
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advances in mass spectrometry based single-cell metabolomics
Kyle D.
Duncan
 ,
Jonas
Fyrestam
and
Ingela
Lanekoff
,
Jonas
Fyrestam
and
Ingela
Lanekoff
Department of Chemistry – BMC, Uppsala University, Sweden. E-mail: Ingela.Lanekoff@kemi.uu.se
First published on 23rd October 2018
Abstract
Metabolomics has grown into a prominent field contributing to the molecular understanding of complex biological processes in both health and disease. Furthermore, single-cells are known to display metabolic differences between seemingly homogeneous populations of cells. Single-cell metabolomics attempts to analyze many cellular metabolites from single cells to understand phenotypic heterogeneity, which is a significant challenge due to the low analyte abundances and limited sample volumes. Label-free metabolite detection can be achieved with mass spectrometry, which is capable of simultaneously analyzing hundreds of metabolites. Herein, we review the recent advances in mass spectrometry based single-cell metabolomics, highlighting the current state-of-the-art within the last three years, and identify the challenges to move the field forward.
Kyle Duncan received his PhD in Chemistry from the University of Victoria in 2016 focusing on the development of direct on-line sampling approaches for mass spectrometry. He then joined the Lanekoff group as a postdoctoral fellow at Uppsala University to construct and apply ambient surface sampling mass spectrometry techniques for complex biological samples. Currently Kyle is a researcher in the Lanekoff group interested in improving the sensitivity and selectivity of ambient ionization mass spectrometry methods. |
Jonas Fyrestam obtained his PhD degree in analytical chemistry from the Department of Environmental Science and Analytical Chemistry, Stockholm University, Sweden, in 2018. During his PhD studies he worked with endogenously produced photosensitizers in microorganisms and their susceptibility for visible light. Jonas is currently a postdoctoral research fellow in the Lanekoff group at Uppsala University, Uppsala, Sweden. His research focus is on mass spectrometry imaging using nanospray desorption electrospray ionization (nano-DESI) and its applications. |
Ingela Lanekoff received her PhD degree in analytical chemistry from the University of Gothenburg, Sweden in 2011 with a focus on single-cell analysis. Before starting her own group in 2014, she was a postdoctoral fellow in the group of Julia Laskin at Pacific Northwest National Laboratory, WA, USA. Ingela is currently an Associate Professor in analytical chemistry at the Department of Chemistry-BMC, Uppsala University, Sweden, and her research focuses on the development and application of ambient surface sampling coupled to mass spectrometry for endogenous small molecules in biological systems. |
Introduction
Metabolomics is a field of study that aims to measure a large amount of metabolites at one time. Metabolites are small biomolecules, such as amino acids, sugars, and lipids, which constitute precursors, intermediates, and products in cellular processes.1 Thus, metabolites that are detected in a biological sample provide a “fingerprint” of on-going biological activities. In addition, there is increasing evidence that metabolites directly initiate cellular signaling cascades and modulate diverse biological processes such as epigenetic mechanisms and post-translational modifications.2 As a result, metabolomics datasets can link cellular activities to metabolic pathways and biological mechanisms in health and disease.Analysis of metabolites in biological samples is challenging for several reasons. The metabolome is highly dynamic, hence the presence and concentration of metabolites are rapidly altered due to cellular actions or responses to the surrounding chemical and physical microenvironments. Furthermore, the diversity of metabolite structures is immense, yielding a complex mixture of isomers and isobars for analysis that puts high demands on the selectivity of the analytical technique. In addition, the acquired data are complex and contain features belonging to both known and unknown metabolites. The number of unknown metabolites is still hard to predict, but new metabolites and metabolic pathways are continuously being discovered.3–5 Finally, some metabolites are only found in trace amounts, and since no amplification is possible, analytical instrumentation requires high sensitivity. Fortunately, recent advances in mass spectrometry (MS) technology, such as high mass resolving power and increased sensitivity, in combination with the development of metabolite databases provide an avenue for metabolomics studies.
MS based metabolomics is traditionally performed on a liquid sample of a biological fluid, homogenized tissue, or homogenized cells using a separation technique coupled to MS.2 Such analysis provides the average metabolome of the biological sample using either a targeted or an untargeted approach. Targeted metabolomics is performed using customized methods for preselected metabolites, and provides data with relatively low complexity and high sensitivity for selected metabolites. A targeted approach can validate untargeted metabolomics results generated from measuring a broad range of metabolites in a sample. Untargeted metabolomics without separation prior to MS is typically referred to as shotgun analysis or direct infusion and is the most common approach for single-cell metabolomics.
In this mini-review we focus on the recent advances and developments in MS-based single-cell metabolomics during the last three years in an attempt to present the current strategies and approaches for elucidating active biochemical processes in individual cells. For readers interested in a more comprehensive view of single-cell MS, including techniques for larger biomolecules and subcellular imaging, there are several recent and nicely written review articles to explore.6–13
Single-cell metabolomics
A single cell can be regarded as the smallest functional biological unit and its chemical activities can provide unique insights into biological processes. Different biological processes continuously occur in single cells as they move, divide, communicate, and respond to their individual chemical microenvironments. Even cells with identical genotypes display different chemical phenotypes as a result of cellular dynamics and unique microenvironments.14 Phenotypical differences can be revealed by single-cell metabolomics to unravel the reasons behind cellular heterogeneity and aid in understanding the cause of cellular biochemical activity with implications in health and disease.Single-cell metabolomics offers many opportunities, but also challenges due to the limited sample volume, low analyte amounts, and rapid turnover rates of the cellular metabolome. Due to the limited material available for analysis, most single-cell metabolomics studies are performed using MS in a shotgun-like approach, preferably with high mass resolution. However, it is important to be aware that without separation prior to MS it is difficult to distinguish between isomeric metabolites or spontaneous in-source fragmentations. In addition, matrix effects, i.e. ion suppression or enhancement, are present when multiple analytes are ionized simultaneously, and can result in both random and systematic errors.15 The rapid turnover rates of the metabolome require that the cell stays in its native environment for as long as possible prior to analysis. For example, turnover rates down to 0.3 seconds have been reported for rapidly cycling metabolites.16 Keeping a cell in its native environment is crucial to acquire reliable results from the cellular metabolome instead of results based on cellular responses to chemical and/or physical treatment, such as changing the chemical environment, centrifuging, or poking the cell. In addition, the cellular metabolism should ideally be quenched immediately upon sampling to stop enzymatic activities. Quenching is typically performed by combining an organic solvent, cold, heat, an acid, or a base to immediately denature enzymes and stop metabolic activity that will otherwise continue to convert metabolites after sampling.15
In addition to being sensitive and non-invasive with rapid quenching and analysis, the ideal technique for single-cell metabolomics should enable the analysis of a large number of individual cells in a high-throughput fashion. This is essential to determine the technical variability and find biological variations linked to biochemical processes. Furthermore, high-throughput is of exceptional importance since the metabolome will vary in time and between single cells even if they have the same genotype. In addition, MS is a destructive technique making replicate analysis of low volume samples a challenge. Many techniques for single-cell metabolomics are currently in their infancy and therefore reports on the detection of highly abundant and easily ionizable compounds, such as phospholipids, are common. Despite the challenges associated with single-cell metabolomics, recent progress in the field, as described in this review, provides great hope for future successes in detecting and identifying metabolites in single cells, establishing their effect on phenotypes, and their roles in metabolic pathways and physiology.
Analysis of entire single cells
Single-cell analysis entails very small sample volumes that contain low absolute quantities of analytes. To maximize the amount of material for analysis, several single-cell MS techniques are developed to sample the entire content of one cell in one sampling event.Ambient ionization of single cells
Several ambient approaches for the analysis of the entire contents of one cell on a surface have been recently developed, as schematically summarized in Fig. 1. Bergman and Lanekoff employed nanospray desorption electrospray ionization (nano-DESI) MS to measure lipids and metabolites from single human cheek cells deposited on glass slides.17 In nano-DESI, a continuously flowing liquid bridge is maintained between two capillaries, a solvent delivery capillary and a nanospray capillary for subsequent ionization, in front of the mass spectrometer. By placing the liquid bridge in contact with a single cell, the entire cell contents are desorbed into the liquid in a few seconds as evidenced by the appearance and subsequent loss of [M + K]+ species. Using this approach, 48 species including phosphatidylcholine (PC), phosphatidylethanolamine (PE), sphingomyelin (SM), plasmalogen, amino acids, creatine, spermidine, and carnitines were identified by tandem mass spectrometry (MS/MS). Additionally, the total amount of PC lipids in one cheek cell was quantified by incorporating a PC standard into the nano-DESI solvent.17 In a similar approach to nano-DESI, Sun et al. employed a single probe for metabolite screening in single marine dinoflagellate cells (S. trochoidea).18 In the single probe, the solvent delivery and nanospray capillary are connected by a pulled tip dual-bore quartz capillary. A continuously flowing solvent creates a microdroplet between the capillaries, which extracts analytes that are subsequently entrained to the mass spectrometer for detection. Over 550 metabolites from single dinoflagellate cells were tentatively identified. Furthermore, the metabolome was compared between cells cultivated in darkness or under light using partial least squares discriminant analysis. The study identified metabolic pathways for polyketides, porphyrins, chlorophylls, terpenoids, and limonene to be significantly altered under dark conditions.18 In a different study, Pan et al. incorporated dicationic reagents into a single probe solvent to complex negatively charged lipids and metabolites extracted from single HeLa cells, enabling them to be monitored in positive mode.19 Over 190 negatively charged metabolites, such as adenosine monophosphate (AMP) and folic acid, were tentatively assigned by accurate mass with the use of dicationic ion pairing reagents, and several metabolites were verified with MS/MS.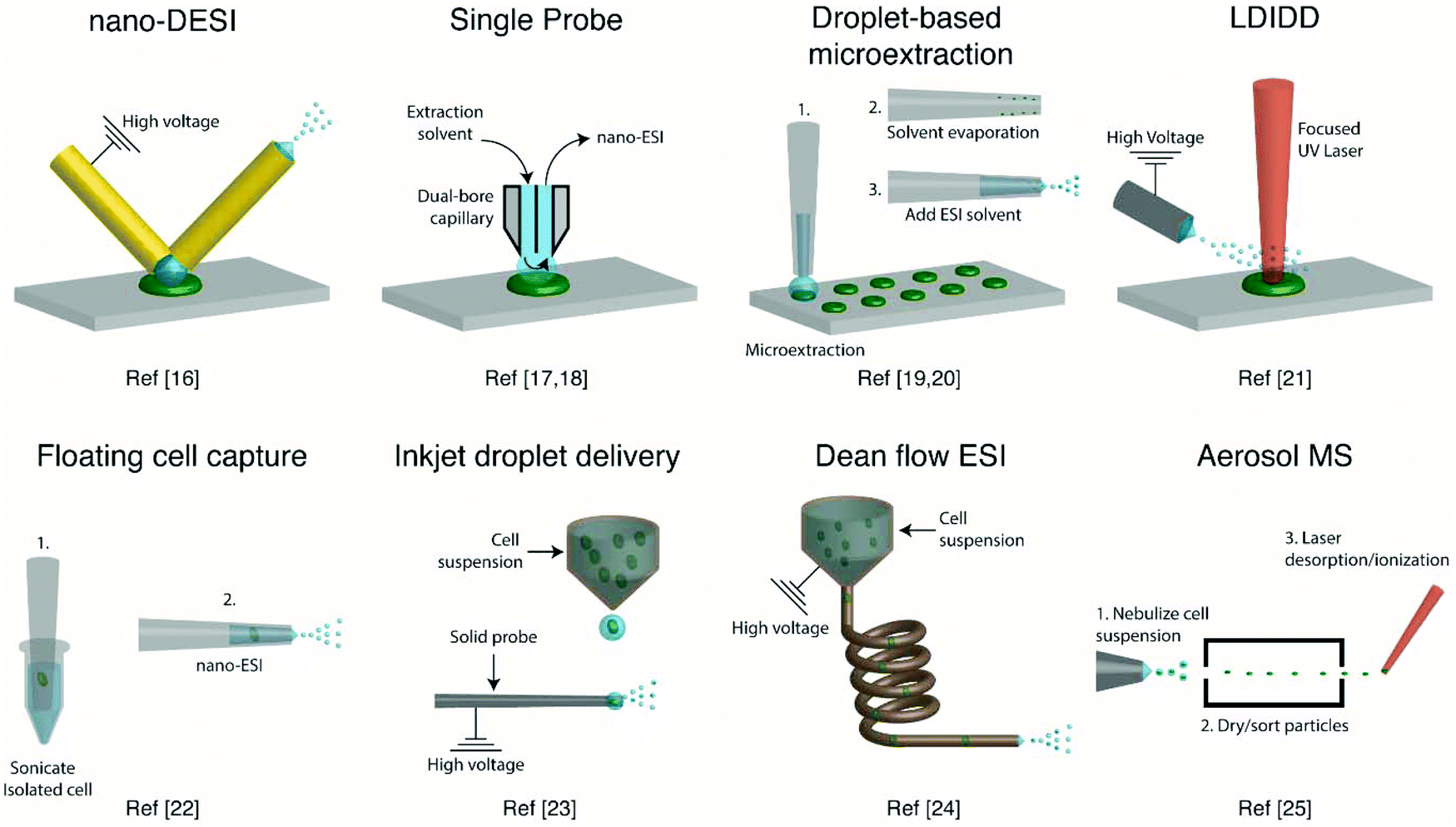 | ||
| Fig. 1 Representative schematics of ambient ionization MS techniques for profiling metabolites from single cells. | ||
A different method for liquid extraction integrating droplet-based microextraction with electrospray ionization (ESI) MS was presented by Zhang et al.20 In this method, a single pulled capillary tip with a small suspended liquid droplet (<2 nL) is lowered onto a single cell such that the microdroplet covers the cell area and extracts the endogenous content into the capillary. The extraction solvent, which can be altered for selectivity, is then evaporated followed by addition of 20–100 pL of electrospray amenable solvent to re-dissolve the cellular contents to facilitate ionization and MS detection. With this method adenosine triphosphate (ATP), adenosine diphosphate (ADP), AMP, and reduced and oxidized glutathione were detected from single human breast cancer cells (MCF-7) and identified using MS/MS. The authors argue that ESI-MS of extracted metabolites (Fig. 2a) provides superior detection over spraying pure cytoplasm (Fig. 2b), since matrix suppression is highly apparent without analyte extraction. A subsequent study employed pulsed direct current ESI-MS liquid microextraction to allow more spectra to be recorded from each cell.21 Using this approach, the authors were able to acquire over 600 MS/MS spectra from a single cell. It was found that the number of unsaturated phospholipids increased in glioblastoma cells compared to normal astrocyte cells.22
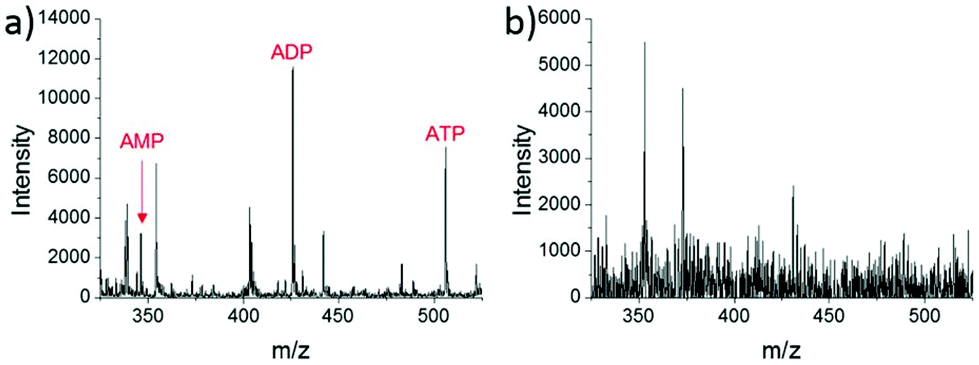 | ||
| Fig. 2 Mass spectra of single MCF-7 cells obtained by: (a) microextraction followed by nano-ESI-MS; and (b) nano-ESI-MS of pure cytoplasm. AMP, adenosine monophosphate; ADP, adenosine diphosphate; and ATP, adenosine triphosphate. Reproduced from ref. 20 with permission from Springer Nature, copyright 2016. | ||
A different route for desorbing material from single cells on a surface is to use laser desorption. In laser desorption/ionization droplet delivery (LDIDD) MS a focused UV laser is directed towards the cell surface to desorb/ionize the cell components. The plume of the material is then picked up by electrosprayed liquid droplets and subsequently delivered to the MS. The combination of laser desorption/ionization and droplet delivery increased the signal for Rhodamine B by ∼7 times, when compared to each modality alone.23 Lee et al. used LDIDD-MS to monitor phospholipids and fatty acids in HEK 293T cells. The study suggests that apoptotic cells alter their composition of PC species. Specifically, they found that PC species with shorter acyl chains increase in apoptotic cells compared to healthy cells. In addition, the study monitored exocytosed metabolites from living PC12 cells and found phenethylamine and tyramine to be secreted.23 Measurements from single cells fixed on a substrate can allow cell analysis by multiple modalities (e.g., optical and MS), as the position of the cells can be recorded. However, not all cells are adherent and thus methods for single-cell metabolomics of cells in suspension are also required.
To allow for single-cell metabolomics of cells in suspension, several recently developed techniques aim to isolate and directly analyze single cells from bulk solution. Hiyama and coworkers demonstrated a method of capturing a single floating white blood cell from a human blood sample into a nanoelectrospray capillary using a micro-manipulator and piston syringe.24 Electrospray compatible solvent was subsequently introduced behind the cell, and the assembly was sonicated prior to mass spectrometric analysis to induce cell lysis. The sonication step enabled the detection of several tentatively identified amino acids, catecholamines, fatty acids, monoacylglycerols, and triacylglycerols from single white blood cells, and a few metabolites were identified by MS/MS. However, while ESI-MS of a single isolated cell provides detailed chemical information, manual cell capturing is laborious and limits the throughput. A method with an increased throughput was developed by Chen et al.25 This method uses an inkjet printer to deliver droplets containing single cells from a cell suspension onto a charged tungsten electrode. When the cell hits the tungsten probe the cell content is ionized via ESI. Careful positioning and experimental control of the inkjet droplet delivery system enabled 486 pL droplets to be delivered onto the 2 μm tungsten probe. This method provided single-cell MS analysis with a duty cycle of ∼0.05 Hz, including the necessary washing steps of the probe between cell samples. Phospholipids were readily observed, and the method enabled four different types of cells to be classified using principal component analysis (PCA).25
Another method developed to improve the throughput for single-cell measurements from cell suspensions uses a Dean flow assisted ordering system to introduce single cells to an ESI source.26 A flow through a curved tube facilitates the formation of Dean vortexes that function as micro-stirrers and separate particles to an equilibrium position. The ordered cells were subsequently ejected at a maximum rate of 2 Hz and subjected to ESI to ionize cell constituents. Lipid profiles, in particular PC and PE, obtained from three types of human tumor cells were used to successfully separate each cell type by PCA. Recently, a study showed the applicability of aerosol time-of-flight MS towards single-cell metabolomics for algal cell suspensions.27 The method works by nebulizing a cell suspension and sorting the cell containing aerosol particles with an aerodynamic lens. Particles between 1 and 10 μm are transmitted through two continuous wave lasers calibrated with polystyrene latex spheres to determine the size of the single cells. Then, a pulsed UV laser desorbs/ionizes the contents of each individual cell, and mass spectra are recorded in both positive and negative modes using a TOF mass analyzer. This approach for single-cell MS analysis shows great promise, as the size in addition to the positive and negative mode mass spectrum is recorded for each cell. Furthermore, the cells were analyzed with a relatively high duty cycle of ∼50 Hz, which is beneficial for robust statistical analysis. By sampling single algal cells (Chlamydomonas reinhardtii) grown under either nitrogen-rich or nitrogen-limited conditions in a time-resolved manner, the authors reported an increase of dipalmitic acid sulfolipid sulfoquinovosyldiacylglycerol under nitrogen limited conditions. However, the method remains to be limited by the quality and mass resolution of the mass spectra obtained with a TOF mass analyzer.
Sampling of metabolites from single cells under ambient conditions requires very little sample preparation, which can reduce the perturbance of cells from their natural state. Additionally, many of the recent advances focus on increasing the method throughput to obtain a higher statistical robustness with the aim of differentiating technical over biological variability. However, as most methods incorporate some variations of ESI, matrix effects such as ionization suppression hinder the observation of less abundant metabolite species and continue to make quantification of metabolites from single cells a challenge.
Matrix assisted laser desorption/ionization of single cells
Matrix assisted laser desorption/ionization (MALDI) MS, in which a laser ablates a sample surface area covered with a light absorbing low-molecular-weight chemical matrix, is a well-established technique for single-cell analysis.16,28 In MALDI, the selection of the matrix for the analysis of metabolites is of utmost importance. The matrix must facilitate ionization through charge transfer for the species of interest, while yielding few but well characterized peaks that do not interfere with endogenous species sampled from cells. Additionally, the matrix needs to form small enough crystals to maintain single-cell resolution.29 Recent studies using MALDI-MS for single-cell metabolomics can be divided into two categories: (i) MS imaging experiments performed at single-cell resolution, where the whole surface is systematically sampled; and (ii) single-cell profiling experiments, where laser pulses are directed towards cell specific coordinates.There are a few recent studies using MALDI imaging for single-cell metabolite profiling that used high spatial resolution to target cellular and sub-cellular metabolite localizations. Kompauer et al. demonstrated an atmospheric pressure MALDI system with 1.4 μm lateral resolution, which was used to image P. caudatum single-cell organisms with subcellular resolution.30 With 4-hydroxycinnamic acid as the matrix, the authors report the tentative identification of around 220 species, such as diacylglycerol (DG) and PC species, with distinct subcellular resolution (Fig. 3). Another study utilized 3D MALDI-MS imaging to map the distribution of lipids in a fertilized zebrafish embryo at the one-cell stage.31 Dueñas and coworkers sectioned zebrafish embryos at 10 μm thickness followed by freeze-drying prior to matrix application and MS analysis. Several phospholipid species with different distributions throughout the embryo were identified, including PE species that were found to localize on the center of the blastodisc. While sub-cellular imaging can reveal unique insights into metabolite localizations, studying the cellular heterogeneity requires a large amount of cells to be analyzed. Yang et al. applied MALDI-MS imaging to monitor the metabolite heterogeneity from single cells cultured on indium tin oxide (ITO) coated glass slides.32 The cells were freeze-dried to be compatible with the vacuum environment and rinsed to remove excess salt prior to analysis. Using automated data analysis software, spectra from fully ablated cells were discriminated from the spectra obtained from partially ablated cells or multiple cells. Over 650 distinct peaks were detected from single cells in the mass range of 525–775 Da. This setup enabled the investigation of the cellular heterogeneity between control and lipopolysaccharide (LPS) treated macrophage cells. It was found that signals of phospholipids with low degrees of unsaturation increased after LPS treatment while those of highly unsaturated phospholipids decreased. Additionally, LPS treatment was found to alter the expression of selected phospholipids heterogeneously, a result that would be overlooked in population studies.32 An alternative to using automated data analysis software to select cellular locations is to distribute cells in an systematic order. Xie et al. presented a method of isolating and ordering single cells on a ITO coated glass slide using a fabricated polydimethylsiloxane (PDMS) microchip.33 The optimized PDMS microchip had holes of 40 μm in diameter, 100 μm apart, and was placed on top of an ITO coated glass slide. Cell suspensions were then added to the microarray cast and the cells were allowed to adhere to the surface of the slide. The results indicated a single-cell occupancy rate of 25% per microwell. Following the removal of the PDMS microchip, the cells were washed, covered with a matrix, and subjected to MALDI-MS imaging. Eight PC species from single human lung cancerous cells (A549) were identified using MS/MS and the authors foresee this becoming a high throughput technique for single-cell analysis.33
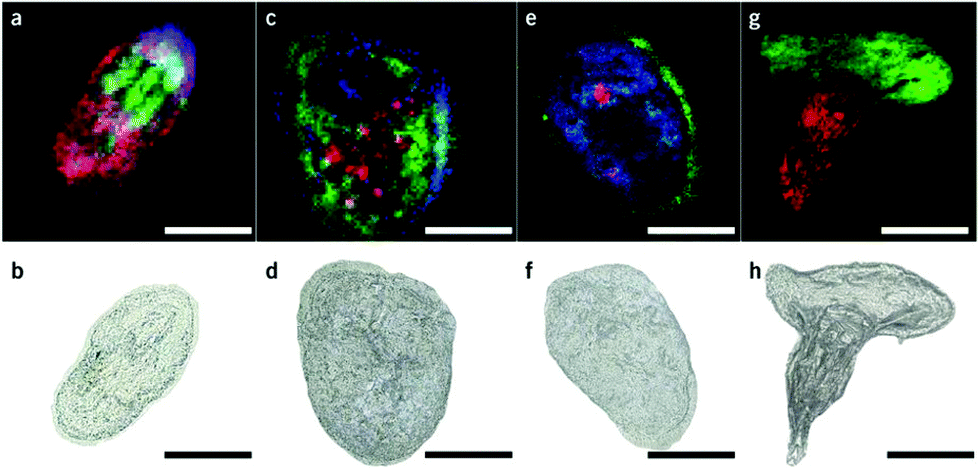 | ||
| Fig. 3 Single-cell AP-MALDI-MSI measurements of P. caudatum or P. caudatum together with a Rotifera and corresponding optical images. (a, b) RGB ion image obtained with a 3 μm step size (a) and the corresponding optical image before MSI measurement of P. caudatum (b). [DG(31:0) + NH4]+ (m/z 572.5240; red), [PC(34:1) + Na]+ (m/z 782.5668; green), and [ceramide(d35:2) + H]+ (m/z 550.5195; blue) are superimposed. (c, d) RGB ion image obtained with a 3 μm step size (c) and the corresponding optical image before MSI measurement of P. caudatum (d). [Phosphatidylserine(32:1) + H-H2O]+ (m/z 716.4903; red), [monoacylglycerol(22:1) + NH4]+ (m/z 430.3885; green), and [(Lys3Ala) + H]+ (m/z 474.3417; blue) are superimposed. (e, f) RGB ion image with a 2 μm step size (e) and the corresponding optical image before MSI measurement of P. caudatum. [DG(38:1) + NH4]+ (m/z 668.6187; red), [(LysAsnArgLeu/Ile) + H-2H2O]+ or [(LysArgValGln) + H-2H2O]+ (m/z 494.3209; green), and [PC(P-30:0) + H]+ (m/z 690.5432; blue) are superimposed. (g, h) RGB ion image obtained with a 2 μm step size of P. caudatum being devoured by Rotifera (g) and the corresponding optical image before MSI measurement (h). [PE(35:2) + H]+ (m/z 730.5377; red) and [PC(34:1) + Na]+ (m/z 782.5668; green) are superimposed. Scale bars, 100 μm. Reproduced from ref. 30 with permission from Springer Nature, copyright 2017. | ||
Profiling single cells by their positional coordinates could further increase the throughput, as less of the surface area is analyzed. This was employed by Krismer et al. for high throughput MALDI-MS screening of single algal cells using microscale sample preparation.34 They used a stainless steel microarray slide, containing 1430 wells, 300 μm in diameter and a center-to-center distance of 720 μm, and loaded each well with droplets containing single cells using a spotting robot. The cell metabolism was then quenched by immersing the microarray into liquid nitrogen, which also facilitates cell lysis due to ice crystal formation. Concurrent monitoring of the cell-loaded microarray with confocal fluorescence microscopy verified successful single-cell loading prior to matrix application and MALDI analysis of cellular locations. Fig. 4 displays the mass spectrum obtained from a single algal cell and a bulk cell spectrum, highlighting some of the molecular species detected.34 The authors highlight that the single-cell and bulk-cell spectra show the same peak composition, but differ in the relative abundance of detected species due to the cell heterogeneity throughout the bulk cell mixture. This method was later applied to study phenotypic variations in isoclonal and genetically diverse C. reinhardtii algal cells.35 Twenty-six metabolites, such as chlorophyll and triacylglycerol, were monitored in thousands of single cells. By culturing cells under different nitrogen conditions, the authors concluded that genetically diverse cultures showed higher plasticity in their phenotypic response compared to isoclonal cultures, hinting towards the importance of the phenotype for biological function.35 To increase the throughput for MALDI analysis of single cells spread on a glass slide, an automated approach for optical imaging was used to determine cell coordinates followed by localized MALDI-MS.36 For optical imaging, cell nuclei were stained with a fluorescent tag and the fluorescence image provided relative coordinates for each cell compared to the etched marks on the glass slide. The MALDI laser was directed to each cell specific coordinate for MS profiling.
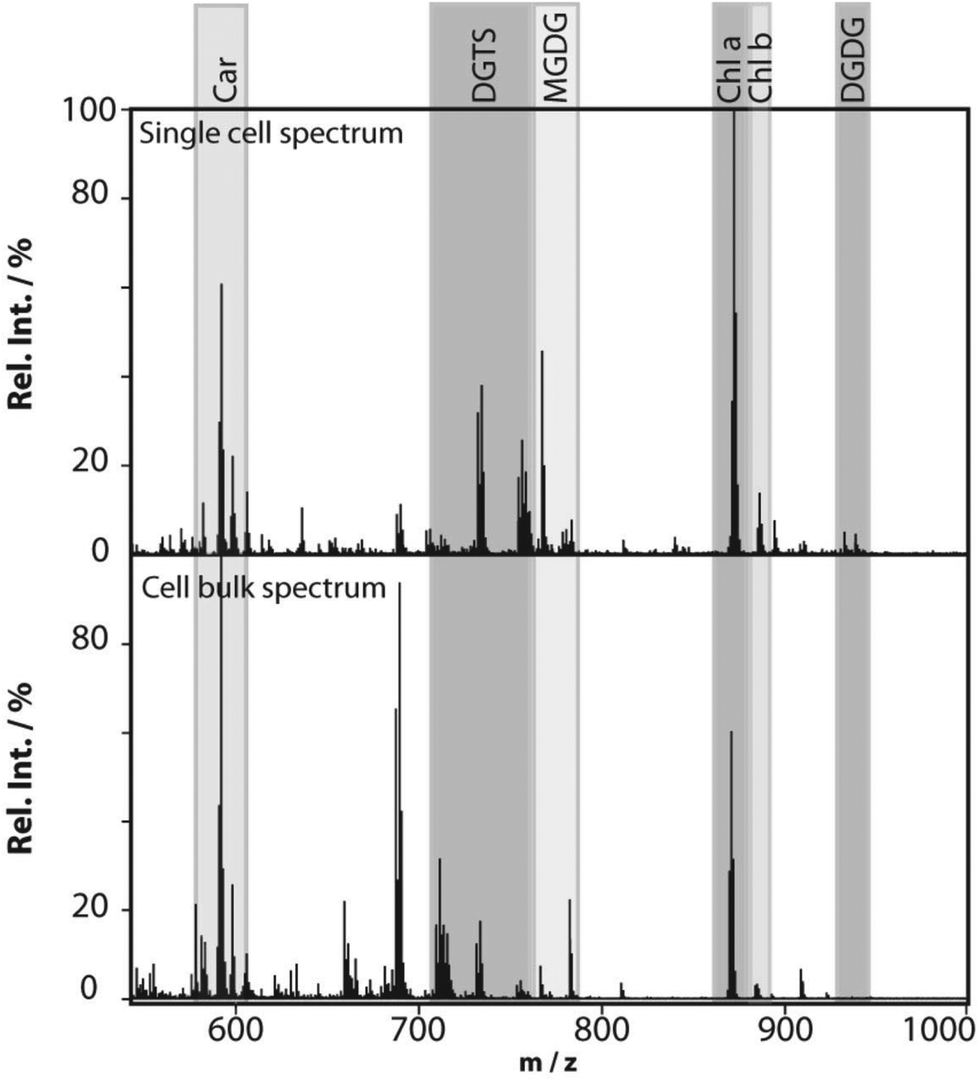 | ||
| Fig. 4 MALDI mass spectra obtained from a single Chlamydomonas reinhardtii cell and from cells in bulk. The single-cell mass spectrum is background corrected for better visibility of the peaks in the lower-mass region. This was achieved by subtracting the spectrum of an empty spot from the single-cell spectrum. Rel. Int., relative intensity; Car, carotenoids; DGTS, diacylglyceryl-N,N,N-trimethylhomoserine; MGDG, monogalactosyl-diacylglycerols; Chl, chlorophyll; and DGDG, digalactosyl-diacylglycerols. Reproduced from ref. 34 with permission from American Society for Microbiology, copyright 2015. | ||
MALDI continues to be one of the foremost techniques used for single-cell MS. Approaches for single-cell MALDI-MS range from subcellular imaging to higher throughput analysis enabling the detection of biological variability in single cell populations. However, as with the ambient MS techniques, most studies observe the highly abundant species. Therefore, innovation leading to higher metabolome coverage in single-cell MALDI-MS will continue to be a point of interest.
Partial cell analysis
Although analysis of intact cells has been the most common approach, analysis of specific subcellular parts, e.g. the cytosol, mitochondria, etc., has gained increasing attention in recent years. This interest correlates with the increasing potential for ambient ionization techniques in detecting metabolites in living cells with little or no sample preparation. Partial cell analysis enables localized metabolomics and can facilitate repeated sampling of one cell without removing it from its environment. Two different ambient sampling strategies for partial cell analysis have been developed: solid probe microextraction and capillary microsampling.Solid probe microextraction
The main principle of solid probe microextraction is that a finely etched probe, usually made out of a metal, is introduced into a cell through the cell membrane or wall. The cell contents will adhere to the surface of the probe, which is subsequently rinsed and analyzed by MALDI or ESI-MS. In one study, Fu et al. constructed a solid probe from a 0.17 mm copper wire.37 By corroding the copper wire in HCl at an elevated temperature, for 5 min to 72 h, different corrosion levels were obtained. Higher degrees of corrosion of the copper wire correlated with the increased porosity and surface area of the probe. After sampling, the probe tip was rinsed with matrix solution and the solute was plated for MALDI-MS analysis. The response factor of oligosaccharides in red onion epithelial cells (Allium cepa) to an internal standard showed that the corroded copper probe yielded 6 times higher signals compared to an untreated probe. Localized sampling suggests that oligosaccharides are less abundant in the outer epidermal cells, and that anthocyanin derivatives are highly abundant in the outer epidermal cells.37Solid phase microextraction (SPME) probes are based on a fiber that is coated with a polymer or an absorbent and can be used to increase the selectivity by targeted extraction. If the partition coefficient of an analyte is high for the coated fiber, enrichment of analytes is possible and can lower the limit of detection in subsequent MS analysis. To date only two research articles are available where SPME fibers have been used for single-cell analysis.38,39 Piri-Moghadam et al. used a 5 μm SPME fiber coated with polypyrrole as a solid phase and detected flavonoids and quercetin from A. cepa, which was not possible with non-coated tips.39 These studies suggest that solid probes for single-cell analysis can selectively trap and enrich metabolites by various retention mechanisms. There is a large variety of absorbents available to trap analytes of interest depending on their physical properties, potentially yielding unique results with lower matrix effects and detection limits. One advantage of using solid probes is the lack of clogging that can be seen with capillary microsampling strategies. However, relatively long extraction times (>60 s) can be needed depending on the size of the cell and the extraction rate of the analytes. Currently, method are under development and require smaller probe sizes to minimize the disturbance of system equilibrium and cell damage caused from membrane penetration or leaking of the intracellular content.39,40 Overcoming these issues will ensure great potential in single-cell metabolomics.
Capillary microsampling
In capillary microsampling, thin glass capillaries or nano-ESI tips with outer diameters of 130 to 5000 nm are inserted into a cell to withdraw small volumes of intracellular content. Experimental details for variations of capillary microsampling techniques discussed in this mini-review are summarized in Table 1. Aspirated volumes normally range from sub pL to nL depending on the cell-type and region. ESI of the content is facilitated using different strategies to add an electrospray solvent and apply a high voltage to the tip positioned in front of the mass spectrometer. In some cases the sample is placed on a target plate and subsequently analyzed with MALDI-MS.| Method | Microsampling capillary | Ionization electrode | Diameter | Sampling volume | Aspiration method | Ionization | Cell type | Ref. |
|---|---|---|---|---|---|---|---|---|
| Live single-cell MS | Metal coated glass | Metal coating | 1 μm | >100 pL | Air pressure | ESI (−) | Vicia faba | 40 |
| Metal coated glass | Metal coating | 1 μm | N/A | Air pressure | ESI (−) | HepG2 | 43 | |
| Metal coated glass | Metal coating | 1 μm | N/A | Air pressure | ESI (+), ESI (−) | WBC, circulating tumor cells | 23 | |
| Metal coated glass | Metal coating | N/A | N/A | Air pressure | ESI | PC 12, T-cell, B-cell | 44 | |
| Glass | Internal platinum wire | ∼1 μm | 0.1–0.3 pL | Air pressure | ESI (−) | HepG2/C3A | 45 and 46 | |
| Pressure driven microsampling | Quartz | — | 600 nm | 8 nL | Air pressure | MALDI | Allium cepa | 47 |
| Quartz | Internal titanium wire | 2–5 μm | 0.5–442.5 pL | Ionic liquid/silicone oil | ESI (−) | Solanum lycopersicum L | 48 | |
| Quartz | — | Sub μm | ∼200 pL | Silicone/engine oil | ESI (+), ESI (−) | Solanum lycopersicum L | 49 | |
| Theta pipette | Double barrel glass | Conductive tape/internal metal wire | ∼1 μm | 0.2–10 nL | Perfluorodecalin | ESI (+) | Allium cepa | 50 |
| Double barrel glass | Internal platinum wire | ∼1 μm | Several nL | Perfluorodecalin | ESI (−) | Allium cepa | 51 | |
| Fluidic force microscopy | Pyramidal tip of silicon nitride | — | 400 nm | 0.8–2.7 pL | Mineral oil | MALDI | HeLa | 52 |
| Electroosmotic probe | Borosilicate glass | Internal platinum wire | <1 μm | 2–5 pL | Electroosmosis | ESI (+) | Allium cepa | 54 |
| Borosilicate glass | Internal platinum wire | 130 nm | fL range | Electroosmosis | ESI (+), ESI (−) | Allium cepa, neuron, HeLa | 55 |
Live single-cell MS is a technique that was initially developed for the analysis of living single cells from plants,41–43 but has recently been used for metabolomics studies of human cells (HepG2).44 In live single-cell MS, an optical microscope is used to target an individual cell for analysis. A sharp metal coated nanospray microcapillary tip (∼1 μm in diameter) penetrates the cell wall or membrane and withdraws a portion of the intracellular content. Following the addition of a solvent for enhancing the signal in ESI, the capillary is repositioned to the front of the MS inlet. In one study, mitochondria in living HepG2 cells were visualized with a fluorescent probe and selectively captured and analyzed using live single-cell MS.44 The authors tentatively annotated metabolites from many classes including amino acids, metabolites of the citric acid cycle, fatty acids, and sterols. One steroid, identified as telocinobufagin by MS/MS, was only detected in mitochondria and not in the cytosol, which suggests its importance in mitochondrial metabolism.44 The combination of live single-cell MS with a micro-well array has enabled the collection of samples inside and outside of the cells to investigate its application for single-cell secretomics.45 Adrenal pheochromocytoma (PC-12) cells were trapped in a well and treated with KCl, causing secretion of epinephrine, dopa and dopamine. As in most partial cell metabolomic studies, the method selectivity arises through high resolution MS and accurate mass assignment. While MS/MS confirmation of analytes is desirable, the limit of detection is sometimes too high. However, the use of ion mobility separation (IMS) with capillary microsampling enables ions to be separated by their different collision cross-sections.43,46,47
By coupling microsampling to ion mobility MS, Zhang and Vertes were able to identify 76 metabolites, including amino acids, nucleotides, and phospholipids, in single human hepatocytes (HepG2/C3A) using a combination of their accurate mass (±15 mDa) and their collision cross-section (±5 Å) (Fig. 5).46 The combination of IMS and capillary microsampling improved signal-to-noise ratios and facilitated identification. Furthermore, the adenylate energy charge was observed after exposing the cells to an insecticide (rotenone), known to inhibit ATP production and result in the accumulation of reactive oxygen species. After 5 h exposure to the insecticide, the metabolic signature changed drastically and the ATP levels were significantly reduced compared to controls. In addition, the ratio of reduced glutathione to oxidized glutathione significantly decreased when the cells were exposed to H2O2, suggesting a cellular response to oxidative stress.46 Zhang et al. combined fluorescence microscopy with capillary microsampling to distinguish the metabolic differences of subpopulations corresponding to the mitotic phases in single cells (HepG2/C3A).47 The mitotic phases were determined by staining cellular DNA and capillary microsampling IMS-MS was used to track the 83 tentatively assigned metabolites, including amino acids, nucleotides and nucleotide sugars. Significant differences were found between the mitotic phases and the results indicated that the metaphase is a high energy state compared to other phases.47
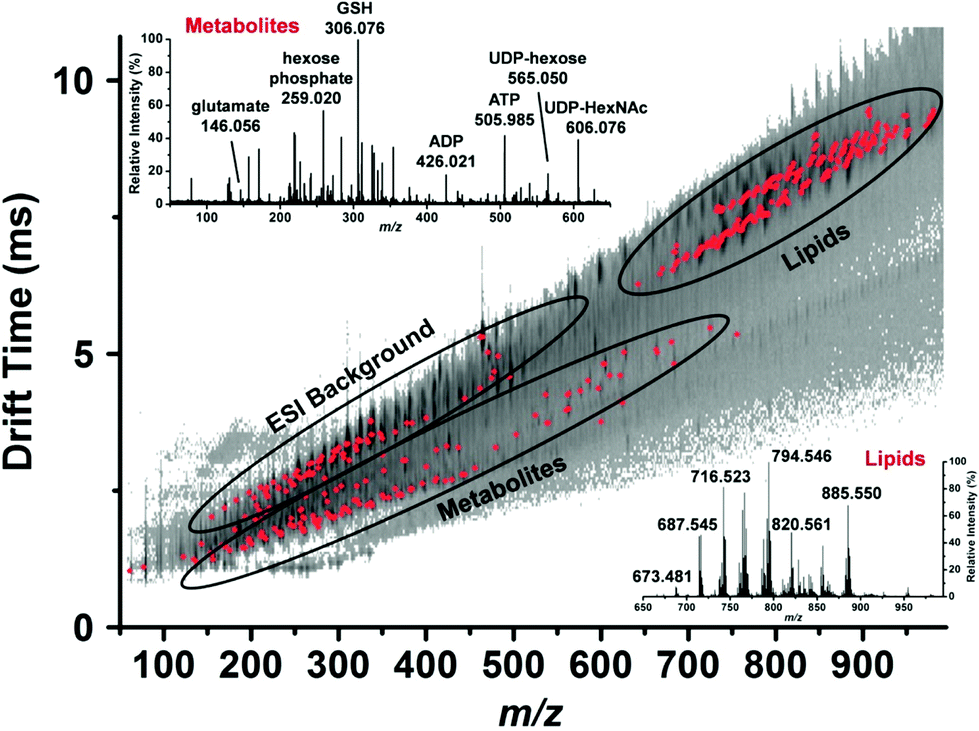 | ||
| Fig. 5 Representative drift time vs. m/z plot of the identified metabolites and lipids (red dots) in a single human hepatocyte. Lipids are well separated from small metabolites and shown by ellipses. Mass spectra from each domain are shown in the insets and some major peaks labeled. GSH, reduced glutathione; ADP, adenosine diphosphate; ATP, adenosine triphosphate; UDP-hexose, uridine diphosphate hexose and UDP-HexNAc, uridine diphosphate N-acetylhexosamine. Reproduced from ref. 46 with permission from American Chemical Society, copyright 2015. | ||
One of the main challenges with capillary microsampling for single-cell analysis has been to withdraw very small volumes with high accuracy and precision. Saha-Shah et al. employed nanopipettes with a tip diameter of 600 nm to collect <10 nL volumes of sample from single-cells of A. cepa.48 These nanopipettes could reproducibly collect fluids by controlling the pressure actuation and the aspirated volume correlated with the applied pressure down to capillary inner diameters of 200 nm. A gas tight syringe plunger was used to apply negative pressure for aspiration and positive pressure for dispensing. Metabolites were characterized by MALDI-MS by dispensing ∼8 nL of the cytoplasm onto target plates, tentatively identifying hexose-oligosaccharides. However, differences in the viscosity of the sample and the air-filled plug between the piston and the capillary tip can substantially affect the accuracy of the sampled volume.48
To increase the accuracy of the sampled volume, the capillary can be filled with a hydrophobic oil and connected to a pressure sensor, known as pressure driven microsampling. Exact sample volumes are calculated from optical images and the geometry of the capillary. Pressure driven microsampling using silicone oil has been used for sampling the cytosol of single cells from tomato plants, enabling the detection of sugars, amino acids etc.49,50 Since the sampled volume was known, the concentration of some metabolites in the cytosol could be determined by external calibration.49,50 Nanopipettes have more recently been refined for pressure driven microsampling using double-barrel nanopipettes (theta pipettes) for repeated single-cell analysis.51 Both barrels were filled with perfluorodecalin (PFD), a liquid that is immiscible in both aqueous and organic solvents. A theta pipette was then inserted into a single A. cepa cell. By applying a negative pressure on one barrel, the cytoplasm was aspirated into the pipette. A positive pressure was then applied on the second barrel, creating an outward droplet of PFD at the tip. This was aspirated again by applying a negative pressure in the first barrel. Repeating this procedure multiple times created segments of sample plugs in the capillary between immiscible plugs of PFD. This strategy enabled sampling of multiple single cells, one per cycle, for subsequent ESI-MS analysis. Red flavonoids were only found in red onion cells while hexose-oligosaccharides were found in both red and white cells.51 The theta pipette further enabled on-line derivatization or ion pairing of the analytes to decrease detection limits for targeted analytes. Using a PFD filled theta pipette with added reagent solution, Saha-Shah et al. performed derivatization of the cis-diols in oligosaccharides with phenylboronic acid to target oligosaccharides. In addition, acid catalyzed degradation enhanced signals for low abundant flavonoids and anthocyanins, such as rutin and quercetin glycoside.52 This strategy yielded a more targeted analysis with improved limits of detection and lower matrix effects for the selected compounds.
To minimize cell perturbation, fluidic force microscopy (FluidFM) has recently been used to withdraw down to 0.8 pL intracellular volumes from mammalian cells (HeLa) by using a 400 nm wide triangular aperture for subsequent analysis by MALDI-MS.53 The sampling and dispensing procedure is illustrated in Fig. 6. An atomic force microscope with a microfluidic probe was placed on top of an inverted optical microscope to localize green fluorescent protein (GFP) expressing cells. By using the force controlled approach, the probe was inserted into a cell and maintained in the same location by keeping the force constant. Negative pressure, maintained by mineral oil, enabled sampling rates between 300 and 500 fL min−1. At zero pressure the probe was retracted out of the cell and the sampled material was plated on a MALDI target plate for analysis. FluidFM enabled the detection and tentative identification of 20 metabolites, such as phosphorylated compounds, sugars, amino acids, and glutathione based on measured m/z. Previous investigations on cell viability after cytoplasmic aspiration concluded that 82% of HeLa cells survived an aspiration of 4.0 pL of cytoplasm, while 100% cell death was seen when 4.5 pL was aspirated.54 This suggests that if less than 4 pL are withdrawn the cell integrity will be maintained during the FluidFM procedure.
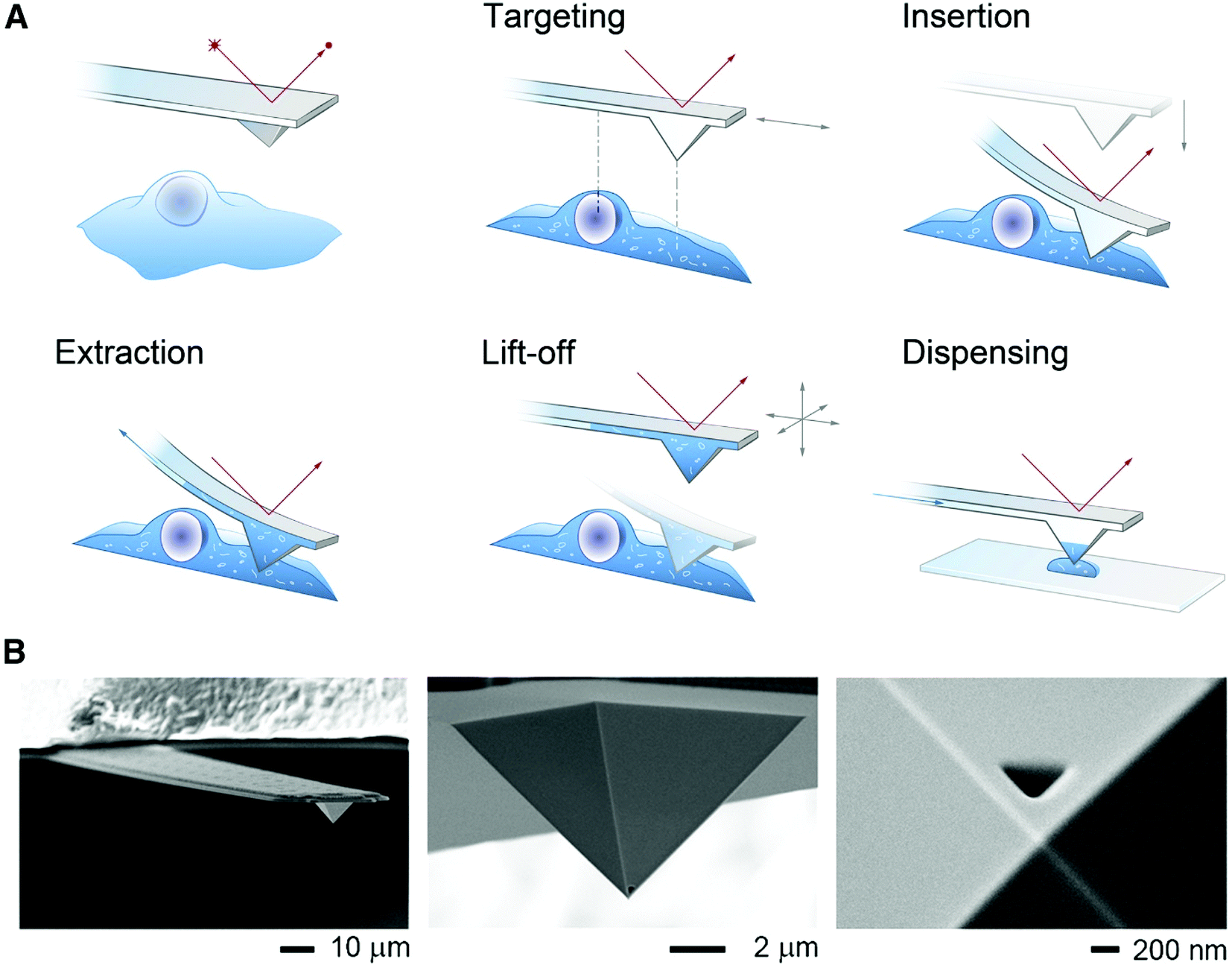 | ||
| Fig. 6 (A) Schematic of the FluidFM-based extraction workflow. The cantilever is monitored by a red laser and targeting a cell is performed with an inverted microscope. By aligning the probe directly above an area of interest the probe can be inserted into the cell by force microscopy and a preset force. Sampling of the intracellular content is performed by applying a negative pressure through the microchannel. After the desired volume is withdrawn, the probe is lift off, and the extract can be plated on a glass slide or a MALDI target plate by applying an overpressure through the channel. (B) Scanning electron micrographs of a FluidFM probe with a 400 nm aperture. Reproduced from ref. 54 with permission from Elsevier, copyright 2016. | ||
Another technique for sampling accurate and small volumes is to use an electroosmotic probe. The electroosmotic probe has enabled the analysis of metabolites in single cells from A. cepa.55 The probe consists of an electrode inserted into a pipette filled with a hydrophobic electrolyte. For sampling, an additional electrode is placed in the buffer surrounding the cells and a voltage is applied. The sampled volume is a function of the applied voltage and can be readily scaled down to sample smaller volumes. Furthermore, by altering the voltage polarity it is possible to switch between extraction and injection. Yin et al. used an electroosmotic probe to withdraw down to 2 pL of the cytoplasm from A. cepa cells.55 By repositioning the pipette in front of a mass spectrometer and applying a positive voltage to the capillary, 50 metabolites were detected out of which 9, including glucose, disaccharide, and glucosides, were identified with MS/MS. Addition of an internal standard (glucose-d2) enabled the quantification of glucose in the onion cell.55 Recently, an electroosmotic probe with an outer dimension of 130 nm was reported.56 The probe enabled tentative identification of fructans and glucosides from single A. cepa cells and 19 components, including amino acids and adenosines, from single HeLa cells. The small diameter of the capillary further enabled the analysis of the cell body, axon, and dendrite from a living primary cultured rat hippocampal neuron. To ensure cell viability throughout analysis, intracellular calcium was monitored using a fluorescent probe (Fluo-3). Several tentatively identified metabolites were detected in the neuron, and high levels of pyroglutamic acid and glutamic acid were found in the axon compared to the cell body and dendrite. The authors conclude that while further control of sampling volumes is needed the gain of subcellular information can help in the understanding of cellular function.56
Capillary microsampling holds a promising future for quantitative single-cell analysis since it allows its user to sample a well-defined volume, a fundamental requirement for quantitative MS. However, this procedure can be relatively invasive due to the capillary size, limiting sampling to larger cells. Recent developments in the FluidFM technology have made it possible to sample as low as 0.1 pL of intracellular content from HeLa cells, while keeping the cells in their original environment and preserving cell viability.54 In addition, capillary microsampling MS has revealed cellular energy and redox state distributions in single cells, metabolic alterations due to environmental stimuli or mitotic stages, and selective sampling of organelles and their unique metabolites.
Separation based MS techniques for single-cell metabolomics
The vast majority of single-cell MS methodologies directly introduce cellular contents into the mass spectrometer to facilitate analysis from small sample volumes. However, simultaneous ionization of all cellular contents, including phospholipids and salts, can introduce matrix effects such as ionization suppression, which can hinder the observation of low abundance analytes. The added dimension of separation (e.g., chromatography or electrophoresis) before MS analysis can reduce matrix effects, separate isomeric and isobaric compounds, and improve detection limits for less abundant compounds. Yu et al. developed a targeted liquid chromatography (LC) ESI-MS/MS method for detecting glutathione, a suggested biomarker of cell health, at the single-cell level.57 Cell suspensions were serially diluted, and samples that statistically contained 3 cells were lysed by sonication. The combination of monobromobimane for derivatizing glutathione and reversed phase LC separation enabled the detection of glutathione from 3 human erythrocyte cells with a relative standard deviation of 26%. The authors reason that the obtained detection limits are low enough to observe glutathione from a single cell.57Another separation method that can be used for single-cell separation is capillary electrophoresis (CE) coupled to ESI-MS. CE-MS was used by Onjiko et al. to monitor cellular metabolites effecting the development of a 16 cell Xenopus laevis embryo.58 Individual cells were manually dissected from the embryo and dissolved in 100 μL of methanol. Cell contents were then pre-concentrated and reconstituted in 10 μL of methanol/water, whereby ∼10 nL was hydrodynamically loaded onto the CE separation capillary. This single-cell CE-MS method enabled the identification of 10 differentially enriched metabolites, such as spermidine, creatine, leucine, and GABA, along the left-right axis of an 8-cell embryo.58 Despite the rich information obtained, the method throughput was low due to manual cell dissection and preconcentration. Therefore, the same group developed an in situ microprobe to directly sample the contents of living single cells in the X. laevis embryo.59 The microprobe was inserted into cells, and 0.02% of the cellular volume was aspirated into the tip of the capillary. Using an automated stage setup, the cellular extract was then dispensed into a vial containing 5 μL of solvent, which was subsequently injected onto the CE separation column for CE-MS analysis. Using this method, ∼230 chemical features were detected, including 70 known metabolic species such as amino acids, acetylcholine, acetylcarnitine, glutathione, and hypoxanthine. The authors argue that extraction after capillary microsampling increases the method sensitivity over manual dissection due to reduced chemical interference and ionization suppression caused by culture media and salts. Additionally, the material can be resampled and the results suggest that capillary microsampling reduces oxidative stress from sampled cells, when compared to manual dissection. Overall, this is a promising method for single-cell metabolomics, which can provide detailed electropherograms containing extensive metabolomic data with an automated extraction platform.59 In an effort to increase automation and throughput for single-cell CE-MS experiments, Li et al. presented a microchip electrophoresis MS platform which lyses single cells, and separates and detects the cell contents by CE-MS.60 The cells were lysed between two gold electrodes to which 250 V was applied. Each run consisted of 1 s for cell injection, 1 s for cell lysis, and 58 s for microchip CE separation and MS detection. This enabled the analysis of 50 PC-12 cells in 50 minutes. The authors applied this method for studying dopamine and glutamate release following KCl induced neuronal depolarization in 600 single PC-12 cells and found that dopamine was quickly released while glutamic acid was released at a much slower pace.60 Single-cell metabolomics can be further enhanced by combining optically guided MALDI and CE-MS. Comi et al. developed a system that allowed for single cells to first be localized optically, then analyzed by MALDI-MS followed by CE-MS. Sampling for CE-MS was performed by directing a liquid microjunction extraction device to selected cell coordinates for extraction and subsequent CE-MS.61 Due to the low sample consumption of MALDI the device enabled CE characterization of amino acid profiles and dopamine of α and β cells from rat pancreatic islets. The addition of separation to single-cell MS will undoubtedly continue to enable the identification of a greater number of cellular metabolites and attempt to quantify and increase the robustness of single-cell MS assays.
The next step: analyte identification and result interpretation
The plethora of creative metabolite sampling techniques for single cells in combination with recent developments in MS instrumentation enables hundreds of ions to be detected in one experiment. The next challenge is to accurately annotate the detected m/z values, and several free and commercially available databases have been developed for MS and MS/MS data.4,62,63 For example, human metabolome database (HMDB) (http://www.hmdb.ca) and MELTIN (metlin.scripps.edu) both enable batch searches of precursor ions, which is beneficial for single-cell MS data without chromatographic peaks. These databases are continuously being updated with new metabolites and, as of July 2018, METLIN holds more than one million entries from different organisms and HMDB includes 114![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 entries of metabolites specifically found in the human body. Both METLIN and HMDB include hyperlinks to other databases, such as KEGG and PubChem, and are freely accessible on-line. However, despite the ease with which an m/z can be putatively assigned using accurate mass and databases, it is important to note that additional measures should be taken to further establish the identity of the metabolite. Addition of separation modalities, such as CE or IMS, can provide further confidence in metabolite annotation. Nonetheless, unless there is high confidence in the metabolite annotation it is difficult to draw biological conclusions.
100 entries of metabolites specifically found in the human body. Both METLIN and HMDB include hyperlinks to other databases, such as KEGG and PubChem, and are freely accessible on-line. However, despite the ease with which an m/z can be putatively assigned using accurate mass and databases, it is important to note that additional measures should be taken to further establish the identity of the metabolite. Addition of separation modalities, such as CE or IMS, can provide further confidence in metabolite annotation. Nonetheless, unless there is high confidence in the metabolite annotation it is difficult to draw biological conclusions.
Metabolomics is essential for understanding the physiological roles of metabolites and increase the knowledge with functional and mechanistic biological studies. However, this requires accurately identified metabolites and as metabolomics is still a field in development many detected m/z values do not match any entries found in databases. A recent comparison showed that the overlap of entries between metabolite databases is relatively low, suggesting that several databases need to be queried for extensive assessment of a dataset.62 The lack of annotation for detected m/z values can also be due to isotopic peaks, unexpected adduct formations, spontaneous in-source degradation,64 or unknowns.4 Unknowns can be known metabolites that do not exist in current databases, actual unknown metabolites, or epimetabolites.4,5 The probability of unknowns is relatively high for metabolites since the structural diversity is large and lacks common building blocks, such as amino acids or nucleotides found in proteins or genetic code, respectively. Additionally, there are no complete lists of metabolites for any organism, which makes full identification of an organism's metabolome currently impossible.65
Moving beyond metabolite identification and towards establishing physiological roles of metabolites in metabolic pathways is a great challenge that needs informatics approaches. However, with continued efforts in building databases of tools for MS based metabolite annotation and increased data sharing there is great potential for the current bottleneck to be eliminated to accommodate the increasing interest in single-cell metabolomics.
Perspective and future directions
Continuous efforts in MS based single-cell metabolomics is moving the field towards more sensitive techniques with higher throughput, quantitative abilities, and decreased technical variability. Nevertheless, single-cell techniques in their infancy will frequently report data for abundant species, with an aim for further developments to improve detection limits. Current limitations of single-cell metabolomics for biologically relevant conclusions include relatively low throughput for methods to distinguish technical variability from biological variability, a need for higher sensitivity for low abundant metabolites or metabolites with low ionization efficiencies, and the need for improvements in software/databases to identify metabolites of interest and provide a biological relevance to the results. In addition, many studies currently use signal intensities to approximate biological abundances of metabolites between sample cohorts, with no method of quantification. Because a complex chemical mixture is analyzed, the obtained results may be skewed by matrix effects influencing detectability and quantification. Ultimately, combining metabolomics with other techniques will bring us closer to true metabolite identities and a mechanistic understanding of metabolism. Currently, there is no method for single-cell metabolomics that is perfect in all aspects, however, as described in this review, methods providing important and unique information on the cellular metabolome are continuously developed and optimized.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Funding for this work was provided by the Swedish Foundation for Strategic Research and the Swedish Research Council.References
- S. Emara, S. Amer, A. Ali, Y. Abouleila, A. Oga and T. Masujima, Adv. Exp. Med. Biol., 2017, 965, 323–343 CrossRef CAS PubMed.
- C. H. Johnson, J. Ivanisevic and G. Siuzdak, Nat. Rev. Mol. Cell Biol., 2016, 17, 451 CrossRef CAS PubMed.
- N. Zamboni, A. Saghatelian and G. J. Patti, Mol. Cell, 2015, 58, 699–706 CrossRef CAS PubMed.
- D. A. Dias, O. A. H. Jones, D. J. Beale, B. A. Boughton, D. Benheim, K. A. Kouremenos, J.-L. Wolfender and D. S. Wishart, Metabolites, 2016, 6, 46 CrossRef PubMed.
- M. R. Showalter, T. Cajka and O. Fiehn, Curr. Opin. Chem. Biol., 2017, 36, 70–76 CrossRef CAS PubMed.
- Y. Yang, Y. Huang, J. Wu, N. Liu, J. Deng and T. Luan, TrAC, Trends Anal. Chem., 2017, 14–26 CrossRef.
- T. J. Comi, T. D. Do, S. S. Rubakhin and J. V. Sweedler, J. Am. Chem. Soc., 2017, 139, 3920–3929 CrossRef CAS PubMed.
- L. Zhang and A. Vertes, Angew. Chem., Int. Ed., 2018, 57, 4466–4477 CrossRef CAS PubMed.
- J. Nunez, R. Renslow, J. B. Cliff 3rd and C. R. Anderton, Biointerphases, 2017, 13, 03B301 CrossRef PubMed.
- K. Wu, F. Jia, W. Zheng, Q. Luo, Y. Zhao and F. Wang, J. Biol. Inorg. Chem., 2017, 22, 653–661 CrossRef CAS PubMed.
- J. S. Fletcher, Biointerphases, 2015, 10, 18902 CrossRef PubMed.
- H.-M. Bergman, K. D. Duncan and I. Lanekoff, Encycl. Anal. Chem, 2018 Search PubMed.
- M. Qi, M. C. Philip, N. Yang and J. V. Sweedler, ACS Chem. Neurosci., 2018, 9, 40–50 CrossRef CAS PubMed.
- L. Armbrecht and P. S. Dittrich, Anal. Chem., 2017, 89, 2–21 CrossRef CAS PubMed.
- W. Lu, X. Su, M. S. Klein, I. A. Lewis, O. Fiehn and J. D. Rabinowitz, Annu. Rev. Biochem., 2017, 86, 277–304 CrossRef CAS PubMed.
- L. Zhang and A. Vertes, Angew. Chem., Int. Ed., 2018, 4466–4477 CrossRef CAS PubMed.
- H. M. Bergman and I. Lanekoff, Analyst, 2017, 142, 3639–3647 RSC.
- M. Sun, Z. Yang and B. Wawrik, Front. Plant Sci., 2018, 9, 571 CrossRef PubMed.
- N. Pan, W. Rao, S. J. Standke and Z. Yang, Anal. Chem., 2016, 88, 6812–6819 CrossRef CAS PubMed.
- X.-C. Zhang, Z.-W. Wei, X.-Y. Gong, X.-Y. Si, Y.-Y. Zhao, C.-D. Yang, S.-C. Zhang and X.-R. Zhang, Sci. Rep., 2016, 6, 24730 CrossRef PubMed.
- X.-C. Zhang, Q. Zang, H. Zhao, X. Ma, X. Pan, J. Feng, S. Zhang, R. Zhang, Z. Abliz and X. Zhang, Anal. Chem., 2018, 90(16), 9897–9903 CrossRef CAS PubMed.
- X.-C. Zhang, Q. Zang, H. Zhao, X. Ma, X. Pan, J. Feng, S. Zhang, R. Zhang, Z. Abliz and X. Zhang, Anal. Chem., 2018, 90(16), 9897–9903 CrossRef CAS PubMed.
- J. K. Lee, E. T. Jansson, H. G. Nam and R. N. Zare, Anal. Chem., 2016, 88, 5453–5461 CrossRef CAS PubMed.
- E. Hiyama, A. Ali, S. Amer, T. Harada, K. Shimamoto, R. Furushima, Y. Abouleila, S. Emara and T. Masujima, Anal. Sci., 2015, 31, 1215–1217 CrossRef CAS PubMed.
- F. Chen, L. Lin, J. Zhang, Z. He, K. Uchiyama and J. M. Lin, Anal. Chem., 2016, 88, 4354–4360 CrossRef CAS PubMed.
- Q. Huang, S. Mao, M. Khan, L. Zhou and J.-M. Lin, Chem. Commun., 2018, 54, 2595–2598 RSC.
- J. F. Cahill, T. K. Darlington, C. Fitzgerald, N. G. Schoepp, J. Beld, M. D. Burkart and K. A. Prather, Anal. Chem., 2015, 87, 8039–8046 CrossRef CAS PubMed.
- T. J. Comi, T. D. Do, S. S. Rubakhin and J. V. Sweedler, J. Am. Chem. Soc., 2017, 139, 3920–3929 CrossRef CAS PubMed.
- H.-M. Bergman, K. D. Duncan and I. Lanekoff, Encycl. Anal. Chem, 2018 Search PubMed.
- M. Kompauer, S. Heiles and B. Spengler, Nat. Methods, 2017, 14, 90–96 CrossRef CAS PubMed.
- M. E. Dueñas, J. J. Essner and Y. J. Lee, Sci. Rep., 2017, 7, 14946 CrossRef PubMed.
- B. Yang, N. H. Patterson, T. Tsui, R. M. Caprioli and J. L. Norris, J. Am. Soc. Mass Spectrom., 2018, 29, 1012–1020 CrossRef CAS PubMed.
- W. Xie, D. Gao, F. Jin, Y. Jiang and H. Liu, Anal. Chem., 2015, 87, 7052–7059 CrossRef CAS PubMed.
- J. Krismer, J. Sobek, R. F. Steinhoff, S. R. Fagerer, M. Pabst and R. Zenobi, Appl. Environ. Microbiol., 2015, 81, 5546–5551 CrossRef CAS PubMed.
- J. Krismer, M. Tamminen, S. Fontana, R. Zenobi and A. Narwani, ISME J., 2016, 11, 988 CrossRef PubMed.
- T.-H. Ong, D. J. Kissick, E. T. Jansson, T. J. Comi, E. V. Romanova, S. S. Rubakhin and J. V. Sweedler, Anal. Chem., 2015, 87, 7036–7042 CrossRef CAS PubMed.
- Q. Fu, J. Tang, M. Cui, J. Xing, Z. Liu and S. Liu, J. Mass Spectrom., 2016, 51(1), 62–68 CrossRef CAS PubMed.
- J. Deng, Y. Yang, M. Xu, X. Wang, L. Lin, Z. P. Yao and T. Luan, Anal. Chem., 2015, 87(19), 9923–9930 CrossRef CAS PubMed.
- H. Piri-Moghadam, F. Ahmadi, G. A. Gómez-Ríos, E. Boyacı, N. Reyes-Garcés, A. Aghakhani, B. Bojko and J. Pawliszyn, Angew. Chem., Int. Ed., 2016, 55, 7510 CrossRef CAS PubMed.
- N. Reyes-Garcés, E. Gionfriddo, G. A. Gómez-Ríos, M. N. Alam, E. Boyacı, B. Bojko, V. Singh, J. Grandy and J. Pawliszyn, Anal. Chem., 2018, 90, 302–360 CrossRef PubMed.
- T. Shimizu, S. Miyakawa, T. Esaki, H. Mizuno, T. Masujima, T. Koshiba and M. Seo, Plant Cell Physiol., 2015, 56(7), 1287–1296 CrossRef CAS PubMed.
- K. Yamamoto, K. Takahashi, H. Mizuno, A. Anegawa, K. Ishizaki, H. Fukaki, M. Ohnishi, M. Yamazaki, T. Masujima and T. Mimura, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3891–3896 CrossRef CAS PubMed.
- T. Fujii, S. Matsuda, M. L. Tejedor, T. Esaki, I. Sakane, H. Mizuno, N. Tsuyama and T. Masujima, Nat. Protoc., 2015, 10, 1445–1456 CrossRef CAS PubMed.
- T. Esaki and T. Masujima, Anal. Sci., 2015, 31, 1211–1213 CrossRef CAS PubMed.
- H. Fujita, T. Esaki, T. Masujima, A. Hotta, S. H. Kim, H. Noji and T. M. Watanabe, RSC Adv., 2015, 5, 16968–16971 RSC.
- L. Zhang and A. Vertes, Anal. Chem., 2015, 87, 10397–10405 CrossRef CAS PubMed.
- L. Zhang, C. J. Sevinsky, B. M. Davis and A. Vertes, Anal. Chem., 2018, 90(7), 4626–4634 CrossRef CAS PubMed.
- A. Saha-Shah, A. E. Weber, J. A. Karty, S. J. Ray, G. M. Hieftje and L. A. Baker, Chem. Sci., 2015, 6, 3334–3341 RSC.
- T. Nakashima, H. Wada, S. Morita, R. Erra-Balsells, K. Hiraoka and H. Nonami, Anal. Chem., 2016, 88, 3049–3057 CrossRef CAS PubMed.
- Y. Gholipour, R. Erra-Balsells and H. Nonami, Environ. Control Biol., 2017, 55, 41–51 CrossRef.
- A. Saha-Shah, C. M. Green, D. H. Abraham and L. A. Baker, Analyst, 2016, 141, 1958–1965 RSC.
- A. Saha-Shah, J. A. Karty and L. A. Baker, Analyst, 2017, 142, 1512–1518 RSC.
- O. Guillaume-Gentil, T. Rey, P. Kiefer, A. J. Ibáñez, R. Steinhoff, R. Brönnimann, L. Dorwling-Carter, T. Zambelli, R. Zenobi and J. A. Vorholt, Anal. Chem., 2017, 89(9), 5017–5023 CrossRef CAS PubMed.
- O. Guillaume-Gentil, R. V. Grindberg, R. Kooger, L. Dorwling-Carter, V. Martinez, D. Ossola, M. Pilhofer, T. Zambelli and J. A. Vorholt, Cell, 2016, 166, 506–516 CrossRef CAS PubMed.
- R. Yin, V. Prabhakaran and J. Laskin, Anal. Chem., 2018, 90(13), 7937–7945 CrossRef CAS PubMed.
- M. Xu, R. Pan, Y. Zhu, D. Jiang and H.-Y. Chen, Analyst, 2018 10.1039/c8an00483h.
- J. Yu, C. Li, S. Shen, X. Liu, Y. Peng and J. Zheng, Rapid Commun. Mass Spectrom., 2015, 29, 681–689 CrossRef CAS PubMed.
- R. M. Onjiko, S. A. Moody and P. Nemes, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6545–6550 CrossRef CAS PubMed.
- R. M. Onjiko, E. P. Portero, S. A. Moody and P. Nemes, Anal. Chem., 2017, 89, 7069–7076 CrossRef CAS PubMed.
- X. Li, S. Zhao, H. Hu and Y. M. Liu, J. Chromatogr. A, 2016, 1451, 156–163 CrossRef CAS PubMed.
- T. J. Comi, M. A. Makurath, M. C. Philip, S. S. Rubakhin and J. V. Sweedler, Anal. Chem., 2017, 89(14), 7765–7772 CrossRef CAS PubMed.
- M. Vinaixa, E. L. Schymanski, S. Neumann, M. Navarro, R. M. Salek and O. Yanes, TrAC, Trends Anal. Chem., 2016, 78, 23–35 CrossRef CAS.
- T. Kind, H. Tsugawa, T. Cajka, Y. Ma, Z. Lai, S. S. Mehta, G. Wohlgemuth, D. K. Barupal, M. R. Showalter, M. Arita and O. Fiehn, Mass Spectrom. Rev., 2018, 37, 513–532 CrossRef CAS PubMed.
- Y.-F. Xu, W. Lu and J. D. Rabinowitz, Anal. Chem., 2015, 87, 2273–2281 CrossRef CAS PubMed.
- M. R. Viant, I. J. Kurland, M. R. Jones and W. B. Dunn, Curr. Opin. Chem. Biol., 2017, 36, 64–69 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |