 |
33 |
41 |
 |
| | Scheme 10 β-Hydride elimination from the tin amido-borane to give a tin hydride and tBuN(H)BH2. | |
2.2 B–P bond formation
The dehydrocoupling of phosphine-boranes would be expected to be easier than amine-boranes due to the weaker P–H bond relative to the N–H bond. The strong Lewis acid B(C6F5)3 was found to be a good catalyst for the dehydrocoupling of phosphine-boranes to form polymeric compounds. 0.5 mol% B(C6F5)3 was found to catalyse the dehydrocoupling of PhPH2BH3 to form poly(phenylphosphino)boranes [PhPH–BH2]n in quantitative yield after 3 days at 20 °C or after 3 h at 90 °C (Scheme 13).33 In this reaction B(C6F5)3 was found to be much more active than the transition metal Rh(I) catalyst (RhI) reported around the same time which afforded the polymer in 100% yield after 15 h at 110 °C using the same catalyst loading.34 In addition the dehydrocoupling of phosphine-borane PH3BH3 was also achieved. PH3 and BH3 were bubbled into solution of B(C6F5)3 (ca. 5 mol%) and the resulting solution heated to 70 °C. The 31P and 11B NMR spectra showed the formation of oligomeric and polymeric poly(phosphinoboranes) H3P(BH2PH2)nBH3 species (Scheme 13). The mechanism likely follows that identified for other B(C6F5)3 reactions in which the most hydridic E–H bond is activated by the Lewis acidic boron atom (Section 2.3).33
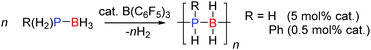 |
| | Scheme 13 Dehydrocoupling of phosphine-boranes using B(C6F5)3. | |
2.3 Si–E bond formation
Hetero-dehydrocoupling to form Si–E bonds has also been an area of significant interest given the diversity of applications of compounds such as silicones and silazanes ranging from construction materials through to elastomers and medical applications (vide infra). In many of these cases the outcome of Si–E (E = N, O, S) bond forming reactions using Lewis acids is strongly dependent upon the coupling reaction since the lone pairs on amines, alcohols and thiols can act as efficient Lewis bases thereby shutting down catalyst activity: for example, with small sterically unencumbered primary alkyl alcohols (EtOH and nPrOH) catalyst ‘poisoning’ occurs due to the strongly Lewis basic nature of the lone pair on O which favours formation of strong adducts with the Lewis acid. However, for the less basic thiols, more weakly basic alcohols such as catechol (π-conjugation of the O–lone pair to the aromatic ring) or secondary amines (in which the steric bulk inhibits formation of a Lewis acid–base pair) reactivity is observed in which the rate of reaction was dependent upon the concentration of the oxygen-donor. Conversely the weaker nature of the B–S bond (compared to B–O) means that Lewis acid-catalysed reactions in the presence of thiols reveal a higher activity than the corresponding alcohol due to the poorer Lewis basic nature of sulfur resulting in more free borane for Si–H activation.
2.3.1 Si–O bond formation.
The chemical inertness of the Si–O bond furnishes a range of chemically stable siloxanes (silicones) which occur in many aspects of everyday life; the inertness and oxygen permeability of polysiloxanes (Me2SiO)n favours their use in medical applications ranging from contact lenses to surgical implants, whilst they are also used in building materials such as water-repellent silicone sealants inter alia.35 Si–OC coupling has also gained interest for its applications in silicone-grafted ethers. Conventionally precious metal catalysts such as rhodium and platinum have been employed in the dehydrocoupling of alcohols with hydrosilanes. However, these catalysts suffer from the disadvantage that they are frequently intolerant to CC or C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C functional groups often resulting in sequential dehydrocoupling/hydrogenation reactions. However the strong Lewis acid B(C6F5)3 has been found to be catalytically active in the dehydrocoupling of alcohols with silanes to form Si–O bonds and has the potential to support a wider range of substrates. These reactions were found to occur in high yields at room temperature using 2 mol% catalyst (eqn (3)).36
C functional groups often resulting in sequential dehydrocoupling/hydrogenation reactions. However the strong Lewis acid B(C6F5)3 has been found to be catalytically active in the dehydrocoupling of alcohols with silanes to form Si–O bonds and has the potential to support a wider range of substrates. These reactions were found to occur in high yields at room temperature using 2 mol% catalyst (eqn (3)).36| |
 | (3) |
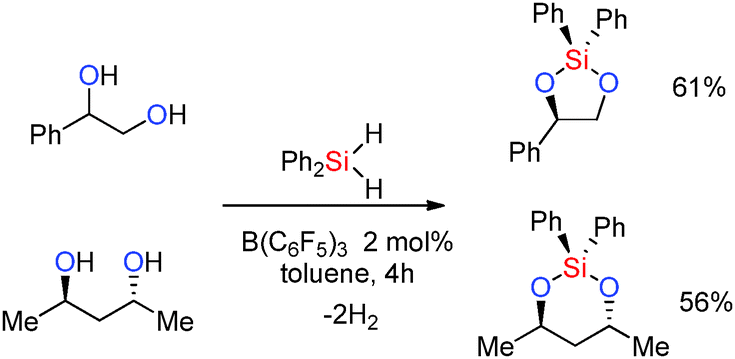
