
Chemical analysis in saliva and the search for salivary biomarkers – a tutorial review
Kamonwad
Ngamchuea
,
Korbua
Chaisiwamongkhol
,
Christopher
Batchelor-McAuley
and
Richard G.
Compton
 *
*
Department of Chemistry, Physical & Theoretical Chemistry Laboratory, University of Oxford, South Parks Road, Oxford, OX1 3QZ, UK. E-mail: richard.compton@chem.ox.ac.uk; Fax: +44(0) 1865275410; Tel: +44(0) 1865275 957
First published on 3rd November 2017
Abstract
Biomarkers refer to analytes that can be used in the diagnosis of diseases or disorders. In saliva, there are many components that are potential biomarkers, and increasing research has focussed on the development of saliva as a diagnostic fluid. This review summarizes the existing uses of salivary biomarkers and highlights the importance of the choice of saliva collection as well as the storage procedures. A case study on the effect of collection tools on the concentrations of one of the potential biomarkers, glutathione, is highlighted. Moreover, molecular diagnosis requires reliable measurement assays. This review presents electroanalytical methods for the detection of salivary biomarkers. It further reviews approaches that can be taken to improve the selectivity of electroanalytical assays without the use of biologically selective materials, notably without the use of enzymes or antibodies.
1 Introduction
Saliva is a complex bodily fluid consisting of ca. 99% water, inorganic and organic substances and a variety of proteins such as enzymes, mucus and glycoproteins.1 It is secreted by three major pairs of salivary glands and other minor mucous glands surrounded by capillaries.2 Many blood components enter the saliva via either active transport requiring chemical energy from adenosine triphosphate (ATP) or passive diffusion (no energy required; movement down a concentration gradient) through cell membranes. Accordingly, the presence, or the change in concentrations, of salivary components may be used in the diagnosis of diseases and the analysis of the state of health. Examples of commercial uses of saliva tests include HIV detection and steroid hormone monitoring.This review focuses on salivary biomarkers i.e. diagnostically useful salivary components. Saliva testing offers advantages over blood analysis in part due to its non-invasive nature, the ease of collection, storage and shipment of specimens, as well as the possibility of collecting multiple samples. Compared with other non-invasive procedures saliva overcomes the problems of the intrusion of privacy of urine and limited access to sweat and tears. Areas where saliva biomarkers may become particularly advantageous include point-of-care analysis, mass detection, epidemiologic surveillance, situations where the sample volume is limited e.g. in newborns and cases where self-monitoring may be desirable. For example in psychiatry, most diseases have persistent life-long symptoms and long-term monitoring is needed. Many neurological disorders can currently be assessed by behaviour observations as definitive causes and molecular markers are not known. Saliva tests may be able to offer alternative methods to aid the diagnosis of such diseases and disorders. Other potential uses of saliva are the detection of drugs of abuse and doping substances in forensics and sports.
A large number of publications have reported the search for saliva biomarkers. Some focus on the development of measurement assays, while others study the correlations between the presence/absence or the levels of saliva biomarkers with specific diseases/disorders. These two areas form the basis of this review. The review starts with a short description of the functions of saliva and how salivary components play important roles in maintaining the normal functions of the body. Through understanding their functions, it is possible to investigate the potential of salivary components as biomarkers for specific diseases and disorders. Details of saliva composition are provided as a starting point for analytical chemists wanting to develop analytical assays for applications in saliva diagnosis. Also, available recipes for artificial saliva are provided for use in the initial stage in the development of new assays. In working with real saliva samples, the importance of the careful selection of collection and storage procedures is highlighted. The review then summarizes the existing diagnostic uses of salivary biomarkers and any correlation with blood plasma and other bodily fluids. Examples of successful case histories and the important factors needed to be considered in the search for new biomarkers are then discussed. The review closes with the report of electrochemical assays for diagnostic uses. In particular, methods for improving the selectivity of the assays without the use of biologically selective molecules are considered.
2 Functions of saliva
There are five major functions of saliva: lubrication and protection, buffering, maintaining tooth integrity, antibacterial activity, and taste and digestion (Fig. 1).3 “Lubrication and protection” preserves the oral tissues against irritating agents. The lubricating effects also help mastication, speech, and swallowing processes. The main lubricating compounds in saliva are mucins which are complex protein molecules.4 “Buffering action” is the second role of saliva. Whole saliva contains three major components contributing to the buffer capacity including bicarbonate, phosphate, and protein buffer.5 Enzymes such as carbonic anhydrase also participate in controlling the pH of saliva.6 This buffer capacity allows saliva to maintain a relatively constant pH (normal pH range 6.7–7.3) against the acids produced by micro-organisms and consumed through the diet.7 The third main function of saliva is “maintaining tooth integrity” by balancing a demineralization and a remineralization process. Demineralized processes happen when acids diffuse into the enamel resulting in crystalline dissolution and occur in a critical pH range (pH 5–5.5) for the development of caries. Dissolved mineral diffuses from the tooth into the saliva. The mineral loss may be recovered to the tooth structure from ions dissolved in the saliva by a remineralization process.8 “Anti-bacterial activity” is another important function of saliva. Saliva contains several immunologic and non-immunologic proteins with anti-bacterial properties. Among the immunologic components of saliva, Secretory immunoglobulin A (IgA) is the largest proportion which can neutralize viruses and bacterial, and enzyme toxins. The non-immunologic salivary proteins also act as anti-bacterial agents; for example lysozyme can hydrolyze the cell wall of some bacteria. A fifth function of saliva is “taste and digestion”. For a tasting function, the main role of saliva includes the transport of taste substances to and the protection of the taste receptors.9 The salivary enzyme amylase has an early role in the total digestion process by breaking down starch into sugars.10 | ||
| Fig. 1 Main functions of saliva. | ||
3 Components of human saliva and artificial saliva recipes
The composition of saliva varies during the day and between individuals.11 Attempts to report saliva constituents and concentrations mainly use average compositions that can be found in healthy subjects. Components of authentic human saliva and a comparison of the normal range of the concentrations between saliva and other biological fluids are summarized in Table 1. Briefly, saliva is composed of a variety of ions including sodium, potassium, calcium, bicarbonate, thiocyanate and phosphate. Also found in saliva are low molecular weight organic substances such as uric acid and lactate, immunoglobulins, enzymes, and mucins and some important hormones such as cortisol.| Real saliva composition | Normal range | Ref. | ||
|---|---|---|---|---|
| Saliva | Other biological fluids | |||
| a U mL−1: enzymatic activity per unit (mL) of saliva. | ||||
| 1. Inorganic compounds | Na+ | 20–80 mmol L−1 | Plasma 145 mmol L−1 | 11 |
| K+ | 20 mmol L−1 | 4 mmol L−1 | ||
| Ca2+ | 1–4 mmol L−1 | 2.2 mmol L−1 | ||
| Cl− | 30–100 mmol L−1 | 120 mmol L−1 | ||
| HCO3− | 15–80 mmol L−1 | 25 mmol L−1 | ||
| Phosphate | 4 mmol L−1 | 1.2 mmol L−1 | ||
| Mg2+ | 0.2 mmol L−1 | 1.2 mmol L−1 | ||
| SCN− | 2 mmol L−1 | <0.2 mmol L−1 | ||
| NH3 | 3 mmol L−1 | 0.05 mmol L−1 | ||
| 2. Organic compounds (non-protein and lipids) | Uric acid | 3.38 ± 0.21 mg dL−1 | Serum 6.31 ± 0.24 mg dL−1 | 21–23 |
| 217.2 ± 110.3 mol L−1 | ||||
| 0.1–7.5 mg dL−1 | ||||
| Bilirubin | 0.5–5.0 μmol L−1 | Serum 0.2–1.2 mg dL−1 | 24 | |
| Creatinine | 0.12 ± 0.06 mg dL−1 | Serum 0.89 ± 0.17 mg dL−1 | 25 | |
| 0.05–0.2 mg dL−1 | Serum 0.6–1.5 mg dL−1 | |||
| Glucose | 91.3 ± 10.1 mg dL−1 | Plasma 80–120 mg dL−1 | 22 and 26 | |
| 4–13 mg dL−1 | ||||
| Cholesterol | 0.02–5.46 μmol L−1 | Serum <5 mmol L−1 | 27 | |
| Lactate | 0.3–1.8 mM | Serum 0.5–1.0 mM | 28 and 29 | |
| 0.1 to 2.5 mmol L−1 | ||||
| 3. Protein/polypeptide compounds | a-Amylase | 19–308 U mL−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
Serum 0.05–0.125 U mL−1a |
23 and 30 |
| 93 ± 62 U L−1a |
||||
| 2.64 ± 1.8 mg mL−1 | ||||
| Albumin | 0.2 ± 0.1 mg mL−1 | Serum 3.5–5.5 g dL−1 | 31 | |
| Secretory-IgA | 80–717 mg dL−1 | Serum 70–400 mg dL−1 | 23 and 32 | |
| 124.3–333.5 μg mL−1 | ||||
| Mucin group | MUC5B: 2.4 ± 1.7 U mL−1 | Serum 9.9 ± 0.8 ng ml−1 | 31 and 33 | |
| 1.19 ± 0.17 mg mL−1 | ||||
| Lysozyme | 3–50 μg mL−1 | Serum 7.4 ± 1.8 mg mL−1 | 23, 32 and 34 | |
| 59.7 to 1062.3 μg ml−1 | Serum 4–9 μg mL−1 | |||
| Total proteins | 7.1–223.2 mg dL−1 | Serum 6–8 g dL−1 | 23 and 31 | |
| 0.9 ± 0.2 mg mL−1 | ||||
| 4. Hormones | Cortisol | 3.5–27.0 mg dL−1 | Serum 2–25 mg dL−1 | 35 |
| Testosterone | 32–55 pg mL−1 | Serum 320–600 ng dL−1 | 36 | |
| Progesterone | Luteal phase 436 ± 34 pmol L−1 | Serum male: <1 ng mL−1 | 37 | |
| Follicular phase 22.1 ± 2.7 pmol L−1 | Serum female: 0.1–20 ng mL−1 | |||
| Estrogen (Estradiol) | Luteal phase 20.6 ± 0.4 pmol L−1 | Serum male: 15–60 pg mL−1 | 37 | |
| Serum female: 15–370 pg mL−1 | ||||
In research studies, the use of natural saliva is limited as it is difficult to collect and sterilize a large amount of saliva. Moreover, saliva samples vary in composition and properties.12 Accordingly, synthetic saliva was developed for use in in vitro studies. A basic requirement of synthetic saliva is to react in the system or with the test material in the same manner as that of authentic saliva. However, it is well understood that an exact duplication of human saliva properties is impossible owing to the inconsistent and unstable nature of authentic saliva which varies during the day and within every individual as mentioned earlier.13 Many synthetic saliva recipes can be found in the literature as reviewed by Leung et al.13 who collected several formulae published between 1931 and 1997.
This review presents five different synthetic saliva recipes as developed for different purposes. The selected artificial saliva formulae are frequently used by others with or without modification of the proposed recipe. As shown in Table 2, some of these formulae include exclusively inorganic compounds which are used for in vitro studies,14–18 whereas others include some organic compounds and proteins.12,19 AFNOR, Fusayama–Meyer and SAGF recipes include only inorganic compositions which are more or less similar to an average composition of human saliva. The composition was also chosen based on the similarity of experimental results obtained from artificial saliva and from authentic human saliva.16 The proposed recipes were employed in the study of electrochemical behaviour and corrosion resistance of materials in dental applications.14–18 The organic components and some proteins are responsible for saliva viscosity and as such might influence the diffusion rates of other solutes, and thus some reaction rates. Note that the viscosity of human whole saliva is ca. 1.30 relative to distilled water.20 For saliva samples that have been centrifuged and filtrated to remove viscous components, the viscosity reduces to being close to that of distilled water. In developing artificial saliva for chemical and electrochemical research, there is no attempt to simulate the viscosity of authentic saliva owing to the practical difficulty of obtaining suitable materials. Therefore, most electrochemical studies included only inorganic compositions of artificial saliva. For biological studies, organic components and proteins are necessary to mimic the medium as closely as possible for the use of in vitro model systems to study dental biofilms. For example, in the cultural studies of dental plaque reported by Shellis,12 the first step in an artificial saliva preparation was the extraction of salivary glycoproteins from bovine submandibular glands.
