
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantification of pharmaceutical peptides in human plasma by LC-ICP-MS sulfur detection
Laura Hyrup
Møller
a,
André
Macherius
b,
Thomas Hesselhøj
Hansen
b,
Hanne Mørck
Nielsen
a,
Claus
Cornett
a,
Jesper
Østergaard
a,
Stefan
Stürup
a and
Bente
Gammelgaard
*a
aUniversity of Copenhagen, Department of Pharmacy, Denmark. E-mail: bente.gammelgaard@sund.ku.dk
bUniversity of Copenhagen, Department of Plant and Environmental Sciences, Denmark
First published on 1st July 2016
Abstract
A method for quantification of a pharmaceutical peptide in human plasma was developed using gradient elution LC-ICP-MS. A membrane desolvation (MD) system was applied to remove organic solvents from the eluent prior to the detection as SO+ in the dynamic reaction cell (DRC) of the ICP-DRC-MS instrument and subsequent quantification by post-column isotope dilution (IDA). Plasma proteins were precipitated prior to analysis. Analytical figures of merit including linearity, precision, LOD, LOQ and accuracy were considered satisfactory for analysis of plasma samples. The selectivity of the developed method was demonstrated for five pharmaceutically relevant peptides: desmopressin, penetratin, substance P, PTH (1-34) and insulin. Preliminary experiments on an ICP-MS/MS system using oxygen to reduce the effect of organic solvents were also performed to compare sensitivity. The results of the study demonstrated that LC-ICP-MS post-column IDA may constitute a valuable additional tool in quantification of non-labelled peptides in the early drug development offering absolute quantification without need of species specific standards.
Introduction
The interest in peptides and proteins as drugs and excipients is continuously increasing in biomedical research. The number of peptide therapeutics reaching preclinical and clinical trials increases every year. Peptides are favorable as therapeutics as they offer good efficacy, safety and tolerability due to their composition of amino acids.1 During drug development, parameters such as stability, pharmacokinetics and metabolism must be monitored; therefore, sensitive and selective analytical methods are required. Quantitative analysis of peptides in biological matrices is challenging due to the similarity of pharmaceutical peptides to endogenous compounds. Peptide quantification has predominantly been performed by fluorescence detection (FLD) exploiting fluorophores naturally present in the peptide sequence or by adding a fluorescent label.2 Additionally, immunoassays, e.g. ELISA, are widely used for quantification of peptides and proteins.3 Nowadays, identification and quantification is predominantly performed by liquid chromatography mass spectrometry (LC-MS). Analysis of peptides by LC-MS in complex biological matrices such as plasma demands thorough sample preparation and species specific peptide standards for absolute quantification.4The large potential of inductively coupled plasma mass spectrometry (ICP-MS) in quantitative analysis of biomolecules has already been highlighted in a number of reviews.5–7 Hyphenated to a separation technique, ICP-MS is a powerful technique for quantitative analysis of biomolecules offering low limits of detection, a wide dynamic range and in principle species independent sensitivity, thus, quantification using a single inorganic standard is possible. The premise of using ICP-MS for detection is the presence of an ICP-MS detectable element in the peptide structure such as sulfur (S), phosphorus (P) or selenium (Se), otherwise an ICP-MS detectable label must be inserted. Introduction of elemental labels as metal-chelating moieties or metal affinity tags has been widely reported.5,8 Labelling with metals ensure high sensitivity and selectivity, but the drawback of these approaches is alteration of the structure and physicochemical properties of the peptide. Furthermore, the stoichiometry of the label and the molecule must be controlled and known for quantitative purposes. Unequivocal stoichiometry is achieved using heteroelement tags, which are covalently bound in the peptide structure. Recently, we introduced Se as an elemental detection label for quantification of peptides.9 By exchange of the S-containing amino acid methionine (Met) with the Se-containing analogue selenomethionine (SeMet) during peptide synthesis, we introduced a label causing minimal alteration of the native peptide structure and physicochemical properties.10 The most attractive approach would therefore be to monitor a heteroelement in the native structure of the peptide. S is present in the natural occurring amino acids Met and cysteine (Cys) and thus constituting a versatile label in the majority of biomolecules, but measuring S in complex biological matrices by ICP-MS is not