The reaction times (a few hours to several days) reflect the potential of the alcohol to poison the Lewis-acid through (reversible) adduct formation. Thus primary aliphatic alcohols ROH gave the silylated product cleanly albeit with longer reaction times of ca. 1–6 days at room temperature although accelerated rates were observed when heating the reaction to 60 °C [thereby labilising the ROH·B(C6F5)3 adduct] or by using higher catalyst loadings (8 mol%).36 With secondary and tertiary alcohols much faster reactions were observed with the products being formed in just 30 min to 1 h (see ESI,† Table S1). Reactions with dihydrosilanes were also possible and allowed for two dehydrocoupling reactions to occur for each silane molecule. Thus the reactions of a 1,2- or 1,3-diol with diphenylsilane resulted in elimination of two equivalents of dihydrogen and the formation of cyclic siloxanes in moderate yields (Scheme 14).36
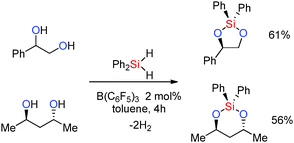 |
| | Scheme 14 Catalytic dehydrocoupling of 1,2- and 1,3-diols with diphenylsilane. | |
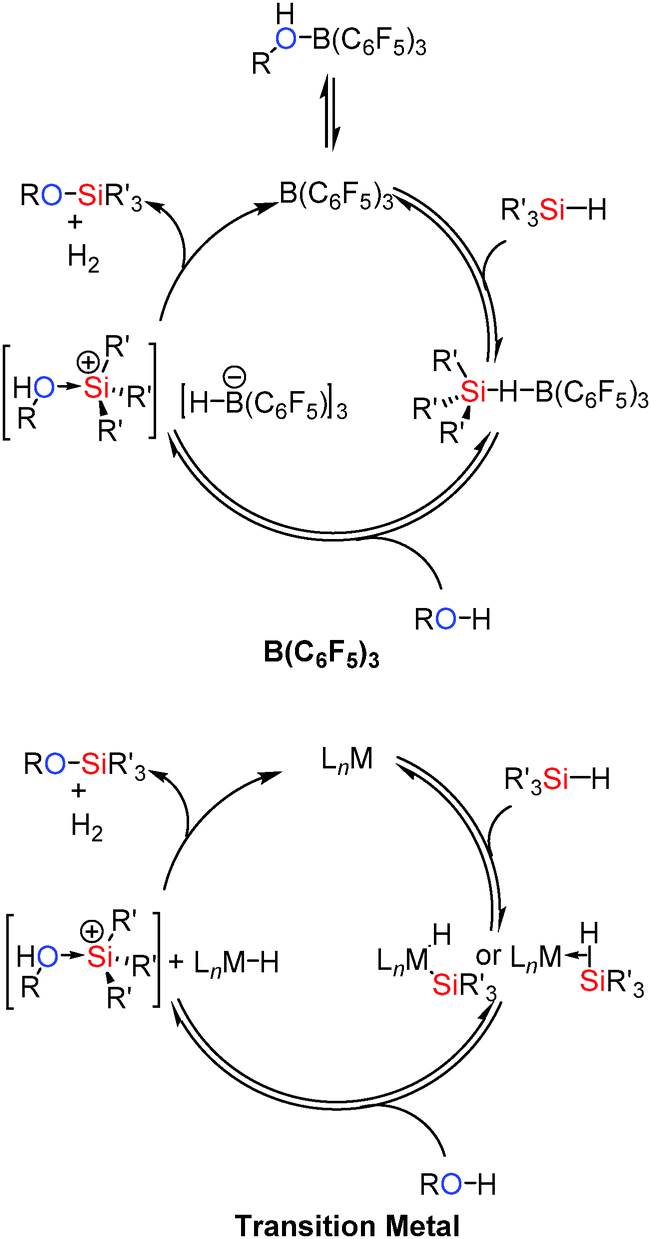
The mechanism for the reaction is thought to be related to that in the catalytic hydrosilation of carbonyls using B(C6F5)3.37 Thus the borane activates the silane Si–H bond via formation of a bridging hydride. Removal of electron density from the Si–H bond activates the Si to nucleophilic attack from the alcohol yielding [R(H)O–SiR3′]+ and [HB(C6F5)3]−. This can then readily liberate H2 to give the coupled product and regenerate B(C6F5)3 (Scheme 15). Since the lone pairs on the alcohol (or other functional groups present) may interact with the catalyst, dissociation of the alcohol from the [R(H)O–B(C6F5)3] adduct to regenerate free B(C6F5)3 is necessary before it can interact with the silane. Thus more basic and less sterically hindered primary alcohols react more slowly; an observation that has also been seen in some transition metal catalysed systems.38 These mechanisms have been found to be similar to those observed for transition metal catalysed Si–O bond forming dehydrocoupling reactions in that Si–H bond activation occurs in the initial step of the reaction.39 In the case of the transition metals this is via oxidative addition or by formation of a σ-complex40 which is then activated to nucleophilic attack from the alcohol to give a trialkylsubstituted silylium cation as an alcohol adduct. This acidic species can then protonate the metal hydride [similar to the boron hydrido species [(C6F5)3B–H]−] eliminating H2 and regenerating the catalyst (Scheme 15).36
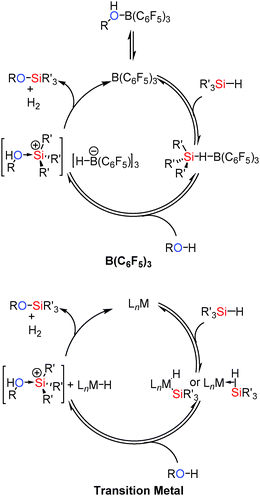 |
| | Scheme 15 Similarities between B(C6F5)3 (top) and transition metal (bottom) catalysed Si–O coupling. | |
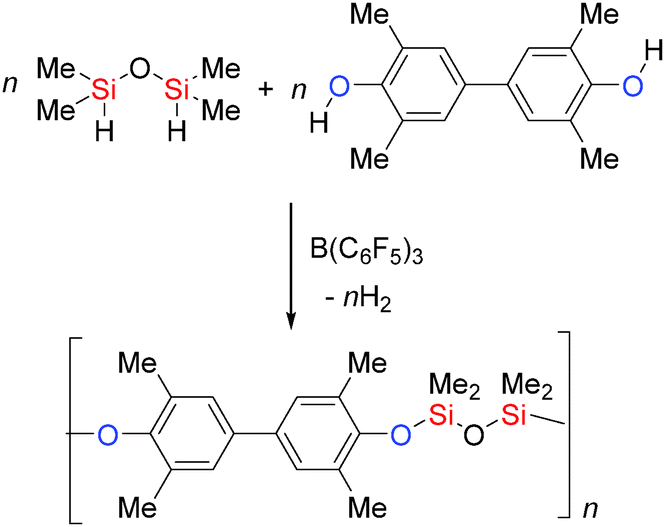
B(C6F5)3 has been employed in the synthesis of the Si–O containing polymers polyaryloxysilanes and siloxanes. The rapid dehydrocoupling of bis-phenols with dihydrosilanes was achieved using low catalyst loadings (0.1–0.025 mol%) to yield high molecular weight Si–O polymers and with higher catalyst loadings (2–5 mol%) lower molecular weight polymers were obtained (Scheme 16).41
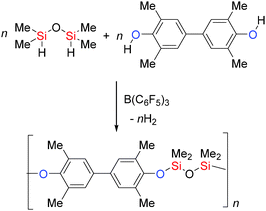 |
| | Scheme 16 Dehydrocoupling to generate Si–O containing polymers. | |
The dehydrocoupling of phenols with the silane Et3SiH using a phosphonium catalyst [(C6F5)3PF][B(C6F5)4]42 (Scheme 17) has also been studied (see ESI,† Fig. S1 and Table S2). The phosphonium ion is a very strong phosphorus centred Lewis acid. Whilst B(C6F5)3 described above owes its high Lewis activity to the presence of a vacant p-orbital in combination with three electron withdrawing C6F5 groups, the electrophilic phosphonium ion is highly electrophilic due to the presence of electron withdrawing groups in combination with the cationic charge resulting in a low lying P–F σ*-orbital. This catalyst leads to rapid quantitative conversion to ROSiEt3 products in just 2 h using C6H5OH or (o-Me)2C6H3OH and in 3 h using p-MeC6H4OH.42 When the alcohol contains a strongly electron-withdrawing group, e.g. C6F5OH, then slower reaction rates are observed taking a day to reach completion. With the phenol p-MeOC6H4OH demethylation and silylation of the methoxy group was observed in addition to the dehydrocoupling of the alcohol. In this reaction the former process could be prevented by adding one equivalent of the silane. Finally the dehydrocoupling of the carboxylic acid p-C8H17(C6H4)CO2H with Et3SiH was also possible giving full conversion to the ester p-C8H17(C6H4)CO2–SiEt3 in less than one hour.42 The mechanism follows that described in Section 2.3.3 for Si–N bond formation via dehydrocoupling in which a hypervalent trigonal bipyramidal phosphorus atom is postulated in Si–H bond activation.
