
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid negative enrichment of circulating tumor cells from whole blood in a 3D-printed monolithic device†
Chia-Heng
Chu
 a,
Ruxiu
Liu
a,
Tevhide
Ozkaya-Ahmadov
a,
Mert
Boya
a,
Brandi E.
Swain
a,
Jacob M.
Owens
b,
Enerelt
Burentugs
c,
Mehmet Asim
Bilen
de,
John F.
McDonald
fg and
A. Fatih
Sarioglu
*aegh
a,
Ruxiu
Liu
a,
Tevhide
Ozkaya-Ahmadov
a,
Mert
Boya
a,
Brandi E.
Swain
a,
Jacob M.
Owens
b,
Enerelt
Burentugs
c,
Mehmet Asim
Bilen
de,
John F.
McDonald
fg and
A. Fatih
Sarioglu
*aegh
aSchool of Electrical and Computer Engineering, Georgia Institute of Technology, Atlanta, GA, USA. E-mail: sarioglu@gatech.edu
bWallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology, Atlanta, GA, USA
cSchool of Chemical & Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA, USA
dDepartment of Hematology and Medical Oncology, Emory University School of Medicine, Atlanta, GA, USA
eWinship Cancer Institute, Emory University, Atlanta, GA, USA
fSchool of Biological Sciences, Georgia Institute of Technology, Atlanta, GA, USA
gParker H. Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GA, USA
hInstitute for Electronics and Nanotechnology, Georgia Institute of Technology, Atlanta, GA, USA
First published on 20th September 2019
Abstract
Isolation and analysis of circulating tumor cells (CTCs) from blood samples present exciting opportunities for basic cancer research and personalized treatment of the disease. While microchip-based negative CTC enrichment offers both sensitive microfluidic cell screening and unbiased selection, conventional microchips are inherently limited by their capacity to deplete a large number of normal blood cells. In this paper, we use 3D printing to create a monolithic device that combines immunoaffinity-based microfluidic cell capture and a commercial membrane filter for negative enrichment of CTCs directly from whole blood. In our device, stacked layers of chemically-functionalized microfluidic channels capture millions of white blood cells (WBCs) in parallel without getting saturated and the leuko-depleted blood is post-filtered with a 3 μm-pore size membrane filter to eliminate anucleated blood cells. This hybrid negative enrichment approach facilitated direct extraction of viable CTCs off the chip on a membrane filter for downstream analysis. Immunofluorescence imaging of enriched cells showed ∼90% tumor cell recovery rate from simulated samples spiked with prostate, breast or ovarian cancer cells. We also demonstrated the feasibility of our approach for processing clinical samples by isolating prostate cancer CTCs directly from a 10 mL whole blood sample.
Introduction
Reliable isolation and analysis of circulating tumor cells (CTCs) from blood samples of cancer patients present an opportunity not only for clinical management of the disease1,2 but also for understanding the biology of the metastatic process at the cellular and molecular level.3–5 On the other hand, the rarity of tumor cells in the blood and intra- and interpatient tumor heterogeneity make identification of tumor cells a challenging process.6,7 Various laboratory techniques, including size-based separation,8,9 density gradient centrifugation,10 scanning cytometry,11 and magnetophoretic separation,12,13 have been applied to differentiate CTCs from hematological cells based on the contrast in their physical and chemical properties. In fact, the U.S. Food and Drug Administration (FDA) approved the clinical use of CellSearch®, which employs immunomagnetic labeling of CTCs against the epithelial cell adhesion molecule (EpCAM), an epithelial marker that is not expressed by normal blood cells.14 While these batch processes typically utilize existing laboratory instrumentation and involve commonplace sample preparation protocols, they may not be ideally suited for the detection of CTCs because of the fact that they inherently rely on stochastic processes for CTC detection and use multi-step protocols, which reduces the sensitivity required to detect rare CTCs.15Microfluidic CTC isolation techniques offer distinct advantages over batch processes as they can be engineered to deterministically screen a blood sample in a controlled microenvironment with higher sensitivity and specificity.16 In addition, the ability of microfluidic platforms to utilize various modalities for cell discrimination allows biophysical17–24 or biochemical23–30 isolation strategies, some of which do not have macroscale analogs. Among these strategies, immunoaffinity-based discrimination of CTCs23,31–39 is the most common and arguably the most relevant in terms of clinical assessment of tumor cells. Microfluidic platforms are routinely used to isolate EpCAM-positive CTCs either through functionalized surfaces40,41 or through immunomagnetic labeling.42–44 While the combination of the sensitivity of microfluidics with the specificity of antigen–antibody interaction is highly effective, antigen-dependent capture of CTCs leads to a biased population and fails to accommodate heterogeneity among tumor cells and dynamic cell expression due to epithelial to mesenchymal transition.45,46
Negative enrichment techniques directly target white blood cells (WBCs) through their well-characterized membrane antigens (e.g., CD45) and selectively depletes them off blood to enrich CTCs.47–52 In this process, WBCs are typically immunolabeled with antibody-functionalized magnetic beads and pulled away from the sample in the downstream under a magnetic field gradient.23,24,47–50 The other approach is to capture WBCs out of the sample through immunoaffinity in a device functionalized with antibodies against WBC markers.51,52 While it potentially offers label-free negative enrichment of WBCs, this approach suffers from several technological hurdles. First, label-free microfluidic devices for leukocyte depletion are currently not practical for processing clinical samples because the limited surface area of a typical antibody-functionalized microfluidic device is quickly saturated by captured leukocytes. Second, sample preparation such as pre-lysing of the red blood cells (RBCs) is typically required to remove the RBCs from the collected blood sample, but this process drastically increase the sample volume through dilution and is also prone to losing valuable tumor cells during sample manipulation.53
In this paper, we address the aforementioned shortcomings of immunoaffinity-based negative enrichment of CTCs with a microfluidic chip by employing additive manufacturing54,55 to create a 3D microfluidic device that provides sufficient active surface area to deplete WBCs directly from clinically relevant volumes of whole blood. To eliminate the need for a sample preparation step for lysing RBCs, the device is integrated with a membrane micropore filter in the downstream of the immunodepletion stage. The membrane filter is used to retain all nucleated cells (tumor cells and potential WBCs that eluded immunocapture) in the immunodepleted sample and dispose of RBCs and platelets (Fig. 1a). We also demonstrate the clinical feasibility of our device by isolating prostate cancer CTCs directly from a 10 mL of patient's blood sample.