straightforward. The ionization efficiency of S in the argon plasma is limited due to the high first ionization energy (10.357 eV). Furthermore, detection of the main S isotope (32S, 94.99% (ref. 11)) is hampered by the formation of polyatomic interferences in the plasma, mainly dimers of oxygen (16O2+). Different instrumental setups and approaches to overcome these interferences have been developed. One approach is to use a quadrupole based collision/reaction cell ICP-MS instrument. By using xenon as collision gas polyatomic interferences are removed by collision induced dissociation and allows for measurement of 32S.12 The main approach for collision/reaction cell instruments is to use oxygen as reaction gas for the formation of sulfur oxide (32S16O+) which is less interfered as compared to the S isotopes.13 The application of high resolution sector field (SF) instruments facilitates the separation of the different masses of 32S+ (m/z = 31.9898) and 16O16O+ (m/z = 31.9721) can be separated at a resolution of 4000 (m/Δm).14,15 Recently, ICP-MS/MS was introduced offering improved removal of polyatomic interferences and further reduction of the background level. The LOD of this instrument has thus been reported to be lowered to the ng L−1 range for S.16,17
Measurement of S in biological matrices demands hyphenation of a separation method to the ICP-MS in order to separate analytes from natural occurring S-species in the matrix. Peptide separation is most often performed by reversed-phase liquid chromatography (RP-LC) and gradient elution using either acetonitrile or methanol.18 The continuous change in organic solvent load from the eluent is not readily compatible with ICP-MS as it may cool the plasma, change ionization and thus potentially change sensitivity. Exposure to very high concentrations of organic solvent may even extinguish the plasma. Furthermore, the introduction of organic solvents may cause carbon build-up on the cones and decrease sensitivity over time.19,20 By application of a membrane desolvation (MD) system the organic solvent is removed from the eluent prior to introduction to the ICP-MS and up to 100% methanol or acetonitrile at flow rates of 200 μL min−1 are tolerated.21 Optimization of the MD is only possible for a single concentration level of organic solvent and the removal of eluent may therefore not be constant throughout the gradient, causing problems in species unspecific calibration. Isotope dilution analysis (IDA) can be applied to overcome this problem.22 The technique is based on measurement of the isotope ratios in a sample spiked with a solution enriched with a minor natural abundant isotope of the element. In species unspecific IDA, also known as post-column IDA, the enriched isotope is added to the chromatographic eluent after separation of the sample.23 The technique and the equations used in calculations have been presented in a tutorial review by Rodriguez-Gonzáles.24 Sulfur post column IDA has been applied in several studies regarding protein quantification by ICP-MS.22,25,26 The number of quantitative studies of peptides or proteins in complex biological matrices using ICP-MS detection of sulfur is very limited. Konz et al. developed a method for quantification of the peptide hepcidin in human urine by post column IDA27 but, the majority of studies does not report on using IDA for quantification.28–30
The aim of this study was to examine if it is possible to quantify therapeutically relevant levels of sulfur-containing pharmaceutical peptides in human plasma samples by LC-ICP-MS and post-column IDA.
Experimental
Instrumentation
The 34S enriched isotope standard was prepared by dissolving 20 mg elemental 34S in 3 mL sub boiled nitric acid (65%) and digested in a microwave until a clear and colorless solution was obtained. The solution was diluted with purified water to 20 mL. The total S concentration of the solution was determined by reversed isotope dilution using a PlasmaCAL elemental standard of S.
Procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 of TCA and sample. The samples were centrifuged at 14000g for 15 min at room temperature. The supernatant was diluted 1 + 1 in eluent (10% v/v ACN, 0.1% v/v TFA) prior to analysis.
:5 of TCA and sample. The samples were centrifuged at 14000g for 15 min at room temperature. The supernatant was diluted 1 + 1 in eluent (10% v/v ACN, 0.1% v/v TFA) prior to analysis.
Sample preparation by ultrafiltration was executed in 50% human plasma diluted in purified water by addition of TFA to a final concentration of 0.5% and ultrafiltration through 500 μL centrifugal filter units, Amicon® Ultra with a semi-permeable membrane of molecular weight cut-off 30 kDa. The ultra filtrates were diluted 1 + 1 in eluent (10% v/v ACN, 0.1% v/v TFA) prior to analysis.
Results and discussion
Method development
The aim of this study was to examine if it is possible to quantify pharmaceutical S-containing peptides in human plasma by LC-ICP-MS using the cell-penetrating peptide Pen as a model compound. Reversed phase gradient separation using methanol or acetonitrile is the preferred method for separation of peptides.4 In order to overcome the challenges arising from introduction of organic solvents to the ICP-MS, a membrane desolvation (MD) system (Aridus II, Cetac) was applied. The use of the MD system limits the flow rate of the separation method, as only nebulizers with a flow rate up to 200 μL min−1 are available for sample introduction. Furthermore, the MD extends the distance between the HPLC column and the ICP-MS causing peak broadening. Narrow peaks and excellent resolution is therefore demanded for the separation when a MD system is applied.Recently we developed an LC-ICP-MS method applying a linear methanol gradient (0.1% v/v acetic acid, 0.05% v/v TFA) for separation of a Se-labelled analogue of Pen and its degradation products during cell uptake studies.9 Separation of S-containing peptides in human plasma is more challenging due to the presence of large amounts of endogenous S compounds in the sample matrix. In order to separate low molecular weight S compounds and S-containing proteins in plasma, a multi-step acetonitrile (0.1% v/v TFA) gradient was developed for separation of Pen from S-containing plasma constituents. The column temperature was set to 60 °C to obtain narrow peaks and improve resolution. Satisfactory peak shape and separation of Pen from plasma components were obtained.
Quantification by post column isotope dilution analysis
The species independent sensitivity of LC-ICP-MS is lost when gradient elution is applied, as the shift in organic content in the eluent changes the sensitivity during elution of the species. Thus, the instrument sensitivity of early eluting species will differ from later eluting species. In order to compensate for these variations in sensitivity as well as drift of the instrument signal, post column IDA was employed for absolute quantification (details are described in the Experimental section). The mass bias factor (F) was estimated and applied in correction of the experimental isotope ratios of 32S/34S. Subsequent to correction, the isotope dilution equation24 was applied in the entire chromatogram to obtain the mass flow data and the peak areas were integrated. An example of a mass flow chromatogram obtained from MD-ICP-DRC-MS analysis of blank plasma and plasma spiked with Pen is shown in Fig. 1.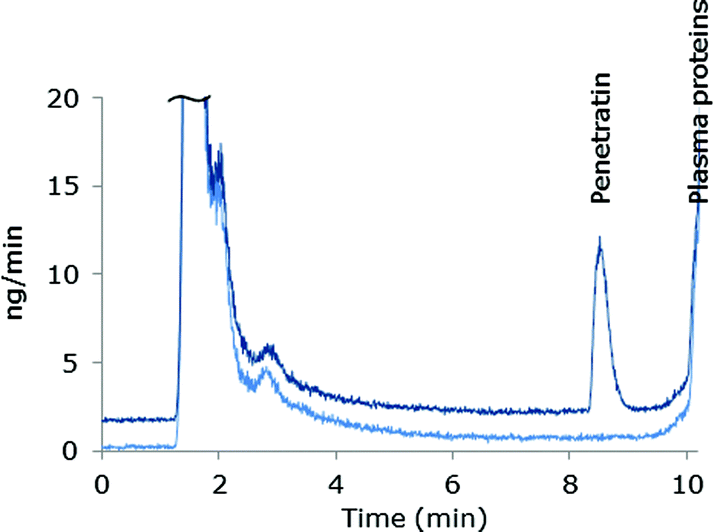 | ||
| Fig. 1 Mass flow chromatograms from LC-MD-ICP-DRC-MS analysis of 50% plasma samples. Light blue: blank plasma, offset by −1.2 ng min−1. Dark blue: 50 μM Pen spiked in 50% plasma prior to sample pre-treatment. | ||
Sample preparation
Plasma proteins should be removed prior to LC-ICP-MS analysis, but it is important that the analyte remains in the sample with a satisfactory and reproducible recovery of analyte. Working with peptide-containing samples can be a challenge due to unspecific adsorption of peptides to surfaces and other constituents of the matrix e.g. proteins. Adsorption is often dependent on the specific side chains on the amino acids of the peptide and may be reduced by choosing conditions which affects the equilibrium constant between the peptide and surfaces or between the peptide and proteins which it may adsorb to.31 Pen contains 16 amino acids (RQIKIWFQNRRMKWKK-NH2) of which 7 are basic residues, and thus have a net charge of +8 at physiological pH. In order to reduce ionic interactions of the peptide with the surroundings, negatively charged residues were reduced by acidifying samples during sample pretreatment. Two different approaches for removal of plasma proteins were pursued; precipitation and ultrafiltration. TCA (10% v/v, volume ratio 1:5) was chosen as precipitation reagent, ensuring acidic conditions. A protein precipitation efficiency above 99% has been reported for TCA.32 To determine the recovery of peptide after precipitation, spiked plasma samples were precipitated and the areas from the mass flow chromatograms compared to the areas of plasma samples spiked after protein precipitation. Recovery was determined at two concentration levels, 10 μM and 40 μM Pen; recoveries of 70.6 ± 6.2% (n = 3) and 69.2 ± 4.0% (n = 3) were obtained, respectively. Chromatograms of plasma samples showed that some S-containing protein still remained after precipitation with 10% TCA. For ultrafiltration, a semipermeable membrane with a molecular weight cut-off (MWCO) value of 30 kDa was used and the plasma acidified (0.5% v/v TFA) prior to filtration. S-containing plasma proteins were almost completely removed by ultrafiltration, but the recovery of 10 μM Pen was only 32.4 ± 9.0%. Ultrafiltration is attractive due to the almost complete removal of proteins, but recovery and repeatability of Pen were not satisfactory. Therefore, protein precipitation with 10% v/v TCA was chosen for further experiments. The remaining protein content after precipitation is a drawback, but as Pen was clearly separated from the protein residues, this was accepted.