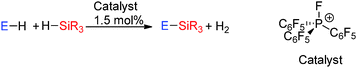 |
| | Scheme 17 Dehydrocoupling of alcohols and thiols with silanes using a phosphonium ion catalyst (the [B(C6F5)4]− counterion is not shown). | |
2.3.2 Si–S bond formation.
The weaker basicity of the thiol RSH inhibits poisoning of the B(C6F5)3 catalyst and even relatively unencumbered thiols such as nPrSH can be successfully used in hetero-dehydrocoupling reactions. For example reactions of RSH (R = nPr, p-tol) with tertiary silanes using low B(C6F5)3 loadings of 0.01–0.50 mol% gave quantitative yields of the product at room temperature in less than 75 min (Scheme 18).43
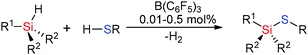 |
| | Scheme 18 Dehydrocoupling of silanes with thiols. | |
Selectivity for Si–X bond formation over Si–Si bond cleavage was observed in some cases in the hetero-dehydrocoupling reactions of disilanes with alcohols and thiols to form Si–O and Si–S bonds using catalytic amounts of B(C6F5)3. Less sterically encumbered sym-dihydridodisilanes [R2SiH]2 also undergo dehydrocoupling reactions with thiols R′SH (R′ = nPr, p-tol) using catalytic amounts of B(C6F5)3 to give [R2SiSR′]2 (Scheme 19).43 In these reactions disilanes bearing smaller R-groups showed faster reactions with the bulky disilane [iPr2SiH]2 showing no reactivity suggesting that a key step may be the activation of the Si–H bond by the Lewis acid catalyst. In the case of 1,2-benzene dithiol as the starting material an intermolecular reaction took place affording a C2S2Si2 heterocyclic ring (Scheme 19).43 These reactions were extremely fast, for example, the reaction of 1,2-benzenedithiol with [Me2SiH]2 using 0.004 mol% B(C6F5)3 gave the product in just 30 minutes.43 The hetero-dehydrocoupling of silanes with thiols also takes place in an analogous fashion to that between alcohols and silanes when using the fluorophosphonium catalyst, [(C6F5)3PF][B(C6F5)4] (Scheme 17).42 The formation of Si–S bonds from the dehydrocoupling of thiols with Et3SiH generally formed the dehydrocoupled product quantitatively in less than one hour (see ESI,† Table S3). Importantly, the dehydrocoupling of thiols using the phosphonium catalyst was found to be faster than that for the dehydrocoupling with alcohols presumably due to the weaker S–H bond compared to O–H. As with the dehydrocoupling of the alcohol C6F5OH, the corresponding thiol C6F5SH also resulted in slightly slower reaction rates taking 1 week to reach completion at ambient temperatures but could be accelerated by heating the reaction to 100 °C to give complete conversion to C6F5S–SiEt3 in 3 h.42 Mechanistically, the hetero-dehydrocoupling of Si–H and E–H (E = N, O, S) is believed to occur via the same process which is discussed in Section 2.3.3 for Si–N bond formation.
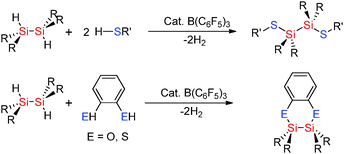 |
| | Scheme 19 Dehydrocoupling of dihydridodisilanes with thiols and alcohols. | |
The Lewis acidic borane B(C6F5)3 in the presence of thiols has been found to be selective in the Si–H bond cleavage of poly(phenyl)silane to generate thiol substituted polysilanes by dehydrocoupling reactions.43 When poly(phenyl)silane and nPrSH or p-tolSH were mixed with a catalytic amount of B(C6F5)3 (5 mol%), release of H2 was observed resulting in solid polymers in which hetero-dehydrocoupling (to form Si–S bonds) occurred (Scheme 20). The degree of Si–H to Si–S dehydrocoupling in the polysilane polymer was estimated by NMR spectroscopy to be 15–40% with both νSi–H bands and νSi–S bands being observed in the IR spectrum.44
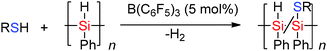 |
| | Scheme 20 Hetero-dehydrocoupling to form Si–Si and Si–S bonds. | |
2.3.3 Si–N bond formation.
Si–N bonds are usually synthesised by the aminolysis of chlorosilanes.45 However alternative methods such as dehydrocoupling which do not release HCl are of interest. The dehydrogenic Si–N coupling between hydrosilanes and anilines, carbazoles and indoles have all been catalysed using B(C6F5)3 (Scheme 21, also see ESI,† Table S4).46 Here too the potential for the N lone pair to coordinate to the Lewis acid catalyst can potentially prove problematic for less sterically hindered primary amines (cf. alcohols in Section 2.3.1) but B(C6F5)3 appears active towards a wide range of amines. For example, in the dehydrocoupling of diphenylamines the 5 mol% and 1 mol% loading of B(C6F5)3 in the dehydrocoupling of bis(4-tolyl)amine with Ph2MeSiH led to the formation of the dehydrocoupled product in 95% and 73% yield respectively.46 Carbazoles have also been used as substrates giving the Si–N products in 83–97% yields using 1 mol% catalyst. In addition, a variety of silanes could be used in the reaction. Thus Et3SiH or (Me2SiH)2O could be used in the dehydrocoupling with carbazole or bis(4-tolyl)amine respectively. Silylation reactions of primary anilines including electron deficient anilines was also very effective at 60–70 °C to give the products in 88–97% yield.46 The dehydrocoupling of diamine, pyrrole and indole derivatives were also examined. Whilst pyrroles were unreactive, diamines and indoles were active in the intermolecular heterodehydrocoupling with silanes. Indoles were selectively transformed into 1-silylated indoline products via a dehydrocoupling/hydrogenation reaction.46
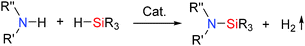 |
| | Scheme 21 Si–N coupling using B(C6F5)3 or the fluorophosphonium catalyst [(C6F5)3PF][B(C6F5)4]. | |
The Si–N dehydrocoupling reactions performed using 1.5 mol% of the fluorophosphonium catalyst [(C6F5)3PF]+ described earlier (Section 2.3.1) were also performed (Scheme 21, also see ESI,† Table S5) and found to be dependent upon the silane used.42 Thus, increased reaction times were necessary to give the products in appreciable yields when using bulky silanes such as Ph3SiH or PhMe2SiH and, with even more sterically encumbered silanes (e.g.iPr3SiH), no reaction was observed. The best results were therefore observed using the smaller silane Et3SiH.42
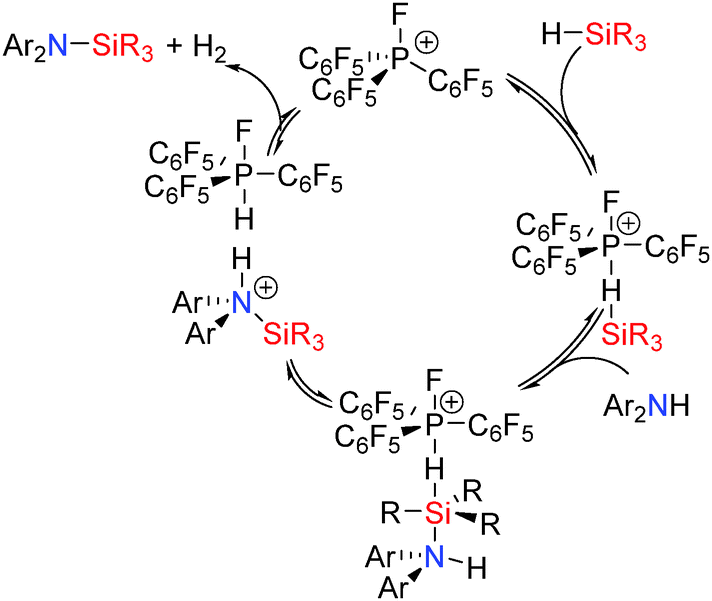
The mechanism for the dehydrocoupling (Scheme 22) is proposed to be similar to that described for the Lewis acid B(C6F5)3 and transition metals systems reported earlier and involves initial interaction of the Si–H bond with the fluorophosphonium [(C6F5)3PF]+ Lewis acid. This activates the Si–H bond to attack from the Lewis basic coupling partner (N, S, O) yielding a hypervalent silicon species containing hydridic (Si–Hδ−) and protic (E–Hδ+) hydrogen atoms which readily release H2 generating the dehydrocoupled product and regenerating the catalyst.42 This is consistent with the observation that sterically encumbered silanes were less effective in the reaction in which a hypervalent silicon species is formed. Additionally, small and/or basic amines (e.g.iPr2NH or aniline) are not as active in the dehydrocoupling reaction with Et3SiH due to interaction of the Lewis basic amine with the catalyst in competition with the silane similar to that observed for B(C6F5)3 with less sterically bulky alcohols or amines. This competitive process was supported by the observation that the reaction was accelerated when excess silane was added whilst addition of an excess of amine diminished the reaction rate.42
 |
| | Scheme 22 Catalytic dehydrocoupling using phosphonium catalysts (the [B(C6F5)4]− counterion is not shown). | |
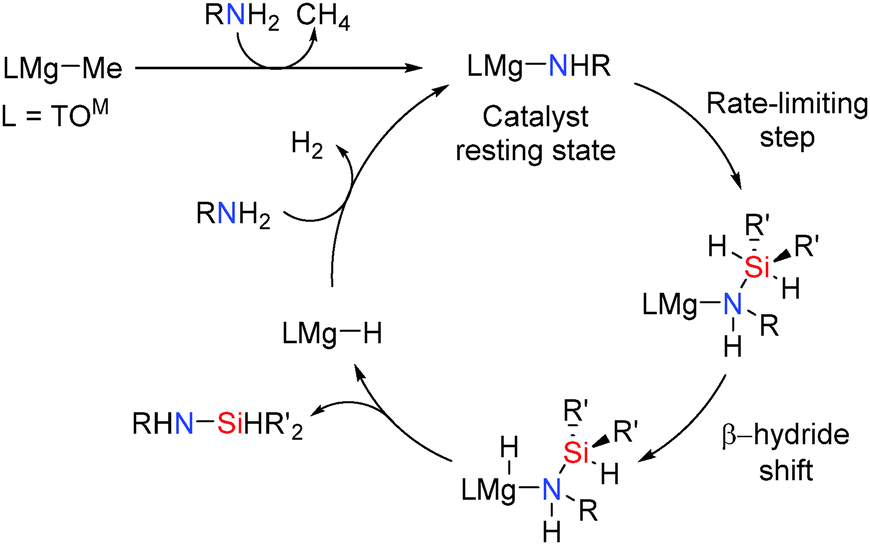
