
Size-tunable rhodium nanostructures for wavelength-tunable ultraviolet plasmonics†
Xiao
Zhang
a,
Pan
Li
a,
Ángela
Barreda
b,
Yael
Gutiérrez
b,
Francisco
González
b,
Fernando
Moreno
b,
Henry O.
Everitt
 *cd and
Jie
Liu
*a
*cd and
Jie
Liu
*a
aDepartment of Chemistry, Duke University, Durham, NC 27708, USA. E-mail: j.liu@duke.edu
bOptics Group, Department of Applied Physics, University of Cantabria, Avda de Los Castros, s/n 39005 Santander, Spain
cDepartment of Physics, Duke University, Durham, NC 27708, USA. E-mail: everitt@phy.duke.edu
dC.M. Bowden Laboratory, Army Aviation & Missile RD&E Center, Redstone Arsenal, AL 35898, USA
First published on 7th October 2015
Abstract
Polydisperse rhodium nanoparticles have recently shown promise for ultraviolet (UV) plasmonics, but controlling the size and morphology of metal nanoparticles is essential for tuning surface plasmon resonances. Here we report the use of slow-injection polyol methods to synthesize monodisperse Rh nanocubes with unprecedentedly large sizes and slightly concave faces. The associated local surface plasmon resonances (LSPRs) red-shifted with increasing sizes in the UV region from deep UV to around 400 nm, consistent with numerical simulations. UV illumination of p-aminothiophenol attached to the Rh nanocubes generated surface-enhanced Raman spectra and accelerated photo-decomposition, and these enhancements were largest for nanocubes whose LSPR was resonant with the UV laser. The lack of a native oxide coating, the precise control of nanocube size and morphology demonstrated here, and the ability to tune the surface plasmon resonance from the deep UV to near UV spectral region, make rhodium a compelling choice for UV plasmonic applications.
Conceptual insightsThis work introduces size-tunable and localized surface plasmon resonance (LSPR) energy-tunable Rh nanostructures to the field of ultraviolet (UV) plasmonics. It could be considered as the counterpart of Au and Ag in visible plasmonics in the UV spectrum range. The research of UV plasmonics is limited by the stability of currently studied materials, including aluminum and gallium. Rhodium, a stable noble metal without surface oxide, provides appealing model system to investigate the morphology–LSPR relationship in the UV region. The precise size control and tunability of sizes as well as the associate resonant energies of Rh nanostructures demonstrated here represents a major advance in understanding the fundamental principles of UV plasmonics. Additionally, the enhanced photo-decomposition through the excitation of UV LSPRs in Rh nanostructures manifests the photocatalytic capability of concentrated electromagnetic fields and resulted hot electrons, which could provide an access to chemical transformations unavailable previously. The tunability and photocatalytic activity of UV LSPRs in Rh nanostructures pave the way for both understanding the fundamentals in UV plasmonics and developing practical applications that utilize these uniquely concentrated high energy electromagnetic waves. |
Excitations of collective electron oscillations in metal nanostructures by external electromagnetic fields concentrate light in sub-wavelength spaces.1,2 Localized surface plasmon resonances (LSPRs) are the manifestation of these concentrated electromagnetic fields near the surfaces of plasmonic nanoparticles, and their ability to enhance the optical response of nearby analytes has been exploited for many applications including surface-enhanced spectroscopy, optoelectronics, and photocatalysis.3–9 Tuning the size and shape of the metal nanoparticles is a well-established approach for controlling their plasmonic properties, since these parameters determine the boundary conditions in Maxwell's equations. Over the last decade, tremendous efforts have been devoted to developing plasmonic nanoparticles made of Au, Ag, and Cu, but their resonant frequencies are limited to the visible and near-infrared (NIR) regions.10–13 Ag possesses excellent optical properties but is oxidized upon exposure to air and moisture, deteriorating its plasmonic performance. Therefore, Au nanoparticles, with stability against different environments, are widely used as plasmonic nanostructures in the visible and NIR regions.
Interest is growing for ultraviolet (UV) plasmonics, driven by applications such as label-free detection of biomolecules, so metal nanostructures that exhibit plasmonic effects in the UV region are needed.14,15 Currently, Al16–20 and Ga21–23 are the most studied materials with UV plasmonic responses and are attractive because of their wide availability and compatibility with CMOS processing. However, Al and Ga nanostructures suffer from fast oxidation in aerobic and aqueous environments, which changes the size and shape of the metal nanostructures as well as deteriorates their plasmonic properties, especially for applications where direct surface contact is necessary. A stable, noble metal with UV plasmonic effects is needed to serve as the UV counterpart to Au and promote research into UV plasmonics. Rhodium, a noble metal with superior stability against various environments,24 has been demonstrated to exhibit LSPRs in the UV region. Rh multipods, synthesized by Xia and colleagues, was the first experimental demonstration of UV plasmonic Rh nanostructures.25 We recently employed similar Rh multipods in a quantitative spectroscopic study and observed surface-enhanced Raman scattering, fluorescence, and photo-degradation of p-aminothiophenol (PATP) upon irradiation of UV laser, all in good agreement with numerical simulations.26,27 In addition, no perceivable change in morphology of individual Rh multipods was observed after the treatment with nitric acid, proving the excellent stability of Rh nanoparticles.26 Indeed, the stability and tunable UV LSPRs of Rh nanostructures make them a compelling system for studying structure–property relationships and establishing the principles of UV plasmonics.