Selectivity
The selectivity of the chromatographic method was examined for a number of pharmaceutically relevant biomolecules: desmopressin, Pen, substance P, PTH (1-34) and insulin (Table 1). The species were all satisfactorily separated by extension of the 38% v/v ACN isocratic part of the gradient after elution of Pen. Chromatograms of the peptides are shown in Fig. 2. It must be noted that the developed method for Pen is not directly applicable to any peptide, as the properties of all peptides are different, the sample pre-treatment must be examined and optimized for each peptide of interest.| Peptide | M w | No. of amino acids | No. of S |
|---|---|---|---|
| Desmopressin | 1069.22 | 9 | 2 |
| Penetratin | 2245.74 | 16 | 1 |
| Substance P | 1347.63 | 11 | 1 |
| PTH (1-34) | 4117.74 | 34 | 2 |
| Insulin | 5807.6 | 110 | 6 |
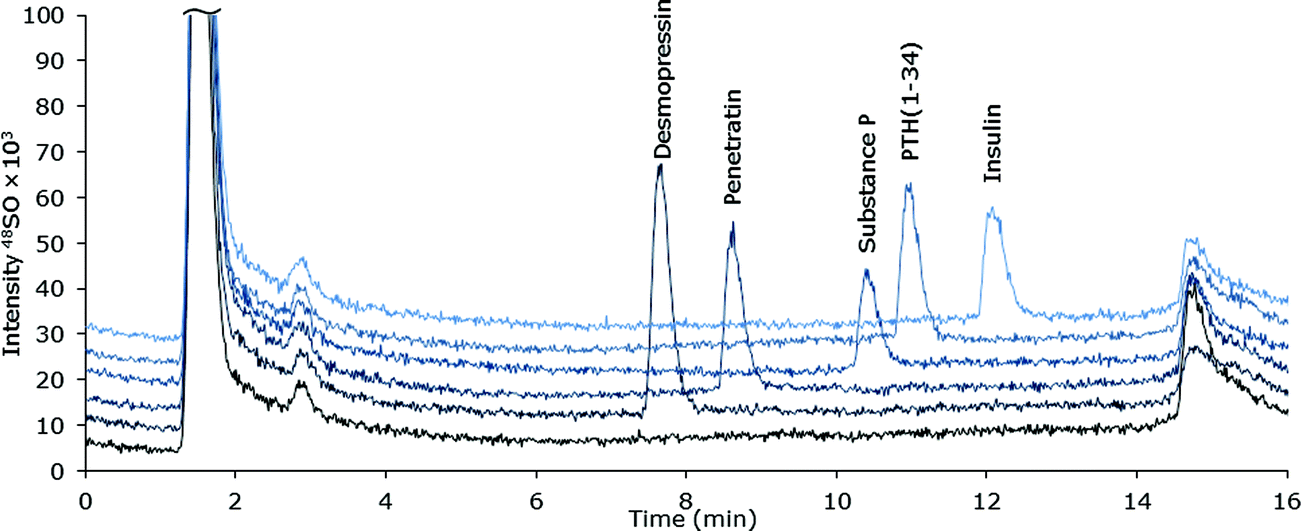 | ||
| Fig. 2 MD-LC-ICP-DRC-MS chromatograms of S-containing peptides and protein (∼10 μM S) spiked into ultra-filtrated human plasma. | ||
Analytical figures of merit
The chromatographic method was validated for Pen using the DRC instrument equipped with a MD system (MD-ICP-DRC-MS) and quantification by post column IDA. Oxygen was applied as reaction gas resulting in measurement of 32S16O+ at m/z 48. The analytical figures of merit are summarised in Table 2.Linearity was examined in order to demonstrate a linear response in the applied concentration range. Pen was spiked into 50% v/v human plasma followed by protein precipitation reaching a final concentration of 0.5–15 μM Pen (15–450 μg S L−1). Linearity was established with correlation coefficients of 0.999. Precision was determined at two concentration levels (0.7 μM and 3.5 μM). For both concentrations the precision were considered satisfactory.