Group 2 catalysts have also been found to promote Si–N coupling. The azametallacyclopropane Ca(η2-Ph2CNPh)(hmpa)3 was the first alkaline earth catalyst to be described in catalytic Si–N dehydrocoupling.47 Subsequently more extensive studies into the activity of alkaline earth pre-catalysts and the mechanisms of dehydrocoupling were made. For example, magnesium catalysts have also been found to be active in the formation of Si–N bonds via dehydrocoupling reactions.48 The 4-coordinate magnesium pre-catalyst TOMMgMe [TOM = tris(4,4-dimethyl-2-oxazolinyl)phenylborate] (Scheme 23) was found to be active in the cross-dehydrocoupling of N–H and Si–H bonds to give Si–N bonded products with the release of hydrogen. Using 5 mol% catalyst the Si–N silazane product from the reactions between amines and silanes could be formed in high yields after 24 h at room temperature (see ESI,† Table S6).48 The catalytic dehydrocoupling of hydrazine N2H4, with silanes (Et3SiH, (C3H5)Me2SiH and BnMe2SiH) was also possible giving the mono-silylhydrazines R3SiNHNH2. Whilst the reaction with (C3H5)Me2SiH goes to completion after 7 h to give (C3H5)Me2SiNHNH2 the reactions with Et3SiH or BnMe2SiH were much slower.48 Selective mono-silylation of ammonia was also possible (Scheme 23). This is interesting because the N–H proton is not acidic (pKa of 41 in DMSO) and thus single N–H activation of ammonia is difficult. In addition, multiple silylation might also be expected since once one N–H bond is replaced by an N–Si bond the remaining N–H bonds become more acidic thereby rendering mono-silylation difficult and potentially resulting in multiple silylation reactions. In the reaction of ammonia with BnMe2SiH or (C3H5)Me2SiH using 5 mol% TOMMgMe the mono-silylated products BnMe2SiNH2 and (C3H5)Me2SiNH2 were formed exclusively after 15 h and 5 h respectively.48
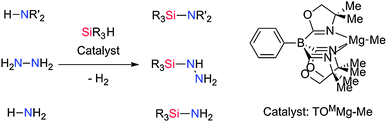 |
| | Scheme 23 Structure of the catalyst TOMMgMe and Si–N coupling reactions. | |
The mechanism for this reaction is shown in Scheme 24. The catalyst resting state in these reactions was observed to be the magnesium amide TOMMgNHR (R = Pr, iPr, Ph) which is formed rapidly upon reaction of the amine with the pre-catalyst TOMMgMe. Mechanistic studies suggested that the reactions involve nucleophilic attack of the metal amide onto the silicon centre to form a five-coordinate silicon intermediate. This is the rate limiting step which is then followed by a β-hydride type transfer from the silicon atom to the magnesium centre (a similar process to that seen in the dehydrocoupling of amine boranes using aluminium hydrides described earlier). This then generates the metal hydride and the silazane product. This mechanism is consistent with the observation that more nucleophilic amines react faster in the rate determining step and secondly that changing the amine concentration had no effect on the rate of the reaction even at high amine concentrations showing a zeroth-order rate law in amine concentration.48 The rate law for the reaction was found to be:
| Rate = k[amine]0[cat]1[silane]1 |
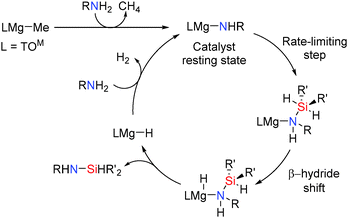 |
| | Scheme 24 Proposed mechanism for magnesium catalysed Si–N bond formation. | |
Alkaline earth amido complexes [M{N(SiMe3)2}2]2 (M = Mg, Ca or Sr) were also found to be active in dehydrogenative Si–N bond formation when using a 5 mol% loading of the pre-catalyst.49 These reactions were possible for a variety of amines with the calcium pre-catalysts being the more active than the strontium and magnesium counterpart, consistent with their enhanced Lewis acidity (high charge:radius ratio). The steric and electronic properties of the amine and silane were also found to be influential in the extent and rate of formation of the silazane product. In contrast to that found for the TOMMgMe pre-catalyst described above, the rate of the dehydrocoupling reactions involving PhMeSiH2 and tBuNH2 using [M{N(SiMe3)2}2]2 (M = Mg, Ca) were found to be dependent upon the concentration of amine and not the silane. The zero order dependence upon the concentration of silane suggests that in this case the silane plays no role in the rate-limiting step.49 The reactions with magnesium and calcium could be described using the rate equation:
| Rate = k[amine]1[cat]1[silane]0 |
The strontium catalysts showed a different rate law being second order depending upon both the concentrations of amine and silane suggesting that perhaps Si–H/Mg–N σ-bond metathesis step to give a Sr–H bond is the rate determining step.49
| Rate = k[cat]2[amine]1[silane]1 |
Importantly, the order of the overall reaction, the method of activation of the pre-catalyst and the rate limiting step vary depending upon the alkaline earth metal used and are dependent upon the radius of the M2+ ion and the consequent charge density at the group 2 metal centre.
2.3.4 Si–C bond formation.
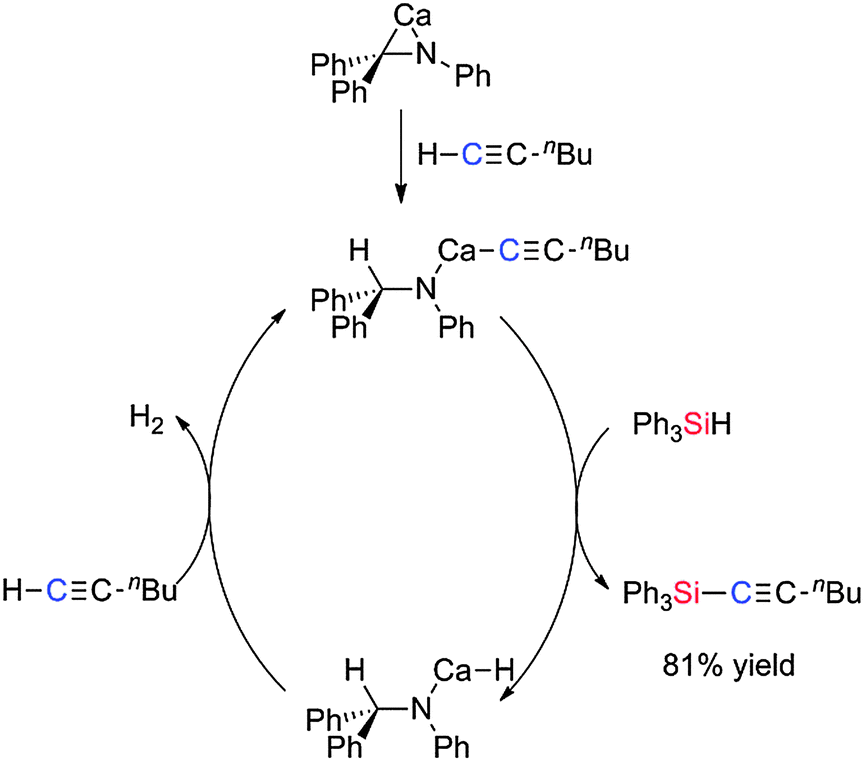
Si–C coupling between silanes and the terminal acetylene C4H9CCH has been achieved using the azametallacyclopropane Ca(η2-Ph2CNPh)(hmpa)3.47 The reaction of 1-hexyne with Ph3SiH yielded the expected product Ph3Si–CC–C4H9 in 81% yield using a 5 mol% loading of the catalyst. With Ph(Me)SiH2 a mixture of mono- and di-alkynyl products were formed with more of the mono-alkynyl product being formed with higher silane/alkyne ratios. The active catalyst in the reaction is formed from the reaction of the azametallacyclopropane with the terminal alkyne to form the calcium acetylide complex which can then react with the silane generating the calcium hydride species and the Si–C coupled product. Reaction of the Ca–H species with a molecule of alkyne regenerates the Ca–acetylide complex with release of hydrogen (Scheme 25).47
 |
| | Scheme 25 Si–C coupling using a calcium catalyst. | |
3. General mechanistic aspects of main group catalysed dehydrocoupling
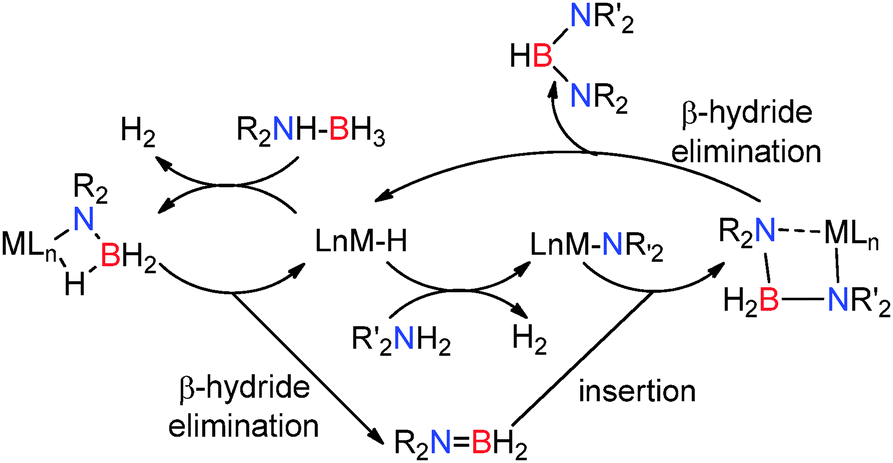
An examination of this varied group of dehydrocoupling processes discussed in Section 2 reveal a number of common features which lead to several generic catalytic cycles which we can separate into two categories: (i) Lewis-acid catalysed processes and (ii) s-block and p-block metal-catalysed reactions implementing Lewis acidic metals coupled to strongly basic substituents e.g. metal hydrides, metal-alkyls and metal-amides all of which have the potential to activate the substrate by deprotonation.
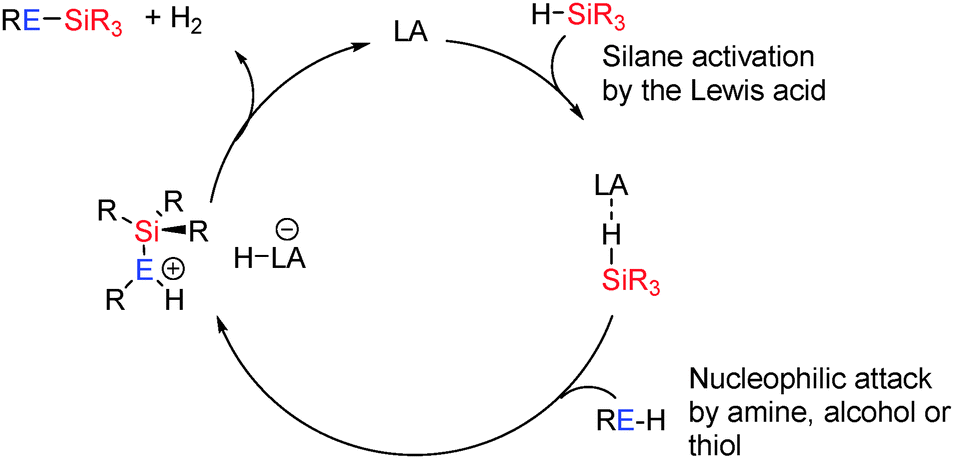
3.1 Main group Lewis-acid catalysed dehydrocoupling
The most common Lewis-acid used in these reactions is B(C6F5)3 which is a strong but sterically hindered Lewis acid. The [(C6F5)3PF]+ cation can also be considered Lewis acidic by virtue of the strongly electron-withdrawing perfluoroaryl groups and the potential for phosphorus to adopt a 5-coordinate hypervalent state. Both species can be deactivated in the presence of strong Lewis-bases (alcohols, amines, thiols) through Lewis acid/Lewis base adduct formation. However, the steric demands at the Lewis acidic B or P+ centre typically make such adduct formation reversible particularly at elevated temperatures or, for sterically demanding substrates, impossible. These Lewis acidic centres appear to activate the E–H bonds bearing hydridic hydrogens (e.g. Si–H) making the heteroatom E itself more susceptible to nucleophilic attack by a lone pair on ROH, R2NH, RSH etc. This is then followed by H2 elimination to generate the E–E′ bond (Scheme 26).
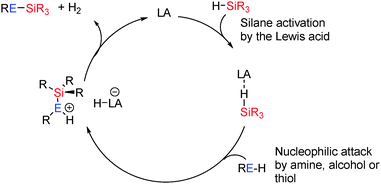 |
| | Scheme 26 General mechanism for Lewis acid catalysed dehydrocoupling. | |
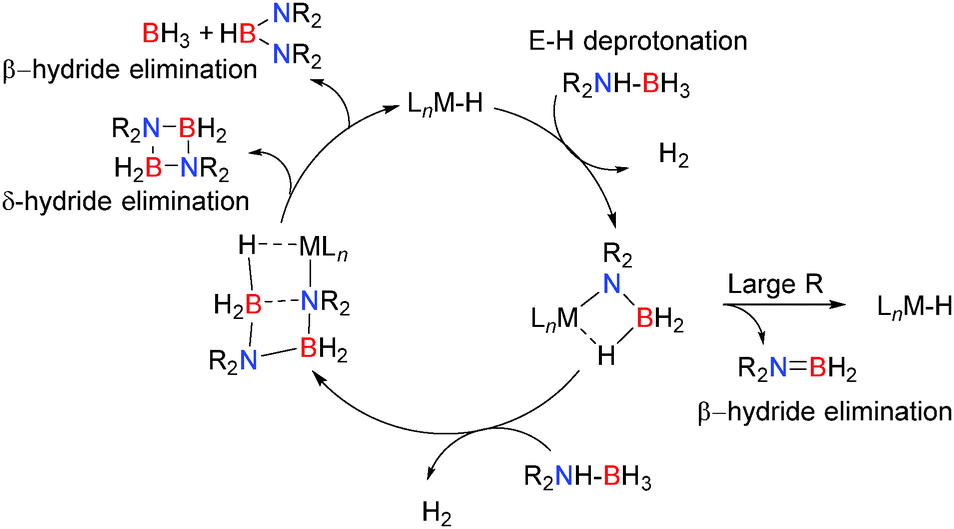
3.2 Basic s- and p-block Lewis-acid catalysed dehydrocoupling
Here the reaction chemistry appears somewhat different but common themes are already emerging in both alkaline earth and group 13 chemistry. Typically the catalyst contains one or more M–H, M–R or M–NR2 groups which are strongly basic and deprotonate the E–H bonds bearing protic hydrogens e.g. N–H in the initial step of the reaction. In the case of amine-borane dehydrocoupling, sterically demanding groups on the substrate favour a close M⋯H–B contact which leads to formation of R2NBH2via a β-hydride elimination. For less sterically demanding groups, the larger angle accommodated at nitrogen disfavours β-hydride elimination and a bimolecular reaction becomes favoured in which the terminal boron atom becomes activated to nucleophilic attack and a chain growth process occurs to form M–NR2BH2NR2BH3. In most cases the resultant six-membered intermediate can be generated which then favours a δ-hydride elimination, regenerating the active metal-hydride catalyst (Scheme 27).
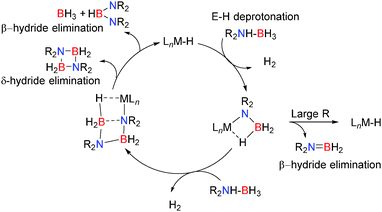 |
| | Scheme 27 General mechanism for Lewis acid catalysed dehydrocoupling. | |
4. Conclusions
In the last two decades we have witnessed a remarkable revolution in the area of main group catalysed dehydrocoupling reactions including both s- and p-block metals as well as non-metal catalysed processes. In many cases these main group catalysts reveal remarkable selectivity in product formation, often affording a single major product. Whilst such reactivity initially appeared diverse, common themes are now becoming apparent in their reactivity and mechanistic pathways which should favour more tailored approaches to not only optimise catalyst activity but also control the outcome of such dehydrocoupling reactions.
Future work in the area of main group catalysis should focus on the development of new types of E–E′ bond-forming reactions and the optimisation of catalyst activity. With regard to the latter, unlike transition metal chemistry, it should be noted that there have been no previous systematic attempts to vary the ligand set in order to optimise activity of main group metal catalysts. As is already apparent from studies so far, changing the steric demands of the ligands in the pre-catalysts is one obvious way of modifying both the kinetics and selectivity. However, a further way of enhancing activity in the future is to control the Lewis acidity of the metal centres, and increase redox stability. This would lead to greater polarisation of metal-bonded intermediates and potentially longer catalyst life-times.
References
- E. M. Leitao, T. Jurca and I. Manners, Nat. Chem., 2013, 5, 817–829 CrossRef CAS PubMed , and references therein.
- T. R. Hogness, T. L. Wilson and W. C. Johnson, J. Am. Chem. Soc., 1936, 58, 108–112 CrossRef CAS.
- C. H. Lewis, H. C. Kelly, M. B. Giusto and S. Johnson, J. Electrochem. Soc., 1961, 108, 1114–1118 CrossRef CAS.
- N. N. Greenwood and R. Greatrex, Pure Appl. Chem., 1987, 59, 857–868 CrossRef CAS.
-
Chemistry of the elements, ed. N. N. Greenwood and A. Earnshaw, Butterworth–Heinemann, Oxford, 2nd edn, 1997 Search PubMed.
- Compiled E–H bond enthalpies drawn from multiple sources.
- L. S. Kassel, J. Am. Chem. Soc., 1932, 54, 3949–3961 CrossRef CAS.
- K. Tamaru, M. Boudart and H. Talor, J. Phys. Chem., 1955, 59, 801–805 CrossRef CAS.
- C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279–293 RSC , and references therein.
- T. J. Clark, K. Lee and I. Manners, Chem. – Eur. J., 2006, 12, 8634–8648 CrossRef CAS PubMed , and references therein.
- R. Waterman, Chem. Soc. Rev., 2013, 42, 5629–5641 RSC.
- G. Wolf, J. Baumann, F. Baitalow and F. P Hoffmann, Thermochim. Acta, 2000, 343, 19–25 CrossRef CAS.
- R. J. Less, R. L. Melen and D. S. Wright, RSC Adv., 2012, 2, 2191–2199 RSC , and references therein.
- D. J. Liptrot, M. S. Hill, M. F. Mahon and D. J. MacDougall, Chem. – Eur. J., 2010, 16, 8508–8515 CrossRef CAS PubMed.
- P. Bellham, M. S. Hill, D. J. Liptrot, D. J. MacDougall and M. F. Mahon, Chem. Commun., 2011, 47, 9060–9062 RSC.
- M. S. Hill, M. Hodgson, D. J. Liptrot and M. F. Mahon, Dalton Trans., 2011, 40, 7783–7790 RSC.
- H. Wadepohl, U. Kohl, M. Bittner and H. Koeppel, Organometallics, 2005, 24, 2097–2105 CrossRef CAS.
- J. Spielmann, G. Jansen, H. Bandmann and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 6290–6295 CrossRef PubMed.
- J. Spielmann, M. Bolteb and S. Harder, Chem. Commun., 2009, 6934–6936 RSC.
- A. G. M. Barrett, M. R. Crimmin, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2007, 26, 4076–4079 CrossRef CAS.
- J. Spielmann and S. Harder, J. Am. Chem. Soc., 2009, 131, 5064–5065 CrossRef CAS PubMed.
- P. Bellham, M. S. Hill, G. Kociok-Köhn and D. J. Liptrot, Chem. Commun., 2013, 49, 1960–1962 RSC.
- P. C. Keller, Inorg. Synth., 1977, 17, 30 CrossRef CAS.
- H. J. Cowley, M. S. Holt, R. L. Melen, J. M. Rawson and D. S. Wright, Chem. Commun., 2011, 47, 2682–2684 RSC.
- M. M. Hansmann, R. L. Melen and D. S. Wright, Chem. Sci., 2011, 2, 1554–1559 RSC.
- R. J. Less, H. R. Simmonds and D. S. Wright, Dalton Trans., 2014, 43, 5785–5792 RSC.
- C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424–9434 CrossRef CAS PubMed.
- R. L. Melen, Dalton Trans., 2014, 42, 16449–16465 RSC.
- K. A. Erickson, D. S. Wright and R. Waterman, J. Organomet. Chem., 2014, 751, 541–545 CrossRef CAS.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed , and references therein.
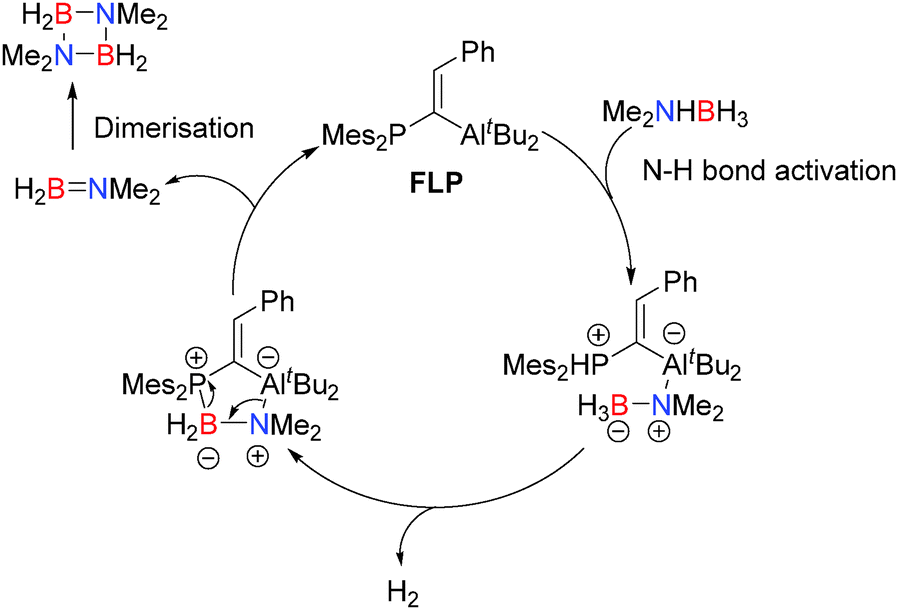
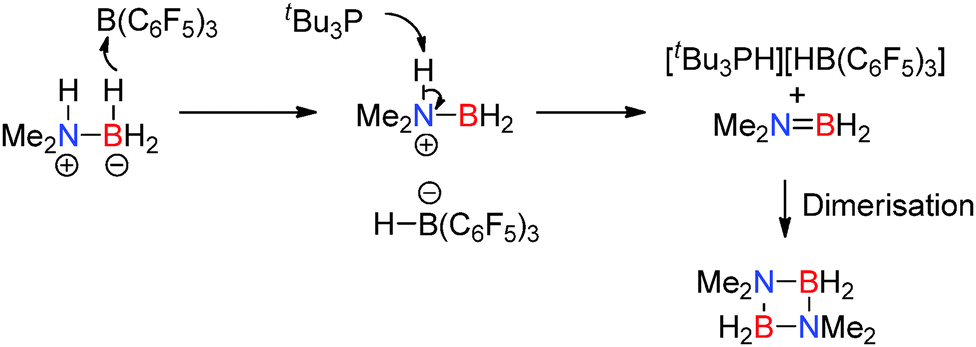
- C. Appelt, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2013, 52, 4256–4259 CrossRef CAS PubMed.
- A. J. M. Miller and J. E. Bercaw, Chem. Commun., 2010, 46, 1709–1711 RSC.
- J.-M. Denis, H. Forintos, H. Szelke, L. Toupet, T.-N. Pham, P.-J. Madec and A.-C. Gaumont, Chem. Commun., 2003, 54–55 RSC.
- H. Dorn, R. A. Singh, J. A. Massey, J. M. Nelson, C. A. Jaska, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2000, 122, 6669–6678 CrossRef CAS.
- S. C. Shit and P. Shah, Natl. Acad. Sci. Lett., 2013, 36, 355–365 CrossRef CAS.
- J. M. Blackwell, K. L. Foster, V. H. Beck and W. E. Piers, J. Org. Chem., 1999, 64, 4887–4892 CrossRef CAS PubMed.
- For example see: D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS PubMed.
- X. Luo and R. H. Crabtree, J. Am. Chem. Soc., 1989, 111, 2527–2535 CrossRef CAS.
- For example see: T. B. Gregg and A. R. Cutler, Organometallics, 1994, 13, 1039–1043 CrossRef.
- R. H. Crabtree, Angew. Chem., Int. Ed., 1993, 32, 789–805 CrossRef.
- J. Cella and S. Rubinsztajn, Macromolecules, 2008, 41, 6965–6971 CrossRef CAS.
- M. Pérez, C. B. Caputo, R. Dobrovetsky and D. W. Stephan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10917–10921 CrossRef PubMed.
- D. J. Harrison, D. R. Edwards, R. McDonald and L. Rosenberg, Dalton Trans., 2008, 3401–3411 RSC.
- P. T. K. Lee, M. K. Skjel and L. Rosenberg, Organometallics, 2013, 32, 1575–1578 CrossRef CAS.
- R. Fessenden and J. S. Fessenden, Chem. Rev., 1961, 61, 361–388 CrossRef CAS , and references therein.
- L. Greb, S. Tamke and J. Paradies, Chem. Commun., 2014, 50, 2318–2320 RSC.
- F. Buch and S. Harder, Organometallics, 2007, 26, 5132–5135 CrossRef CAS.
- J. F. Dunne, S. R. Neal, J. Engelkemier, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2011, 133, 16782–16785 CrossRef CAS PubMed.
- M. S. Hill, D. J. Liptrot, D. J. MacDougall, M. F. Mahon and T. P. Robinson, Chem. Sci., 2013, 4, 4212–4222 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cs00521c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a