| Artificial saliva compositions | Concentration (g L−1) | |||||
|---|---|---|---|---|---|---|
| AFNOR14,15 | Fusayama–Meyer16 | SAGF17,18 | Klimek19 | Shellis12 | ||
| a Bacto-Mucin Bacteriological. b Somogyi's unit/L is a measure of the level of activity of amylase in blood serum. One Somogyi unit is defined as the amount of amylase required to produce the equivalent of 1 mg of glucose when acting on a standard starch solution under a defined condition.38 | ||||||
| 1. Inorganic compounds | NaCl | 6.70 | 0.40 | 0.13 | 0.58 | — |
| KCl | 1.20 | 0.40 | 0.96 | 1.27 | 1.16 | |
| Na2HPO4 | 0.26 | — | — | 0.34 | 0.375 | |
| KH2PO4 | 0.20 | — | 0.66 | 0.33 | 0.35 | |
| NaHCO3 | 1.50 | 0.10 | 0.63 | — | 0.54 | |
| KSCN | 0.33 | — | 0.19 | 0.16 (NaSCN) | 0.22 | |
| CaCl2·2H2O | — | 0.69 | 0.23 | 0.17 | 0.21 | |
| Na2S·9H2O | — | 0.005 | — | — | — | |
| Urea | — | 1.00 | 0.20 | 0.20 | 0.17 | |
| NaH2PO4·H2O | — | 0.69 | — | — | — | |
| NH4Cl | — | — | — | 0.16 | 0.233 | |
| Na2SO4·10H2O | — | — | 0.76 | — | — | |
| MgCl2·6H2O | — | — | — | — | 0.043 | |
| Sodium citrate | — | — | — | — | 0.013 | |
| 2. Organic compounds | Ascorbic acid | — | — | — | 0.002 | — |
| Glucose | — | — | — | 0.03 | — | |
| Uric acid | — | — | — | — | 0.0105 | |
| Creatinine | — | — | — | — | 0.0001 | |
| Choline | — | — | — | — | 0.013 | |
| Mixture of vitamins | — | — | — | — | 0.0008 | |
| 3. Protein/polypeptide compounds | Mucin | — | — | — | 2.70a | — |
| Glycoprotein | — | — | — | — | 2.5 | |
| Alpha amylase | — | — | — | — | 3 × 105 Somogyi's unit L−1b |
|
| Albumin | — | — | — | — | 0.025 | |
| Mixture of amino acids | — | — | — | — | 0.041 | |
4 Saliva collection and storage
The successful measurement of target analytes in saliva requires appropriate collection, processing, and storing under suitable conditions.39 Saliva samples can be easily collected from humans. Collection of saliva can be generally categorized into two groups: whole saliva collection and individual gland collection. Whole saliva is simple to collect and does not need to be performed by specialists but can be done by study subjects or patients. Unstimulated whole saliva can be readily obtained by ‘passive drooling’ or ‘spitting’ directly into a container.40,41Stimulated whole saliva can be collected through mechanical stimulation by chewing inert materials such as parafilm and/or gustatory stimulation with acid i.e. 0.01 M citric acid. Briefly, for citric acid use, cotton soaked with citric acid was placed on the back of the tongue for a period of time i.e. 30 s and then was removed. The stimulated whole saliva was then collected into a container.42 Note that the use of citric acid lessens the pH of samples to below 3; so it is apparent that the use of citric acid stimulation is not suitable for obtaining samples for pH measurement in saliva. Moreover, citric use may also have an effect on the measurement of some analytes such as testosterone and electrolyte concentrations.43,44 There are some devices that facilitate the stimulated collection of whole saliva. Most of them comprise cotton based material for saliva adsorption and a conical tube for the centrifugation and retrieval of collected saliva. Note however that cotton based sampling may affect some of the salivary composition levels because of the binding of analytes to cotton.45 In order to avoid the binding of analytes to the collection devices, non-cotton based materials such as polystyrene and polyester are preferable.46Herein, we present a case study from the authors’ laboratory to demonstrate the effects of the collection tools on the concentration of glutathione (GSH) detected. In particular, the differences between cotton and polystyrene swabs are investigated using standard glutathione solutions and real saliva samples. The importance of glutathione and its potential as a biomarker will be discussed later in the following section.
In the case of standard solutions, 1 μM of GSH in phosphate buffer pH 7.0 was used. First, the GSH solution with initial volumes of 245, 980 or 1960 μL was exposed to a cotton Salivette® swab. The solution was left on the cotton swab for a varied time between 0 and 60 min. The soaked cotton swab was then centrifuged at the supplier recommended speed of 1000g for 1 min. The extracted liquid, whose volume was smaller than the initial volume, was then subjected to GSH measurement by the modified Tietze method.47 The amount of GSH recovered from the swab is quantified by the percentage (%) recovery defined as:
| % recovery = (cf − ci)/ci × 100%, | (1) |
Fig. 2a shows that the recovery of GSH from cotton swabs is always lower than 100%, even in the case that the Salivettes are centrifuged immediately after collection. Fig. 2a also demonstrates the decrease of glutathione recovery as a function of exposure time (t), with only ca. 20% of GSH recovered in the worst case (245 μL initial volume and 60 min exposure time). In real applications where there is unavoidable varying delay in the transfer of samples from collection points such as clinic or hospital to the analysis laboratory, the delay can give rise to significant errors.
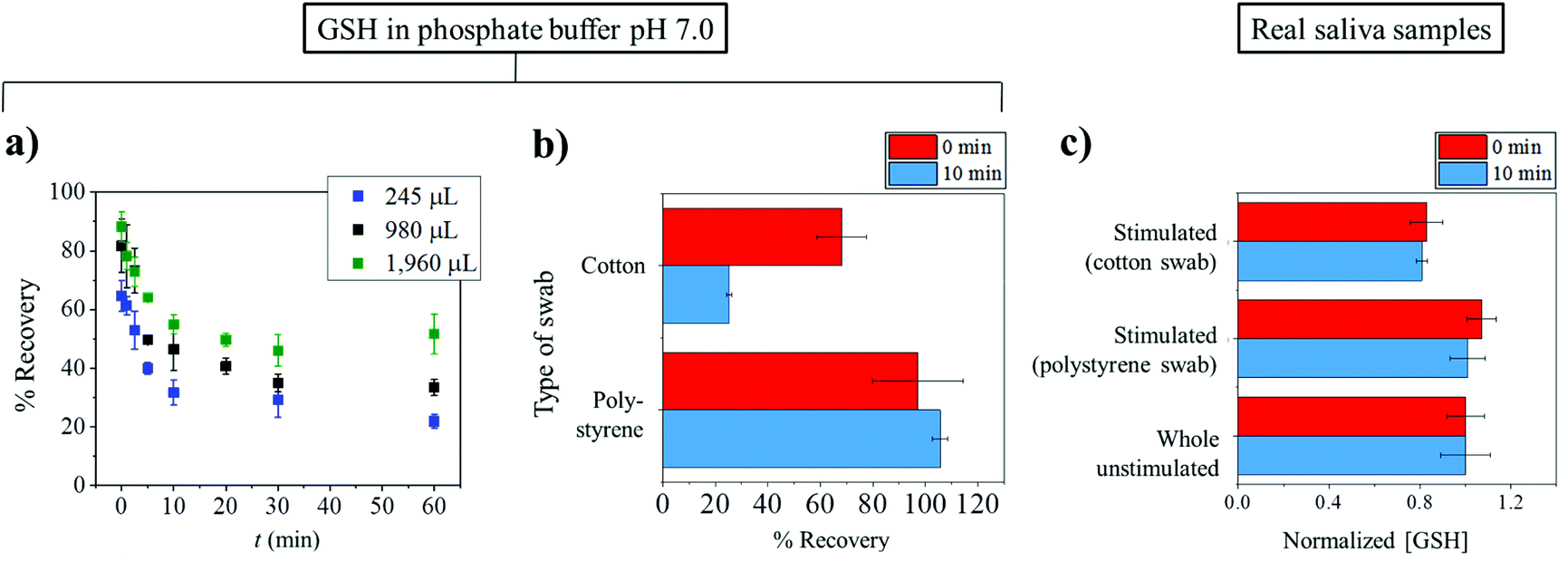 | ||
| Fig. 2 The effects of collection tools on the recovery of glutathione. (a) The amount of glutathione recovered from cotton swabs as a function of time between collection and centrifugation retrieval of the samples. (b) A bar graph showing the significantly higher recovery of glutathione from synthetic polystyrene Salivette® as compared with cotton Salivette® at two different exposure times (t) of 0 min (red) and 60 min (blue). (c) Concentrations of glutathione in real saliva samples: whole unstimulated saliva collected by spitting directly into a container and stimulated saliva collected by cotton Salivette® and polystyrene Salivette® at two different exposure times (t) of 0 min (red) and 10 min (blue). | ||
From Fig. 2a, it can also be seen that the percentage recovered from the cotton swabs also depends on the initial volumes used. The dependency on the initial volume makes it not possible to correct for the loss of GSH, especially in saliva collection experiments where initial volumes are unknown. Consequently, the use of a cotton-based tool induces random errors to saliva analysis especially for substances which have high affinity to bind to cotton. As a comparison to the cotton Salivette®, synthetic polystyrene Salivette® was studied. In contrast to cotton, polystyrene yields ca. 100% recovery of the material (Fig. 2b), suggesting that polystyrene may be a better alternative for saliva collection.
The effect of the collection tools on the concentrations of glutathione detected was further investigated in real saliva samples. The subjects were given the cotton and polystyrene swabs to chew for 1 min. The stimulated saliva samples were then extracted from the swabs by centrifuging at 1000g for 1 min. Whole unstimulated saliva collected by spitting was also studied in comparison. As glutathione concentrations vary from sample to sample, the results are presented as ‘normalized GSH concentration’ where the GSH concentration in whole unstimulated saliva is normalized to 1, and the GSH levels in stimulated saliva collected from the same subject are scaled accordingly. When collecting stimulated saliva using the cotton Salivette®, the glutathione measured was 81 ± 2% (t = 0 min) and 83 ± 7% (t = 10 min) of that detected in unstimulated saliva (Fig. 2c). On the other hand when polystyrene Salivette was used, the levels of GSH detected were 101 ± 8% (t = 0 min) and 107 ± 6% (t = 10 min) of the whole unstimulated saliva level (Fig. 2c). Comparable results in whole unstimulated saliva and stimulated saliva obtained using polystyrene Salivette® further evidence that the polystyrene swab is a suitable saliva collection tool for glutathione analysis. However, note that this may not be the case for some other analytes as they may be able to adsorb onto polystyrene, and hence the effect of the collection tools on the recovery of analytes should be studied prior to any real saliva applications. It is of note that in some cases, the levels of analytes in unstimulated and stimulated saliva may not be expected to be the same even if the material is not lost to the collection tool. This difference between the stimulated and unstimulated saliva may occur when the analyte's concentration is affected by the change in flow rates or the different contributions of salivary glands.48
In the above, we have reviewed the available methods for whole saliva collection and the potential problems associated with the use of cotton-based collection tools; next, we will discuss individual gland collection. Individual gland saliva can be obtained by glandular duct cannulation (a thin tube inserted into a duct to obtain saliva) or by using specific collecting devices to the ducts of interest. For example, pure parotid saliva may be collected by the use of a metal or acrylic cup placed over Stenson's duct.49 However, from a practical viewpoint, these sampling procedures are slow and complex, and require a specialist for collecting the specimens. For these reasons, these collection methods are not widely used.
It is also important to use an appropriate container for the target analytes. For example, if the target analytes are steroid hormones, such as cortisol, low-affinity plastic containers should be used to prevent protein binding to the walls of collection tubes.50 For these reasons, the nature of the compounds of interest must be always taken into account when designing sampling approaches.
After collection, saliva samples should be rapidly processed or suitably stored. Some components are stable in saliva for a long time, while others have very short lifetimes. Inorganic compounds are relatively stable at room temperature. Czégény et al.51 reported that calcium and magnesium contents showed good stability for at least 1 week when the samples were stored at room temperature. Moreover, calcium and phosphate were found to be stable when stored at −20 °C for up to two months.52 On the other hand, the cortisol (steroid hormone) level decreased by ca. 10% per month in samples stored at room temperature.53 Cortisol is stable in saliva for up to three months at 5 °C and up to one year at −20 °C and −80 °C. Proteins also showed rapid degradation within 30 min after sample collection at room temperature.54 Storing the samples at 4 °C and the use of protease inhibitors can help reduce degradation.54
An alternative approach to preserve target analytes in saliva samples is the pre-analytical treatment of the samples prior to storage. For example, snap-freezing of saliva with glycerol in liquid nitrogen is a method for inhibiting bacterial protease activity from degrading some proteins in saliva. The method was used by Nurkka et al.55 for storing saliva before testing anticapsular IgA concentrations. Briefly, the saliva specimen was mixed with an equal volume of 80% glycerol in H2O and dipped into liquid nitrogen. This approach yielded an antibody concentration ca. 50% higher than that in specimens temporarily stored at 4 °C (whether or not the sample was stored with protease inhibitors). Another possible method for pre-treatment is to mix saliva with enzyme inhibitors, stabilizing substances and/or sodium azide (NaN3) to delay bacterial growth. Gröschl et al.56 demonstrated that added sodium azide stabilized the concentrations of salivary steroids (cortisol and progesterone). However, sodium azide can interfere with the activity of horseradish peroxidase which is a common component in enzyme-based saliva immunoassays.57 Therefore if pre-treatment is used, any interferences caused by added compounds to the measurement assays and to other salivary components have to be thoroughly investigated.