Although Rh multipods exhibit UV plasmonic effects, most synthetic methods produce polydisperse mixtures of tripods and tetrapods whose sizes are difficult to adjust controllably, preventing plasmonic properties from being tailored for specific applications.25,26,28 Syntheses of Rh nanocubes, tetrahedra, icosahedra, and hollow frames with sizes mainly smaller than 15 nm have been reported in the literature.29–35 We reproduced some of these syntheses, and as expected, no LSPR features were experimentally observed at energies less than 4.8 eV, which was consistent with our numerically simulated results that the resonant energies of these small Rh nanostructures are in the deep UV region (Fig. S1, ESI†). Hence, new synthetic methods are needed that controllably produce larger, monodisperse Rh nanostructures with LSPRs in UVA and UVB regions (280–400 nm, 4.4–3.1 eV).
In this work, we report unseeded (no pre-synthesized Rh seeds added) and seeded (pre-synthesized Rh seeds added) slow-injection polyol methods to synthesize size-tunable, monodisperse Rh nanocubes (NCs) with wavelength-tunable LSPRs in the UV region. The cubic shape was chosen due to the sharp corners to better concentrate electromagnetic fields and the well understood mechanism of shape control, so we can focus on the tunability of sizes and associated LSPR energies. We achieve size control of large Rh NCs through the slow injection of Rh precursor solution, which ensures a low concentration of Rh atoms and a high amount of Rh precursor in the reaction mixture. The plasmonic properties of the Rh nanocubes were studied by UV-vis extinction spectroscopy and correlated with numerical modeling using finite element-based methods.36 The LSPR frequencies red-shifted as the size of Rh nanocubes increased in a manner that was in good agreement with simulations. Accelerated photochemical decomposition of PATP attached to Rh NCs was observed by surface-enhanced Raman spectroscopy (SERS), and the sample whose LSPR was most resonant with the UV laser wavelength exhibited the fastest decomposition rate. To our knowledge, this is the first report of Rh nanostructures with tunable UV plasmonic properties. Understanding the relationship of Rh nanostructure size and morphology to UV plasmonic effects and enhancements could promote new avenues of research in photodetection, photochemistry, and photocatalysis.
The size of the Rh nanoparticles is primarily determined by the ratio of Rh seeds to injected Rh precursor. To prepare large Rh NCs, the concentration of generated Rh atoms must be maintained at low supersaturation to minimize the formation of seeds while injecting enough Rh precursor into the reaction mixture to grow those few seeds to larger sizes. To achieve these two requirements, an unseeded polyol method was used to synthesize Rh nanocubes (see detailed experimental methods, ESI†). Briefly, KBr dissolved in ethylene glycol (EG) was held at 160 °C, into which RhCl3 and polyvinylpyrrolidone (PVP) solutions were separately injected at a slow rate by a two-channel syringe pump. The Rh precursor, RhCl3, was reduced immediately by EG upon addition into the hot reaction mixture, as observed from the color change (colorless to brown). The slow injection rate ensured that the RhCl3 in each droplet of the precursor solution was consumed before the next precursor droplet was added, thereby maintaining a low concentration of Rh atoms throughout the synthesis. Under these conditions, small Rh seeds form during the addition of the first few drops of the Rh precursor, and subsequently added precursor deposits Rh atoms on the surface of existing seeds rather than forming new seeds. By controlling the injection time t, and thus the total volume of precursor solution added to the reaction mixture, Rh NCs with edge lengths between 10 and 27 nm were successfully synthesized. Transmission electron microscope (TEM) images of monodisperse Rh NCs with edge lengths of 15 nm (t = 20 min), 21 nm (t = 50 min) and 27 nm (t = 120 min) are shown in Fig. 1b–d. These Rh NCs have slightly concave surfaces as observed by TEM, consistent with the growth mechanism in which Rh atoms first deposit on the corners of the seeds and then migrate to the edges and faces.37
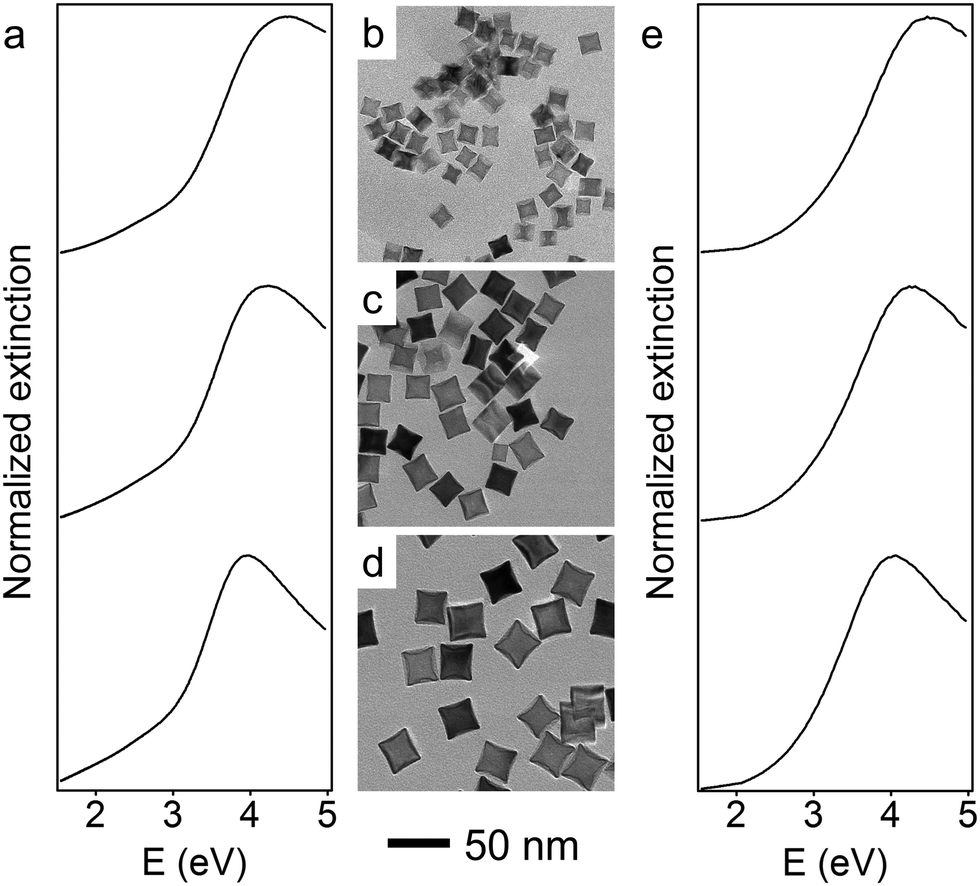 | ||
| Fig. 1 UV-vis extinction spectra (a: experimental; e: theoretical) of monodisperse Rh NCs in ethanol, with corresponding TEM images from unseeded slow-injection methods of average edge lengths of 15 (b), 21 (c), and 27 nm (d), respectively. | ||
The optical properties of these Rh NCs were studied by UV-vis extinction spectroscopy and compared with numerical simulations (Fig. 1a and e). The UV-vis extinction spectra were measured, and the LSPR peaks for 15, 21, and 27 nm Rh NCs dispersed in ethanol were found at 4.51 eV (275 nm), 4.23 eV (293 nm) and 3.96 eV (313 nm), respectively. The resonant energy increasingly red-shifted with increasing size, in good agreement with predictions from a finite element electromagnetics simulation in which a linearly polarized plane wave illuminated a perfect structured Rh NC immersed in ethanol (Fig. 3). The spectral extinction cross-section was calculated as the sum of the absorption and scattering spectral cross-sections (see theoretical simulation for more details, ESI†).
To red-shift the LSPR of Rh NCs to lower UV energies, even larger Rh NCs must be synthesized. One approach to synthesize larger NCs would be to continue increasing the amount of Rh precursor in the unseeded method, but this would be unnecessarily time-consuming since the amount of precursor required, and thus the time required, grows cubically with edge length. Moreover, the prolonged time could affect the shape control of nanoparticles due to parasitic side reactions or the dilution of other reagents (Fig. S2, ESI†). Instead, a more time-efficient, seeded method was developed to obtain larger Rh NCs.38 The reaction mixture containing 27 nm Rh NCs was directly used as a seed solution without any post-synthesis purification or washing treatment. Larger NCs grew as the RhCl3 solution was injected into EG containing a predetermined volume of this seed solution held at 160 °C. To help maintain the cubical shape of the Rh NCs, KBr was co-dissolved with PVP and injected to the reaction mixture along with the RhCl3 solution. By tuning the amount of precursor added, Rh NCs with edge lengths between 27 and 59 nm were synthesized within 2 h using this seeded method.
Representative TEM images of these larger Rh NCs are shown in Fig. 2b–d. These NCs are uniform in size and have slightly concave faces as well. Only a few small Rh NCs were observed from TEM, indicating successful prevention of secondary nucleation during the seeded synthesis. The optical properties of these Rh NCs were also investigated by extinction spectroscopy and compared with the numerical simulation (Fig. 2a and e). The experimentally measured LSPR peaks of 39, 47, and 59 nm Rh NCs were found to be 3.65 eV (340 nm), 3.44 eV (360 nm) and 3.25 eV (381 nm), respectively, increasingly red-shifted toward the lower edge of UV region with increasing size (Fig. 3).
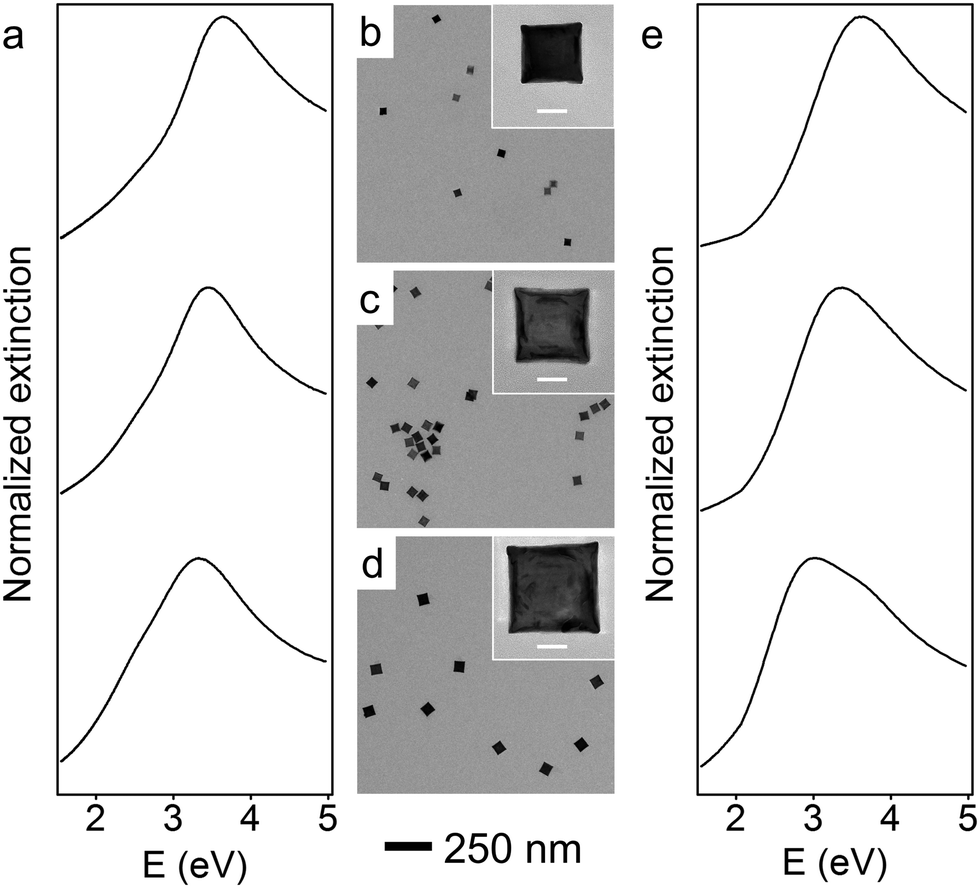 | ||
| Fig. 2 UV-vis extinction spectra (a: experimental; e: theoretical) of monodisperse Rh NCs in ethanol, with corresponding TEM images from seeded slow-injection methods of average edge lengths of 39 (b), 47 (c), and 59 nm (d), respectively. The scale bars for inset images are 20 nm. | ||
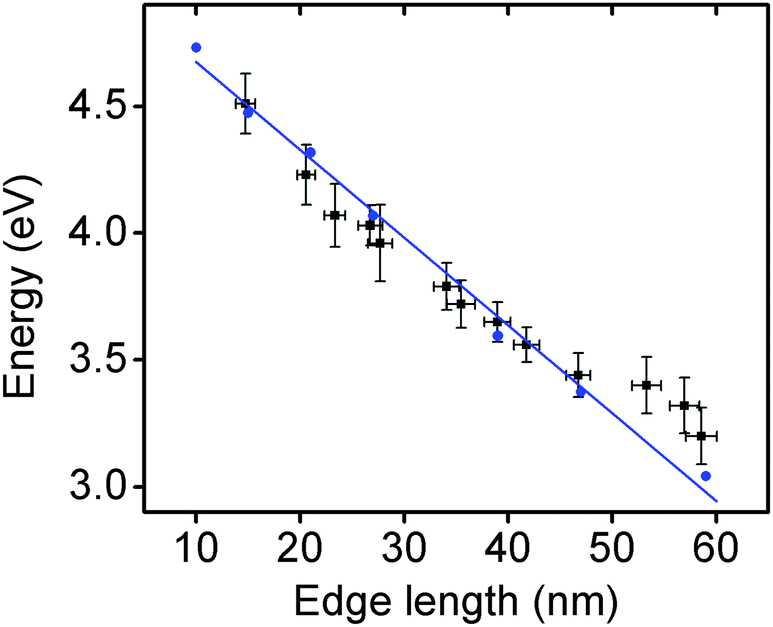 | ||
| Fig. 3 The relationship of edge lengths and LSPR energies from experimental measurements (black squares) and numerical simulations (blue circles) of Rh NCs in ethanol. The blue line is a linear fitting of the simulated results. The x-error bars of experimental data are the standard deviations of the edge lengths of Rh NCs measured by TEM in each sample. The y-error bars are the peak widths at which 99% of maximum intensity in extinction spectra occurs. | ||
The size distribution of these Rh NCs was measured using more than 50 nanoparticles for each sample (Fig. S3, ESI†). The narrow distribution of particle size (<±1.5 nm) could be attributed to the separation of seed formation and time-controlled nanocrystal growth in both unseeded and seeded methods. Seeds formed in the early stage of synthesis, whether formed in situ (unseeded) or intentionally added (seeded), experience the same opportunity to receive new Rh atoms from subsequent addition of Rh precursor, thereby growing into NCs with uniform sizes. By tuning the amount of precursor added to the reaction mixture, the sizes of the produced nanocubes and the associated LSPR energies can be precisely controlled with an excellent match to numerical predictions (Fig. 3). The Rh NC suspension is also stable after stored in ambient condition for several months. Recently, a hydrothermal method was reported to synthesize concave Rh NCs with edge length from 5 to 25 nm. The slow-injection polyol methods in this work, however, offer wider size tunability in a more time-efficient manner.39 Rh NCs with edge lengths larger than 90 nm and LSPRs in the visible region were synthesized, indicating that the LSPR energies of Rh NCs could be further red-shifted with increasing sizes (Fig. S4, ESI†). The size uniformity was not as good as the samples discussed here, and we are currently working on improving the size uniformity.
Examination of high-resolution TEM (HRTEM) imagery of a Rh NC with edge length of 26.0 nm, captured along [100] zone axis (Fig. S5, ESI†), reveals that fringes parallel to the faces possess a lattice spacing of 1.91 Å, corresponding to the {200} planes of face-centered cubic Rh. The contrast along the edge emphasizes the concave faces of these Rh NCs. The simulated near field intensity distribution |E|2 for resonant illumination at the surface of a 27 nm Rh NC is depicted in Fig. 4. Large enhancements of incident radiation intensity are observed at the NC edges, reaching factors as high as 700 at the NC corners. Comparing the enhancements of the edges parallel and perpendicular to the polarization of the incident light reveals a minimum |E|2 of ∼10 occurs at the edge center for parallel polarization.
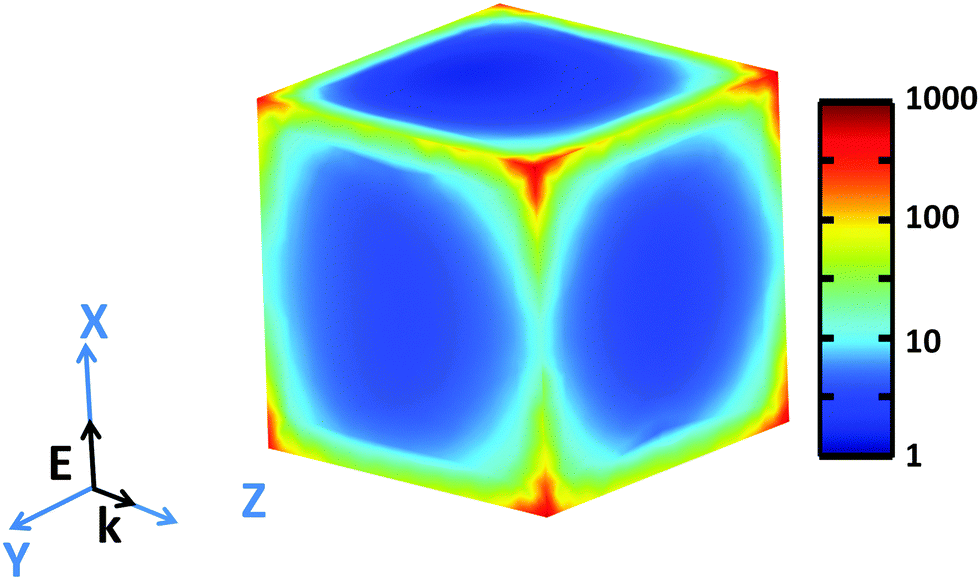 | ||
| Fig. 4 Simulated near-field intensity distribution |E|2 at the surface of a 27 nm Rh NC at the resonant energy 3.96 eV, along with the coordinates used for the finite element modeling. The light is polarized along the X-axis and propagating in Z-axis in the simulation. | ||
The concentrated high-energy fields and hot electrons from the excitation of UV LSPR in Rh NCs could be important sources for photochemical reactions, in much the same way that Au, Ag, and Cu have been used for visible light applications.9,40–43 To probe the efficacy of photochemical reactions accelerated by Rh NCs, UV photo-degradation of a Raman active molecule, PATP, was comparatively studied neat and attached to the surfaces of 21, 39, and 59 nm Rh NCs using Raman spectroscopy with a 325 nm wavelength He–Cd laser. The intensity of the C–C stretch mode of PATP near 1590 cm−1, proportional to the amount of PATP under constant field intensity, was used to quantify the reaction kinetics.44–47
As shown in Fig. 5a and b, the neat PATP sample exhibits a slow decay of the Raman signal with increasing exposure to UV light, while the signal from PATP attached to the surfaces of 39 nm Rh NCs exhibits a much faster decay under identical experimental conditions. Assuming that the photocatalytic reaction is first order on PATP, an exponential decay model is used to fit the Raman data, and the results are shown in Fig. 5c. The model fit the data well, and the rate constants for these four samples are extracted. The neat PATP sample shows the slowest decay with a rate constant of 0.020 s−1. The 21, 39, and 59 nm Rh NC samples have much higher rate constants of 0.66, 0.84, and 0.51 s−1, respectively. The measured LSPR energy of 21, 39, and 59 nm Rh NCs in ethanol is 4.23, 3.65, and 3.25 eV, respectively. Since the LSPR energy of Rh nanoparticles on Si substrates blue-shifts ∼0.2 eV compared to the case in ethanol, the 39 nm Rh NCs on a Si substrate are most resonant with the 325 nm (3.81 eV) laser, and attached PATP decomposed fastest. Conversely, the 21 and 59 nm Rh NCs are non-resonant with the excitation, and the attached PATP decomposed more slowly but still faster than the neat PATP under otherwise identical conditions.26 The observation that PATP attached to 39 nm Rh NCs shows the fastest decay (4 times faster than neat PATP) validates the expected enhancement of photochemical reactions by the excitation of a UV LSPR in Rh nanoparticles. To estimate the enhancement factor, consider that the monolayer of PATP attached to the surfaces of Rh NCs, which together cover ∼3% of the Si substrate (Fig. S6b, ESI†), exhibits a Raman signal comparable to that of the neat PATP with thickness ∼70 nm (as determined by AFM imaging). Since the thickness of PATP monolayer was 0.7 nm,48 the SERS enhancement factor could be volumetrically estimated to be about 400. The very weak Raman signal from a Si substrate without Rh NCs following the same PATP coating procedure (Fig. S6a, ESI†) also proves that the enhancement of Raman signal and accelerated photo-decomposition of PATP indeed come from the excitation of LSPR in Rh NCs.
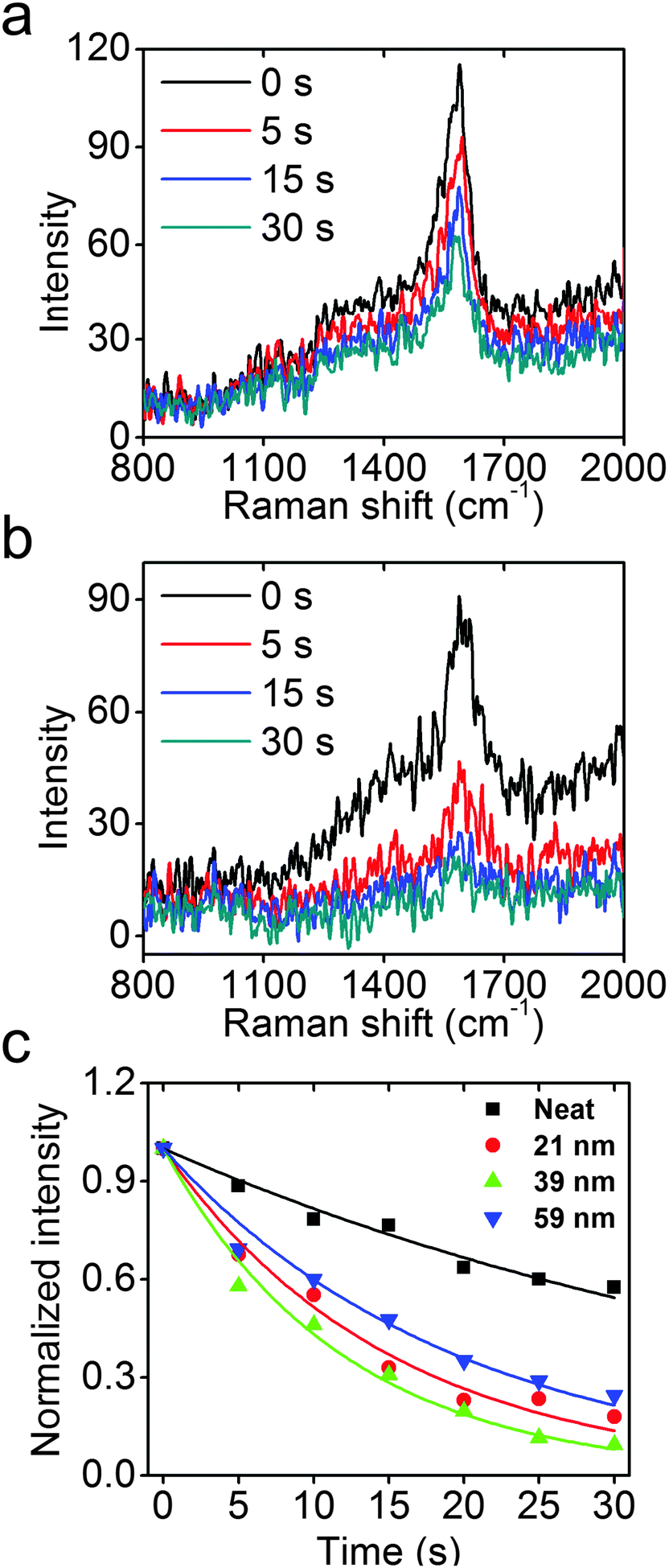 | ||
| Fig. 5 Raman spectra of neat PATP (a) and PATP with 39 nm Rh NCs (b) at representative times (each spectrum integrated over five seconds). (c) Summary of 1590 cm−1 Raman signal decay for PATP neat and attached to 21, 39, and 59 nm Rh NCs. | ||
Conclusion
In conclusion, uniform Rh NCs with edge lengths ranging from 10 to 59 nm have been successfully and controllably synthesized with unseeded and seeded slow-injection methods. UV-vis extinction spectra of these Rh NCs revealed the existence of LSPRs in the UV region that red-shifted with increasing size in a manner consistent with numerical simulations. The sizes and associated resonant energies of Rh NCs can be precisely controlled by the amount of precursor added to the reaction. These UV plasmonic Rh NCs were employed in the photochemical decomposition of PATP under UV illumination and exhibited accelerated reaction rates that were fastest when the LSPR coincided with the laser wavelength. The precise control of particle morphology and LSPR wavelength of noble metal Rh NCs, and the associated field-enhanced spectroscopic and photochemical processes, could prove transformative to the nascent field of UV plasmonics.Acknowledgements
This work has been partially supported by the Paul M. Gross Fellowship from the Department of Chemistry at Duke University, NSF (CHE-1213469) and MICINN (Spanish Ministry of Science and Innovation) through project FIS2013-45854-P. The authors also acknowledge the support from Duke University SMIF (Shared Materials Instrumentation Facilities). A. Barreda wishes to thank the University of Cantabria for a research contract.References
- S. A. Maier, M. L. Brongersma, P. G. Kik, S. Meltzer, A. a. G. Requicha and H. A. Atwater, Adv. Mater., 2001, 13, 1501–1505 CrossRef CAS
.
- J. A. Schuller, E. S. Barnard, W. Cai, Y. C. Jun, J. S. White and M. L. Brongersma, Nat. Mater., 2010, 9, 193–204 CrossRef CAS PubMed
- K. A. Willets and R. P. Van Duyne, Annu. Rev. Phys. Chem., 2007, 58, 267–297 CrossRef CAS PubMed
- S. Lal, N. K. Grady, J. Kundu, C. S. Levin, J. B. Lassiter and N. J. Halas, Chem. Soc. Rev., 2008, 37, 898–911 RSC
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed
- M. W. Knight, H. Sobhani, P. Nordlander and N. J. Halas, Science, 2011, 332, 702–704 CrossRef CAS PubMed
- A. Marimuthu, J. Zhang and S. Linic, Science, 2013, 339, 1590–1593 CrossRef CAS PubMed
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed
- S. Linic, U. Aslam, C. Boerigter and M. Morabito, Nat. Mater., 2015, 14, 567–576 CrossRef CAS PubMed
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS
- B. Wiley, Y. Sun, B. Mayers and Y. Xia, Chem. – Eur. J., 2005, 11, 454–463 CrossRef CAS PubMed
- K.-S. Lee and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 19220–19225 CrossRef CAS PubMed
- X. Lu, M. Rycenga, S. E. Skrabalak, B. Wiley and Y. Xia, Annu. Rev. Phys. Chem., 2009, 60, 167–192 CrossRef CAS PubMed
- J. M. McMahon, G. C. Schatz and S. K. Gray, Phys. Chem. Chem. Phys., 2013, 15, 5415–5423 RSC
- J. M. Sanz, D. Ortiz, R. Alcaraz de la Osa, J. M. Saiz, F. González, A. S. Brown, M. Losurdo, H. O. Everitt and F. Moreno, J. Phys. Chem. C, 2013, 117, 19606–19615 CAS
- N. Mattiucci, G. D'Aguanno, H. O. Everitt, J. V. Foreman, J. M. Callahan, M. C. Buncick and M. J. Bloemer, Opt. Express, 2012, 20, 1868–1877 CrossRef PubMed
- M. W. Knight, L. Liu, Y. Wang, L. Brown, S. Mukherjee, N. S. King, H. O. Everitt, P. Nordlander and N. J. Halas, Nano Lett., 2012, 12, 6000–6004 CrossRef CAS PubMed
- G. Maidecchi, G. Gonella, R. Proietti Zaccaria, R. Moroni, L. Anghinolfi, A. Giglia, S. Nannarone, L. Mattera, H.-L. Dai, M. Canepa and F. Bisio, ACS Nano, 2013, 7, 5834–5841 CrossRef CAS PubMed
- M. W. Knight, N. S. King, L. Liu, H. O. Everitt, P. Nordlander and N. J. Halas, ACS Nano, 2014, 8, 834–840 CrossRef CAS PubMed
- M. J. McClain, A. E. Schlather, E. Ringe, N. S. King, L. Liu, A. Manjavacas, M. W. Knight, I. Kumar, K. H. Whitmire, H. O. Everitt, P. Nordlander and N. J. Halas, Nano Lett., 2015, 15, 2751–2755 CrossRef CAS PubMed
- P. Albella, B. Garcia-Cueto, F. González, F. Moreno, P. C. Wu, T.-H. Kim, A. Brown, Y. Yang, H. O. Everitt and G. Videen, Nano Lett., 2011, 11, 3531–3537 CrossRef CAS PubMed
- S. R. C. Vivekchand, C. J. Engel, S. M. Lubin, M. G. Blaber, W. Zhou, J. Y. Suh, G. C. Schatz and T. W. Odom, Nano Lett., 2012, 12, 4324–4328 CrossRef CAS PubMed
- Y. Yang, J. M. Callahan, T.-H. Kim, A. S. Brown and H. O. Everitt, Nano Lett., 2013, 13, 2837–2841 CrossRef CAS PubMed
-
S. D. Cramer, B. S. Covino and C. Moosbrugger, ASM Handbook: Corrosion: Materials, ASM International, Materials Park, OH, 10th edn, 2005 Search PubMed
- N. Zettsu, J. M. McLellan, B. Wiley, Y. Yin, Z.-Y. Li and Y. Xia, Angew. Chem., Int. Ed., 2006, 45, 1288–1292 CrossRef CAS PubMed
- A. M. Watson, X. Zhang, R. Alcaraz de la Osa, J. M. Sanz, F. González, F. Moreno, G. Finkelstein, J. Liu and H. O. Everitt, Nano Lett., 2015, 15, 1095–1100 CrossRef CAS PubMed
- R. Alcaraz de la Osa, J. M. Sanz, A. I. Barreda, J. M. Saiz, F. González, H. O. Everitt and F. Moreno, J. Phys. Chem. C, 2015, 119, 12572–12580 CAS
- S. M. Humphrey, M. E. Grass, S. E. Habas, K. Niesz, G. A. Somorjai and T. D. Tilley, Nano Lett., 2007, 7, 785–790 CrossRef CAS PubMed
- J. D. Hoefelmeyer, K. Niesz, G. A. Somorjai and T. D. Tilley, Nano Lett., 2005, 5, 435–438 CrossRef CAS PubMed
- Y. Zhang, M. E. Grass, S. E. Habas, F. Tao, T. Zhang, P. Yang and G. A. Somorjai, J. Phys. Chem. C, 2007, 111, 12243–12253 CAS
- Y. Zhang, M. E. Grass, J. N. Kuhn, F. Tao, S. E. Habas, W. Huang, P. Yang and G. A. Somorjai, J. Am. Chem. Soc., 2008, 130, 5868–5869 CrossRef CAS PubMed
- Y. Zhang, M. E. Grass, W. Huang and G. A. Somorjai, Langmuir, 2010, 26, 16463–16468 CrossRef CAS PubMed
- A. J. Biacchi and R. E. Schaak, ACS Nano, 2011, 5, 8089–8099 CrossRef CAS PubMed
- S. Xie, H. Zhang, N. Lu, M. Jin, J. Wang, M. J. Kim, Z. Xie and Y. Xia, Nano Lett., 2013, 13, 6262–6268 CrossRef CAS PubMed
- A. J. Biacchi and R. E. Schaak, ACS Nano, 2015, 9, 1707–1720 CrossRef CAS PubMed
-
COMSOL Multiphysics 4.4, http://www.comsol.com, accessed May 5, 2015 Search PubMed
- H. Zhang, W. Li, M. Jin, J. Zeng, T. Yu, D. Yang and Y. Xia, Nano Lett., 2011, 11, 898–903 CrossRef CAS PubMed
- C. Gao, J. Goebl and Y. Yin, J. Mater. Chem. C, 2013, 1, 3898–3909 RSC
- Y. Chen, Q.-S. Chen, S.-Y. Peng, Z.-Q. Wang, G. Lu and G.-C. Guo, Chem. Commun., 2014, 50, 1662–1664 RSC
- P. Christopher, H. Xin, A. Marimuthu and S. Linic, Nat. Mater., 2012, 11, 1044–1050 CAS
- S. Mukherjee, F. Libisch, N. Large, O. Neumann, L. V. Brown, J. Cheng, J. B. Lassiter, E. A. Carter, P. Nordlander and N. J. Halas, Nano Lett., 2013, 13, 240–247 CrossRef CAS PubMed
- S. Linic, P. Christopher, H. Xin and A. Marimuthu, Acc. Chem. Res., 2013, 46, 1890–1899 CrossRef CAS PubMed
- S. Mukherjee, L. Zhou, A. M. Goodman, N. Large, C. Ayala-Orozco, Y. Zhang, P. Nordlander and N. J. Halas, J. Am. Chem. Soc., 2014, 136, 64–67 CrossRef CAS PubMed
- K. T. Carron and L. G. Hurley, J. Phys. Chem., 1991, 95, 9979–9984 CrossRef CAS
- Y. Fang, N.-H. Seong and D. D. Dlott, Science, 2008, 321, 388–392 CrossRef CAS PubMed
- K. Kim, D. Shin, J.-Y. Choi, K. L. Kim and K. S. Shin, J. Phys. Chem. C, 2011, 115, 24960–24966 CAS
- M. Pilo-Pais, A. Watson, S. Demers, T. H. LaBean and G. Finkelstein, Nano Lett., 2014, 14, 2099–2104 CrossRef CAS PubMed
- D. M. Disley, D. C. Cullen, H.-X. You and C. R. Lowe, Biosens. Bioelectron., 1998, 13, 1213–1225 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nh00062a |
| This journal is © The Royal Society of Chemistry 2016 |