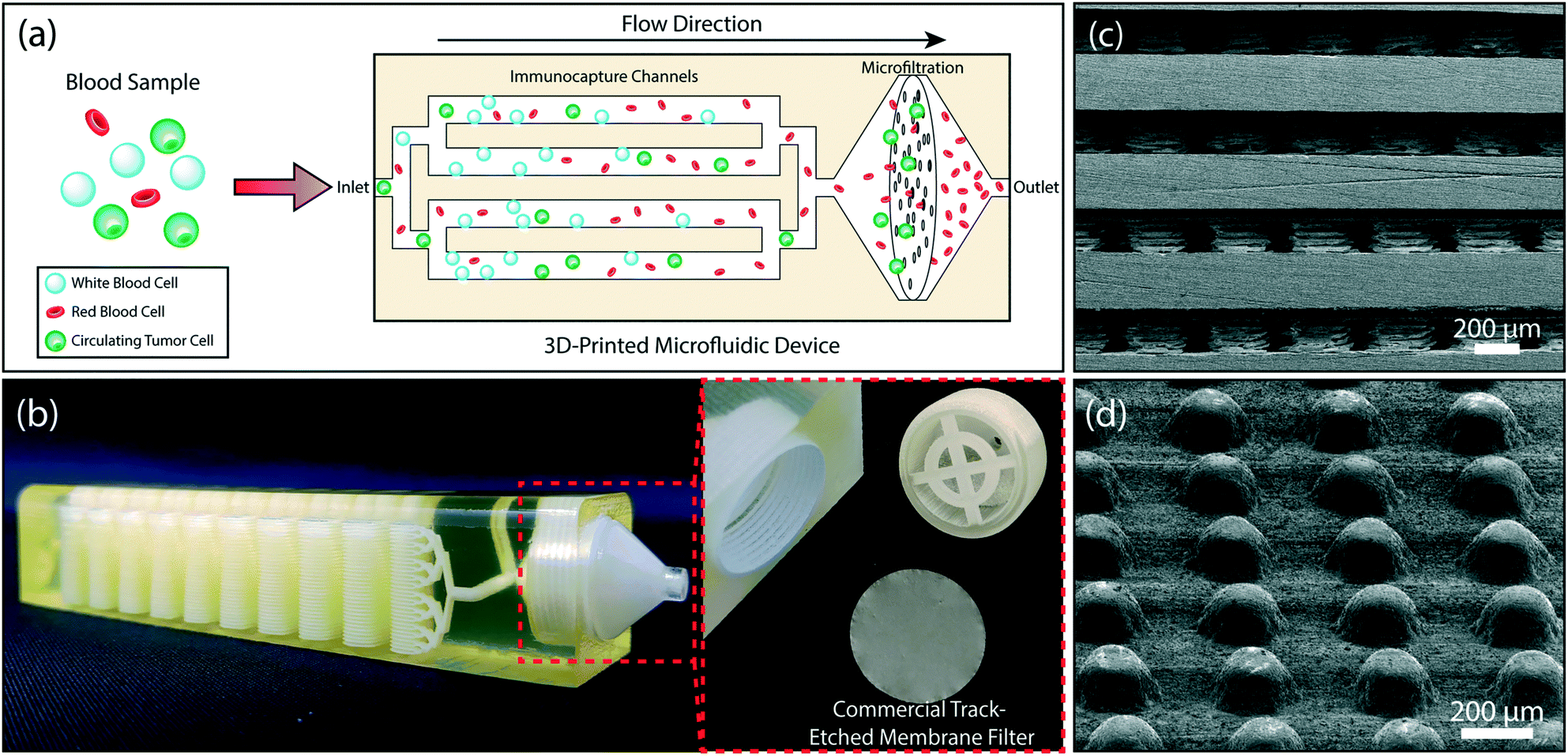 | ||
| Fig. 1 The design of the 3D-printed microfluidic device. (a) A schematic showing the tumor cell enrichment process in the device. Whole blood is introduced to the device. WBCs are captured in the multi-layered immunocapture channels. A membrane filter retains all nucleated cells (including the residual WBCs) and eliminates anucleated blood cells. (b) A photo of the 3D-printed device showing the microfluidic channels with 32 stacked microfluidic layers and the filter holder (right). The membrane filter can be accessed by removing the threaded cap (left). (c) A scanning electron micrograph of the cross-section of the device showing 200 μm-diameter microposts within the microfluidic layers. (d) Arrayed micropillars, within each layer, are shifted by 10 μm from row-to-row to maximize cell-micropost interactions. | ||
Experimental methods
Device design
Our microfluidic device is composed of two sections: a multi-layered immunoaffinity-based leukocyte capture section and a filtration section (Fig. 1b). The device was designed with an overall dimension of 100 mm × 20.5 mm × 19.2 mm. The immunoaffinity capture section is composed of 4–32 stacked microfluidic layers, each with a footprint of a 20 mm × 67 mm. Microfluidic layers are designed to be 175 μm high and are arrayed in the vertical direction with a 475 μm-pitch. Inside the microfluidic layers, 200 μm-diameter microposts (1) increase the cell capture surface area (2) maximize the frequency of interactions between WBCs and the functionalized surface and (3) provide structural support between the stacked microfluidic layers to ensure the device integrity (Fig. 1c). Microposts are separated from each other by 200 μm and are shifted by 10 μm in each row to ensure interaction with WBCs that may follow different streamlines in laminar flow (Fig. 1d). The filtration section of the device serves as a holder for a commercially available track-etched membrane filter with 3 μm-diameter pores and has a compartment with a threaded cap to facilitate straightforward installation and removal of the membrane filter (Fig. 1b). The integration of a commercial membrane filter into our device provides several advantages: first, potential cell loss due to sample handling during filtration is eliminated. Second, using commercially available membrane filters allows us to attain desired pore sizes that are well below a typical 3D printer resolution. Third, the removable membrane filter allows downstream analysis with a microscope or micromanipulation following the enrichment and on-chip staining of the tumor cells. Our device has two inlets, a sample inlet and a buffer inlet, which bypasses the immunodepletion section and is dedicated for washing the membrane filter. Through a network of 3D bifurcating microfluidic channels, the sample is first uniformly distributed into microfluidic layers for immunocapture and then recollected to pass onto the membrane filter. The filtrate (i.e., red blood cells, platelets and serum) along with wash buffer is then discharged from the waste outlet.Device fabrication and assembly
A computer drawing of the microfluidic device was created using SolidWorks (SolidWorks Corp., Waltham, MA) in stereolithography (STL) file format. To reduce the file size, microposts were specifically drawn with hexagonal cross-section with the rationale that they would regardless be printed in a circular form due to the limited printer resolution. The finished STL file was transferred to a ProJet 3510 HD 3D printer (3D Systems, Rock Hill, SC) and the device was printed in VisiJet® M3-X plastic material (3D Systems, Rock Hill, SC) (Fig. 1b). The VisiJet® M3-X was chosen due to its favorable physical properties among other printable materials available from the vendor. Specifically, the VisiJet® M3-X has relatively low opacity and high rigidity (Young's Modulus = 2.168 GPa). Furthermore, the heat distortion temperature of the VisiJet® M3-X is 88 °C, which is well above the 65 °C melting temperature of the sacrificial wax support material (VisiJet® S300). In addition, the cost of the VisiJet® M3-X (∼$5.7 per in3) is comparable to polydimethylsiloxane (PDMS) (∼$4.6 per in3) commonly used to build microfluidic devices with soft lithography. After the device was printed, the wax support material was etched by submerging the device in 65 °C mineral oil (Durvet, Blue Springs, MO) within a 50 mL conical tube and centrifuging at 500 × g for an hour. During the centrifugation, the oil temperature was kept at 65 °C via an external heat gun supplying hot air into the centrifuge. The process continued at room temperature with cleaning the microchannels by centrifuging the device in soapy water (P&G, Cincinnati, OH) and DI water, each for 40 minutes, to remove the residual mineral oil. The dewaxed channels were examined with a scanning electron microscope (SEM) to determine the printed feature size (Fig. 1c and d). The observed surface roughness in our device is due to the specific 3D printing technique (Inkjet 3D Printing) employed by the printer.56 Smoother surfaces could be obtained with a stereolithography-based 3D printer,54 which was not readily available for our use (Fig. S1†).In this work, the size of our device was limited by the size of the centrifugation tube for dewaxing. While we used a proof of principle device with 32 stacked microfluidic layers for our experiments, the printing and dewaxing process we developed could easily be scaled to print devices with higher complexity (Fig. S2†).
Surface functionalization and characterization
To specifically capture WBCs, we functionalized the printed microfluidic channels through a modified version of the functionalization protocol developed by Stott et al.31 Specifically, the microfluidic channels were first treated with 3-mercaptopropyl-trimethoxysilane (MPTMS) (Gelest, Morrisville, PA) mixed with 200 proof ethanol (Thermo Fisher Scientific, Waltham, MA) at 4% v/v ratio. Following 1 h incubation at room temperature, channels were washed with ethanol. Next, a stock solution of N-γ-maleimidobutyryloxy succinimide ester (GMBS) (Pierce Biotechnology, Rockford, IL) in dimethyl sulfoxide (DMSO) (Pierce Biotechnology, Rockford, IL) was prepared at a final concentration of 100 mg mL−1. The device was incubated with 0.28% v/v of GMBS stock solution in ethanol for 30 minutes at room temperature. Residual GMBS was then removed with an ethanol flush and the device was rinsed with phosphate buffered saline (PBS) (Corning Inc., Corning, NY). Next, a 10 μg mL−1 solution of NeutrAvidin (Pierce Biotechnology, Rockford, IL) in PBS was introduced into the device. Following 1 h incubation, the protocol was completed by washing the functionalized device with PBS (Fig. 2a). | ||
| Fig. 2 Surface functionalization of the 3D-printed material and characterization. (a) A schematic showing the (top right) surface functionalization and (lower right) WBC labeling chemistries. (b) Results from the XPS scan of sulfur on the surface of VisiJet® M3-X material before and after the coating of MPTMS. The peak confirms the successful coating. Fluorescence microscope images of (c) NeutrAvidin (FITC) and (d) biotin (Atto 610) on chemically modified VisiJet® M3-X material. Differential fluorescence between the functionalized surface and the exterior control surface confirms the specific surface modification. | ||
To characterize the surface chemistry following the functionalization process, we used X-ray photoelectron spectroscopy (XPS) and fluorescence microscopy. We first verified the attachment of MPTMS molecules with XPS by measuring the amount of sulfur, which is part of the MPTMS molecule. The presence of the sulfur peak from the scan of the coated sample, while no peak could be observed for the uncoated sample, verified the successful coating of the MPTMS on the 3D-printed material (Fig. 2b). To validate the GMBS attachment to the MPTMS, sole use of XPS was not as effective because the GMBS and VisiJet® M3-X material both contained nitrogen, oxygen, and carbon. Therefore, we used fluorophore-conjugated reagents to investigate the GMBS coating and the subsequent functionalization steps. In this process, a section of the 3D-printed material was first selectively coated with MPTMS and GMBS and then FITC-conjugated NeutrAvidin (Thermo Fisher Scientific, Waltham, MA) was applied to the entire surface and washed thoroughly with PBS. Fluorescence signal differentially observed at the MPTMS-coated section compared to the bare sample demonstrated the specific attachment of GMBS to the surface-bound MPTMS (Fig. 2c). Following a similar process, we also verified specific binding of the Atto 610-conjugated biotin (Sigma-Aldrich, Saint Louis, MO) to the NeutrAvidin on the 3D-printed surface (Fig. 2d).
Characterization of the immunocapture of white blood cells
To investigate immunodepletion in our device, we used both CD45+ cell lines and whole blood samples. For cell line experiments, CD45+ Jurkat Clone E61 (ATCC® TIB152™) cells were cultured according to the instructions provided by the manufacturer. Cells were incubated in an RPMI 1640 medium (Corning, Corning, NY) mixed with 10% fetal bovine serum (FBS) (Corning, Corning, NY) at 37 °C in a 5% CO2 ambient. A seeding density >1 × 105 cells per mL was used in subcultures to ensure optimum growth rate. When cells reach 80% confluency, they were stained with CellTracker™ Orange CMRA dye (Molecular Probes, Eugene, OR) according to the protocol provided by the company and resuspended in PBS to reach a final concentration of 7 × 106 cells per mL, to mimic the WBC concentration in blood.For the device characterization, blood was withdrawn from consenting healthy donors according to the protocol approved by the Georgia Institute of Technology Central Institutional Review Board. For the patient blood sample, the blood was collected after consenting the patient according to the protocol approved by the Emory University Institutional Review Board. All experiments with blood samples were approved and performed in compliance with the institutional guidelines of the Georgia Institute of Technology Central Institutional Review Board. All blood samples were collected in tubes with ethylenediaminetetraacetic acid (EDTA). Following a complete blood count (CBC), blood samples were incubated with 500 fg per WBC of biotinylated mouse anti-human CD45 antibodies (SouthernBiotech, Birmingham, AL) for 20 minutes on a rocker to label the WBCs with biotin (Fig. 2a).
The immunocapture efficiency of the chip was calculated by comparing the concentration of target cells in the sample and in the product with a fluorescence microscope. For whole blood samples, Turk's blood diluent (Azer Scientific, Morgantown, PA) was mixed with the product at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v ratio to both lyse the RBCs and stain the nuclei of WBCs for accurate quantification. The number of cells in the product were counted on a Nageotte Chamber (Hausser Scientific, Horsham, PA) using a fluorescence microscope (Nikon Eclipse Ti-E).
:1 v/v ratio to both lyse the RBCs and stain the nuclei of WBCs for accurate quantification. The number of cells in the product were counted on a Nageotte Chamber (Hausser Scientific, Horsham, PA) using a fluorescence microscope (Nikon Eclipse Ti-E).
Measurement of the tumor cell enrichment
To investigate the tumor cell enrichment with our device, we prepared simulated samples by spiking cultured tumor cells into whole blood. Ovarian cancer cell line HeyA8, human breast cancer cell line MDA-MB-231 (ATCC® HTB-26™) and prostate cancer cell line LNCaP (ATCC® CRL-1740™) were cultured according to the manufacturer-provided protocols. HeyA8 and LNCaP cells were grown in RPMI 1640, while Dulbecco's modified Eagle's medium (DMEM) (Corning, Corning, NY) was used to culture MDA-MB-231 cells. All cultures were supplemented with 10% FBS and incubated in 5% CO2 atmosphere at 37 °C. Cells were subcultured every 2–4 days depending on the confluency levels. Prior to spiking, cultured cells were stained with CellTracker™ Orange dye to facilitate optical detection in the final product. Tumor cells were spiked (at a concentration of 3 × 103–4 × 103/mL) into blood withdrawn from healthy donors according to the IRB-approved protocol. Prepared blood samples were stored in EDTA tubes until they were processed through the device within 4 h of phlebotomy.To calculate the tumor cell enrichment factor, we compared the tumor-cell-to-WBC concentration ratio of the sample and the product. To count WBCs and tumor cells in the product, filter-retained cells were immunostained following the blood processing and washing steps. In addition to the nuclear stain with Hoescht 33342 (Thermo Fisher Scientific, Waltham, MA), FITC-conjugated NeutrAvidin was used to label the WBCs that were prelabeled with biotin. After staining, the filter was carefully removed from the filter holder compartment of the device, transferred onto a glass slide and cells are counted with a fluorescent microscope. The viability of tumor cells was determined using 0.4% trypan blue (Thermo Fisher Scientific, Waltham, MA) and live/dead cell assay (ab115347) (Abcam, Cambridge, UK) according to the manufacturer-suggested protocol.
Results and discussion
Optimization of surface functionalization with cell lines
To test the immunocapture in our device, we initially used Jurkat cells with known CD45 expression as a model system. This allowed us to determine the target cell capture efficiency in the absence of steric effects due to RBCs. We tested two different strategies for the cell capture. In the first scheme, we functionalized the device surface with the anti-CD45 antibody (10 μg mL−1) and captured the cells through antibody–antigen interaction. In the second scheme, we labeled Jurkat cells with biotinylated anti-CD45 antibody (500 fg per Jurkat cell) and captured the cells on NeutrAvidin-coated channels via stronger avidin–biotin interaction. To compare the two strategies, we processed matched samples at controlled flow rates (0.125–2.5 mL h−1 per layer) and calculated the capture efficiency by counting the fluorescently labeled cells in the product versus the input (Fig. 3a). Overall, tagged Jurkat cells were found to be captured with higher efficiency (Fig. 3b). In both functionalization strategies, >99% of the Jurkat cells were captured at a low sample flow rate (125 μL h−1 per layer) and virtually no cells could be observed in the product (Fig. 3c). In contrast, a large concentration of non-captured Jurkat cells was observed in the product processed at a high sample flow rate (2.5 mL h−1 per layer) (Fig. 3d). Furthermore, even though ∼90% of Jurkat cells could be captured for both strategies at sample flow rates below 1 mL h−1 per layer, the capture efficiency for labeled Jurkat cells was significantly higher for the faster sample flow rate (2.5 mL h−1 per layer) (Fig. 3b).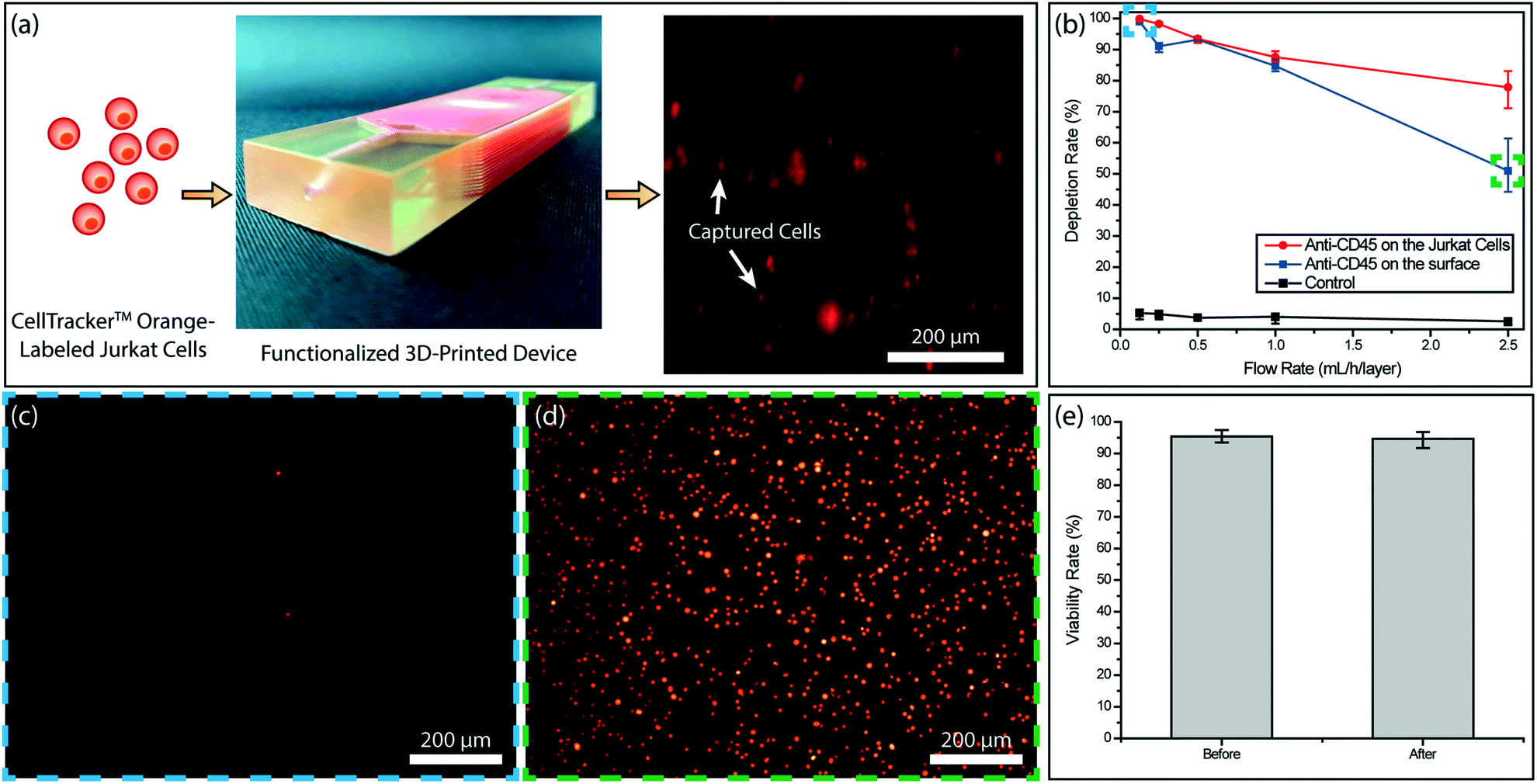 | ||
| Fig. 3 Optimization of the surface functionalization using CD45+ Jurkat cells. (a) Procedure used to test Jurkat cell immunocapture. Fluorescently-labeled Jurkat cells were driven through the functionalized 3D-printed device at a controlled flow rate using a syringe pump. A fluorescence microscope image showing CellTracker™ Orange -labeled Jurkat cells captured in the device. The image was taken on an analytical version of the device, whose walls were thinned down to reduce device opacity. (b) Jurkat cell depletion rate as a function of the sample flow rate for labeled (red curve) and non-labeled (blue curve) Jurkat cells. Representative fluorescence images of the non-captured cells collected in the product for (c) a low sample flow rate (∼125 μL h−1 per layer) and (d) a high sample flow rate (∼2.5 mL h−1 per layer). (e) Measured MDA-MB-231 tumor cell viability before and after processing through the 3D-printed microfluidic device. | ||
Based on results from experiments with Jurkat cells, biotin-labeling of target cells was chosen as the functionalization strategy for blood samples. Besides providing higher immunocapture efficiency, biotin-labeling of target cells is also more practical as it reduces the antibody consumption considering the large device surface that needs to be coated otherwise.
To ensure against potential cytotoxicity of our device on tumor cells, we tested tumor cell viability after they were processed on our device. Using the live/dead assay we observed no notable difference on the viability of MDA-MB-231 tumor cells before and after processing with the 3D-printed device (Fig. 3e).
Immunodepletion of leukocytes from whole blood
We processed whole blood samples collected from consenting healthy donors to characterize immunodepletion performance under different operating conditions (Fig. 4a). In these experiments, 200 μL blood/layer were processed and WBCs were captured by targeting their CD45 antigen, which is expressed by virtually all WBCs at varying levels. In agreement with the previous studies,52 we found WBC capture rate from whole blood to be lower than Jurkat cells due to steric effects by other blood cells. While RBCs can be lysed to improve WBC capture rate, we chose to optimize our device for processing whole blood samples with the goal of minimizing upstream sample manipulation that may introduce cell loss.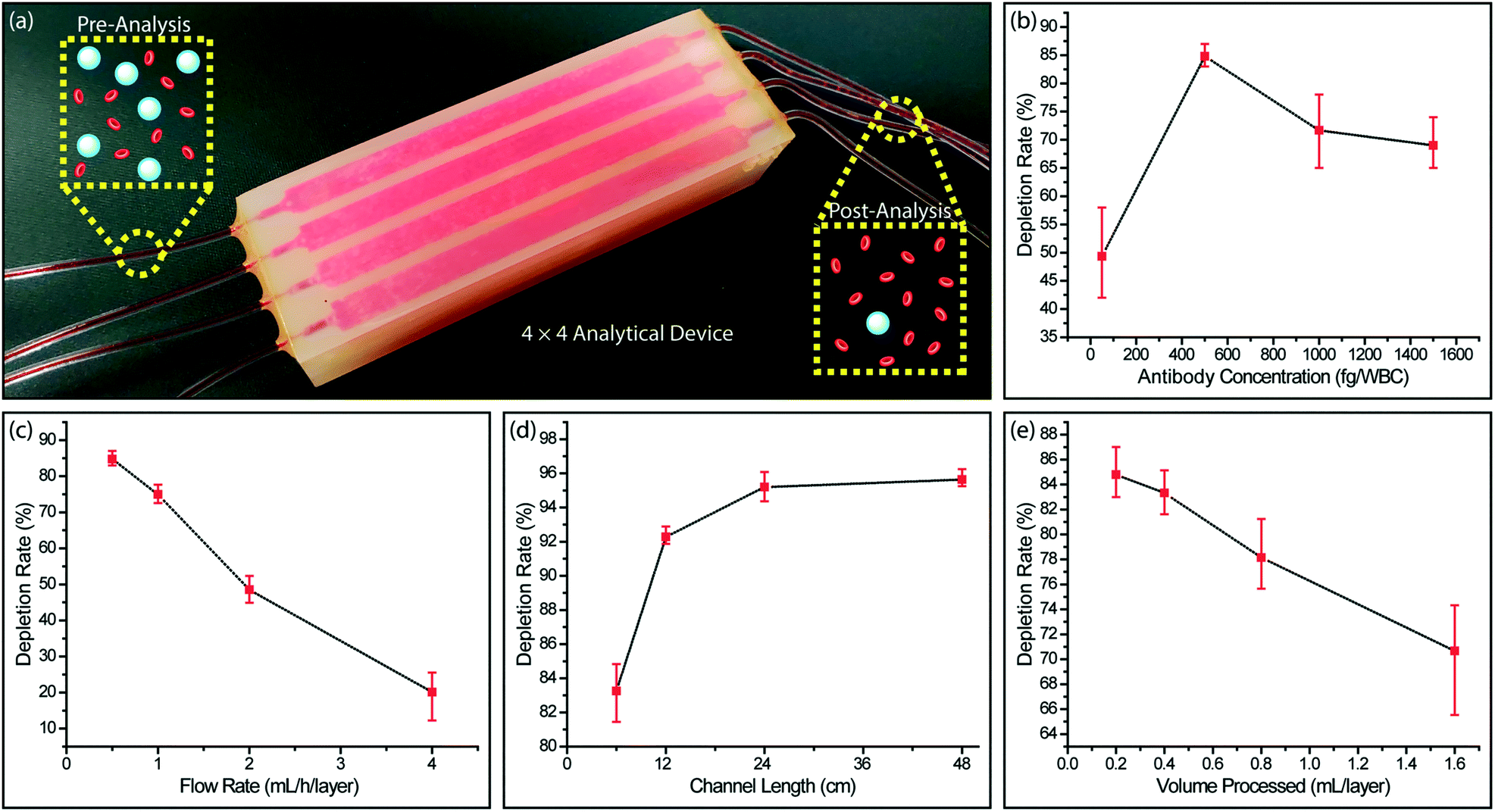 | ||
| Fig. 4 Immunodepletion of leukocytes from whole blood. (a) A photo of the analytical device equipped with 16 (4 × 4) smaller versions of the full device for testing the WBC immunocapture efficiency with whole blood samples. WBC immunocapture rate was measured under different operating conditions by comparing the WBC concentration between the inlets and outlets. Measured whole blood WBC immunocapture rates as a function of (b) the biotinylated anti-CD45 antibody concentration used per WBC, (c) the sample flow rate, (d) the microfluidic channel length and (e) the processed whole blood volume per microfluidic layer. | ||
To optimize the functionalization process, we first measured the WBC capture efficiency at different antibody concentrations for labeling WBCs. We found that increasing the biotinylated anti-CD45 antibody concentration improved the cell capture efficiency only up to a certain point. Higher biotinylated anti-CD45 antibody concentration led to free biotin competing with WBCs for NerutrAvidin binding sites on the device and resulted in lower WBC capture efficiency (Fig. 4b). The optimal concentration of biotinylated anti-CD45 antibody per WBC was found to be 500 fg per WBC, which was used for the subsequent experiments.
Next, we characterized the device performance under different flow rates to optimize the device operating conditions. As expected, the WBC immunocapture efficiency decreased with higher sample flow rate (Fig. 4c). Differently, we can prioritize the capture rate over the throughput in this typical trade-off, as our scalable approach allows the sacrifice in the flow speed to be compensated by parallel running microfluidic layers to achieve a desired sample processing rate. Also considering an excessively low flow speed will lead to non-specific cell attachment and cell sedimentation, we chose 0.5 mL h−1 per layer, where ∼83% of the WBCs were captured, as the optimum flow rate.
We also explored different channel geometries to increase the WBC capture rate from whole blood. Because our feature size was already at the limit of the 3D printer, we extended the channel length through 3D serpentine geometries. We found that by doubling the channel length from 6 cm to 12 cm, the depletion rate for WBCs increased from ∼83% to ∼92% (Fig. 4d). Further increases in the channel length to 24 cm and 48 cm produced tapering WBC depletion rates of ∼95% and ∼96%, respectively and demonstrated a limit for depletion rate enhancement that can be achieved by channel length increases alone.
To investigate the effect of surface saturation on the device performance, we measured cell capture efficiency as a function of processed sample volume. Our device was designed such that immunocaptured WBCs from 550 μL of whole blood per layer will occupy ∼10% of the total active surface area. In agreement with our calculations, we observed that at low sample volumes (200–400 μL per layer), the WBC depletion rate remained at ∼85%, while there was a noticeable decrease in the WBC depletion rate for processing larger sample volumes (800 and 1600 μL per layer) (Fig. 4e). These results showed the importance of providing a substantial capture surface area for the efficient depletion of a large number of WBCs in a blood sample and effectively validated our enrichment approach.
Removal of RBCs and platelets from whole blood
To remove the remaining blood cells following the depletion of WBCs, we used filtration. Considering the significant size contrast between nucleated and anucleated cells in the blood, we chose a filter with a 3 μm-pore diameter to ensure against tumor cell loss. Furthermore, filtration-based CTC enrichment technologies typically use larger pore size (5–10 μm) filters with high CTC recovery rates (>90%), and therefore, we thought a 3 μm membrane filter should minimize potential CTC loss in post-filtration.57–59 Through spiked cell experiments, we characterized a commercial 3 μm-pore membrane filter (Whatman plc, Maidstone, United Kingdom) separately from our 3D-printed device. In our experiments, we processed 200 μL aliquots of whole blood samples each spiked with ∼4 × 103 fluorescently labeled MDA-MB-231 breast cancer cells per milliliter of blood under different filtration pressures. Filtration pressure was set by pneumatically introducing a diluting buffer (PBS) into the filter holder, while the blood was concurrently driven by a syringe pump at a flow rate of 2 mL h−1. Under low buffer pressure (<50 mbar), the filter got clogged due to RBCs not being cleared efficiently and eventually forming a cake layer on the filter (Fig. 5a). In contrast, we achieved a clog-free operation at buffer pressures of 50 mbar and higher with no visible caking of RBCs on the filter.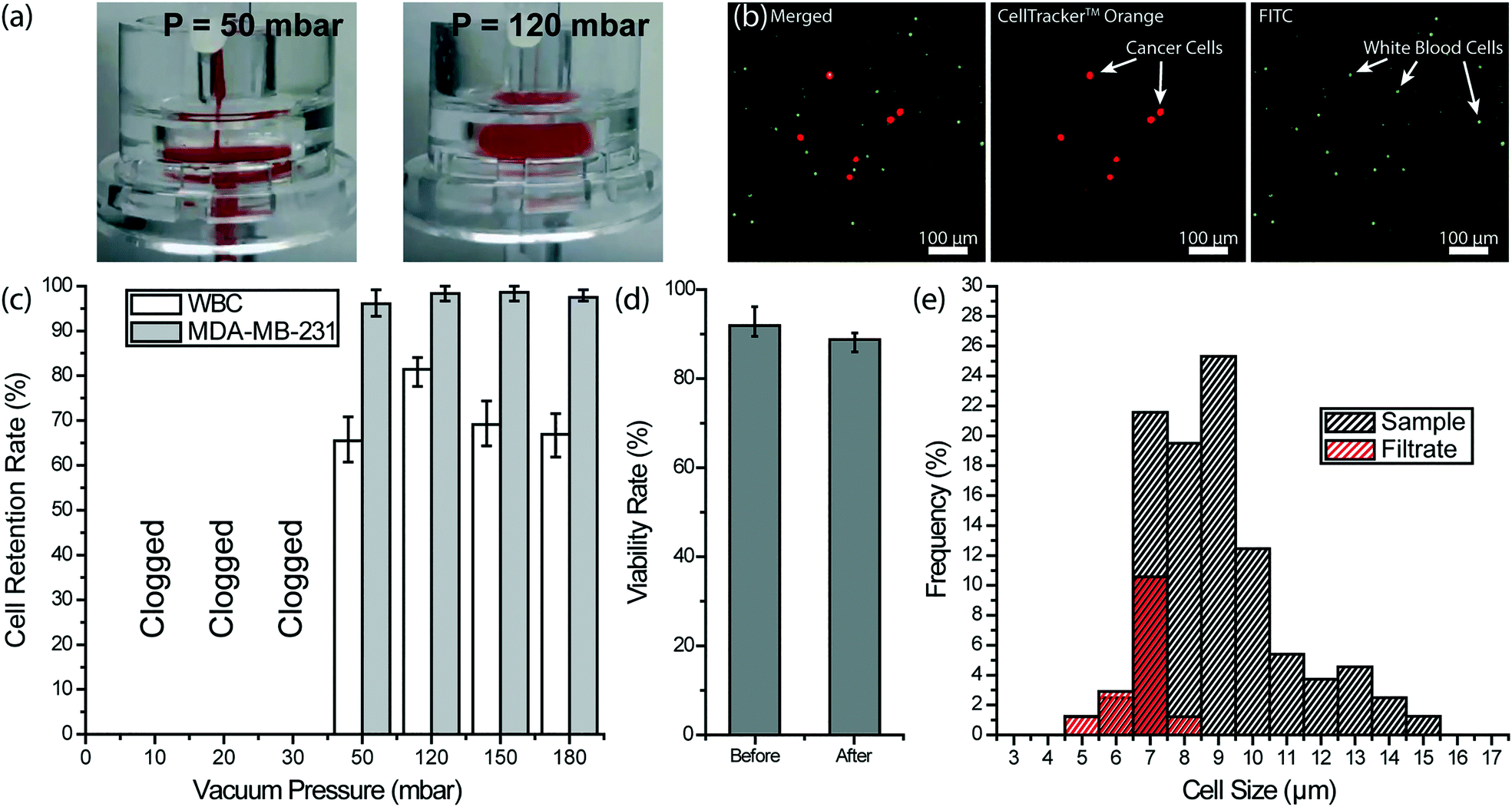 | ||
| Fig. 5 Removal of RBCs and platelets from whole blood with a membrane filter. (a) Images of a 3 μm-pore membrane filter operated under different buffer pressures to recover spiked cancer cells from whole blood. (left) When the filter was operated at 50 mbar, a cake layer was formed due to RBC accumulation. (right) At 120 mbar, RBCs squeezed through pores more efficiently and no cake layer could be observed. (b) Fluorescence microscope images of nucleated cells retained on the filter. The MDA-MB-231 breast cancer cells were pre-stained with CellTracker™ Orange and the WBCs were labeled with FITC-conjugated anti-CD45 antibody. (c) Measured retention rates for both spiked MDA-MB-231 breast cancer cells and the WBCs under different vacuum pressures. (d) Measured MDA-MB-231 tumor cell viability rate before and after processing through the membrane filter under 120 mbar. (e) Measured cell size distribution of the WBCs in the sample and the filtrate at 120 mbar vacuum pressure showing that no WBCs larger than 8 μm were able to pass through the membrane filter. | ||
To optimize the filter operating conditions for maximizing tumor cell enrichment, we measured retention rates for both tumor cells and WBCs. For retention rate measurements, WBCs on the filter were immunostained with a color different than pre-labeled tumor cells and counts for both cells were compared with the original counts at the inlet (Fig. 5b). Among different buffer pressures tested, 120 mbar was determined to deliver optimum results such that virtually all tumor cells, as well as ∼81% of WBCs, were retained on the filter (Fig. 5c). At 50 mbar, partial caking on the filter led to a pressure build-up and tumor cell loss. At the high end, pressures >120 mbar did not change the tumor cell retention rate notably but resulted in a lower WBC retention rate indicating a higher stress on retained cells. The viability of the tumor cells, retained on the filter under 120 mbar, was measured using live/dead assay and no notable effect was observed (Fig. 5d). Moreover, an analysis of the WBC cell size in the filtrate collected at 120 mbar showed that cells with >8 μm diameter are not likely to squeeze through the pores (Fig. 5e). Compared to WBCs ranging in size from 5 to 15 μm in the blood sample, WBCs passing through the 3 μm-pore size filter ranged between 5 to 8 μm. In addition, virtually all (∼96%) of 8 μm diameter WBCs and half (∼52%) of 7 μm diameter WBCs were retained on the filter. Therefore, we concluded the size range of cells that could pass through the 3 μm-pore membrane filter overlaps with the reported CTC size range only for very small CTCs.23,60,61 Having said that, we believe post-filtration cell loss can further be reduced by employing a smaller pore size and through further optimization of the processing conditions.
Circulating tumor cell enrichment from patient blood in the monolithic device
Combining the immunoaffinity based WBC depletion and post-filtration processes that were separately characterized, we tested a 3D-printed microfluidic device with an embedded micropore filter. In assembling the final device, previously characterized 3 μm-pore size commercial filter was placed in the dedicated filter holder printed as part of the microfluidic device and sealed with an O-ring to prevent leakage. The device was operated under previously identified optimal conditions. Specifically, the blood sample was premixed with 500 fg per WBC biotinylated anti-CD45 antibody, driven at a flow rate of 0.5 mL h−1 per layer using a syringe pump and with a blood volume of ∼300 μl per layer and PBS was supplied from the dedicated buffer inlet of the 3D-printed device and driven under 120 mbar vacuum by an electronic pressure regulator. Because the buffer line bypassed the immunocapture chambers within the device and directly drained onto the membrane filter, it did not interfere with the WBC depletion process (Fig. 6a). Moreover, driving the blood sample via syringe pump served two purposes: first, the sample flow rate, which is crucial for the immunocapture, could be kept constant independent of pressure fluctuations in the filtration section. Second, any potential backflow from the pneumatically driven filtration section into the immunocapture was eliminated.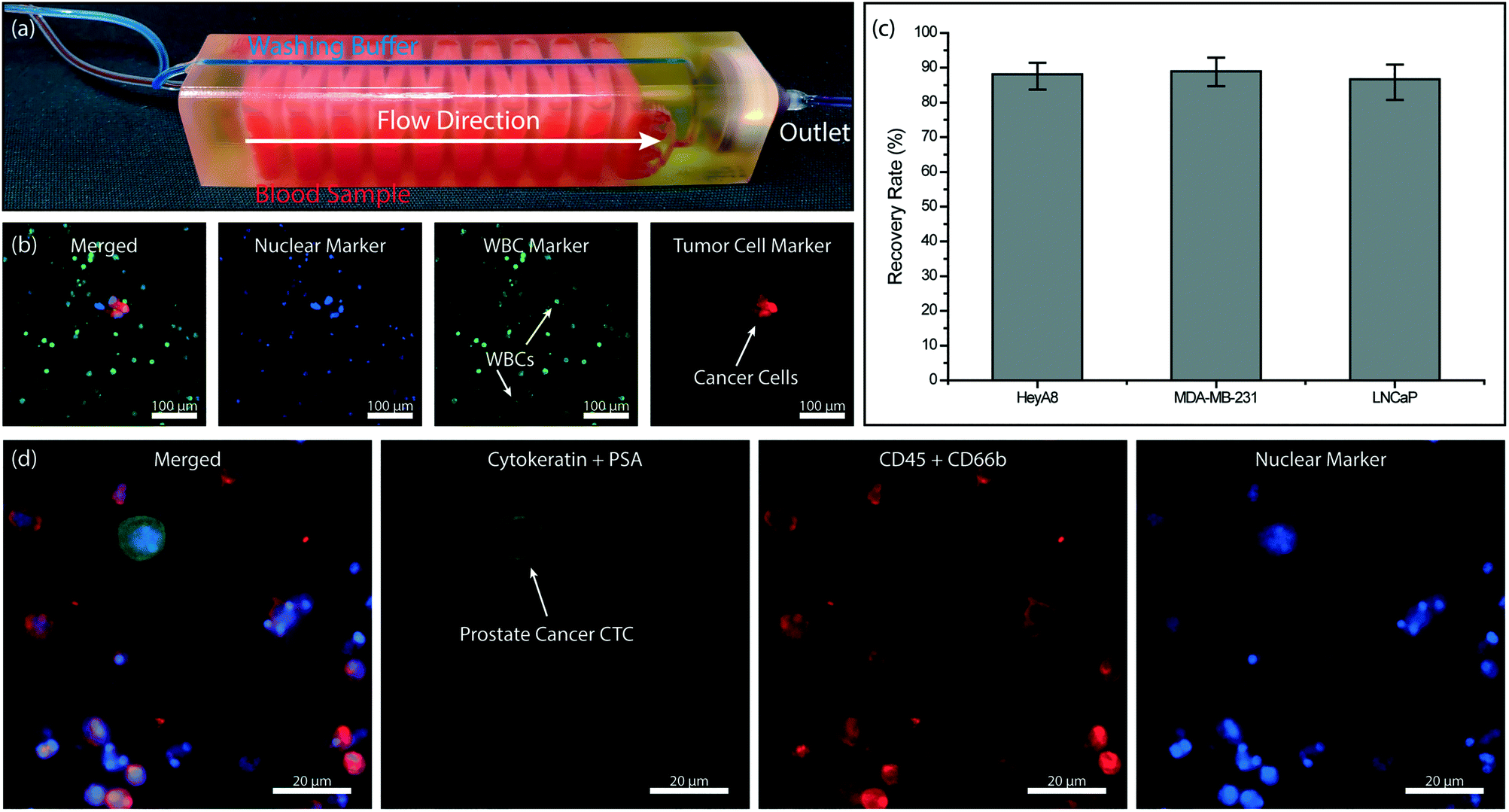 | ||
| Fig. 6 Tumor cell enrichment from whole blood using the full device. (a) A photo of the full 3D-printed device. The device is filled with red- and blue-colored dyes to visualize dedicated sample and buffer paths within the device, respectively. (b) Fluorescence microscope images of the nucleated cells retained on the membrane filter. Hoechst 33342 dye was used to stain the cell nucleus. (c) Measured recovery rates for ovarian (HeyA8), breast (MDA-MB-231) and prostate (LNCaP) cancer cells spiked into whole blood samples. (d) Fluorescence microscope images of the prostate cancer CTC retained on the membrane filter. | ||
We first processed simulated blood samples spiked with fluorescently labeled tumor cells to characterize the performance of our system. To calculate enrichment factors, the product (i.e., cells retained on the filter) was post-stained for WBC and nuclear markers and imaged using fluorescence microscopy (Fig. 6b). To demonstrate the antigen-agnostic enrichment of tumor cells from whole blood, we used cancer cell lines of different cancers, namely breast (MDA-MB-231), prostate (LNCaP) and ovarian (HeyA8) cancers. For all cancer cell types tested, we achieved a ∼90% tumor cell recovery rate (Fig. 6c). The cell loss was found to be due to non-specific adhesion and/or retention of tumor cells in the immunocapture channels from control experiments (Fig. S3†) and can potentially be lowered with optimized blocking protocols and by using different 3D printing techniques and materials.
To demonstrate the feasibility of employing our device for clinical samples, we designed a device for large-volume blood samples and processed a sample collected from a consenting prostate cancer patient according to an IRB-approved protocol. For the clinical sample, we printed a device with 32 immunodepletion layers, each in the form of serpentine channels with an effective path length of ∼40 cm (Fig. 6a). Based on the previously measured 96% leukodepletion and ∼80% membrane retention rates, we estimated ∼60% of pores on the 13 mm-diameter membrane filter to be eventually clogged with a WBC while processing a 10 mL blood sample with a WBC concentration of 5 million cells per mL. Following the processing of 10 mL of patient whole blood sample on our device, the cells on the filter were first fixed with 4% paraformaldehyde (PFA) (Electron Microscopy Sciences, Hatfield, PA), permeabilized with 1% Nonidet-P40 (Thermo Fisher Scientific, Waltham, MA), and post-stained on the filter with their respective markers. Cytokeratin 8/18 (Invitrogen, Carlsbad, CA) and prostate specific antigen Kallikrein 3 (PSA/KLK3) (Cell Signaling, Danvers, MA) were used to label the prostate tumor cells with Alexa Fluor 488 (Invitrogen, Carlsbad, CA) while anti-CD45 (BD Biosciences, San Jose, CA) and anti-CD66b (Biolegend, San Diego, CA) were used to label the WBCs with Alexa Fluor 594. The nuclei of both prostate tumor cells and WBCs were stained with DAPI (Invitrogen, Carlsbad, CA). The recovered prostate tumor cells were identified on the filter with fluorescence microscopy (Fig. 6d).
Conclusions
Depletion of WBCs from blood allows antigen-agnostic and unbiased enrichment of tumor cells. We have developed a monolithic 3D microfluidic device combining immunodepletion and post-filtration for negative enrichment of CTCs directly from whole blood. Taking advantage of the design flexibility afforded by the 3D printing, we created stacked microfluidic layers with an immunocapture surface, large enough to accommodate millions of WBCs from clinically relevant volumes of blood. Post-filtration of leuko-depleted blood within the same device allowed us to retain all nucleated cells, including residual WBCs on a detachable membrane filter, which enabled effortless removal of viable tumor cells off the chip for downstream assays. With the demonstrated feasibility in processing clinical samples, we envision that finer microscale features that will be enabled by ongoing advances in additive manufacturing62 will further improve the performance and ultimately allow our scalable technique to be used for label-free negative depletion of CTCs from whole blood in clinical settings.Author contributions
C. H. C. and A. F. S. designed the research, analyzed the data and prepared the manuscript. R. L. and T. O. A. helped with immuno-functionalization of the devices. M. B. helped with the device characterization for tumor cell enrichment. B. E. S. helped develop the immunostaining protocol for the patient sample. J. M. O. and E. B. helped with manufacturing of devices. M. A. B. provided the prostate cancer patient blood sample for this study. J. F. M. provided some of the cell lines used for the characterization of our devices. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank for all patients and healthy volunteers who contributed blood samples for this study. This work was supported by the start-up funds (to A. F. S.) provided by the School of Electrical and Computer Engineering at the Georgia Institute of Technology, and the seed grant (to A. F. S.) from the Integrated Cancer Research Center funded by the Executive Vice-President for Research Office at the Georgia Institute of Technology. The authors would like to thank the Georgia Tech GVU Center Prototyping Lab staff, especially to David E. Howard, for their help in printing our devices. The authors would also like to thank Prof. Shashi K. Murthy of Northeastern University, and Dr. David Gottfried of the Georgia Institute of Technology for their helpful suggestions about surface modification procedures.References
- G. Lurje, M. Schiesser, A. Claudius Hoffmann and P. M. Schneider, J. Oncol., 2010, 2010, 392652 CrossRef PubMed
.
- S. K. Arya, B. Lim and A. R. A. Rahman, Lab Chip, 2013, 13, 1995–2027 RSC
- G. P. Gupta and J. Massagué, Cell, 2006, 127, 679–695 CrossRef CAS PubMed
- S. L. Stott, R. J. Lee, S. Nagrath, M. Yu, D. T. Miyamoto, L. Ulkus, E. J. Inserra, M. Ulman, S. Springer, Z. Nakamura, A. L. Moore, D. I. Tsukrov, M. E. Kempner, D. M. Dahl, C. L. Wu, A. J. Iafrate, M. R. Smith, R. G. Tompkins, L. V. Sequist, M. Toner, D. A. Haber and S. Maheswaran, Sci. Transl. Med., 2010, 2, 25ra23 Search PubMed
- I. Baccelli, A. Schneeweiss, S. Riethdorf, A. Stenzinger, A. Schillert, V. Vogel, C. Klein, M. Saini, T. Bäuerle, M. Wallwiener, T. Holland-Letz, T. Höfner, M. Sprick, M. Scharpff, F. Marmé, H. P. Sinn, K. Pantel, W. Weichert and A. Trumpp, Nat. Biotechnol., 2013, 31, 539–544 CrossRef CAS
- W. J. Allard, J. Matera, M. C. Miller, M. Repollet, M. C. Connelly, C. Rao, A. G. J. Tibbe, J. W. Uhr and L. W. M. M. Terstappen, Clin. Cancer Res., 2004, 10, 6897–6904 CrossRef
- T. A. Yap, D. Lorente, A. Omlin, D. Olmos and J. S. de Bono, Clin. Cancer Res., 2014, 20, 2553–2568 CrossRef CAS
- G. Vona, A. Sabile, M. Louha, V. Sitruk, S. Romana, K. Schütze, F. Capron, D. Franco, M. Pazzagli, M. Vekemans, B. Lacour, C. Bréchot and P. Paterlini-Bréchot, Am. J. Pathol., 2000, 156, 57–63 CrossRef CAS
- S. Zheng, H. Lin, J. Q. Liu, M. Balic, R. Datar, R. J. Cote and Y. C. Tai, J. Chromatogr. A, 2007, 1162, 154–161 CrossRef CAS PubMed
- R. Rosenberg, R. Gertler, J. Friederichs, K. Fuehrer, M. Dahm, R. Phelps, S. Thorban, H. Nekarda and J. R. Siewert, Cytometry, 2002, 49, 150–158 CrossRef CAS PubMed
- R. T. Krivacic, A. Ladanyi, D. N. Curry, H. B. Hsieh, P. Kuhn, D. E. Bergsrud, J. F. Kepros, T. Barbera, M. Y. Ho, L. B. Chen, R. A. Lerner and R. H. Bruce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10501–10504 CrossRef CAS PubMed
- A. H. Talasaz, A. A. Powell, D. E. Huber, J. G. Berbee, K. H. Roh, W. Yu, W. Xiao, M. M. Davis, R. F. Pease, M. N. Mindrinos, S. S. Jeffrey and R. W. Davis, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3970–3975 CrossRef CAS PubMed
- T. E. Witzig, B. Bossy, T. Kimlinger, P. C. Roche, J. N. Ingle, C. Grant, J. Donohue, V. J. Suman, D. Harrington, J. Torre-Bueno and K. D. Bauer, Clin. Cancer Res., 2002, 8, 1085–1091 Search PubMed
- M. C. Miller, G. V. Doyle and L. W. M. M. Terstappen, J. Oncol., 2010, 2010, 617421 Search PubMed
- O. Lara, X. Tong, M. Zborowski and J. J. Chalmers, Exp. Hematol., 2004, 32, 891–904 CrossRef PubMed
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS
- Z. Liu, F. Huang, J. Du, W. Shu, H. Feng, X. Xu and Y. Chen, Biomicrofluidics, 2013, 7, 011801 CrossRef PubMed
- M. G. Lee, J. H. Shin, C. Y. Bae, S. Choi and J. K. Park, Anal. Chem., 2013, 85, 6213–6218 CrossRef CAS
- E. Sollier, D. E. Go, J. Che, D. R. Gossett, S. O'Byrne, W. M. Weaver, N. Kummer, M. Rettig, J. Goldman, N. Nickols, S. McCloskey, R. P. Kulkarni and D. Di Carlo, Lab Chip, 2014, 14, 63–77 RSC
- A. F. Sarioglu, N. Aceto, N. Kojic, M. C. Donaldson, M. Zeinali, B. Hamza, A. Engstrom, H. Zhu, T. K. Sundaresan, D. T. Miyamoto, X. Luo, A. Bardia, B. S. Wittner, S. Ramaswamy, T. Shioda, D. T. Ting, S. L. Stott, R. Kapur, S. Maheswaran, D. A. Haber and M. Toner, Nat. Methods, 2015, 12, 685–691 CrossRef CAS
- M. E. Warkiani, B. L. Khoo, L. Wu, A. K. P. Tay, A. A. S. Bhagat, J. Han and C. T. Lim, Nat. Protoc., 2016, 11, 134–148 CrossRef CAS
- J. M. Park, J. Y. Lee, J. G. Lee, H. Jeong, J. M. Oh, Y. J. Kim, D. Park, M. S. Kim, H. J. Lee, J. H. Oh, S. S. Lee, W. Y. Lee and N. Huh, Anal. Chem., 2012, 84, 7400–7407 CrossRef CAS PubMed
- E. Ozkumur, A. M. Shah, J. C. Ciciliano, B. L. Emmink, D. T. Miyamoto, E. Brachtel, M. Yu, P. Chen, B. Morgan, J. Trautwein, A. Kimura, S. Sengupta, S. L. Stott, N. M. Karabacak, T. A. Barber, J. R. Walsh, K. Smith, P. S. Spuhler, J. P. Sullivan, R. J. Lee, D. T. Ting, X. Luo, A. T. Shaw, A. Bardia, L. V. Sequist, D. N. Louis, S. Maheswaran, R. Kapur, D. A. Haber and M. Toner, Sci. Transl. Med., 2013, 5, 179ra47 Search PubMed
- N. M. Karabacak, P. S. Spuhler, F. Fachin, E. J. Lim, V. Pai, E. Ozkumur, J. M. Martel, N. Kojic, K. Smith, P. Chen, J. Yang, H. Hwang, B. Morgan, J. Trautwein, T. A. Barber, S. L. Stott, S. Maheswaran, R. Kapur, D. A. Haber and M. Toner, Nat. Protoc., 2014, 9, 694–710 CrossRef CAS PubMed
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS PubMed
- G. Liu, X. Mao, J. A. Phillips, H. Xu, W. Tan and L. Zeng, Anal. Chem., 2009, 81, 10013–10018 CrossRef CAS PubMed
- W. Sheng, T. Chen, W. Tan and Z. H. Fan, ACS Nano, 2013, 7, 7067–7076 CrossRef CAS PubMed
- K. Pantel, R. H. Brakenhoff and B. Brandt, Nat. Rev. Cancer, 2008, 8, 329–340 CrossRef CAS
- K. Pantel and C. Alix-Panabières, Trends Mol. Med., 2010, 16, 398–406 CrossRef
- M. Labib, B. Green, R. M. Mohamadi, A. Mepham, S. U. Ahmed, L. Mahmoudian, I. H. Chang, E. H. Sargent and S. O. Kelley, J. Am. Chem. Soc., 2016, 138, 2476–2479 CrossRef CAS PubMed
- S. L. Stott, C. H. Hsu, D. I. Tsukrov, M. Yu, D. T. Miyamoto, B. A. Waltman, S. M. Rothenberg, A. M. Shah, M. E. Smas, G. K. Korir, F. P. Floyd Jr., A. J. Gilman, J. B. Lord, D. Winokur, S. Springer, D. Irimia, S. Nagrath, L. V. Sequist, R. J. Lee, K. J. Isselbacher, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18392–18397 CrossRef CAS PubMed
- G. D. Chen, F. Fachin, M. Fernandez-Suarez, B. L. Wardle and M. Toner, Small, 2011, 7, 1061–1067 CrossRef CAS PubMed
- H. Choi, K. B. Kim, C. S. Jeon, I. Hwang, S. Lee, H. K. Kim, H. C. Kim and T. D. Chung, Lab Chip, 2013, 13, 970–977 RSC
- W. Harb, A. Fan, T. Tran, D. C. Danila, D. Keys, M. Schwartz and C. Ionescu-Zanetti, Transl. Oncol., 2013, 6, 528–538 CrossRef PubMed
- Y. J. Kim, G. B. Koo, J. Y. Lee, H. S. Moon, D. G. Kim, D. G. Lee, J. Y. Lee, J. H. Oh, J. M. Park, M. S. Kim, H. G. Woo, S. I. Kim, P. Kang, W. Choi, T. S. Sim, W. Y. Park, J. G. Lee and Y. S. Kim, Biomaterials, 2014, 35, 7501–7510 CrossRef CAS PubMed
- J. Lu, T. Fan, Q. Zhao, W. Zeng, E. Zaslavsky, J. J. Chen, M. A. Frohman, M. G. Golightly, S. Madajewicz and W. T. Chen, Int. J. Cancer, 2010, 126, 669–683 CrossRef CAS PubMed
- S. Wang, K. Liu, J. Liu, Z. T. F. Yu, X. Xu, L. Zhao, T. Lee, E. K. Lee, J. Reiss, Y. K. Lee, L. W. K. Chung, J. Huang, M. Rettig, D. Seligson, K. N. Duraiswamy, C. K. F. Shen and H. R. Tseng, Angew. Chem., Int. Ed., 2011, 50, 3084–3088 CrossRef CAS PubMed
- J. D. Besant, R. M. Mohamadi, P. M. Aldridge, Y. Li, E. H. Sargent and S. O. Kelley, Lab Chip, 2015, 7, 6278–6285 CAS
- M. Poudineh, P. M. Aldridge, S. Ahmed, B. J. Green, L. Kermanshah, V. Nguyen, C. Tu, R. M. Mohamadi, R. K. Nam, A. Hansen, S. S. Sridhar, A. Finelli, N. E. Fleshner, A. M. Joshua, E. H. Sargent and S. O. Kelley, Nat. Nanotechnol., 2017, 12, 274–281 CrossRef CAS PubMed
- D. Antolovic, L. Galindo, A. Carstens, N. Rahbari, M. W. Büchler, J. Weitz and M. Koch, BMC Biotechnol., 2010, 10, 35 CrossRef PubMed
- S. B. Cheng, M. Xie, J. Q. Xu, J. Wang, S. W. Lv, S. Guo, Y. Shu, M. Wang, W. G. Dong and W. H. Huang, Anal. Chem., 2016, 88, 6773–6780 CrossRef CAS
- R. Jack, K. Hussain, D. Rodrigues, M. Zeinali, E. Azizi, M. Wicha, D. M. Simeone and S. Nagrath, Lab Chip, 2017, 17, 1349–1358 RSC
- P. Chen, Y. Y. Huang, K. Hoshino and X. Zhang, Lab Chip, 2014, 14, 446–458 RSC
- A. A. Powell, A. H. Talasaz, H. Zhang, M. A. Coram, A. Reddy, G. Deng, M. L. Telli, R. H. Advani, R. W. Carlson, J. A. Mollick, S. Sheth, A. W. Kurian, J. M. Ford, F. E. Stockdale, S. R. Quake, R. F. Pease, M. N. Mindrinos, G. Bhanot, S. H. Dairkee, R. W. Davis and S. S. Jeffrey, PLoS One, 2012, 7, e33788 CrossRef CAS PubMed
- S. Kasimir-Bauer, O. Hoffmann, D. Wallwiener, R. Kimmig and T. Fehm, Breast Cancer Res., 2012, 14, R15 CrossRef CAS PubMed
- T. M. Gorges, I. Tinhofer, M. Drosch, L. Röse, T. M. Zollner, T. Krahn and O. von Ahsen, BMC Cancer, 2012, 12, 178 CrossRef CAS PubMed
- J. Jiang, H. Zhao, W. Shu, J. Tian, Y. Huang, Y. Song, R. Wang, E. Li, D. Slamon, D. Hou, X. Du, L. Zhang, Y. Chen and Q. Wang, Sci. Rep., 2017, 15, 42612 CrossRef PubMed
- W. Zhao, Y. Liu, B. D. Jenkins, R. Cheng, B. N. Harris, W. Zhang, J. Xie, J. R. Murrow, J. Hodgson, M. Egan, A. Bankey, P. G. Nikolinakos, H. Y. Ali, K. Meichner, L. A. Newman, M. B. Davis and L. Mao, Lab Chip, 2019, 19, 1860–1876 RSC
- W. Y. Luo, S. C. Tsai, K. Hsieh and G. B. Lee, J. Micromech. Microeng., 2015, 25, 084007 CrossRef
- T. Y. Lee, K. A. Hyun, S. I. Kim and H. I. Jung, Sens. Actuators, B, 2017, 238, 1144–1150 CrossRef CAS
- K. A. Hyun, T. Y. Lee and H. I. Jung, Anal. Chem., 2013, 85, 4439–4445 CrossRef CAS PubMed
- L. Diéguez, M. A. Winter, K. J. Pocock, K. E. Bremmell and B. Thierry, Analyst, 2015, 140, 3565–3572 RSC
- M. M. Ferreira, V. C. Ramani and S. S. Jeffrey, Mol. Oncol., 2016, 10, 374–394 CrossRef CAS PubMed
- N. Bhattacharjee, A. Urrios, S. Kang and A. Folch, Lab Chip, 2016, 16, 1720–1742 RSC
- A. K. Au, W. Huynh, L. F. Horowitz and A. Folch, Angew. Chem., Int. Ed., 2016, 55, 3862–3881 CrossRef CAS PubMed
- J. O'Conner, J. Punch, N. Jeffers and J. Stafford, J. Phys.: Conf. Ser., 2014, 525, 012009 CrossRef
- A. Meunier, J. A. Hernández-Castro, K. Turner, K. Li, T. Veres and D. Juncker, Anal. Chem., 2016, 88, 8510–8517 CrossRef CAS
- F. A. Coumans, G. van Dalum, M. Beck and L. W. Terstappen, PLoS One, 2013, 8, e61774 CrossRef CAS PubMed
- S. Khetani, M. Mohammadi and A. S. Nezhad, Biotechnol. Bioeng., 2018, 115, 2504–2529 CrossRef CAS PubMed
- S. H. Au, J. Edd, D. A. Haber, S. Maheswaran, S. L. Stott and M. Toner, Curr. Opin. Biomed. Eng., 2017, 3, 13–19 CrossRef PubMed
- F. A. Coumans, G. van Dalum, M. Beck and L. W. Terstappen, PLoS One, 2013, 8, e61770 CrossRef CAS PubMed
- A. P. Kuo, N. Bhattacharjee, Y. S. Lee, K. Castro, Y. T. Kim and A. Folch, Adv. Mater. Technol., 2019, 4, 1800395 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc00575g |
| This journal is © The Royal Society of Chemistry 2019 |