5 Biomarkers in saliva – what correlations exist, do not exist
Prior to diagnostic use, the applicability of the biomarkers for a specific disease has to be validated. Ideally, the aetiology of the disease – that is the mechanisms by which the biomarker takes part in the development of diseases or the action of the biomarker during the diseases – should be understood. However, the mechanistic detail is often unknown or not adequately known. Hence, the relevance of the biomarker to a specific disease has to be investigated based on a statistical model.58The potential of a salivary component as a biomarker may be evaluated by the correlation of the presence/absence or the concentrations of the biomarker in saliva samples between a control population and patients. Additionally, it is recommended that the correlation between matched blood plasma and saliva samples is investigated alongside the correlation with diseases. In general, the ratio of the concentrations in plasma to saliva should be approximately constant; otherwise the factors influencing the ratios should be examined.59
Hitherto, the potential of salivary inorganic constituents, anti-oxidants, hormones, antibodies and antigens as biomarkers in the diagnosis of several oral and systemic diseases has been recognized. In oral diseases, saliva has been used to detect oral cavity cancer, dental caries, periodontal disease and oral dryness; see Table 3. In systemic diseases (i.e. diseases that affect the entire body), saliva has been demonstrated to have strong correlations with plasma or serum for many cell components, and hence can act as a mirror reflecting the health states of the body.60 Examples of reported uses of saliva in systemic conditions, diseases, disorders and infections include the monitoring of hormone levels, pregnancy, risks for preterm labour, psychological disorders, neurological disorders, the condition of the immune system, smoking status, virus infections and nutritional status (Table 3).
| Biomarkers | Diagnostic uses | Bodily fluid(s) | Correlation | Detection method | Ref. |
|---|---|---|---|---|---|
| a Prohibited doping substances that have been demonstrated to be detectable in saliva include amphetamine, methamphetamine, norselegiline, ephedrine/norephedrine, ephedrine-d3, selegiline, pentetrazol, crotetamide, cropropamide, sibutramine, codeine/dihydrocodeine, modafinil, ISTD1, cocaine, cocaine-d3, BEG, BEG-d3, EME, and EME-d3.106 Detection methods: FID: flame ionization detector, GC: gas chromatography, GLC: gas–liquid chromatography, HPLC: high performance liquid chromatography, MS: mass spectrometry. | |||||
| Chemicals | |||||
| Cotinine | Cigarette usage | Saliva/plasma | Strong correlation (r = 0.99) | GC | 64 |
| Cigarette usage | Saliva/serum | Strong correlation | HPLC/MS | 65 | |
| Nicotine | Cigarette usage | Saliva/urine | — | Single-drop microextraction/GC-FID | 66 |
| Nicotine skin patch users | Stimulated saliva/plasma | Strong correlation (r = 0.82) | — | 67 | |
| Anabasine | Cigarette usage | Saliva/urine | — | Single-drop microextraction/GC-FID | 66 |
| Nitrates and nitrites | Gastric cancer | Saliva | — | Colorimetric | 68 |
| Periodontal disease | Stimulated/unstimulated saliva | — | Colorimetric | 69 | |
| Caffeine | Caffeine clearance: liver function | Saliva/serum | Strong correlation (r > 0.98) | Enzyme immunoassay | 70 |
| Probe for CYP1A2 | Saliva/plasma /urine | Strong correlation with plasma (r = 0.97) | — | 71 | |
| Drug monitoring | Saliva/plasma | Strong correlation (r = 0.98) | HPLC | 72 and 73 | |
| SCN− | Smoking status | Saliva/plasma/urine | — | Colorimetric | 74 |
| Cystic fibrosis | Saliva | — | Colorimetric | 75 | |
| Anti-oxidants | |||||
| Glutathione (GSH, GSSG) | Head and neck squamous cell carcinoma (GSH) | Stimulated saliva | — | HPLC | 76 |
| Oral squamous Cell carcinoma (GSH) | Unstimulated saliva | — | Colorimetric | 77 | |
| Health monitoring (GSH + GSSG) | Stimulated saliva/whole blood | Weak correlation (r = 0.43) | Colorimetric | 47 | |
| Diabetic/parenteral drug addict (GSH, GSSG) | Stimulated saliva | — | Fluorometric | 78 | |
| Ascorbic acid | Correlation studies | Parotid saliva/plasma/white blood cell (WBC) | Significant correlation with WBC (p < 0.001) | — | 79 |
| Uric acid | Oral cavity cancer | Saliva | — | Colorimetric | 80 |
| Gout | Saliva/blood | — | Colorimetric | 81 | |
| Diabetes | Saliva/serum | — | Colorimetric | 82 | |
| Metabolic syndrome | Saliva | — | Colorimetric | 83 | |
| Therapeutic drugs | |||||
| Anticonvulsants/antiepileptic | Drug monitoring | Saliva/plasma | Strong correlation | GLC, immunoassay | 84–86 |
| Drug monitoring | Saliva/plasma/Cerebrospinal fluid | strong correlation | GC-FID | 87 | |
| Theophylline | Drug monitoring | Stimulated saliva/plasma | Strong correlation (r > 0.9) | HPLC, immunoassay | 88–90 |
| Drug monitoring | Unstimulated (U), stimulated (S) saliva/plasma | Strong correlation (U: r = 0.97) (S: r = 0.88) | Immunoassay | 91 | |
| Acetaminophen (paracetamol) | Drug monitoring | Whole saliva/plasma | Strong correlation (r = 0.95) | Colorimetric | 92 |
| Lithium | Drug monitoring | Saliva/serum | Strong correlation (r = 0.83) | — | 93 |
| Drug monitoring | Stimulated saliva/plasma/RBC | Strong correlation | Flame photometry | 94 | |
| Drugs of abuse | |||||
| Ethanol | Drug monitoring | Saliva/serum, whole blood/urine | Strong correlation | GC-FID, colorimetric | 95–98 |
| Cocaine | Drug monitoring | Saliva/plasma | Significant correlation (p ≤ 0.05) | — | 99 and 100 |
| Heroin | Drug monitoring | Saliva/plasma | — | GC-MS, immunoassay | 101–103 |
| Marijuana | Drug monitoring | Saliva | — | Fluorometric, immunoassay | 104 and 105 |
| Prohibited doping substances | |||||
| Amphetamine, methamphetamine, etc.a | Detection | Saliva/urine | — | GC-MS | 106 |
| Hormones | |||||
| Cortisol | Chronic fatigue syndrome, depression | Saliva/plasma | Strong correlation | Immunoassay | 107 and 108 |
| Evaluation of adrenal function | Saliva/plasma/serum | Strong correlation | Immunoassay | 109 and 110 | |
| Correlation studies | Saliva/serum | Strong correlation (r = 0.89) | Immunoassay | 111 | |
| Aldosterone | Correlation studies | Saliva/plasma | Strong correlation (r = 0.84,112 0.93113) | Dialysis, immunoassay | 112 |
| Testosterone | Correlation studies | Saliva/plasma | Strong correlation (r > 0.7) | Dialysis, immunoassay | 114 and 115 |
| Correlation studies | Saliva/serum | Strong correlation (r > 0.8) | Immunoassay | 116 and 117 | |
| 17-Hydroxyprogesterone | Assessing endocrine function | Saliva/plasma/parotid fluid | Strong correlations (r > 0.9) | Immunoassay | 118 |
| 21-Hydroxylase deficiency | Saliva/serum | Strong correlation (r = 0.93) | Immunoassay | 119 | |
| Progesterone | Correlation during late pregnancy | Saliva/serum | Weak correlation | Immunoassay | 120 |
| Correlation during the menstrual cycle | Saliva/plasma | Significant correlation (r = 0.58, p = 0.001) | — | 121 | |
| Estradiol | Correlation studies in postmenopausal women | Saliva/serum | Strong correlation in users on estrogen therapy (r = 0.81) | Immunoassay | 122 |
| Monitoring follicular stimulation | Saliva/serum | Strong correlation (r = 0.77) | Immunoassay | 123 | |
| Estriol | Correlation during normal pregnancy | Saliva/plasma | Strong correlation (r ≥ 0.79) | — | 124 and 125 |
| Correlation during third trimester pregnancy | Saliva/serum | Strong correlation (r = 0.99) | Immunoassay | 126 | |
| Insulin | Normal and Type I diabetic subjects | Saliva/serum | Strong correlation (r = 0.81) | Immunoassay | 127 |
| Melatonin | Correlation studies | Saliva/serum | Strong correlation (r = 0.95,128r = 0.81129) | Immunoassay | 128 and 129 |
| Circadian phase marker | Saliva/plasma | Strong correlation of time of onset (r = 0.70) | Immunoassay | 130 | |
| Antibodies/antigens | |||||
| HIV antibody | HIV infection | Saliva/serum | Immunoassay | 131 and 132 | |
| Immunoglobulin A (IgA) | Systemic sicca syndrome and rheumatoid arthritis | Saliva/serum | Strong correlation (p < 0.001) | Immunoassay | 133 |
| pH | |||||
| pH | Dental caries | Saliva | — | — | 134 |
It has also been shown that therapeutic drugs and drugs of abuse can be detected in saliva, the levels of which exhibit strong correlations with those in plasma or serum. In addition to providing an alternative choice of specimen for drug monitoring, the ability to detect therapeutic drugs in saliva allows the studies of pharmacokinetics and facilitates the individualization of pharmacotherapy as multiple non-invasive saliva samples can be readily collected. The saliva tests of drugs of abuse and prohibited doping substances provide convenient, accessible and lower cost methods for vehicle checkpoints, quarantine stations, sport competitions and in other screening tests. Table 3 provides examples of the current diagnostic uses of salivary biomarkers, together with any correlations with serum, plasma and other bodily fluids.
Note that the correlation between two sets of data may be determined by the Pearson correlation coefficient (r) and/or the p-value (p). The former measures the linear relationship between two random variables.61 The positive value of r means that when the value of one parameter increases, it is likely the value of the other parameter will also increase. The higher the magnitude of r (|r| ≤ 1), the greater probability that this is true. For example, when r = 0: no correlation, r = +0.5: moderately positive correlation, r = +0.8: strong positive correlation and r = +1: perfectly positive correlation. In Table 3, all correlations presented refer to positive correlations. Alternatively, the correlation may be assessed by the p-value which is based on hypothesis testing.62,63 First, a null hypothesis (H0) is assumed. For example in the correlation studies, the null hypothesis is that there is no correlation between the two sets of data. The p-value quantifies the possibility that the null hypothesis is true. The lower the p-value, the more likely that we can reject the null hypothesis. If the p-value is smaller than the designated significant level (α) of usually 0.05, H0 is rejected with less than 5% chance that this is the incorrect rejection of H0. In other words, usually when p < 0.05, the correlation is considered to be statistically significant. Note the difference between strong correlation which is based on ‘r’ and significant correlation which is based on ‘p.’
6 Searches for biomarkers – what is needed and case histories
Biomarkers may be classified into three different categories based on their origins and mechanisms of action: exposure, effect (response) and susceptibility.135 Any substances coming or arising from a source outside the organism or cell are referred to as ‘exposure.’ ‘Effect’ is a change in a biological system corresponding to impairment in the organism or cell, and is potentially related to diseases. The ability of the system to defend itself from toxic events is known as ‘susceptibility.’ For example, antioxidants reflect a type of susceptibility that responds to reactive oxidizing species and prevents oxidation processes that may otherwise damage important organisms or cells.A compound, ion or metabolite is considered a good biomarker when it is representative of a specific condition of the target cell, disease or the health state of an individual. In other words, it should not be affected by other factors unrelated to the disease or condition under study. For ease of detection, biomarkers should be present and measurable in samples which can be readily collected, and easily processed and stored. The concentrations of the biomarker in the normal control population should be well-examined and preferably invariant in the control group such that any small change can be detected with reliability.135
For diagnosis, there need to be reliable assays for the detection of the biomarkers of interest. Before any of the developed assays can be used in the real diagnosis of diseases, the following have to be considered: the precision, accuracy, reproducibility, specificity, selectivity and sensitivity of the assays. The timescale of the measurements, costs and ease of use are also important factors. In the applications of the assays to real samples, collection methods, storage and preparation of the samples for analysis have to be optimized. The comparisons between different methods and the same methods performed by different laboratories have to be in agreement. The above-mentioned parameters are summarized in Fig. 3.
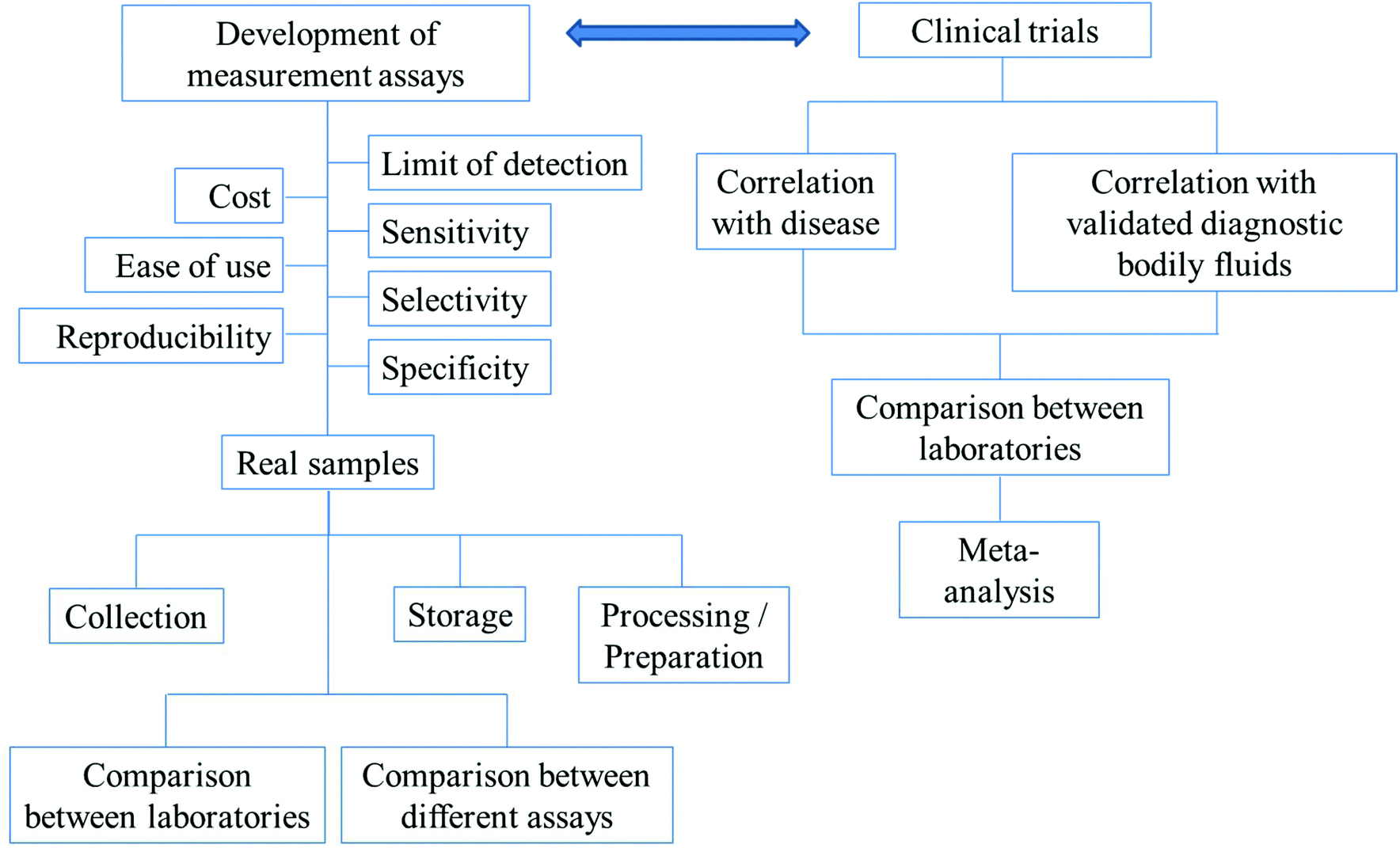 | ||
| Fig. 3 The outlines of factors/parameters needed to be considered in the development of new analytical assays for diagnostic applications and the procedure of the search for new biomarkers. | ||
Fig. 3 further outlines the important parameters that need to be considered in the search for new biomarkers. In the search for a new biomarker, the correlation between the disease/disorder and the presence or the concentrations of the biomarkers has to be directly investigated. For salivary biomarkers, the correlation with disease may be studied along with the correlation between saliva and blood plasma or serum levels. The statistical significance or the accuracy of the diagnosis of the biomarker in question is recommended to be assessed using (i) sensitivity: the proportion of positive tests in the individuals in doubt of being diseased and (ii) specificity: the proportion of negative tests in individuals without the disease.136 Notably, misdiagnosis as false positives is less likely to cause damage than false negatives. Individuals whose test results are positive will undergo further examination, while false negatives could lead to the disease not being detected and necessary treatments may be significantly delayed.
In the following, two successful case histories of saliva diagnostic tests are given as examples: cortisol and HIV antibody detection. Both have been developed into commercial products. In particular, the HIV saliva tests have been approved by the Food and Drug Administration (FDA) for home use and for use at point-of-care sites. The importance of sample collection and storage is highlighted. Then the on-going research into glutathione as a biomarker for psychological disorders will be reviewed.
Cortisol
Cortisol is a lipid-soluble steroid hormone. The saliva cortisol test has been used to assess adrenal hormone function and psychological disorders such as depression. The cortisol level in saliva reflects the unbound level in blood plasma, which is indicative of the activity of the hormone.107–111 Cortisol enters saliva by passive intracellular diffusion. Its level is independent of the saliva flow rate.137 Consequently, saliva may be used as a reliable diagnostic sample for cortisol monitoring. Cortisol can be detected via different techniques, mainly chromatography and immunoassays based on luminescence, optical or electrical detection.138 In the commercial salivary cortisol test kits such as those by Salimetrics® and Oxford Biomedical Research®, an enzyme-linked immunosorbent assay (ELISA) based on horseradish peroxidase is employed. There are different means as to how saliva can be collected and stored as described in section 4. Cotton tools are known to alter the level of cortisol in saliva, while the synthetic Salivette® gives a reported recovery yield of 99.8%.139 Hence, synthetic Salivettes are recommended for saliva collection for cortisol tests. Note also that cortisol levels change throughout the day (circadian rhythm).140 For the development of any new biomarker, the time of sample collection, the method of collection and sample storage need to be optimized.HIV
The diagnosis to identify individuals infected by human immunodeficiency virus (HIV) is one of the examples where the advantage of understanding the aetiology is realized. Instead of directly detecting the virus, the HIV antibody which is also present in saliva can be detected. The diagnosis of HIV infection via saliva testing is a much safer method than blood analysis as the level of the antigen is very low and unlikely to be transmitted through saliva contact, reducing the risk of transmission during tests.141 Independent studies by Major et al.132 and Frerichs et al.131 reported a strong correlation of the appearance of HIV antibodies in saliva and serum samples. The results were consistent between laboratories. Major et al.132 reported complete concordance on the presence of HIV antibodies between matched saliva and serum samples, while Frerichs et al.131 found six disparate findings in over 1000 samples demonstrating the high level of accuracy of the tests. The studies were performed under rigorous control of the measurement methods with three different enzyme linked immunosorbent assays (ELISAs) following the WHO Confirmatory Strategy III.131 The numbers of subjects studied were significantly large, such that, any risks of misdiagnosis have been carefully assessed. In addition to serum, plasma and saliva, the tests of antibody have been shown to be effective in whole blood, dried blood spots and urine.142Glutathione
Turning to the on-going research into glutathione as a potential biomarker for psychological disorders, the development of measurement assays and the possibility of saliva glutathione as a biomarker are discussed in this section.The search for biomarkers is particularly important in psychiatry. For most psychological disorders, there is no reliable biomarker which can be used in the diagnosis, and patients may face life-long symptoms. Biomarkers which can be used to monitor the disorders are thus potentially desirable. Here, we focus on bipolar disorder (BD). Different from the case of HIV discussed above, the pathophysiology of BD is not clearly known. From the symptoms alone, it is also difficult to distinguish between BD and other disorders such as major depressive disorder. A hypothesis that BD is related to oxidative stress has been proposed.143 As a major antioxidant, glutathione becomes one of the potential biomarkers for BD and an increasing number of research studies have focussed on this compound.
A number of assays have been developed for the selective and specific measurements of reduced (GSH) and oxidized (GSSG) glutathione. The most commonly used method for glutathione measurement to date is the Tietze enzymatic assay which measures the concentration of total glutathione (GSH + GSSG). The Tietze assay is based on the use of an enzyme glutathione reductase (GR) for the high selectivity of the assay, and the reaction between Ellman's reagent (DTNB) and GSH to produce a yellow product which can be detected using UV-visible spectrophotometry. The assay is summarized in Fig. 4.144
 | ||
| Fig. 4 The Tietze enzymatic assay for the determination of total glutathione concentrations. GSH: reduced glutathione, GSSG: oxidized glutathione, GR: glutathione reductase, NADPH: nicotinamide adenine dinucleotide phosphate, DTNB: 5,5′-dithiobis(2-nitrobenzoic acid), TNB2−: 5-thio-2-nitrobenzoate. | ||
The enzyme reduces GSSG to GSH at the beginning of the assay. Therefore, to measure the concentration of either GSH or GSSG, a further step of using a masking reagent is required. Tietze144 proposed the use of N-ethylmaleimide (NEM) as a masking agent. NEM completes the reaction with GSH almost instantaneously, but any excess NEM has to be removed from the system as it will inhibit the activity of the enzyme (glutathione reductase) used in the measurement assay, and furthermore the compound is highly toxic. NEM removal can require a lot of effort and take a long time.144,145 The work of Griffith146 suggests the alternative use of 2-vinylpyridine (2VP) as a masking reagent. 2VP is less toxic than NEM, and does not need to be removed from the system. However, 2VP takes ca. one hour for the reaction to go to completion. This one-hour period can significantly alter the result as will be further discussed below.
Shaik and Mehvar147 suggested the use of 1-methyl-4-vinyl-pyridinium trifluoromethane sulfonate as a masking reagent, while some of the present articles148 recommend benzoquinone as an alternative. Both reported the complete reactions almost instantly (<2 min). Ngamchuea et al.148 further suggest a quick route to remove the masking agents remaining in the samples simply by adding a non-glutathione thiol. The methods have been demonstrated to have high sensitivity and selectivity, and have been successfully applied to real samples such as whole blood, blood plasma and saliva. Other means of measuring glutathione include chromatographic methods (HPLC, GC),149,150 capillary electrophoresis,151,152 nanoparticle-based colorimetric tests,153 and chemiluminescence.154 Electrochemical techniques have also been reported.155–158 It can be said that at this stage, the assays for glutathione measurements are well-developed. The next step requires progress in clinical studies.
Rosa et al.159 investigated the correlations between total (GSH + GSSG), reduced (GSH) and oxidized (GSSG) glutathione in blood plasma from 50 bipolar and 50 control subjects, using the Tietze assay and the 2VP masking agent. Rosa et al.159 observed lower both total and GSH levels in BD patients than in controls (p ≤ 0.05). Other factors such as smoking showed no sign of escalating the level of oxidative stress in BD patients,159 although some studies reported the effects of smoking on glutathione levels in healthy control populations.160 Raffa et al.143 and Gawryluk et al.161 studied the correlations between glutathione and BD in red blood cells (40 controls and 30 BD; Tietze assay and NEM) and the post-mortem prefrontal cortex (12 controls and 14 BD; Tietze assay and 2VP), respectively. Both found the levels of total and reduced glutathione to be lower in BD patients.
Tuncel et al.162 also studied the plasma total glutathione levels in 18 BD and 18 control subjects using the Tietze assay. They observed that the plasma total glutathione levels are higher in BD patients than in the control group, but the results are not statistically significant (p = 0.51). Lagopoulos et al. (51 controls and 53 BD; 1H-MRS)163 and Godlewska et al. (11 controls and 13 BD; 1H-MRS)164 found no significant difference in GSH levels in the cortex between BD patients and the control groups.
There are many possible explanations for the discrepancy of the results discussed above. The sample sizes in BD studies, in the range of 11–50 samples, may be relatively small, giving rise to high possibilities that the population is not representative of the inhomogeneous population. Moreover, correlations between glutathione levels in different biological fluids/compartments do not necessarily exist.47 For example, Kand'ar et al.165 reported no correlation between the levels of reduced (GSH) and oxidized (GSSG) glutathione in whole blood and blood plasma collected from healthy participants. Similarly, there are reports suggesting that there is no correlation between the concentrations of total glutathione (GSH + GSSG) in blood plasma as compared with epithelial lining fluid166 and seminal plasma.167
There are also different types of BD (e.g. types I and II). The levels of glutathione may be different at different stages of the disease, or during depression and mania. The effects of treatment, medication and other influences such as smoking and alcohol consumption may have to be considered. For plasma samples, different preparation procedures have been shown to significantly alter the measured glutathione concentrations.168 In plasma samples, glutathione concentrations also change at rapid rates (half-life of ca. 30 min) due to the activity of the γ-glutamyltransferase enzyme and metal-catalyzed oxidation of GSH to GSSG.148 On the other hand, salivary glutathione displays significantly greater stability. At the temperature of −20 °C, saliva GSH has been demonstrated to be stable for 11 days,169 while at room temperature, saliva GSH is stable for at least 90 min.148 The stability of saliva GSH offers a great advantage in diagnostic/clinical applications in addition to the non-invasive nature of saliva collection.
Potential problems in the measurements of biomarkers in saliva
Saliva is a viscous fluid due to the presence of mucopolysaccharides and mucoproteins. The difficulty in measuring precise volumes of viscous saliva samples can decrease the accuracy of analytical measurements. Importantly, most potential biomarkers are present in saliva at low concentrations. Any error in volume may give rise to large errors in the concentrations measured. Due to the low concentrations, assays with high sensitivity and low limit of detection are required, in addition to high selectivity and specificity. The components of saliva may depend on the area in the oral cavity in which the sample is collected as well as the collection methods (unstimulated vs. stimulated). If stimulation is used, the effects of the collection tools (e.g. cotton pads, polyester swabs, paraffin wax) and/or the chemicals (e.g. citric acid) have to be carefully assessed, as previously discussed in section 4 and in the case of cortisol. Oral diseases such as gingivitis, change in pH and other potential interferences such as smoking, fasting and hydration level can also influence the levels of the biomarkers of interest.170There are relatively large variations in metabolite concentrations between different individuals. Calcium and phosphate concentrations in unstimulated whole saliva vary significantly between individuals (ca. 4-fold and 6-fold ranges of calcium and phosphate concentrations, respectively) although the samples are collected at the same time of the day and have similar pH and buffer capacity.171 The marked inter-individual variation is likely because many salivary components play multifunctional roles and, in some cases, have overlapping functions.172 For example, glutathione, ascorbic acid and uric acid are all antioxidants. Amylases, cystatins, histatins, mucins and peroxidases are all involved in anti-bacterial activity in the oral cavity. Diurnal fluctuation, seasons (temperature) and other intra-individual inconsistency may also have to be considered. Accordingly, single salivary analysis could be unreliable. Multiple saliva samples and follow-up tests are desirable.
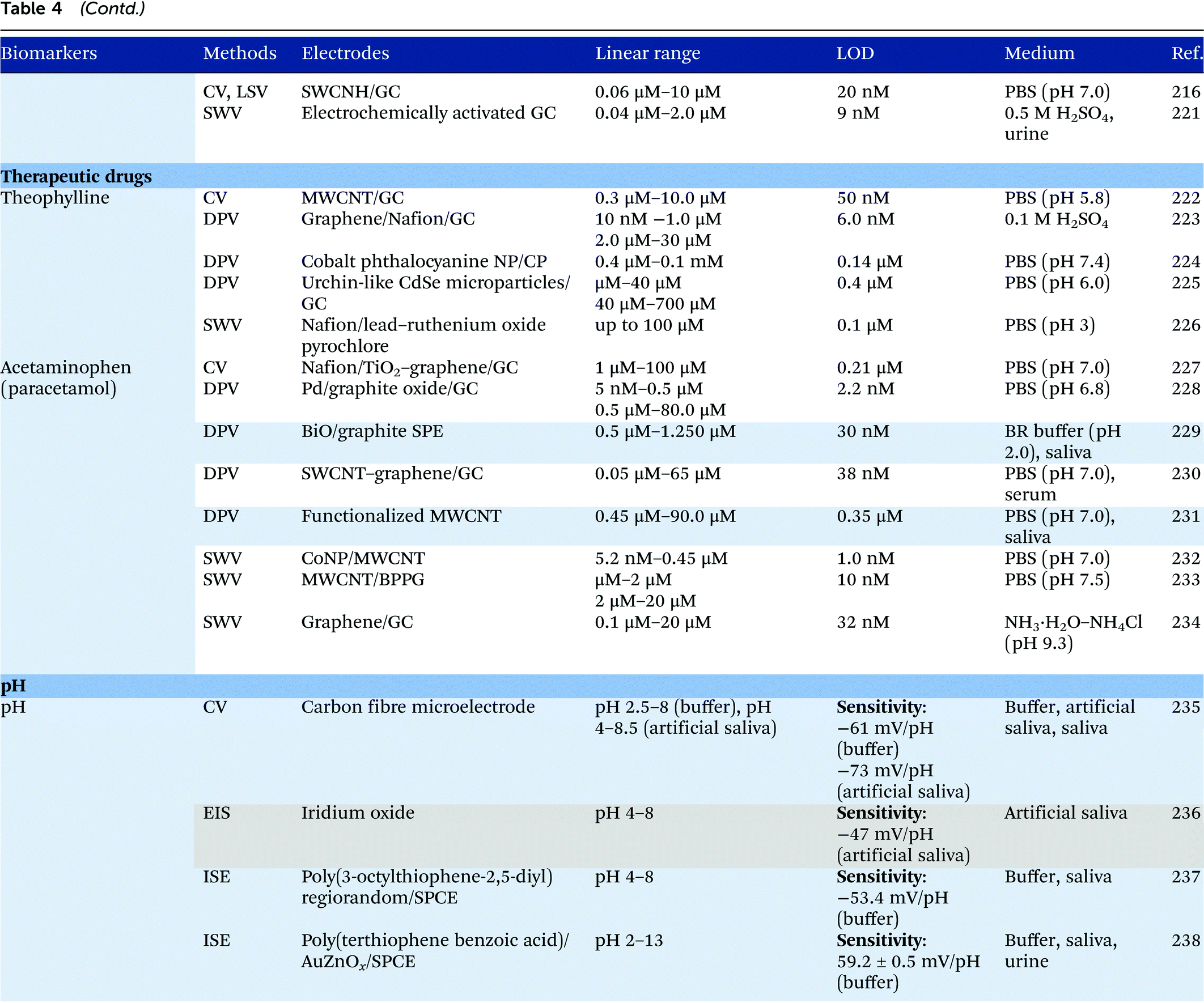
7 Electroanalysis in saliva
Electrochemical measurements provide simple, rapid and inexpensive methods for the detection of analytes and often exhibit high sensitivity over linear ranges. A variety of electrochemical techniques are available for the detection of analytes including potentiometric, voltammetric and amperometric measurements. Techniques which minimize the contribution of capacitative currents such as square-wave voltammetry and differential pulse voltammetry may also be used to improve the sensitivity and limit of detection of the measurements. Table 4 provides examples of the reported electrochemical methods in the detection of some salivary biomarkers listed in Table 3. Note that methods that involve the use of biologically selective materials are not included. For methods that use the same technique, only some of the methods that reported lower limits of detection or methods that have been validated in biological samples are presented in Table 4.| The methods highlighted in grey and blue have been validated in artificial saliva and real saliva, respectively. Methods: AMP: amperometry, CA: chronoamperometry, CE: capillary electrophoresis, CV: cyclic voltammetry, DPV: differential pulse voltammetry, EIS: electrochemical impedance spectroscopy, ISE: ion-selective electrode, LSV: linear sweep voltammetry, MPA: multiple-pulse amperometry, SWV: square wave voltammetry. Electrodes: BDD: boron-doped diamond, BPPG: basal plane pyrolytic graphite, CC: carbon ceramic, CIL: carbon ionic liquid, CNF: carbon nanofiber, CP: carbon paste, EPPG: edge plane pyrolytic graphite, GC: glassy carbon electrode, MWCNT: multi-walled carbon nanotube, NP: nanoparticle, SPE: screen-printed electrode, SWCNT: single-walled carbon nanotube, SWCNH: single-walled carbon nanohorn.a Not stated in the text – values are taken from calibration curves present in the papers. |
|---|

|
|
Despite the many advantages, electrochemical assays are relatively less common in diagnostic and clinical uses. It can also be seen from Table 4 that in many cases, the developed assays have not been tested with real biological samples. The lack of such validations may be one of the reasons that the applications of electroanalysis in diagnostic fields are relatively limited. The difficulty of electrochemical measurement in bio-samples lies in the selectivity and specificity of the methods as there are many potential interferences in real samples.
To improve the selectivity and specificity of the electrochemical assays, biologically selective substances such as enzymes, antibodies or antigens may be used. Using the diagnosis of cancers as an example, cancer antibodies can be immobilized on an electrode surface. Cancer antigens such as α-fetoprotein in the samples can then be detected by antibody–antigen recognition.173 In biosensors and immunosensors, it is important to consider the conductivity of the electrode after modification, the stability of the antibody receptors, and that they retain the bio-activities. The major disadvantages of such sensors could be the effort required in the production of enzymes, antigens or antibodies, the fabrication of the modified electrodes and questions over the reusability of the electrodes.
The selectivity of electrochemical assays can also be improved without the need for bio-selective molecules. Compounds which carry the same functional groups or have similar structures are likely to undergo electrochemical reactions at similar potentials and hence be indistinguishable in the resulting electrochemical responses. They may however have different reaction kinetics. The use of different scan rates in voltammetric measurements may allow the separation of the peaks and the elimination of interferences, for example, dopamine, ascorbic acid and uric acid, all of which are important metabolites in biological fluids. The measurement of the concentrations of individual species poses difficulties as they have similar oxidation potentials. However, it has been shown that the detection of dopamine in the presence of ascorbic acid or uric acid in the presence of ascorbic acid can be achieved by fast scan voltammetry at bare electrodes because of the differences in their electrochemical kinetics.174
Alternatively, the use of small organic or inorganic molecules as redox mediators may allow otherwise electrochemically inert species to be detected. Different reaction mechanisms or rates of reactions with the mediators offer the simultaneous detection of different species. For example, thiol-containing amino acids do not undergo oxidation nor reduction in the potential window that is easily achieved in aqueous media. It was shown that O-benzoquinone undergoes fast reaction with thiols via two possible mechanisms: 1,4-Michael addition and electrocatalytic reaction, with the former being the main pathway.175O-Benzoquinone exhibits a clear electrochemical response in the suitable potential window, and hence can be used as a redox mediator in the detection of thiol species. The reactions between O-benzoquinone and thiols form adducts as a result of the 1,4-addition reaction. Because the products are different for the different thiols, they display voltammetric peaks at distinct potentials and thus allow the simultaneous detection of different anti-oxidants such as cysteine, homocysteine, glutathione and ascorbic acid by simple voltammetry measurement without the need for deoxygenation or further sample preparation.175 In the case that the resulting adducts give rise to similar reduction potentials, different scan rates can be used to further increase the selectivity.
In addition to the change in scan rates, the modification of electrodes by conducting porous layers can be employed to improve the selectivity of the electrochemical sensors. The formation of thin porous layers on the electrode surface changes the mass transport regimes of the redox-active species to the electrode from planar (linear) diffusion to ‘thin layer’ behaviour. As a result, the voltammetric peaks are shifted and compounds which originally give rise to voltammetric peaks at the same or similar potentials may now show separated peaks, provided that the values of electrochemical parameters such as the formal potential (Ef), the standard electrochemical rate constant (k0) and transfer coefficient (α) are different. For example, Henstridge et al.176 modified the surface of glassy carbon electrodes with multiwalled carbon nanotubes. The modified electrodes were able to detect dopamine in the presence of interferences such as uric acid, serotonin and ascorbic acid, which would otherwise display voltammetric peaks at similar potentials at bare glassy carbon electrodes.
For systems that undergo inner-sphere electron transfers or processes that involve adsorption of the redox-active species to the electrode surface, the change in the electrode materials or morphology can offer improvement of selectivity. For example, carbon electrodes with different structures (e.g. glassy carbon, edge/basal plane pyrolytic graphite, carbon fiber and carbon nanomaterials) have been shown to yield different electron transfer kinetics.177,178 Ion-selective electrodes have also been developed for the detection of specific ions by converting the activity of the ion into a potential such as allows the detection of calcium and potassium.179,180 Nanoparticles and nanomaterials in sensors have been demonstrated to display enhanced sensitivity as well as increase the selectivity of the electrochemical detection.181 The development of various forms of portable electrode platforms such as screen-printed electrodes,182–184 microwire electrodes,158,185 as well as stretchable and wearable electrodes186 also facilitates the uses of electrochemistry in bio-sensing.
8 Conclusions
Although saliva contains various potential biomarkers that exhibit strong correlations with the concentrations in blood plasma or serum, the use of saliva as a diagnostic fluid has to date been relatively limited despite offering diverse opportunities. This review facilitates the recognition of these biomarker compounds, as well as the great advantages of saliva over blood and other bodily fluids, in order to encourage the future development of measurement assays for the detection of salivary components. In particular, we note that even when assays are available for the detection of certain analytes, they have often not been applied to real or even artificial saliva samples. By providing essential knowledge of saliva, this review encourages the validation and the application of assays in saliva specimens.In the collection of saliva for analysis, a large number of research studies rely on cotton-based collection tools. However, we have demonstrated the potential interferences of cotton with the concentrations of analytes and highlighted that it is of critical importance to validate the applicability of the tool prior to analysis. Appropriate methods for storing the saliva specimens also need to be evaluated for different analytes under study.
The list of potential salivary biomarkers is presented in Table 3. However, only a small number of electroanalytical assays are available for the detection of such analytes although electrochemistry offers many advantages especially in terms of quick and inexpensive measurements. User-friendly, portable electrochemical devices can be readily developed. One of the reasons for the lack of electrochemical applications in this field is likely the problems of poor selectivity of the assays without the use of enzymes or antibodies. In this review, we have presented different approaches that can be employed to improve the selectivity of the electroanalytical measurements and to encourage the use of electroanalytical techniques in diagnostic applications.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
KN and KC receive funding from the Royal Thai government. This project is supported by funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007–2013)/ERC Grant Agreement no. [320403]. We thank Prof. Philip J. Cowen and Clare Williams for saliva collection and very helpful discussions.References
- V. de Almeida Pdel, A. M. Gregio, M. A. Machado, A. A. de Lima and L. R. Azevedo, J. Contemp. Dent. Pract., 2008, 9, 72–80 Search PubMed
.
- J. L. Chicharro, A. Lucía, M. Pérez, A. F. Vaquero and R. Ureña, Sports Med., 2012, 26, 17–27 CrossRef
- M. D. Kaplan and B. J. Baum, Dysphagia, 1993, 8, 225–229 CrossRef CAS PubMed
- B. L. Slomiany, V. L. N. Murty, J. Piotrowski and A. Slomiany, Gen. Pharmacol., 1996, 27, 761–771 CrossRef CAS PubMed
- A. Bardow, D. Moe, B. Nyvad and B. Nauntofte, Arch. Oral Biol., 2000, 45, 1–12 CrossRef CAS PubMed
- J. Kivelä, S. Parkkila, A.-K. Parkkila, J. Leinonen and H. Rajaniemi, J. Physiol., 1999, 520, 315–320 CrossRef
- H. Islas-Granillo, S. A. Borges-Yañez, C. E. Medina-Solís, C. A. Galan-Vidal, J. J. Navarrete-Hernández, M. Escoffié-Ramirez and G. Maupomé, West Indian Med. J., 2014, 63, 758–765 CAS
-
B. S. Manjunatha, Textbook of Dental Anatomy and Oral Physiology, Jaypee Brothers, Medical Publishers Pvt. Limited, 2012 Search PubMed
- H. Mese and R. Matsuo, J. Oral Rehabil., 2007, 34, 711–723 CrossRef CAS PubMed
- S. J. Moss, J. Esthet. Restor. Dent., 1995, 7, 197–203 CrossRef CAS
- J. K. M. Aps and L. C. Martens, Forensic Sci. Int., 2005, 150, 119–131 CrossRef CAS PubMed
- R. P. Shellis, Arch. Oral Biol., 1978, 23, 485–489 CrossRef CAS PubMed
- V. W. H. Leung and B. W. Darvell, J. Dent., 1997, 25, 475–484 CrossRef CAS PubMed
- K. Elagli, M. Traisnel and H. F. Hildebrand, Electrochim. Acta, 1993, 38, 1769–1774 CrossRef CAS
- F. C. Giacomelli, C. Giacomelli and A. Spinelli, J. Braz. Chem. Soc., 2004, 15, 541–547 CrossRef CAS
- J.-M. Meyer, Corros. Sci., 1977, 17, 971–982 CrossRef CAS
- B. Levallois, Y. Fovet, L. Lapeyre and J. Y. Gal, Dent. Mater., 1998, 14, 441–447 CrossRef CAS PubMed
- J.-Y. Gal, Y. Fovet and M. Adib-Yadzi, Talanta, 2001, 53, 1103–1115 CrossRef CAS PubMed
- J. Klimek, E. Hellwig and G. Ahrens, Caries Res., 1982, 16, 156–161 CrossRef CAS PubMed
- H. NordbÖ, S. Darwish and R. S. Bhatnagar, Eur. J. Oral Sci., 1984, 92, 306–314 CrossRef
- K. Shibasaki, M. Kimura, R. Ikarashi, A. Yamaguchi and T. Watanabe, Metabolomics, 2012, 8, 484–491 CrossRef CAS
- M. Soukup, I. Biesiada, A. Henderson, B. Idowu, D. Rodeback, L. Ridpath, E. G. Bridges, A. M. Nazar and K. G. Bridges, Diabetol. Metab. Syndr., 2012, 4, 14–14 CrossRef CAS PubMed
- O. Hershkovich and R. M. Nagler, Arch. Oral Biol., 2004, 49, 515–522 CrossRef CAS PubMed
- E. De Corso, S. Baroni, S. Agostino, G. Cammarota, G. Mascagna, A. Mannocci, M. Rigante and J. Galli, Ann. Surg., 2007, 245, 880–885 CrossRef PubMed
- R. Venkatapathy, V. Govindarajan, N. Oza, S. Parameswaran, B. Pennagaram Dhanasekaran and K. V. Prashad, Int. J. Nephrol., 2014, 2014, 6 Search PubMed
- S. Kumar, S. Padmashree and R. Jayalekshmi, Contemp. Clin. Dent., 2014, 5, 312–317 CrossRef PubMed
- S. Karjalainen, L. Sewón, E. Soderling, B. Larsson, I. Johansson, O. Simell, H. Lapinleimu and R. Seppänen, J. Dent. Res., 1997, 76, 1637–1643 CrossRef CAS PubMed
- R. Segura, C. Javierre, J. L. Ventura, M. A. Lizarraga, B. Campos and E. Garrido, Br. J. Sports Med., 1996, 30, 305–309 CrossRef CAS PubMed
- C. G. J. Schabmueller, D. Loppow, G. Piechotta, B. Schütze, J. Albers and R. Hintsche, Biosens. Bioelectron., 2006, 21, 1770–1776 CrossRef CAS PubMed
- A. L. Mandel, C. Peyrot des Gachons, K. L. Plank, S. Alarcon and P. A. S. Breslin, PLoS One, 2010, 5, e13352 Search PubMed
- A. Almståhl, M. Wikström and J. Groenink, Oral Microbiol. Immunol., 2001, 16, 345–352 CrossRef
- V. Ng, D. Koh, Q. Fu and S.-E. Chia, Clin. Chim. Acta, 2003, 338, 131–134 CrossRef CAS
- S. Kejriwal, R. Bhandary, B. Thomas and S. Kumari, J. Clin. Diagn. Res., 2014, 8, ZC56–ZC60 Search PubMed
- H. Tomita, S. Sato, R. Matsuda, Y. Sugiura, H. Kawaguchi, T. Niimi, S. Yoshida and M. Morishita, Lung, 1999, 177, 161–167 CrossRef CAS PubMed
- L. Manetti, G. Rossi, L. Grasso, V. Raffaelli, I. Scattina, S. Del Sarto, M. Cosottini, A. Iannelli, M. Gasperi, F. Bogazzi and E. Martino, Eur. J. Endocrinol., 2013, 168, 315–321 CrossRef CAS PubMed
- M. Yasuda, S. Honma, K. Furuya, T. Yoshii, Y. Kamiyama, H. Ide, S. Muto and S. Horie, J. Mens Health, 2008, 5, 56–63 CrossRef
- Y. C. Lu, G. R. Bentley, P. H. Gann, K. R. Hodges and R. T. Chatterton, Fertil. Steril., 1999, 71, 863–868 CrossRef CAS PubMed
- M. Somogyi, J. Biol. Chem., 1938, 125, 399–414 CAS
-
B. S. Henson and D. T. Wong, in Oral Biology: Molecular Techniques and Applications, ed. G. J. Seymour, M. P. Cullinan and N. C. K. Heng, Humana Press, Totowa, NJ, 2010, pp. 21–30, DOI:10.1007/978-1-60761-820-1_2
- C. Golatowski, M. Gesell Salazar, V. M. Dhople, E. Hammer, T. Kocher, N. Jehmlich and U. Völker, Clin. Chim. Acta, 2013, 419, 42–46 CrossRef CAS PubMed
- M. Navazesh and S. K. S. Kumar, J. Am. Dent. Assoc., 2008, 139, 35S–40S CrossRef PubMed
- J. C. J. Kjeilen, P. Brodin, H. Aars and T. Berg, Acta Physiol. Scand., 1987, 131, 169–175 CrossRef PubMed
- D. A. Granger, E. B. Schwartz, A. Booth, M. Curran and D. Zakaria, Psychoneuroendocrinology, 1999, 24, 567–579 CrossRef CAS PubMed
- D. A. Froehlich, R. M. Pangborn and J. R. Whitaker, Physiol. Behav., 1987, 41, 209–217 CrossRef CAS PubMed
- E. A. Shirtcliff, D. A. Granger, E. Schwartz and M. J. Curran, Psychoneuroendocrinology, 2001, 26, 165–173 CrossRef CAS PubMed
- R. L. Hodinka, T. Nagashunmugam and D. Malamud, Clin. Diagn. Lab. Immunol., 1998, 5, 419–426 CAS
- K. Ngamchuea, C. Batchelor-McAuley, P. J. Cowen, C. Williams, L. M. Goncalves and R. G. Compton, Analyst, 2016, 141, 4707–4712 RSC
- R. Haeckel, Ann. N. Y. Acad. Sci., 1993, 694, 128–142 CrossRef CAS PubMed
- M. Navazesh, Ann. N. Y. Acad. Sci., 1993, 694, 72–77 CrossRef CAS PubMed
- K. Hanrahan, A. M. McCarthy, C. Kleiber, S. Lutgendorf and E. Tsalikian, Appl. Nurs. Res., 2006, 19, 95–101 CrossRef PubMed
- Z. S. Czégény, J. L. Chicharro, P. Fernández, A. Gutiérrez and C. Cámara, Biol. Trace Elem. Res., 2001, 79, 131–137 CrossRef
- F. I. Daniel, L. Lima and C. R. d. Santos, Braz. J. Pharm. Sci., 2016, 52, 679–684 CrossRef
- A. H. Garde and Å. M. Hansen, Scand. J. Clin. Lab. Invest., 2005, 65, 433–436 CrossRef CAS PubMed
- D. Esser, G. Alvarez-Llamas, M. P. de Vries, D. Weening, R. J. Vonk and H. Roelofsen, Biomarker Insights, 2008, 3, 25–27 CrossRef CAS PubMed
- A. Nurkka, J. Obiero, H. Kayhty and J. A. G. Scott, Clin. Vaccine Immunol., 2003, 10, 357–361 CrossRef CAS
- M. Gröschl, R. Wagner, M. Rauh and H. G. Dörr, Steroids, 2001, 66, 737–741 CrossRef
- G.-L. S. Whembolua, D. A. Granger, S. Singer, K. T. Kivlighan and J. A. Marguin, Horm. Behav., 2006, 49, 478–483 CrossRef CAS PubMed
- G. F. Read, Ann. N. Y. Acad. Sci., 1993, 694, 146–160 CrossRef CAS PubMed
- J. T. Wilson, Ann. N. Y. Acad. Sci., 1993, 694, 48–61 CrossRef CAS PubMed
- I. D. Mandel, Ann. N. Y. Acad. Sci., 1993, 694, 1–10 CrossRef CAS PubMed
- K. H. Zou, K. Tuncali and S. G. Silverman, Radiology, 2003, 227, 617–622 CrossRef PubMed
-
T. A. Lang and M. Secic, How to Report Statistics in Medicine: Annotated Guidelines for Authors, Editors, and Reviewers, American College of Physicians, 2006 Search PubMed
-
K. M. Ramachandran and C. P. Tsokos, Mathematical Statistics with Applications, Elsevier Science, 2009 Search PubMed
- M. Jarvis, P. Primatesta, B. Erens, C. Feyerabend and A. Bryant, Nicotine Tob. Res., 2003, 5, 349–355 CrossRef CAS PubMed
- J. T. Bernert, J. E. McGuffey, M. A. Morrison and J. L. Pirkle, J. Anal. Toxicol., 2000, 24, 333–339 CrossRef CAS PubMed
- F. Kardani, A. Daneshfar and R. Sahrai, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 2857–2862 CrossRef CAS PubMed
- J. E. Rose, E. D. Levin and N. Benowitz, Ther. Drug Monit., 1993, 15, 431–435 CrossRef CAS PubMed
- D. Forman, S. Al-Dabbagh and R. Doll, Nature, 1985, 313, 620–625 CrossRef CAS PubMed
- G. A. Sanchez, V. A. Miozza, A. Delgado and L. Busch, Nitric Oxide, 2014, 36, 31–35 CrossRef CAS PubMed
- A. Wahllnder, S. Mohr and G. Paumgartner, J. Hepatol., 1990, 10, 129–137 CrossRef
- J. A. Carrillo, M. Christensen, S. I. Ramos, C. Alm, M. L. Dahl, J. Benitez and L. Bertilsson, Ther. Drug Monit., 2000, 22, 409–417 CrossRef CAS PubMed
- E. Zylber-Katz, L. Granit and M. Levy, Clin. Pharmacol. Ther., 1984, 36, 133–137 CrossRef CAS PubMed
- H. N. Alkaysi, M. S. Salem and Y. M. El-Sayed, J. Clin. Pharmacol. Ther., 1988, 13, 109–115 CrossRef CAS PubMed
- T. F. Maliszewski and D. E. Bass, J. Appl. Physiol., 1955, 8, 289–291 CAS
- L. Minarowski, D. Sands, A. Minarowska, A. Karwowska, A. Sulewska, M. Gacko and E. Chyczewska, Folia Histochem. Cytobiol., 2008, 46, 245–246 CAS
- G. Almadori, F. Bussu, J. Galli, A. Limongelli, S. Persichilli, B. Zappacosta, A. Minucci, G. Paludetti and B. Giardina, Head Neck, 2007, 29, 648–654 CrossRef PubMed
- A. R. Shivashankara and M. K. Prabhu, Biomed. Res., 2011, 22, 355–359 CAS
- C. Arana, A. Cutando, M. J. Ferrera, G. Gomez-Moreno, C. V. Worf, M. J. Bolanos, G. Escames and D. Acuna-Castroviejo, J. Oral Pathol. Med., 2006, 35, 554–559 CrossRef CAS PubMed
- J. F. Bates, R. E. Hughes and R. J. Hurley, Arch. Oral Biol., 1972, 17, 1017–1020 CrossRef CAS PubMed
- J. Giebultowicz, P. Wroczynski and D. Samolczyk-Wanyura, J. Oral Pathol. Med., 2011, 40, 726–730 CrossRef CAS PubMed
- B. Owen-Smith, J. Quiney and J. Read, Lancet, 1998, 351, 1932 CrossRef CAS
- M. Zloczower, A. Z. Reznick, R. O. Zouby and R. M. Nagler, Antioxid. Redox Signaling, 2007, 9, 765–773 CrossRef CAS PubMed
- M. Soukup, I. Biesiada, A. Henderson, B. Idowu, D. Rodeback, L. Ridpath, E. G. Bridges, A. M. Nazar and K. G. Bridges, Diabetol. Metab. Syndr., 2012, 4, 14 CrossRef CAS PubMed
- J. J. McAuliffe, A. L. Sherwin, I. E. Leppik, S. A. Fayle and C. E. Pippenger, Neurology, 1977, 27, 409–413 CrossRef CAS PubMed
- C. Knott and F. Reynolds, Ther. Drug Monit., 1984, 6, 35–41 CrossRef CAS PubMed
- R. F. Goldsmith and R. A. Ouvrier, Ther. Drug Monit., 1981, 3, 151–157 CrossRef CAS PubMed
- D. Schmidt and H. J. Kupferberg, Epilepsia, 1975, 16, 735–741 CrossRef CAS PubMed
- S. P. Galant, Arch. Pediatr. Adolesc. Med., 1977, 131, 970 CrossRef CAS
- M. Aviram, A. Tal, Z. Benzvi and R. Gorodischer, Pediatrics, 1987, 80, 894–897 CAS
- G. Levy, E. F. Ellis and R. Koysooko, Pediatrics, 1974, 53, 873–876 CAS
- C. Knott, M. Bateman and F. Reynolds, Br. J. Clin. Pharmacol., 1984, 17, 9–14 CrossRef CAS PubMed
- H. Wade, D. L. McCoubrie, D. M. Fatovich, J. Ryan, S. Vasikaran and F. F. Daly, Clin. Toxicol., 2008, 46, 534–538 CrossRef CAS PubMed
- R. Perry, M. Campbell, D. M. Grega and L. Anderson, J. Clin. Psychopharmacol., 1984, 4, 199–202 CAS
- J. L. Evrard, P. Baumann, R. Pera-Bally and L. Peters-Haefeli, Acta Psychiatr. Scand., 1978, 58, 67–79 CrossRef CAS PubMed
- L. C. Degutis, Acad. Emerg. Med., 2004, 11, 885–887 CrossRef PubMed
- W. Gubala and D. Zuba, Pol. J. Pharmacol., 2002, 54, 161–165 CAS
- G.-c. Tu, B. Kapur and Y. Israel, Alcohol.: Clin. Exp. Res., 1992, 16, 222–227 CrossRef CAS
- M. E. Bates and C. S. Martin, J. Stud. Alcohol, 1997, 58, 531–538 CrossRef CAS PubMed
- E. J. Cone, K. Kumor, L. K. Thompson and M. Sherer, J. Anal. Toxicol., 1988, 12, 200–206 CrossRef CAS PubMed
- L. K. Thompson, D. Yousefnejad, K. Kumor, M. Sherer and E. J. Cone, J. Anal. Toxicol., 1987, 11, 36–38 CrossRef CAS PubMed
- C. W. Gorodetzky and M. P. Kullberg, Clin. Pharmacol. Ther., 1974, 15, 579–587 CrossRef CAS PubMed
- W.-L. Wang, W. D. Darwin and E. J. Cone, J. Chromatogr. B: Biomed. Sci. Appl., 1994, 660, 279–290 CrossRef CAS PubMed
- A. J. Jenkins, J. M. Oyler and E. J. Cone, J. Anal. Toxicol., 1995, 19, 359–374 CrossRef CAS PubMed
- J. L. Valentine and P. Psaltis, Anal. Lett., 1979, 12, 855–866 CrossRef CAS
- R. S. Niedbala, K. W. Kardos, D. F. Fritch, S. Kardos, T. Fries, J. Waga, J. Robb and E. J. Cone, J. Anal. Toxicol., 2001, 25, 289–303 CrossRef CAS PubMed
- S. Strano-Rossi, C. Colamonici and F. Botre, Anal. Chim. Acta, 2008, 606, 217–222 CrossRef CAS PubMed
- P. Strickland, R. Morriss, A. Wearden and B. Deakin, J. Affective Disord., 1998, 47, 191–194 CrossRef CAS PubMed
- K. Vedhara, J. Miles, P. Bennett, S. Plummer, D. Tallon, E. Brooks, L. Gale, K. Munnoch, C. Schreiber-Kounine, C. Fowler, S. Lightman, A. Sammon, Z. Rayter and J. Farndon, Biol. Psychol., 2003, 62, 89–96 CrossRef PubMed
- J.-P. Kahn, D. R. Rubinow, C. L. Davis, M. Kling and R. M. Post, Biol. Psychiatry, 1988, 23, 335–349 CrossRef CAS PubMed
- R. F. Vining, R. A. McGinley, J. J. Maksvytis and K. Y. Ho, Ann. Clin. Biochem., 1983, 20(Pt 6), 329–335 CrossRef CAS PubMed
- U. Teruhisa, H. Ryoji, I. Taisuke, S. Tatsuya, M. Fumihiro and S. Tatsuo, Clin. Chim. Acta, 1981, 110, 245–253 CrossRef
- J. D. Few, P. J. W. Wallis and V. H. T. James, Clin. Endocrinol., 1986, 24, 119–126 CrossRef CAS PubMed
- R. McVie, L. S. Levine and M. I. New, Pediatr. Res., 1979, 13, 755–759 CrossRef CAS PubMed
- F. S. Khan-Dawood, J. K. Choe and M. Y. Dawood, Am. J. Obstet. Gynecol., 1984, 148, 442–445 CrossRef
- T. Ohzeki, B. Manella, C. Gübelin-De Campo and M. Zachmann, Hormone Res., 1991, 36, 235–237 CrossRef CAS PubMed
- S. G. Johnson, G. F. Joplin and J. M. Burrin, Clin. Chim. Acta, 1987, 163, 309–318 CrossRef CAS
- C. Wang, S. Plymate, E. Nieschlag and C. A. Paulsen, J. Clin. Endocrinol. Metab., 1981, 53, 1021–1024 CrossRef CAS PubMed
- R. F. Walker, I. A. Hughes and D. Riad-Fahmy, Clin. Endocrinol., 1979, 11, 631–637 CrossRef CAS PubMed
- M. Zerah, S. Y. Pang and M. I. New, J. Clin. Endocrinol. Metab., 1987, 65, 227–232 CrossRef CAS PubMed
- L. A. Perry, N. Wathen and T. Chard, Horm. Metab. Res., 1987, 19, 444–447 CrossRef CAS PubMed
- J. K. Choe, F. S. Khan-Dawood and M. Yusoff-Dawood, Am. J. Obstet. Gynecol., 1983, 147, 557–562 CrossRef CAS PubMed
- L. J. Tivis, M. D. Richardson, E. Peddi and B. Arjmandi, Prog. Neuropsychopharmacol. Biol. Psychiatry, 2005, 29, 727–732 CrossRef CAS PubMed
- L. D. Belkien, J. Bordt, P. Möller, R. Hano and E. Nieschlag, Fertil. Steril., 1985, 44, 322–327 CrossRef CAS PubMed
- J. J. Evans, A. R. Wilkinson and D. R. Aickin, Clin. Chem., 1984, 30, 120–121 CAS
- M. S. Preti, S. Lodi, P. Busacchi, M. Filicori and C. Flamigni, Steroids, 1984, 43, 469–479 CrossRef CAS PubMed
- N. Kundu, N. Novak and L. P. Petersen, Steroids, 1983, 41, 145–153 CrossRef CAS PubMed
- J. Pasic and J. C. Pickup, Diabetes Care, 1988, 11, 489–494 CrossRef CAS PubMed
- M.-L. Laakso, T. Porkka-Heiskanen, A. Alila, D. Stenberg and G. Johansson, J. Pineal Res., 1990, 9, 39–50 CrossRef CAS PubMed
- R. Nowak, I. C. McMillen, J. Redman and R. V. Short, Clin. Endocrinol., 1987, 27, 445–452 CrossRef CAS PubMed
- A. Voultsios, D. J. Kennaway and D. Dawson, J. Biol. Rhythms, 1997, 12, 457–466 CrossRef CAS PubMed
- R. R. Frerichs, M. T. Htoon, N. Eskes and S. Lwin, Lancet, 1992, 340, 1496–1499 CrossRef CAS
- C. J. Major, S. E. Read, R. A. Coates, A. Francis, B. J. McLaughlin, M. Millson, F. Shepherd, M. Fanning, L. Calzavara and D. MacFadden,
et al.
, J. Infect. Dis., 1991, 163, 699–702 CrossRef CAS PubMed
- K. B. Elkon, A. E. Gharavi, B. M. Patel, G. R. Hughes and A. Frankel, Clin. Exp. Immunol., 1983, 52, 75–84 CAS
- D. J. Bradshaw and P. D. Marsh, Caries Res., 1998, 32, 456–462 CrossRef CAS PubMed
- E. K. Silbergeld, Ann. N. Y. Acad. Sci., 1993, 694, 62–71 CrossRef CAS PubMed
- D. B. P. Goodman, Ann. N. Y. Acad. Sci., 1993, 694, 78–85 CrossRef CAS PubMed
- C. Kirschbaum and D. H. Hellhammer, Psychoneuroendocrinology, 1994, 19, 313–333 CrossRef CAS PubMed
- A. Kaushik, A. Vasudev, S. K. Arya, S. K. Pasha and S. Bhansali, Biosens. Bioelectron., 2014, 53, 499–512 CrossRef CAS PubMed
- M. Groschl and M. Rauh, Steroids, 2006, 71, 1097–1100 CrossRef PubMed
- J. Guechot, J. Fiet, P. Passa, J. M. Villette, B. Gourmel, F. Tabuteau, G. Cathelineau and C. Dreux, Horm. Res., 1982, 16, 357–364 CrossRef CAS PubMed
- D. W. Archibald and G. A. Cole, AIDS Res. Hum. Retroviruses, 1990, 6, 1425–1432 CrossRef CAS PubMed
-
A. D. Harries, D. Maher, S. Graham and S. T. Initiative, W. H. O. D. o. HIV/AIDS., W. H. O. D. o. Child and A. Health and Development, TB/HIV: A Clinical Manual, World Health Organization, 2004 Search PubMed
- M. Raffa, S. Barhoumi, F. Atig, C. Fendri, A. Kerkeni and A. Mechri, Prog. Neuropsychopharmacol. Biol. Psychiatry, 2012, 39, 371–375 CrossRef CAS PubMed
- F. Tietze, Anal. Biochem., 1969, 27, 502–522 CrossRef CAS PubMed
- H. Güntherberg and J. Rost, Anal. Biochem., 1966, 15, 205–210 CrossRef
- O. W. Griffith, Anal. Biochem., 1980, 106, 207–212 CrossRef CAS PubMed
- I. H. Shaik and R. Mehvar, Anal. Bioanal. Chem., 2006, 385, 105–113 CrossRef CAS PubMed
- K. Ngamchuea, C. Batchelor-McAuley and R. G. Compton, Anal. Chem., 2017, 89, 2901–2908 CrossRef CAS PubMed
- D. J. Reed, J. R. Babson, P. W. Beatty, A. E. Brodie, W. W. Ellis and D. W. Potter, Anal. Biochem., 1980, 106, 55–62 CrossRef CAS PubMed
- B. A. Neuschwander-Tetri and F. J. Roll, Anal. Biochem., 1989, 179, 236–241 CrossRef CAS PubMed
- V. Serru, B. Baudin, F. Ziegler, J. P. David, M. J. Cals, M. Vaubourdolle and N. Mario, Clin. Chem., 2001, 47, 1321–1324 CAS
- C. Carru, A. Zinellu, G. Mario Pes, G. Marongiu, B. Tadolini and L. Deiana, Electrophoresis, 2002, 23, 1716 CrossRef CAS PubMed
- Z. Chen, Z. Wang, J. Chen, S. Wang and X. Huang, Analyst, 2012, 137, 3132–3137 RSC
- H.-Y. Han, Z.-K. He and Y.-E. Zeng, Microchim. Acta, 2006, 155, 431–434 CrossRef CAS
- J. C. Harfield, C. Batchelor-McAuley and R. G. Compton, Analyst, 2012, 137, 2285 RSC
- P. T. Lee, L. M. Goncalves and R. G. Compton, Sens. Actuators, B, 2015, 221, 962–968 CrossRef CAS
- S. A. Zaidi and J. H. Shin, Anal. Methods, 2016, 8, 1745–1754 RSC
- K. Ngamchuea, C. Lin, C. Batchelor-McAuley and R. G. Compton, Anal. Chem., 2017, 89, 3780–3786 CrossRef CAS PubMed
- A. R. Rosa, N. Singh, E. Whitaker, M. de Brito, A. M. Lewis, E. Vieta, G. C. Churchill, J. R. Geddes and G. M. Goodwin, Psychol. Med., 2014, 44, 2409–2418 CrossRef CAS PubMed
- J. P. Richie Jr., L. Skowronski, P. Abraham and Y. Leutzinger, Clin. Chem., 1996, 42, 64–70 CAS
- J. W. Gawryluk, J. F. Wang, A. C. Andreazza, L. Shao and L. T. Young, Int. J. Neuropsychopharmacol., 2011, 14, 123–130 CrossRef CAS PubMed
- O. K. Tuncel, G. Sarisoy, B. Bilgici, O. Pazvantoglu, E. Cetin, E. Unverdi, B. Avci and O. Boke, Psychiatry Res., 2015, 228, 688–694 CrossRef PubMed
- J. Lagopoulos, D. F. Hermens, J. Tobias-Webb, S. Duffy, S. L. Naismith, D. White, E. Scott and I. B. Hickie, J. Psychiatr. Res., 2013, 47, 412–417 CrossRef CAS PubMed
- B. R. Godlewska, S. W. Yip, J. Near, G. M. Goodwin and P. J. Cowen, Psychopharmacology, 2014, 231, 327–332 CrossRef CAS PubMed
- R. Kand'ar, P. Zakova, H. Lotkova, O. Kucera and Z. Cervinkova, J. Pharm. Biomed. Anal., 2007, 43, 1382–1387 CrossRef PubMed
- A. M. Cantin, S. L. North, R. C. Hubbard and R. G. Crystal, J. Appl. Physiol., 1987, 63, 152–157 CAS
- F. R. Ochseedorf, R. Buhl, A. Bastlein and H. Beschmann, Hum. Reprod., 1998, 13, 353–359 CrossRef
- D. P. Jones, J. L. Carlson, P. S. Samiec, P. Sternberg, V. C. Mody, R. L. Reed and L. A. S. Brown, Clin. Chim. Acta, 1998, 275, 175–184 CrossRef CAS
- E. Emekli-Alturfan, A. Yarat, E. Caliskan-Ak, R. Pisiriciler, B. Kuru and U. Noyan, J. Clin. Lab. Anal., 2013, 27, 261–266 CrossRef CAS PubMed
- L. von Knorring and H. Mörnstad, Neuropsychobiology, 2008, 15, 146–154 CrossRef PubMed
- M. J. Larsen, A. F. Jensen, D. M. Madsen and E. I. F. Pearce, Arch. Oral Biol., 1999, 44, 111–117 CrossRef CAS PubMed
- M. J. Levine, Ann. N. Y. Acad. Sci., 1993, 694, 11–16 CrossRef CAS PubMed
- C. B. Jacobs, M. J. Peairs and B. J. Venton, Anal. Chim. Acta, 2010, 662, 105–127 CrossRef CAS PubMed
- C. Hsueh, R. Bravo, A. J. Jaramillo and A. Brajter-Toth, Anal. Chim. Acta, 1997, 349, 67–76 CrossRef CAS
- P. T. Lee, D. Lowinsohn and R. G. Compton, Electroanalysis, 2014, 26, 1488–1496 CrossRef CAS
- M. C. Henstridge, E. J. F. Dickinson, M. Aslanoglu, C. Batchelor-McAuley and R. G. Compton, Sens. Actuators, B, 2010, 145, 417–427 CrossRef CAS
- K. R. Kneten and R. L. McCreery, Anal. Chem., 2002, 64, 2518–2524 CrossRef
- R. L. McCreery, Electroanal. Chem., 1991, 17, 221–374 Search PubMed
- H. D. Schwartz, Clin. Chim. Acta, 1975, 64, 227–239 CrossRef CAS
- S. M. Friedman, S. L. Wong and J. H. Walton, J. Appl. Physiol., 1963, 18, 950–954 CAS
- C. R. Raj, T. Okajima and T. Ohsaka, J. Electroanal. Chem., 2003, 543, 127–133 CrossRef CAS
- P. T. Lee and R. G. Compton, Anal. Sci., 2015, 31, 685–691 CrossRef CAS PubMed
- P. T. Lee, D. Lowinsohn and R. G. Compton, Sensors, 2014, 14, 10395–10411 CrossRef CAS PubMed
- P. T. Lee and R. G. Compton, Sens. Actuators, B, 2015, 209, 983–988 CrossRef CAS
- J. Ellison, C. Batchelor-McAuley, K. Tschulik and R. G. Compton, Sens. Actuators, B, 2014, 200, 47–52 CrossRef CAS
- J. R. Windmiller and J. Wang, Electroanalysis, 2013, 25, 29–46 CrossRef CAS
- S. Berchmans, R. Karthikeyan, S. Gupta, G. E. J. Poinern, T. B. Issa and P. Singh, Sens. Actuators, B, 2011, 160, 1224–1231 CrossRef CAS
- M. F. Alecrim, F. M. Oliveira, T. J. Guedes, C. D. c. Neves, V. A. Mendonça, E. S. Gil, R. M. Verly and W. T. P. dos Santos, Electrochim. Acta, 2016, 222, 331–337 CrossRef CAS
- H. Nian, J. Wang, H. Wu, J. G. Lo, K. H. Chiu, J. G. Pounds and Y. Lin, Anal. Chim. Acta, 2012, 713, 50–55 CrossRef CAS PubMed
- S.-J. Wang, H.-W. Liaw and Y.-C. Tsai, Electrochem. Commun., 2009, 11, 733–735 CrossRef CAS
- C. T. Wu, P. Y. Chen, J. G. Chen, V. Suryanarayanan and K. C. Ho, Anal. Chim. Acta, 2009, 633, 119–126 CrossRef CAS PubMed
- M. J. Sims, N. V. Rees, E. J. F. Dickinson and R. G. Compton, Sens. Actuators, B, 2010, 144, 153–158 CrossRef CAS
- L. Highton, R. O. Kadara, N. Jenkinson, B. Logan Riehl and C. E. Banks, Electroanalysis, 2009, 21, 2387–2389 CrossRef CAS
- Ľ. Švorc, D. M. Stanković and K. Kalcher, Diamond Relat. Mater., 2014, 42, 1–7 CrossRef
- K. Liu, W. Z. Wei, J. X. Zeng, X. Y. Liu and Y. P. Gao, Anal. Bioanal. Chem., 2006, 385, 724–729 CrossRef CAS PubMed
- S. Ward-Jones, C. E. Banks, A. O. Simm, L. Jiang and R. G. Compton, Electroanalysis, 2005, 17, 1806–1815 CrossRef CAS
- M. Badea, A. Amine, G. Palleschi, D. Moscone, G. Volpe and A. Curulli, J. Electroanal. Chem., 2001, 509, 66–72 CrossRef CAS
- S. I. R. Malha, J. Mandli, A. Ourari and A. Amine, Electroanalysis, 2013, 25, 2289–2297 CAS
- J. Davis, K. J. McKeegan, M. F. Cardosi and D. H. Vaughan, Talanta, 1999, 50, 103–112 CrossRef CAS PubMed
- A. C. Torres, M. M. Barsan and C. M. Brett, Food Chem., 2014, 149, 215–220 CrossRef PubMed
- G. A. M. Mersal, Food Anal. Methods, 2011, 5, 520–529 CrossRef
- X. Kan, T. Liu, C. Li, H. Zhou, Z. Xing and A. Zhu, J. Solid State Electrochem., 2012, 16, 3207–3213 CrossRef CAS
- B. Habibi, M. Abazari and M. H. Pournaghi-Azar, Chin. J. Catal., 2012, 33, 1783–1790 CrossRef CAS
- R. N. Goyal, S. Bishnoi and B. Agrawal, J. Electroanal. Chem., 2011, 655, 97–102 CrossRef CAS
- P. Yang, W. Wei and C. Tao, Anal. Chim. Acta, 2007, 585, 331–336 CrossRef CAS PubMed
- J. S. Easow, P. Gnanaprakasam and T. Selvaraju, Res. Chem. Intermed., 2015, 42, 2539–2551 CrossRef
- A. Safavi, N. Maleki, E. Farjami and F. A. Mahyari, Anal. Chem., 2009, 81, 7538–7543 CrossRef CAS PubMed
- J. C. Ndamanisha, J. Bai, B. Qi and L. Guo, Anal. Biochem., 2009, 386, 79–84 CrossRef CAS PubMed
- P. T. Lee, K. R. Ward, K. Tschulik, G. Chapman and R. G. Compton, Electroanalysis, 2014, 26, 366–373 CrossRef CAS
- N. Lawrence, J. Davis and R. G. Compton, Talanta, 2001, 53, 1089–1094 CrossRef CAS PubMed
- F. Ricci, F. Arduini, C. S. Tuta, U. Sozzo, D. Moscone, A. Amine and G. Palleschi, Anal. Chim. Acta, 2006, 558, 164–170 CrossRef CAS
- D. Lowinsohn, P. T. Lee and R. G. Compton, Int. J. Electrochem. Sci., 2014, 9, 3458–3472 Search PubMed
- P. T. Lee and R. G. Compton, Electroanalysis, 2013, 25, 1613–1620 CrossRef CAS
- C. L. Sun, H. H. Lee, J. M. Yang and C. C. Wu, Biosens. Bioelectron., 2011, 26, 3450–3455 CrossRef CAS PubMed
- Z. H. Sheng, X. Q. Zheng, J. Y. Xu, W. J. Bao, F. B. Wang and X. H. Xia, Biosens. Bioelectron., 2012, 34, 125–131 CrossRef CAS PubMed
- S. Zhu, H. Li, W. Niu and G. Xu, Biosens. Bioelectron., 2009, 25, 940–943 CrossRef CAS PubMed
- R. Zhang, G.-D. Jin, D. Chen and X.-Y. Hu, Sens. Actuators, B, 2009, 138, 174–181 CrossRef CAS
- J. C. Fanguy and C. S. Henry, Electrophoresis, 2002, 23, 767–773 CrossRef CAS PubMed
- J. M. Zen and P. J. Chen, Anal. Chem., 1997, 69, 5087–5093 CrossRef CAS
- A. Safavi, N. Maleki, O. Moradlou and F. Tajabadi, Anal. Biochem., 2006, 359, 224–229 CrossRef CAS PubMed
- K. Shi and K.-K. Shiu, Electroanalysis, 2001, 13, 1319–1325 CrossRef CAS
- Y.-H. Zhu, Z.-L. Zhang and D.-W. Pang, J. Electroanal. Chem., 2005, 581, 303–309 CrossRef CAS
- Y. Li, S. Wu, P. Luo, J. Liu, G. Song, K. Zhang and B. Ye, Anal. Sci., 2012, 28, 497–502 CrossRef CAS PubMed
- G. J. Yang, K. Wang, J. J. Xu and H. Y. Chen, Anal. Lett., 2007, 37, 629–643 CrossRef
- H. Yin, X. Meng, H. Su, M. Xu and S. Ai, Food Chem., 2012, 134, 1225–1230 CrossRef CAS PubMed
- J. Zen, Talanta, 1999, 50, 635–640 CrossRef CAS PubMed
- Y. Fan, J. H. Liu, H. T. Lu and Q. Zhang, Colloids Surf., B, 2011, 85, 289–292 CrossRef CAS PubMed
- J. Li, J. Liu, G. Tan, J. Jiang, S. Peng, M. Deng, D. Qian, Y. Feng and Y. Liu, Biosens. Bioelectron., 2014, 54, 468–475 CrossRef CAS PubMed
- B. G. Mahmoud, M. Khairy, F. A. Rashwan and C. E. Banks, Anal. Chem., 2017, 89, 2170–2178 CrossRef CAS PubMed
- X. Chen, J. Zhu, Q. Xi and W. Yang, Sens. Actuators, B, 2012, 161, 648–654 CrossRef CAS
- M. Amiri-Aref, J. B. Raoof and R. Ojani, Colloids Surf., B, 2013, 109, 287–293 CrossRef CAS PubMed
- A. Kutluay and M. Aslanoglu, Anal. Chim. Acta, 2014, 839, 59–66 CrossRef CAS PubMed
- R. T. Kachoosangi, G. G. Wildgoose and R. G. Compton, Anal. Chim. Acta, 2008, 618, 54–60 CrossRef CAS PubMed
- X. Kang, J. Wang, H. Wu, J. Liu, I. A. Aksay and Y. Lin, Talanta, 2010, 81, 754–759 CrossRef CAS PubMed
- K. Chaisiwamongkhol, C. Batchelor-McAuley and R. G. Compton, Analyst, 2017, 142, 2828–2835 RSC
- F. Contu, M. Vega-Arroyo and S. R. Taylor, Int. J. Mater. Sci., 2014, 4, 8–13 CrossRef
- C. Zuliani, G. Matzeu and D. Diamond, Electrochim. Acta, 2014, 132, 292–296 CrossRef CAS
- D.-M. Kim, S. J. Cho, C.-H. Cho, K. B. Kim, M.-Y. Kim and Y.-B. Shim, Biosens. Bioelectron., 2016, 79, 165–172 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2018 |