LOD and LOQ were estimated and absolute LOD of 0.6 pmol S and LOQ of 1.9 pmol S were obtained. The LOD is considerably lower as compared to LODs reported by others using ICP-DRC-MS instruments and oxygen as reaction gas. For proteins, peptides or amino acids in standard solutions, LODs in the range of 2.3–3.1 pmols have been reported.13,33,34 Only a limited number of studies, where hyphenated ICP-MS methods were used for absolute quantification of S in plasma, has been described. Koellensperger et al. obtained an LOD of 62 pmol S in albumin in human plasma using a two dimensional chromatographic system and ICP-DRC-MS.29 The improved LOD obtained in this study is explained by the application of the MD system. In the MD, the sample is heated and all volatile matrix constituents removed through a semi-permeable membrane. As the sample is evaporated in the MD, the total amount of analyte is introduced into the ICP-MS. Using a regular ICP-DRC-MS setup with a spray-chamber and a concentric nebulizer, only 2% of the sample spray reaches the detector.35
The method was developed for Pen, which is not yet part of any approved drug formulation. The relevant plasma concentration is therefore not defined. For a well-known drug compound as human insulin, maximum plasma concentrations of 200 μIU mL−1 (1.2 mM insulin ≈ 7.3 mM S) have been reported after a single dose of 0.1 IU kg−1.36 The developed method would be applicable in a similar experiment as the LOD obtained in this study was 0.12 μM Pen (containing 1 S atom). The species dependent LOD will improve with the presence of multiple S atoms in the peptide. If peptides such as desmopressin (2 S atoms) or insulin (6 S atoms) were quantified, the LOD of the species would improve correspondingly.
As no certified standards are available for Pen, the accuracy of the IDA method was determined using desmopressin (Ph. Eur. CRS). Accurate quantification by IDA requires simultaneous interference free detection of two isotopes of S, in this case 32S and 34S.25 Using oxygen as a reaction gas the interferences are limited by the formation of 32S16O+ and 34S16O+. Possible spectral interferences include 48Ti+, 48Ca+, 36Ar12C+ and 31P16O1H+ which may affect the 32S16O+ signal, while 34S16O+ can be interfered by 50Ti+ if interferences are not removed in the DRC.15
Giner Martinez-Sierra et al. demonstrated applicability of using reaction cell technology and quantification of S (32S16O+) by post column isotope dilution in aqueous solutions of methionine with accuracies of 98% and 99%.22 In our laboratory we have demonstrated accuracy of 98.2% for a certified sulfate standard using ion chromatography with isocratic elution hyphenated to the ICP-DRC-MS and quantification by post column IDA. Thus, the interferences are not considered devastating for the IDA quantification. The influence of matrix constituents from biological samples on IDA quantification is not known.
Application of the MD with ICP-DRC-MS may induce other challenges, as differences in sensitivity between species when using an MD system are well known.21,37 In order to obtain accurate quantification by IDA using a MD system, the isotope spike (34S as sulfate) as well as the analyte (desmopressin or Pen) must be stable in the MD system and exhibit similar sensitivity. This was experimentally confirmed and has also been reported by Feldmann and colleagues who demonstrated equal and stable signals of sulfate and smaller peptides using an MD-system.38
ICP-MS/MS detection
The chromatographic method was transferred to an ICP-MS/MS instrument in order to compare sensitivity. As this instrument was equipped with another LC-system, the ACN-gradient was slightly modified in order to move the Pen peak away from front peak tailing. One of the reasons for applying the ICP-MS/MS instrument was that this system can handle larger organic solvent amounts by addition of oxygen (20% in argon) to the plasma and therefore the more complicated MD set-up was avoided. A mass flow chromatogram is shown in Fig. 3. A comparison of the mass flow chromatograms from LC-MD-ICP-DRC-MS (Fig. 1) and LC-ICP-MS/MS (Fig. 3) reveal that the Pen peak is slightly broader using the ICP-DRC-MS, presumably because of the MD system and the different LC-equipment applied.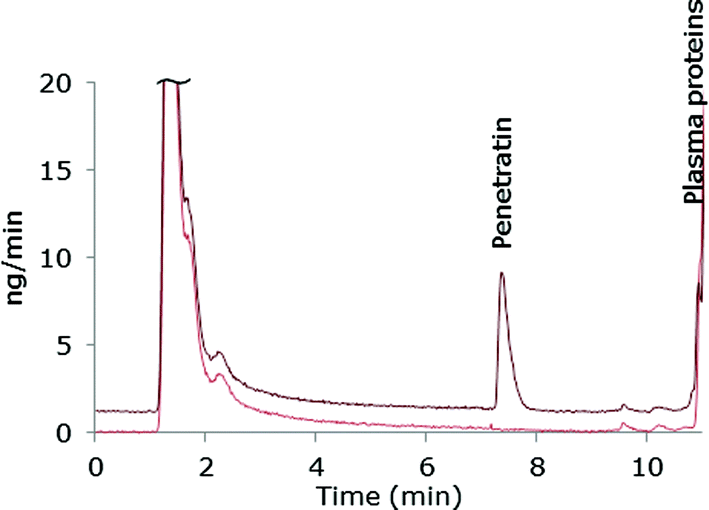 | ||
| Fig. 3 Mass flow chromatogram from LC-ICP-MS/MS. 50 μM Pen spiked in 50% plasma prior to sample pre-treatment and 50% blank plasma. Dark red: Pen in plasma, light red: blank plasma, offset by −1.2 ng min−1. | ||
The main purpose of transferring the developed method to the ICP-MS/MS system was to compare sensitivity with the MD-ICP-DRC-MS set-up. The absolute LOD obtained by the ICP-MS/MS instrument was 0.5 pmol S and the absolute LOQ 1.6 pmol S and thus comparable to the MD-ICP-DRC-MS system.
In theory, interferences are even more efficiently removed in the ICP-MS/MS due to the dual selection of m/z in the first and third quadrupole (Q1 and Q3). When measuring S, m/z 32 and 34 are exclusively selected in Q1 while m/z 48 and 50 interferences are rejected. Only the selected ions reach the reaction cell and react with O2 forming 32S16O+ and 34S16O+. Finally, in the Q3, m/z 48 and m/z 50 are selected while the interferences 16O2+ and 16O18O+ are rejected improving the background level.39 Furthermore less noise was observed in the ICP-MS/MS chromatograms as compared to the MD-ICP-DRC-MS chromatograms. The ICP-MS/MS system was therefore expected to improve the LOD obtained by the ICP-DRC-MS. However, the results demonstrated, that the application of the MD system improved the LOD of the DRC-system to a level comparable to the ICP-MS/MS. Application of the MD system with the ICP-MS/MS might also improve the LOD of this instrument as more sample is introduced and signal suppression from introduction of organic solvent is avoided. However, the MD introduces further complexity to the analytical system and increases the requirements for optimization which is not desirable.
The LOD obtained for the ICP-MS/MS is in accordance with the relatively few studies reported applying ICP-MS/MS hyphenated to a separation technique. Recently, LODs of 0.7 pmol S and 1.3 pmol S were reported for S-containing amino acids by LC-ICP-MS/MS analysis.34 By capillary LC-ICP-MS/MS, an absolute LOD at fmol level has been reported.16,40 To the authors best knowledge, no studies on quantification of exogenous S in human plasma by LC-ICP-MS/MS have yet been published.
Precision of 8.1% and 5.6% were obtained with the ICP-MS/MS system for 0.5 μM Pen and 3.5 μM Pen, respectively. This is slightly worse as compared to the MD-ICP-DRC-MS. As observed from the mass flow chromatograms in Fig. 1 and 3, the intensities of the Pen peaks are comparable, but the noise level was visibly improved using the ICP-MS/MS. The superior precision obtained by the MD-ICP-DRC-MS was therefore unexpected but may be explained by the settings of the data collection, as considerable fewer data points were collected for each chromatographic peak during the LC-ICP-MS/MS analysis as compared to the LC-MD-ICP-DRC-MS analysis.
The accuracy obtained by LC-ICP-MS/MS was, however, only 80.2%. The LC method was developed and thoroughly tested using MD-ICP-DRC-MS and subsequently transferred to the ICP-MS/MS system. Analyte adsorption to surfaces is an important issue in peptide analysis and could be a source of the poorer accuracy as the two systems were completely different. Thus, further experiments should be performed to examine this discrepancy between accuracies in the set-ups.
Conclusion
A reversed phase gradient LC-ICP-DRC-MS method using a membrane desolvation system and post-column IDA was developed for quantification of an S-containing peptide in human plasma. The analytical figures of merit were considered satisfactory for analysis of human plasma samples. The obtained LOD was considerably improved as compared to LODs reported in literature using an ICP-DRC-MS instrument. By transferring the developed method to an ICP-MS/MS system, comparable LOD and improved noise level was achieved, but the accuracy was only 80.2%. Further optimization of the LC-ICP-MS/MS system is thus needed in order to determine the applicability of this method.LC-ICP-MS analysis represents a promising tool for quantification of peptides and small proteins in complex biological matrices. The possibility of quantifying peptides without introduction of a detection label or need of species specific standards is obviously attractive. The selectivity obtained with this method emphasizes the potential as analytical technique in peptide drug development. Compared to UV-detection, ICP-MS offers improved selectivity as only sulfur-containing compounds are detected and may also be valuable for quantification of peptides with limited UV-absorbance. In addition, LC-ICP-MS may offer an alternative method in cases where ELISA is not easily executed or specific antibodies are unavailable.
Acknowledgements
Human insulin and PTH (1-34) were received from the Innovative Medicines Initiative Joint Undertaking under grant agreement no. 115363, resources of which are composed of financial contributions from the European Union's Seventh Framework Programme (FP7/2007–2013) and EFPIA companies in kind contribution.References
- K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122–128 CrossRef CAS PubMed.
- R. Fischer, T. Waizenegger, K. Kohler and R. Brock, Biochim. Biophys. Acta, 2002, 1564, 365–374 CrossRef CAS.
- C. Hempen and U. Karst, Anal. Bioanal. Chem., 2006, 384, 572–583 CrossRef CAS PubMed.
- H. John, M. Walden, S. Schafer, S. Genz and W. G. Forssmann, Anal. Bioanal. Chem., 2004, 378, 883–897 CrossRef CAS PubMed.
- A. Prange and D. Profrock, J. Anal. At. Spectrom., 2008, 23, 432–459 RSC.
- J. Bettmer, B. M. Montes, J. R. Encinar, M. L. Fernandez Sanchez, M. D. F. de la Campa and M. A. Sanz, J. Proteomics, 2009, 72, 989–1005 CrossRef CAS PubMed.
- M. Wang, W. Y. Feng, Y. L. Zhao and Z. F. Chai, Mass Spectrom. Rev., 2010, 29, 326–348 CrossRef CAS PubMed.
- D. Kretschy, G. Koellensperger and S. Hann, Anal. Chim. Acta, 2012, 750, 98–110 CrossRef CAS PubMed.
- L. H. Moller, C. Gabel-Jensen, H. Franzyk, J. S. Bahnsen, S. Sturup and B. Gammelgaard, Metallomics, 2014, 6, 1639–1647 RSC.
- M. L. Hyrup, J. S. Bahnsen, H. M. Nielsen, J. Ostergaard, S. Sturup and B. Gammelgaard, Eur. J. Pharm. Sci., 2015, 67, 76–84 CrossRef PubMed.
- M. Berglund and M. E. Wieser, Pure Appl. Chem., 2011, 83, 397–410 CrossRef CAS.
- D. Schaumloffel, P. Giusti, H. Preud'Homme, J. Szpunar and R. Lobinski, Anal. Chem., 2007, 79, 2859–2868 CrossRef PubMed.
- S. Sturup, L. Bendahl and B. Gammelgaard, J. Anal. At. Spectrom., 2006, 21, 201–203 RSC.
- M. Wind, A. Wegener, A. Eisenmenger, R. Kellner and W. D. Lehmann, Angew. Chem., Int. Ed., 2003, 42, 3425–3427 CrossRef CAS PubMed.
- J. Giner Martinez-Sierra, O. Galilea San Blas, J. M. Marchante Gayon and J. I. Garcia Alonso, Spectrochim. Acta, Part B, 2015, 108, 35–52 CrossRef CAS.
- F. S. Diez, N. Sugishama, E. J. Ruiz and A. Sanz-Medel, Anal. Chem., 2012, 84, 5851–5857 CrossRef PubMed.
- R. S. Amais, C. D. B. Amaral, L. L. Fialho, D. Schiavo and J. A. Nobrega, Anal. Methods, 2014, 6, 4516–4520 RSC.
- I. D. B. van, R. W. Sparidans, J. H. Schellens and J. H. Beijnen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 872, 1–22 CrossRef PubMed.
- K. L. Sutton and J. A. Caruso, J. Chromatogr. A, 1999, 856, 243–258 CrossRef CAS PubMed.
- B. Gammelgaard and B. Packert Jensen, J. Anal. At. Spectrom., 2007, 22, 235–249 RSC.
- L. H. Moller, C. S. Jensen, T. T. T. N. Nguyen, S. Sturup and B. Gammelgaard, J. Anal. At. Spectrom., 2015, 30, 277–284 RSC.
- J. Giner Martinez-Sierra, F. Moreno Sanz, P. Herrero Espilez, R. Santamaria-Fernandez, J. M. Marchante Gayon and J. I. Garcia Alonso, J. Anal. At. Spectrom., 2010, 25, 989–997 RSC.
- L. Rottmann and K. G. Heumann, Fresenius. J. Anal. Chem., 1994, 350, 221–227 CrossRef CAS.
- P. Rodriguez-Gonzáles, J. M. Marchante-Gayon, J. I. Garcia Alonso and A. Sanz-Medel, Spectrochim. Acta, Part B, 2005, 60, 151–207 CrossRef.
- C. Rappel and D. Schaumloffel, Anal. Bioanal. Chem., 2008, 390, 605–615 CrossRef CAS PubMed.
- M. Wang, W. Feng, W. Lu, B. Li, B. Wang, M. Zhu, Y. Wang, H. Yuan, Y. Zhao and Z. Chai, Anal. Chem., 2007, 79, 9128–9134 CrossRef CAS PubMed.
- T. Konz, M. Montes-Bayon, J. Bettmer and A. Sanz-Medel, J. Anal. At. Spectrom., 2011, 26, 334–340 RSC.
- L. Feng, D. Zhang, J. Wang and H. Li, Anal. Methods, 2014, 6, 7655–7662 RSC.
- M. Sulyok, S. Hann, C. G. Hartinger, B. K. Keppler, G. Stingeder and G. Koellensperger, J. Anal. At. Spectrom., 2005, 20, 856–863 RSC.
- A. K. Bytzek, K. Boeck, G. Hermann, S. Hann, B. K. Keppler, C. G. Hartinger and G. Koellensperger, Metallomics, 2011, 3, 1049–1055 RSC.
- K. Maes, I. Smolders, Y. Michotte and E. A. Van, J. Chromatogr. A, 2014, 1358, 1–13 CrossRef CAS PubMed.
- J. Blanchard, J. Chromatogr., 1981, 226, 455–460 CrossRef CAS PubMed.
- S. Hann, G. Koellensperger, C. Obinger, P. G. Furtmuller and G. Stingeder, J. Anal. At. Spectrom., 2004, 19, 74–79 RSC.
- G. Hermann, L. H. Moller, B. Gammelgaard, J. Hohlweg, D. Mattanovich, S. Hann and G. Koellensperger, J. Anal. At. Spectrom., 2016 10.1039/c6ja00039b.
- R. Thomas, in Practical Guide to ICP-MS: A Tutorial for Beginners, CRC Press Taylor and Francis, Boca Raton, 3rd edn, 2013, ch. 3, pp. 11–20 Search PubMed.
- H. E. Scholtz, S. G. Pretorius, D. H. Wessels, C. Venter, M. A. Potgieter and R. H. A. Becker, Acta Diabetol., 2003, 40, 156–162 CrossRef CAS PubMed.
- L. Bendahl and B. Gammelgaard, J. Anal. At. Spectrom., 2005, 20, 410–416 RSC.
- K. Bluemlein, E. M. Krupp and J. Feldmann, J. Anal. At. Spectrom., 2009, 24, 108–113 RSC.
- L. Balcaen, G. Woods, M. Resano and F. Vanhaecke, J. Anal. At. Spectrom., 2013, 28, 33–39 RSC.
- J. Gong, M. J. Solivio, E. J. Merino, J. A. Caruso and J. A. Landero-Figueroa, Anal. Bioanal. Chem., 2015, 407, 2433–2437 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |